

Abstract
Rationale:
Infantile-onset Pompe disease, also known as glycogen storage disease type II, is a progressive and fatal disorder without treatment. Enzyme replacement therapy with recombinant human acid alpha-glucosidase (GAA) enhances survival; however, the best outcomes have been achieved with early treatment.
Patient concerns:
We report a case of a newborn with infantile-onset Pompe disease diagnosed in the first days of life who did not undergo universal neonatal screening. The patient was asymptomatic, with a general physical examination revealing only a murmur. The clinical presentation was dominated by the neonatal detection of hypertrophic cardiomyopathy, without hypotonia or macroglossia.
Diagnoses:
Pompe disease was confirmed in the first week of life by GAA activity in dried blood spots, and a GAA genetic study showed the homozygous mutation p.Arg854X.
Interventions:
Parents initially refused replacement therapy.
Outcomes:
The patient experienced recurrent episodes of ventricular fibrillation during central line placement and could not be resuscitated.
Lessons:
Although Pompe disease is rare, and universal screening has not been established, neonatologists should be alerted to the diagnosis of Pompe in the presence of hypertrophic cardiomyopathy. Diagnosis is achieved in a few days with the aid of dried blood spots.
Keywords: cardiac hypertrophy, case report, echocardiography, glycogen, Pompe
1. Introduction
Pompe disease (PD) (OMIM 232300) is an autosomal recessive glycogen storage disorder caused by deficient activity of the lysosomal enzyme acid alpha-1,4-glucosidase (GAA, EC 3.2.1.20).[1] GAA deficiency leads to glycogen accumulation in lysosomes and the cytoplasm, resulting in tissue destruction. The enzyme is ubiquitous, but the most affected cells are muscle and cardiac tissues.[1] The incidence of PD is 1/40,000 inhabitants but is higher in certain populations, such as African–Americans (1/14,000).[2] Enzyme activity correlates with genotype. The GAA gene is located on chromosome 17q25, and more than 450 mutations of this gene have been found (http://www.pompecenter.nl).
In infants with infantile-onset PD, GAA activity levels are typically <1% of the mean activity in healthy control subjects. This form of PD, regarded as a muscular disorder, presents as a spectrum of features in which symptoms typically present during the first few weeks of life. Hypotonia, progressive weakness, macroglossia, and hepatomegaly are common symptoms.[3,4] The heart is also characteristically affected; an electrocardiogram (EKG) showing high voltages, repolarization abnormalities, and/or short PR intervals should alert clinicians to the possibility of PD.[5] Echocardiography is the leading tool for diagnosis and usually reveals marked myocardial thickening, which can affect either ventricle and can obstruct the ventricular outflow tracts, especially the left. Respiratory muscle involvement can cause respiratory failure. Patients with classic infantile-onset PD rarely survive beyond 1 year of age without treatment;[1] the mean age at death in studies with large patient groups is 6.0 to 8.7 months.[1] Enzyme replacement therapy (ERT) with recombinant human GAA is safe and effective for patients with PD and is the only treatment that has shown increased survival. The best motor outcomes have been achieved when ERT was initiated early. Early diagnosis is therefore essential.[6–8]
The response to ERT is, however, heterogeneous. Treatment effectiveness depends on the patient's age when ERT is started, their pre-existing muscle damage and their baseline cross-reactive immunologic material (CRIM) status. Infants are considered CRIM-positive if they have residual GAA enzyme activity. Exposure to the native GAA enzyme results in the development of immune tolerance to the GAA protein. Consequently, CRIM-negative patients do not have immune tolerance to GAA and mount an antibody response to the native enzyme when it is administered as ERT.[9] These patients tend to respond poorly to ERT, but the response can be significantly improved with immunomodulatory therapy. The most commonly used regimen is rituximab, methotrexate and intravenous immunoglobulin.[4,10] Based on a pooling of clinical studies, 28% of PD cases are infantile onset, approximately 85 and 75% of which are classic infantile onset and CRIM-positive, respectively.[10,11]
We present a case of a newborn with an onset of hypertrophic cardiomyopathy (HCM) in the first days of life who was diagnosed with infantile-onset PD with the aid of dried blood spots.
Ethical approval was not applicable due to the case report. Written consent was obtained from the patient's mother for this report.
2. Case report
A male infant, with a gestational age of 37 weeks and 4 days and a birth weight of 3.640 kg, was transferred to our hospital with a diagnosis of HCM at 48 hours of life. The patient was the third child of nonconsanguineous Nigerian parents and had no family history of disease. A heart murmur was detected during the first hours of life, and an echocardiogram showed severe biventricular hypertrophy.
Upon arrival at our unit, the newborn was asymptomatic and had no dysmorphic features or weakness. A cardiac assessment revealed cardiomegaly, and both an EKG (Fig. 1A) and a 2-dimensional (2D) echocardiogram showed severe biventricular hypertrophy. The 2D echocardiogram also showed a ventricular septal thickness of 11 mm and a left ventricular posterior wall thickness of 8 to 9 mm, which was more significant at the apical level (Fig. 2). The systolic function was normal. The severe biventricular hypertrophy as assessed by the EKG and 2D echocardiogram, along with the short PR interval, raised the suspicion of PD.
Figure 1.
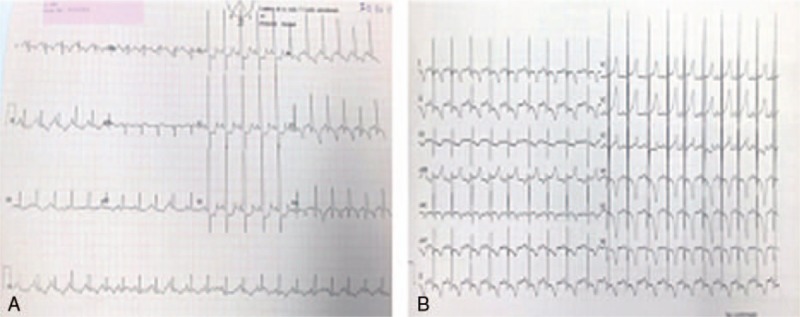
(A) and (B) electrocardiogram (A) at diagnosis and (B) at 3 months of age.
Figure 2.
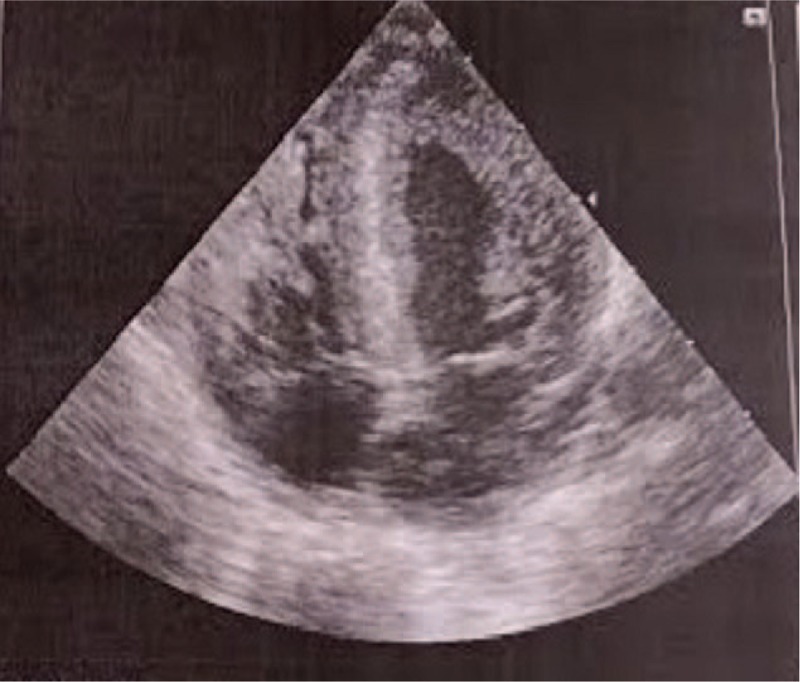
. A 2D echocardiogram with an apical 4-chamber view at diagnosis.
Laboratory tests showed that the only abnormal values were increased creatine phosphokinase (4168 UI/L) and glutamic oxaloacetic transaminase (250 UI/L). No acidosis or hypoglycemia was detected. The results of the brain and abdominal ultrasounds were normal. Dried blood spots were sent to a reference hospital to assess GAA activity, and results in the diagnostic range of PD were obtained at 7 days of life (0.9 μmol/L/h, reference range 1.35–6.0 μmol/L/h at pH 3.8).
A genetic test confirmed a severe homozygous mutation in GAA: exon 18 c. 2560C > T (p.Arg854X). The determination of CRIM status was positive. ERT was indicated; however, after being informed by the multidisciplinary PD team consisting of a neonatologist, expert neurologist, cardiologist and geneticist, the parents refused therapy for the patient. The infant was therefore treated as an outpatient in cardiology and neurology. He was readmitted at 3 months of age with cardiac worsening during an upper respiratory infection 2 months later. At that time, the EKG (Fig. 1B) and 2D echocardiogram showed significantly increased hypertrophy and systolic left ventricular dysfunction. The patient had increasing respiratory failure and was admitted to the prenatal intensive care unit. Treatment with beta-blockers was initiated, consent for ERT was obtained, the medication was administered, and the patient was discharged. A central line placement for the medication was deemed necessary and was scheduled. The patient experienced recurrent episodes of ventricular fibrillation during the procedure and could not be resuscitated.
A postmortem examination showed massive cardiomegaly with severe subsarcolemmal vacuolization of the cardiac muscle, which exhibited prominent glycogen accumulation (Fig. 3). There were also moderate vacuoles due to glycogen accumulation in the skeletal muscle (tongue, proximal esophagus, diaphragm, deltoid, and quadriceps femoris), smooth muscle (urinary bladder and gastrointestinal tract) and in the neurons and glial cells of the nervous system.
Figure 3.
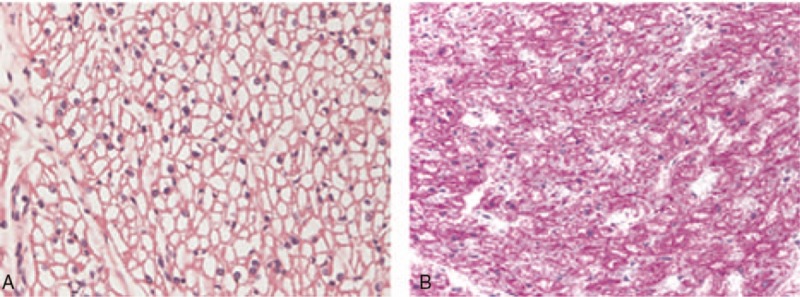
Photomicrography of the myocardium. A (H&E, 400×) and B (PAS, 400×), showing diffuse myocyte vacuolization. H&E = hematoxylin and eosin, PAS = periodic acid–schiff.
3. Discussion
Infantile PD is the most severe form within the spectrum of Pompe phenotypes and is mainly characterized by cardiomyopathy, hypotonia, and respiratory insufficiency. This progressive disorder is fatal after the first year of age without treatment. ERT has been shown to significantly improve survival, quality of life, motor outcomes, and cardiac hypertrophy[12] in most patients. However, the best outcomes have been achieved when treatment was initiated early,[13] before muscle damage occurs.
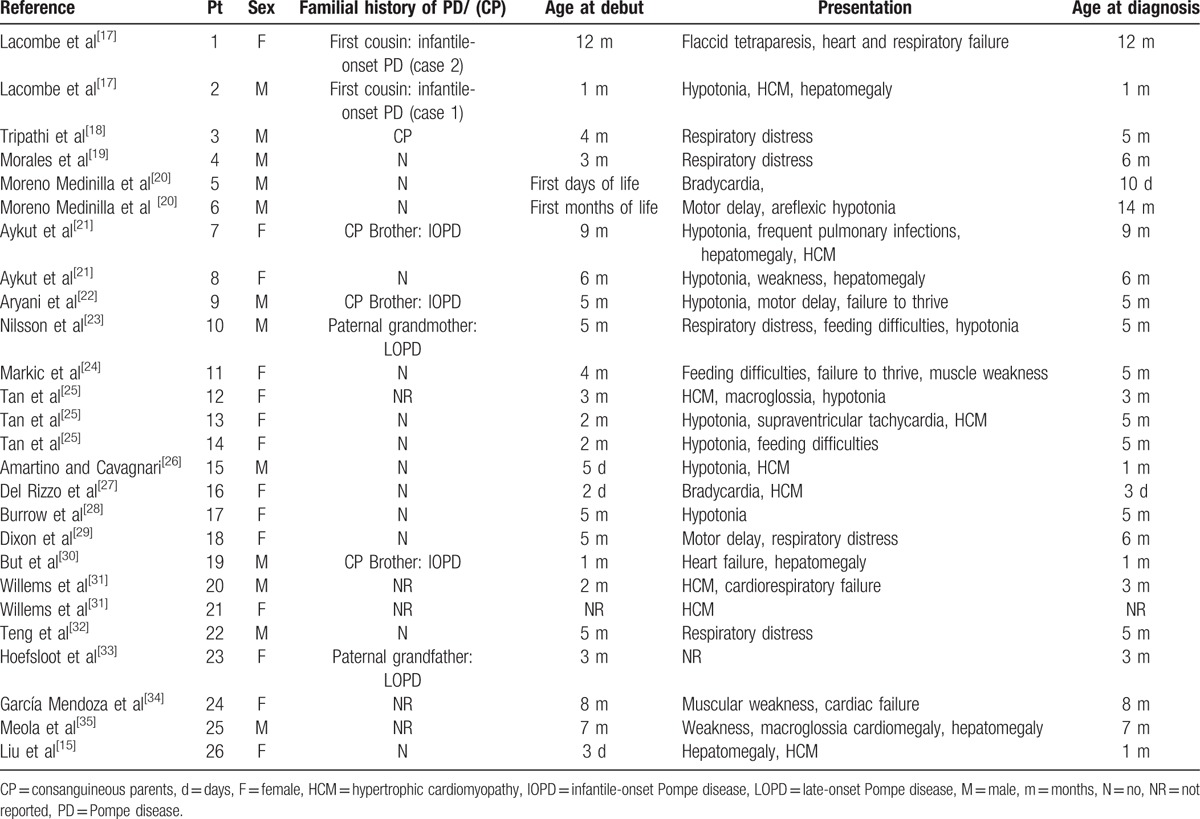
The newborn presented in this case appeared healthy, presenting only a murmur during the physical examination, and the echocardiogram demonstrated significant HCM. The differential diagnosis of HCM in the newborn was performed considering[14] inborn errors of metabolism (storage diseases, fatty acid oxidation disorders and mitochondrial and respiratory chain disorders), congenital syndromes (Noonan syndrome and Beckwith-Wiedemann syndrome), neuromuscular disorders, iatrogenic disorders and gestational diabetes. Our patient presented no abnormal features, hypoglycemia or acidosis. The main diagnostic suspicion was therefore PD, which was confirmed during the first week of life due to the presence of clinical symptoms such as respiratory distress and muscle weakness. Neonatal diagnoses without newborn screening (NBS) have infrequently been achieved.[15] Almost no infants diagnosed before 30 days of age have undergone NBS through the measurement of GAA activity in dried blood spots.[16] However, NBS for PD has not been conducted in most countries due to the high rate of false positives and the lack of clarity on how to follow and manage asymptomatic cases. We reviewed the 26 reported cases of infantile-onset PD without NBS (Table 1),[15,17–35] which had a median age at diagnosis of 5 months. Only 6 cases were confirmed in the first month of life. To the best of our knowledge, this is the second case of a diagnosis in the first days of life without NBS. Abbot et al[36] reported a patient who was prenatally diagnosed (not included in Table 1), based on a positive family history.
Table 1.
Our patient was homozygous for the p.Arg854X mutation, which has been reported as the most frequent GAA sequence variation among patients of African descent African–American with PD,[37] and homozygotes for this mutation have infantile-onset PD.[38,39] The patient was CRIM-positive, a type that tends to respond better to ERT. Despite the improved survival, patients are at high risk of adverse events. Wang et al[40] reported 9 severe anesthetic adverse events in a series of 139 patients undergoing ERT.
The main limitation of our case was the rapid disease progression, despite an early diagnosis, due to the family's initial rejection of ERT. We cannot state that the disease would have been subsequently controlled because the patient had a fatal complication soon after treatment began. Follow-up of these early diagnosed and treated cases will confirm the pertinence of ERT. A multidisciplinary approach is essential due to the special needs of these infants.
In conclusion, it is of paramount importance that frontline neonatologists suspect this rare but life-threatening disease. It is extremely important to consider the diagnosis in regular assessments of newborns and infants with HCM, especially if severe hypertrophy is present.
Footnotes
Abbreviations: CRIM = cross-reactive immunological material, EKG = electrocardiogram, ERT = enzyme replacement therapy, GAA = acid alpha-glucosidase, HCM = hypertrophic cardiomyopathy, NBS = newborn screening, PD = Pompe disease.
Funding: Health Institute Carlos III (Instituto de Salud Carlos III) PI16/00606, AEP 2015
The authors have no conflicts of interest to disclose.
References
- [1]. Lawrence Merrit J. Lysosomal acid alpha-glucosidase deficiency (Pompe disease, glycogen storage disease II, acid maltase deficiency). Rose BD ed. Waltham, MA, UpToDate, 2005. Available at: http://www.uptodate.com/. Accessed January 30, 2016. [Google Scholar]
- [2].Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: literature review and case series. Neurol Clin 2014;32:751–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Van der Ploeg AT, Reuser AJJ. Pompe's disease. Lancet 2008;372:1342–53. [DOI] [PubMed] [Google Scholar]
- [4].Pascual-Pascual SI, Nascimento A, Fernandez-Llamazares CM, et al. Clinical guidelines for infantile-onset Pompe disease. Rev Neurol 2016;63:269–79. [PubMed] [Google Scholar]
- [5].Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733–79. [DOI] [PubMed] [Google Scholar]
- [6].Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr 2015;166:985–91. [DOI] [PubMed] [Google Scholar]
- [7].Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurol 2007;68:99–109. [DOI] [PubMed] [Google Scholar]
- [8].Chakrapani A, Vellodi A, Robinson P, et al. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis 2010;33:747–50. [DOI] [PubMed] [Google Scholar]
- [9].Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chakrapani A, Wraith E, Vellodi A, Reynolds F, Fenton M, Petros A, et al. Guidelines for the management of infantile Pompe disease 2010. Available at: http://www.webarchive.org.uk/wayback/archive/20130325152319/. Accessed February 18, 2016 [Google Scholar]
- [11].Wang Z, Okamoto P, Keutzer J. A new assay for fast, reliable CRIM status determination in infantile-onset Pompe disease. Mol Genet Metab 2014;111:92–100. [DOI] [PubMed] [Google Scholar]
- [12].Kim J, Kim H, Eun LY. Enzyme therapy for hypertrophic cardiomyopathy in non-classical Pompe disease: effectiveness of treatment. Pediatr Int 2017;59:107–8. [DOI] [PubMed] [Google Scholar]
- [13].Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Neurol 2009;11:210–9. [DOI] [PubMed] [Google Scholar]
- [14].Moak JP, Kaski JP. Hypertrophic cardiomyopathy in children. Heart 2012;98:1044–54. [DOI] [PubMed] [Google Scholar]
- [15].Liu Y, Yang Y, Wang B, et al. Infantile Pompe disease: a case report and review of the Chinese literature. Exp Ther Med 2016;11:235–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chien YH, Chiang SC, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics 2008;122:e39–45. [DOI] [PubMed] [Google Scholar]
- [17].Lacombe D, Thambo JB, Fayon M, et al. Recurrence of Pompe disease in first cousins. Genet Couns 2014;26:227–31. [PubMed] [Google Scholar]
- [18].Tripathi SP, Phadke MS, Kerkar PG. Giant heart of classical infantile-onset Pompe disease with mirror image dextrocardia. Circ Cardiovasc Imaging 2015;8:e003637. [DOI] [PubMed] [Google Scholar]
- [19].Morales A, Poling MI, Páez MT, et al. c.1437G>A intron 9 substitution on acid α-glucosidase gene associated with classic infantile-onset Pompe disease phenotype. BMJ Case Rep 2015;2015:bcr2015210688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Moreno Medinilla E, Berzosa López R, Ramírez M, et al. Variabilidad en la presentación clínica de la enfermedad de Pompe infantil: presentación de dos casos y respuesta al tratamiento con enzima recombinante humana. Rev Neurol 2014;59:503–7. [PubMed] [Google Scholar]
- [21].Aykut A, Onay H, Kose M, et al. Two novel mutations in acid α-glucosidase gene in two patients with Pompe disease. J Pediatr Endocrinol Metab 2014;27:1265–7. [DOI] [PubMed] [Google Scholar]
- [22].Aryani O, Manshadi MD, Tondar M, et al. A newly identified c. 1824_1828dupATACG mutation in exon 13 of the GAA gene in infantile-onset glycogen storage disease type II (Pompe disease). Mol Biol Rep 2014;41:6211–4. [DOI] [PubMed] [Google Scholar]
- [23].Nilsson MI, Kroos MA, Reuser AJ, et al. Novel GAA sequence variant c.1211 A>G reduces enzyme activity but not protein expression in infantile and adult onset Pompe disease. Gene 2014;537:41–5. [DOI] [PubMed] [Google Scholar]
- [24].Markic J, Polic B, Stricevic L, et al. Effects of immune modulation therapy in the first Croatian infant diagnosed with Pompe disease: a 3-year follow-up study. Wien Klin Wochenschr 2014;126:133–7. [DOI] [PubMed] [Google Scholar]
- [25].Tan QKG, Cheah SM, Dearmey SM, et al. Low anal sphincter tone in infantile-onset Pompe disease: an emerging clinical issue in enzyme replacement therapy patients requiring special attention. Mol Genet Metab 2013;108:142–4. [DOI] [PubMed] [Google Scholar]
- [26].Amartino HM, Cavagnari BM. Enzyme replacement therapy in the infantile form of Pompe disease: Argentinean experience in a seven-year follow up case. Arch Argent Pediatr 2012;110:323–7. [DOI] [PubMed] [Google Scholar]
- [27].Del Rizzo M, Fanin M, Cerutti A, et al. Long-term follow-up results in enzyme replacement therapy for Pompe disease: a case report. JIMD Rep 2010;33:389–93. [DOI] [PubMed] [Google Scholar]
- [28].Burrow TA, Bailey LA, Kinnett DG, et al. Acute progression of neuromuscular findings in infantile Pompe disease. Pediatr Neurol 2010;42:455–8. [DOI] [PubMed] [Google Scholar]
- [29].Dixon CA, Anderson JB, Ruddy RM, et al. Infantile-onset Pompe disease: a diagnosis not to miss. Pediatr Emerg Care 2010;26:293–5. [DOI] [PubMed] [Google Scholar]
- [30].But WM, Lee SH, Chan AO, et al. Enzyme replacement therapy for infantile Pompe disease during the critical period and identification of a novel mutation. Hong Kong Med J 2009;15:474–7. [PubMed] [Google Scholar]
- [31].Willems J, Petros A, Brierley J. Enzyme replacement therapy for infantile-onset Pompe disease: curse or cure? Neurol 2008;71:380–1. [DOI] [PubMed] [Google Scholar]
- [32].Teng YT, Su WJ, Hou JW, et al. Infantile-onset glycogen storage disease type II (Pompe disease): report of a case with genetic diagnosis and pathological findings. Chang Gung Med J 2004;27:379–84. [PubMed] [Google Scholar]
- [33].Hoefsloot LH, van der Ploeg AT, Kroos MA, et al. Adult and infantile glycogenosis type II in one family, explained by allelic diversity. Am J Hum Genet 1990;46:45–52. [PMC free article] [PubMed] [Google Scholar]
- [34].García Mendoza JJ, Ortega Sánchez A, Acosta Valdez JL, et al. Glycogenosis type II, infantile variant. Report of a case and review of the literature. Arch Inst Cardiol Mex 1986;56:323–6. [PubMed] [Google Scholar]
- [35].Meola G, Scarpini E, Manfredi L, et al. Infantile-acute acid maltase deficiency (Pompe's disease): studies of muscle cultures. Basic Appl Histochem 1983;28:245–55. [PubMed] [Google Scholar]
- [36].Abbott MA, Prater SN, Banugaria SG, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab 2011;104:583–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Becker JA, Vlach J, Raben N, et al. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet 1998;62:991–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Laforêt P, Nicolino M, Eymard PB, et al. Juvenile and adult-onset acid maltase deficiency in France: genotype–phenotype correlation. Neurology 2000;55:1122–8. [DOI] [PubMed] [Google Scholar]
- [39].Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, et al. The genotype–phenotype correlation in Pompe disease. Am J Med Genet 2012;160C:59–68. [DOI] [PubMed] [Google Scholar]
- [40].Wang LY, Ross AK, Li JS, et al. Cardiac arrhythmias following anesthesia induction in infantile-onset Pompe disease: a case series. Paediatr Anaesth 2007;17:738–48. [DOI] [PubMed] [Google Scholar]