

Summary
Immune checkpoint inhibitors (ICIs) targeting cytotoxic T lymphocyte‐associated protein‐4 (CTLA‐4) or programmed cell death protein 1 (PD‐1) receptors have demonstrated remarkable efficacy in subsets of patients with malignant disease. This emerging treatment modality holds great promise for future cancer treatment and has engaged pharmaceutical research interests in tumour immunology. While ICIs can induce rapid and durable responses in some patients, identifying predictive factors for effective clinical responses has proved challenging. This review summarizes the mechanisms of action of ICIs and outlines important preclinical work that contributed to their development. We explore clinical data that has led to disease‐specific drug licensing, and highlight key clinical trials that have revealed ICI efficacy across a range of malignancies. We describe how ICIs have been used as part of combination therapies, and explore their future prospects in this area. We conclude by discussing the incorporation of these new immunotherapeutics into precision approaches to cancer therapy.
Keywords: antibodies, cancer, T cell, tumour immunology
Introduction
There are extensive interactions between tumour cells and the components of the immune system. The process of immune surveillance ensures that aberrant cells with tumorigenic potential can undergo immune destruction before they develop into cancers, and from the very earliest stages of tumour development the tumour microenvironment (TME) contains often‐substantial populations of leucocytes, including various subsets of T cells 1, 2. Effector T cell responses against tumour antigens are induced early during tumour development. Local dendritic cells (DCs) can acquire and process tumour proteins from lysed tumour cells, including mutated versions of normal proteins, and present peptides from these proteins to naive T cells in secondary lymphoid organs. This has the potential of generating potent tumour‐specific effector T cells that could home to the tumour and facilitate selective tumour cell killing. However, tumours evolve diverse mechanisms of immune evasion and immunosuppression to prevent or restrain anti‐tumour T cell responses 3, 4. This includes changing their antigen profile (to make them unrecognizable to effector T cells 5, 6), blocking T cell recruitment 7, 8 and exploiting immunosuppressive leucocytes, such as regulatory T cells (Tregs) and tumour‐associated macrophages 9, 10, 11. In addition, cancer cells and/or the TME can produce molecules that actively inhibit any tumour‐specific T cells that manage to enter the tumour 12, 13, 14. All these processes are potential targets for therapeutic interventions that aim to induce, reinstate or enhance anti‐tumour T cell responses. There have been several exciting recent successes with cancer immunotherapies: principal among these are antibodies targeting the ‘immune checkpoint’ proteins cytotoxic T lymphocyte‐associated protein‐4 (CTLA‐4) or programmed cell death protein 1 (PD‐1).
Enhancing anti‐tumour T cell responses with anti‐CTLA‐4 antibodies
Full activation of naive T cells results in clonal expansion and the development of effector functions. This requires positive signals from several membrane receptors. ‘Signal one’ comes from the T cell receptor (TCR) after it engages cognate antigen displayed on target cell major histocompatibility complex (MHC), but secondary positive signals (co‐stimulation) are also essential, while further signals through cytokine receptors shape effector T cell phenotype 15, 16, 17, 18. Co‐stimulatory signals are delivered principally through CD28 receptors after they bind CD80 and CD86 (also known as B7·1 and B7·2, respectively), which are expressed abundantly on mature DCs. CTLA‐4 also binds to CD80/86 with higher affinity than CD28, but it regulates co‐stimulation negatively 19, 20. CTLA‐4 is induced transiently on activated T cells, peaking 2–3 days after initial activation 19, and is expressed strongly by Treg: naturally occurring Treg (nTreg) are the major cell type constitutively expressing this molecule 9. The receptor CTLA‐4 is reported to transduce negative intracellular signals 21, 22 and enhance T cell motility 23, 24, but perhaps more significant is its ability to outcompete CD28 molecules for CD80/86 binding 19, 24, 25; displace CD28 to distal regions of the immunological synapse 26, 27; and strip CD80/86 from the surface of dendritic cells 28, 29. These immunosuppressive activities prevent or restrain T cell activation, and ensure that autoreactive T cells, or those that bind weakly to antigen, fail to become activated and instead enter a state of unresponsiveness (anergy).
The immunosuppressive properties of CTLA‐4 are evident in Ctla4‐deficient mice, which develop fatal lymphoproliferative disease and multi‐organ failure due to unopposed T cell activation and loss of immunological tolerance 30, 31. These phenotypes are rescued by blocking or deleting CD80/86 or CD28. Moreover, germline heterozygous mutations in CTLA4 in humans are linked to severe immune dysregulation 32, 33, and a CTLA4 variant is associated with early‐onset Crohn's disease and autoimmunity 34. Individuals with CTLA4 mutations have defective Treg function, hyperactivated effector T cells and reduced numbers of circulating B cells 32, 33.
It is well established that anti‐CTLA‐4 monoclonal antibodies can block the immunosuppressive properties of CTLA‐4 and enhance anti‐tumour T cell responses in animal models 35, 36. Blockade induces a reduction in Tregs and an increase in effector T cells within the TME, and simultaneous blockade of CTLA‐4 on these cells can synergize to enhance anti‐tumour responses in animal models 37, 38, 39, 40, 41, 42, 43. Anti‐CTLA‐4 antibodies can also bind Fcγ receptors (FcγRs) to mediate antibody‐dependent cell‐mediated cytotoxicity against CTLA‐4+ Tregs, leading to their selective depletion 40, 41, 42, 43. This is largely dependent upon the presence of atypical, FcγR‐expressing macrophages in the tumour 40, 41, 42, 43 and this mechanism of intratumoural Treg depletion has been identified in humans treated with ipilimumab 44.
The functional effects of anti‐CTLA‐4 antibodies on anti‐tumour responses in a patient depend upon the characteristics of the patient's tumour, the underlying TME and the nature of any previous or concurrent therapies. Predicting responses is challenging and, given the profound immunological consequences of Ctla4 deletion or genetic variation in CTLA4, the potential side effects of administering anti‐CTLA‐4 antibodies may be significant. However, the preclinical research and clinical trial data from CTLA4 blockade has provided a proof‐of‐principle and laid the groundwork for further targeted disruption of other immune checkpoints, such as the programmed cell death‐1/programmed cell death ligand‐1 (PD‐1/PL‐L1) axis.
Dismantling tumour defence by inhibiting PD‐1 activity
PD‐1 is a surface receptor for the cognate ligands PD‐L1 and PD‐L2. It is expressed predominantly by activated T cells, but is also found on other leucocyte subsets, including activated B cells, DCs, monocytes and natural killer (NK) cells 45, 46, 47. PD‐L1 can be found on activated T and B cells, DCs, macrophages and many tissue cells 47, 48, 49, 50, while PD‐L2 appears limited to DCs and macrophages and some stromal cells 47. On effector T cells, PD‐1 ligation causes dephosphorylation of key signalling molecules that lie downstream of the TCR, thereby dampening TCR‐mediated T cell activation 51, 52. It has also been identified recently that PD‐1/PD‐L1 interactions trigger dephosphorylation of CD28 preferentially over the TCR, and is the primary mechanism of T cell suppression 53. PD‐1 also enhances T cell motility to limit T cell/DC interactions 54. PD‐L1 has also been reported to bind to CD80 and might, like CTLA‐4, compete with CD28 55. Deletion of Pd‐1 leads to autoimmune disease 56, 57, but this develops later in life than the more severe, early‐onset disease in Ctla4–/– mice 30, 31. Expression of PD‐L2 in lymphoid organs is thought to help maintain tolerance 51, while PD‐L1, which is up‐regulated in response to interferons, particularly IFN‐γ, protects tissues from excessive immune cell activity 48, 49. This occurs during chronic viral infection when persistent viral antigen exposure causes T cell exhaustion, a form of anergy 58, 59. In this context, blockade of PD‐L1 by administration of anti‐PD‐L1 antibodies causes a resurgence of effector T cell activity 60, predominantly through reactivation of the CD28 signalling pathway 61. Thus, the physiological functions of the PD‐1 pathway are to maintain T cell tolerance and suppress effector T cell responses in peripheral tissues.
Cancers can exploit the PD‐1 pathway, up‐regulating PD‐1 ligands to suppress T cell‐mediated cytolysis. Up‐regulated PD‐1 expression by tumour‐infiltrating lymphocytes is associated with poor outcomes in many human cancers, and correlates with an exhausted T cell phenotype that impedes tumour immunity 62, 63, 64, 65, 66, 67. Early in‐vivo experiments demonstrated that myeloma cells were targeted more effectively when Pd‐1 was absent, and that anti‐PD‐L1 antibodies could suppress myeloma in wild‐type mice 68. In other mouse models, transplantation of Pd‐1–/– T cells 69 or administration of anti‐PD‐L1 antibodies 68, 69 induced regression of established tumours. PD‐L1 expression by infiltrating myeloid cells has also been shown to impair anti‐tumour T cell immunity 70, 71 but, interestingly, this may be due in part to the induction and regulation of peripherally induced Treg subsets (iTreg) by the PD‐L1‐expressing myeloid cells 72. Disruption of this process may represent an additional mechanism underpinning the enhanced anti‐tumour immunity that can be induced by PD‐1 blockade.
Translating efficacy from bench to bedside
CTLA‐4 and PD‐1 provide critical but discrete mechanisms of physiological immunoregulation. A wealth of experimental data shows that antibodies blocking these checkpoints can enhance anti‐tumour immune responses in animal models. Despite some concerns over potential toxicities, this drove the development of humanized monoclonal antibodies targeting these molecules. These novel immunotherapies are referred to as ‘immune checkpoint inhibitors’ (ICIs). These have now been licensed in a number of different tumour types (Fig. 1), and are discussed briefly here.
Figure 1.
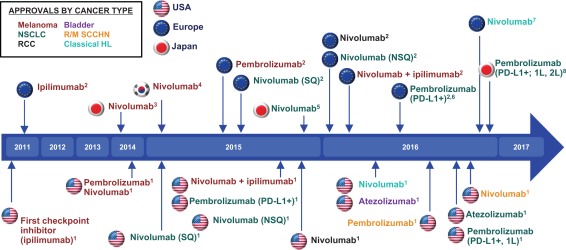
History of checkpoint inhibitors: key milestones. Timeline showing when checkpoint inhibitors were approved for the treatment of specific cancers in the United States, Europe and Japan. Tumour type is indicated by the colour of each immune checkpoint inhibitor (ICI) depicted in the figure, according to the key in the top left. Abbreviations: 1L = first‐line; 2L = second‐line; NSCLC = non‐small‐cell lung cancer; NSQ = non‐squamous; PD‐L1 = programmed death ligand 1; RCC = renal cell carcinoma; R/M = recurrent/metastatic; SCCHN = squamous cell carcinoma of the head and neck; SQ = squamous. 1 US Food and Drug Administration. http://www.fda.gov, accessed 11 November 2016. 2 European Medicines Agency. http://www.ema.europa.eu, accessed 11 November 2016. 3ONO Pharmaceutical Co. Ltd [press release], 4 July 2014. 4ONO Pharmaceutical Co. Ltd [press release]. March 23, 2015. 5 ONO Pharmaceutical Co, Ltd. [press release], 17 December 17, 2015. 6Merck [press release], 27 June 2016, accessed 8 August 2016. 7Bristol‐Myers Squibb Company [press release], 22 November 2016. 8Merck [press release], 10 December 2016.
Anti‐CTLA‐4: licensing ipilimumab for metastatic melanoma
Ipilimumab (Bristol Myers Squibb, New York, NY, USA), an anti‐CTLA‐4 antibody, was licensed for malignant melanoma in 2011 71. This followed a landmark Phase III clinical trial involving approximately 600 patients with previously treated malignant melanoma 73 (Table 1). There were three treatment arms: ipilimumab plus gp100 vaccine, ipilimumab alone or gp100 vaccine alone. The gp100 vaccine targets a protein expressed abundantly by melanomas 74, 75. The treatments containing ipilimumab conferred greatest median overall survival (OS): 10·0 months (ipilimumab/gp100) and 10·1 months (ipilimumab monotherapy) versus 6·4 months (gp100 vaccine alone). Severe immune‐related adverse effects (IAEs) were reported in 10–15% of patients receiving ipilimumab, and were of longer duration in the ipilimumab/gp100 cohort 73. Established protocols for managing immune‐mediated toxicities were adopted from previous trials 76, 77, 78. Subsequent analysis of data from 1861 metastatic melanoma patients in 12 individual clinical trials of ipilimumab showed that OS at 3 years was 20% for previously treated patients and 26% for treatment‐naive patients 79. The survival curve plateaued at approximately 3 years, with evidence in some patients of long‐term progression‐free survival of up to 10 years.
Table 1.
Key clinical trials of immune checkpoint inhibitor (ICIs) in patients with malignant melanoma and their associated survival data
| Trial name and/or Phase [Ref] | Trial drugs (dose) | Patient no. | 1‐year OS rate (%) | 18‐month OS rate (%) | 2‐year OS rate (%) | Median OS (months) |
|---|---|---|---|---|---|---|
| Phase III 73 | Ipi (3 mg/kg) versus gp100 vaccine | 676 | 45·6 versus 25·3 | 33·2 versus 16·3 | 23·5 versus 13·7 | 10·1 versus 6·4 |
| Phase II 76 | Ipi (10 mg/kg) + budesonide versus Ipi (10 mg/kg) | 115 | 55·9 versus 62·4 | n.r. | 40·5 versus 41·7 | 17·7 versus 19·3 |
| Phase II 77 | Ipi (10 versus 3 versus 0·3 mg/kg) | 217 | 48·6 versus 39·3 versus 39·6 | 34·5 versus 30·2 versus 23·0 | 29·8 versus 24·2 versus 18·4 | 11·4 versus 8·7 versus 8·6 |
| Phase II 78 | Ipi (10mg/kg) | 226 | 47·2 | 39·4 | 32·8 | 10·2 |
|
KEYNOTE‐006 Phase III 80, 81 |
Pembro (10mg/kg every 2 weeks) versus Pembro (10mg/kg every 3 weeks) versus Ipi (3mg/kg every 3 weeks) | 834 | 74·1 versus 68·4 versus 58·2 | n.r. | 55·1 versus 55·3 versus 43·0 | n.a. versus n.a. versus 16·0 |
| Phase I 82, 83 | Pembro (1–10 mg/kg) | 104 | 62 | n.r. | 43 | 16·8 |
| Phase III 84 | Niv (3 mg/kg) versus dacarbazine (1000 mg/m2) | 418 | 72·9 versus 42·1 | n.a. | n.a. | n.a. versus 10·8 |
| Phase I 85, 86 | Cohort 1: Niv (0·3 mg/kg) +I pi (3 mg/kg) | 14 | 56 | n.a. | n.a. | 14·8 |
| Cohort 2: Niv (1 mg/kg) + Ipi (3 mg/kg) | 17 | 94 | n.a. | n.a. | N.R. | |
| Cohort 2a: Niv (3 mg/kg) + Ipi (1 mg/kg) | 16 | 89 | n.a. | n.a. | N.R. | |
| Cohort 3: Niv (3 mg/kg) + Ipi ( mg/kg) | 6 | 100 | n.a. | n.a. | N.R. | |
|
CheckMate‐069 Phase II 87, 88 |
Niv (1 mg/kg) + Ipi (3 mg/kg) versus Ipi (3 mg/kg) | 142 | 73·4 versus 64·8 | n.r. | 63·8 versus 53·6 | N.R. |
|
CheckMate‐067 Phase III 89, 90 |
Niv (3 mg/kg) | 316 | n.a. | n.a. | 59 | 37·6 |
| Niv (1 mg/kg) + Ipi (3 mg/kg), then Niv (3 mg/kg) maintenance | 314 | n.a. | n.a. | 64 | n.a. | |
| Ipi (3 mg/kg) | 315 | n.a. | n.a. | 45 | 19·9 | |
|
CheckMate‐064 Phase II 91 |
Niv (3 mg/kg), then Ipi (3 mg/kg) | 68 | 76 | n.a. | n.a. | n.a. |
| Ipi (3 mg/kg), then Niv (3 mg/kg) | 70 | 54 | n.a. | n.a. | 16·9 |
Ipi = ipilimumab; pembro = pembrolizumab; niv = nivolumab; OS = overall survival; n.a. = not available; n.r. = not reported.
PD‐1 blockade moves into the clinic
The safety and efficacy of pembrolizumab (Merck), a monoclonal antibody targeting PD‐1, was demonstrated in patients with ipilimumab‐refractory malignant melanoma. In an expansion cohort of a Phase I trial, 173 patients were treated every 3 weeks with pembrolizumab 92. Equivalent efficacy, as determined by overall response rates (ORR) 93, was observed at both dosing regimens (ORR = 26%). Pembrolizumab was well tolerated: only 12% of patients developed drug‐related grade 3/4 toxicities. Given the promising clinical responses, good safety profiles and the absence of other effective therapies, pembrolizumab was licensed for ipilimumab‐resistant advanced melanoma in 2014 94.
In 2015, Robert and colleagues demonstrated significant improvements in progression‐free survival, OS, response rates and treatment‐related adverse events of pembrolizumab monotherapy compared with ipilimumab in advanced melanoma 80 (Table 1). A total of 834 patients were randomized to either pembrolizumab every 2 weeks, pembrolizumab every 3 weeks or four cycles of ipilimumab every 3 weeks. The 12‐ and 24‐month OS was significantly greater in the pembrolizumab groups, and progression‐free survival at 6 months was 47·3, 46·4 and 26·5%, respectively, with more durable responses seen in the pembrolizumab groups at 7·9 months 81. Lower rates of significant immune‐mediated toxicities were also seen in the pembrolizumab groups.
A Phase I clinical trial of another anti‐PD‐1 monoclonal antibody, nivolumab (Bristol Myers Squibb), was conducted in 296 patients with melanoma, non‐small‐cell lung cancer (NSCLC), renal cell carcinoma (RCC) or other selected treatment‐refractory malignancies 82. Melanoma patients were treated every 2 weeks with increasing doses of 0·1–10 mg/kg and showed overall objective response rates of 28%, with the highest response rate (41%) at a dose of 3 mg/kg. Responses were durable in many responders. Grade 3/4 treatment‐related adverse events occurred in 14% of patients, with IAEs in 6%. Longer‐term follow‐up of melanoma patients showed 1‐ and 2‐year survival rates of 62 and 43%, respectively, with an OS of 16·8 months 83. Long‐term safety evaluation was comparable to the original analysis, with 22% experiencing grade 3/4 treatment‐related adverse events and 5% having grade 3/4 IAEs. Toxicities were not cumulative and occurred almost exclusively in the first 6 months of therapy.
In an open‐label, randomized, Phase III study involving patients with ipilimumab‐refractory melanoma, nivolumab was associated with a higher ORR than chemotherapy (32 versus 11%) 95. Subsequently, nivolumab was compared with dacarbazine in patients with previously untreated metastatic melanoma without proto‐oncogene B‐Raf (BRAF) mutation 84. Nivolumab gave superior 1‐year survival (72 versus 42%), progression‐free survival (5·1 versus 2·2 months), and ORRs (40 versus 13·9%). The survival benefit with nivolumab was observed across prespecified subgroups, including those defined by PD‐L1 status. Grade 3/4 drug‐related adverse events occurred in 11·7% of nivolumab‐treated patients and 17·6% of those receiving dacarbazine.
Combination regimens of ICIs in patients with advanced melanoma
The distinct mechanisms of actions of anti‐PD‐1 and anti‐CTLA‐4 infer that their co‐administration could have enhanced efficacy (Fig. 2). The first Phase I study combining two ICIs, nivolumab and ipilimumab, was undertaken in patients with malignant melanoma and used an initial dose‐escalation trial design to identify safe doses of these drugs when used in combination regimens 85. One mg/kg nivolumab and 3 mg/kg ipilimumab were the maximum doses associated with acceptable levels of adverse events. This regimen yielded objective responses in 21 of 52 patients (40%). Durable responses, ranging from 60·1 to 72·1 weeks, were ongoing in 19 patients at the time of publication. Impressively, 16 patients had tumour reduction ≥ 80% at 12 weeks, including five with a complete response, although treatment‐related grade 3/4 adverse events were reported in 53% of patients. Updated OS data were 82 and 75% for 1‐ and 2‐year survival, respectively (Table 1) 86. Thus, this preliminary trial data indicated that simultaneous targeting of PD‐1 and CTLA‐4 could be tolerated and result in durable responses.
Figure 2.
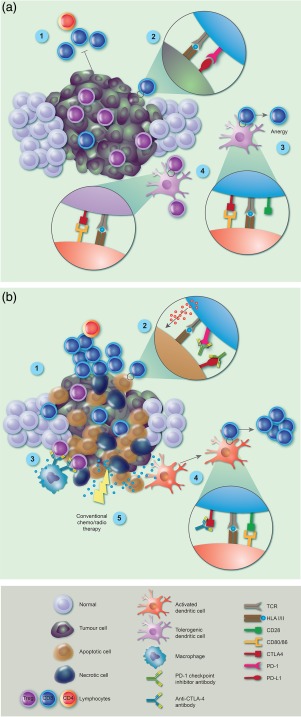
Immune checkpoint inhibitor (ICI) blockade reverses tumour‐mediated immune suppression. (a) Established tumours block immune attack through a variety of mechanisms, including inhibition of tumour‐specific cytotoxic T lymphocyte (CTL) and CD4 T cell activation and function (1). This is driven by tumour over‐expression of programmed cell death protein 1(PD‐L1), interacting with tumour‐specific T cell PD‐1 receptor (2) and T cell anergy induced by tumour‐mediated T cell expression of cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4) inhibitory receptor (4). In addition, tolerogenic dendritic cells (DC) drive regulatory T cell (Treg) induction and expansion via CTLA‐4 (3) and accumulation of Tregs then contributes to the immunosuppressive milieu of the tumour microenvironment (TME). (b) After ICI therapy, there is re‐activation and proliferation of tumour‐specific CTLs via blockade of the PD‐1 axis (1), and return of functional cytotoxicity, resulting in perforin release and tumour cell killing (2). As tumour damage increases, the TME is disrupted allowing macrophages to deplete Tregs via fragment crystalline receptor (FcR) binding of anti‐CTLA‐4 antibody (3). Tumour antigen release is driven by immune lysis of tumour cells which are processed by conventional DC and presented to naive T cells in context of checkpoint inhibition of CTLA‐4, enhancing CTL proliferation and function (4). Tumour damage and antigen release is also supplemented by concomitant use of conventional chemo/radiotherapy, which can reveal new tumour‐associated antigens and contribute to anti‐tumour immune responses (5).
The Checkmate 069 trial compared nivolumab plus ipilimumab, with ipilimumab monotherapy in patients with untreated malignant melanoma (Table 1). Greater objective responses were seen with combination therapy (61 versus 11% with ipilimumab alone), with complete responses in 22% of patients on combination therapy but none with ipilimumab alone 87. One‐ and 2‐year OS was 73·4 and 63·8%, respectively, in the combination cohort, compared with 64.8% and 53.6% with ipilimumab alone 88. Importantly, 2‐year survival was not affected significantly by BRAF mutation or tumour PD‐L1 expression status at diagnosis. A large subsequent Phase III trial (Checkmate 067) compared nivolumab alone, ipilimumab alone and nivolumab plus ipilimumab: median progression‐free survival was 6·9, 2·9 and 11·5 months, respectively 89. Notably, in patients with PD‐L1‐negative tumours, progression‐free survival was greater with combination therapy than either monotherapy. Furthermore, with a minimum follow‐up of 36 months, the median overall survival has not been reached in the combination regimen arm and was 37·6 months in the nivolumab arm of the study compared with 19·9 months with ipilimumab monotherapy [hazard ratio (HR) = 0·55, P < 0·001 for combination versus ipilimumab; HR= 0·65, P < 0·001 for nivolumab versus ipilimumab]. The overall survival at 3 years was 58, 52 and 34% for the combination regimen, nivolumab and ipilimumab monotherapy, respectively. However, treatment‐related grade 3/4 toxicities occurred in 59, 21 and 28% of patients treated with the combination regimen, nivolumab and ipilimumab, respectively 90.
A Phase II trial (Checkmate 064) has also revealed intriguing results of switching between nivolumab and ipilimumab 91. Melanoma patients underwent induction with either nivolumab (3 mg/kg every 2 weeks; six doses) and then ipilimumab (3 mg/kg every 3 weeks; four doses), or the reverse sequence. This was followed by maintenance therapy with nivolumab (3 mg/kg every 2 weeks) in both cohorts. Greater efficacy was observed in the nivolumab followed by ipilimumab cohort, with significantly greater 1‐year OS than the reverse sequence (Table 1). As expected from previous combination trials, the frequency of IAEs was relatively high (43–50%), although this was not affected by the order of drug administration.
Collectively, these trials have allowed ICIs, either alone or in combination, to herald in a new era of immunotherapy for the treatment of malignant melanoma. Impressive durable responses have led to unprecedented improvements for some patients with metastatic disease. Administering ICIs in combination with other therapeutics will no doubt expand their use further in cancer patients.
New therapeutic approaches for non‐small‐cell lung cancer
Anti‐CTLA‐4 therapy has been trialled in patients with NSCLC, but promising outcomes akin to those seen in patients with melanoma have not been forthcoming. One initial trial used tremelimumab, an anti‐CTLA‐4 antibody, but it showed minimal efficacy and a significant side‐effect burden 96 and, when used in combination with chemotherapy, anti‐CTLA‐4 antibodies have consistently demonstrated only modest improvements in responses in NSCLC patients 97, 98. However, antibodies targeting the PD‐1/PD‐L1 axis have shown greater efficacy and marked reductions in side effects, so have largely replaced anti‐CTLA‐4 antibodies as the ICI of choice in NSCLC.
In 2015, nivolumab received Food and Drug Administration (FDA) approval for use in patients with advanced squamous‐cell NSCLC who progressed during or after platinum chemotherapy (Table 2) 94. This followed a Phase III trial (CheckMate‐017) of nivolumab compared with docetaxel chemotherapy in patients with progressive disease following first‐line chemotherapy 99. Median OS was 9·2 months in the nivolumab cohort, compared with 6·0 months in those receiving docetaxel. At 12 months, OS was 42% with nivolumab and 24% with docetaxel. Pretreatment tumour PD‐L1 expression was not predictive of efficacy. A similar Phase III trial (CheckMate‐057) demonstrated considerable improvements in survival for patients with advanced non‐squamous NSCLC treated with nivolumab compared to those receiving docetaxel 100. OS at 12 and 18 months was higher for patients on nivolumab compared with those on docetaxel. An additional Phase II trial (CheckMate‐063) supports the survival benefit of nivolumab, reporting a 1‐year OS of 40·8% 101. Similarly, compared with docetaxel, pembrolizumab showed greater efficacy than docetaxel in advanced NSCLC in a Phase II/III trial (KEYNOTE‐010) 102, 103. Patients with ≥ 1% PD‐L1 expression on the tumour received 2 mg/kg pembrolizumab, 10 mg/kg pembrolizumab or docetaxel, resulting in median OS of 10·4, 12·7 or 8·5 months, respectively. Greater efficacy was seen in patients with high tumour PD‐L1 expression. These results echo findings from a similar Phase I study (KEYNOTE‐001) investigating pembrolizumab in NSCLC 104, while a Phase III trial (KEYNOTE‐024) demonstrated survival benefits, durable responses, increased response rates and reduced treatment‐related adverse events with pembrolizumab compared with platinum‐based chemotherapy in previously untreated patients, with ≥ 50% tumoral PD‐L1 expression 105. This has led to pembrolizumab being licensed for treatment of naive metastatic NSCLC with ≥ 50% PD‐L1 expression 92. In contrast, nivolumab did not improve progression‐free or overall survival compared with chemotherapy in patients (n = 423) with previously untreated stage IV or recurrent NSCLC and with a PD‐1 expression of 5% or more 106. This suggests that robust predictive markers are required to select patients optimally for anti‐PD‐1 therapy, and that the sequence of administration with other standard therapies may also be relevant for optimal treatment strategies.
Table 2.
Key trials of immune checkpoint inhibitor (ICIs) targeting the programmed cell death‐1 (PD‐1) pathway in patients with non‐small‐cell lung cancer (NSCLC)
| Trial name, Phase [Ref] | Disease | Trial drugs (dose) | Patient no. | 1‐year OS rate (%) | Median OS (months) | Objective responses rate (%) | Median PFS (months) | Correlation between tumour PD‐L1 expression and response? | Treatment‐related adverse events (grade ≥ 3) (%) |
|---|---|---|---|---|---|---|---|---|---|
|
CheckMate‐063 Phase III 101 |
Sq NSCLC | Niv (3 mg/kg) | 117 | 40·8 | 8·2 | 14·5 | 1·9 | Yes | 17 |
|
CheckMate‐017 Phase III 99 |
Sq NSCLC | Niv (3 mg/kg) versus docetaxel | 272 | 42 versus 24 | 9·2 versus 6·0 | 20 versus 9 | 3·5 versus 2·8 | n.r. | 7 versus 55 |
|
CheckMate‐057 Phase III 100 |
Non‐Sq NSCLC | Niv (3 mg/kg) versus docetaxel | 582 | 51 versus 39 | 12·2 versus 9·4 | 19 versus 12 | 2·3 versus 4·2 | Yes | 10 versus 54 |
|
KEYNOTE‐001 Phase I 104 |
NSCLC |
Pembro (2–10 mg/kg) |
495 | n.r. | 12·0 | 19·4 | 3·7 | Yes | 9·5 |
|
KEYNOTE‐010 Phase II/III 102, 103 |
>1% PD‐L1 + NSCLC | Pembro (2 mg/kg) versus Pembro (10 mg/kg) versus Docetaxel | 1034 | 43·2 versus 52·3 versus 34·6 | 10·4 versus 12·7 versus 8·5 | n.r. | 3·9 versus 4·0 versus 4·9 | Yes | 13 versus 16 versus 35 |
|
KEYNOTE‐024 Phase II 105 |
>50% PD‐L1 + NSCLC | Pembro (200 mg) versus docetaxel | 305 | n.r. | n.r. | 44·8 versus 27·8 | 10·3 versus 6·0 | High PD‐L1 a prerequisite for inclusion in trial | 26·6 versus 53·3 |
|
POPLAR Phase II 106 |
NSCLC | Atezo (1200 mg) versus docetaxel | 277 | 47·9 versus 37·8 | 12·6 versus 9·7 | 15 versus 15 | 2·7 versus 3·0 | Yes | 40 versus 53 |
| OAK Phase III 107, 108 | NSCLC | Atezo versus docetaxel | 1225 | 55 versus 41 | 13·8 versus 9·6 | 14 versus 13 | 2·8 versus 4·0 | Yes | 37 versus 54 |
| Phase 1b 109 | NSCLC | Durvalumab + tremelimumab | 102 | n.r. | n.r. | 0–23 | n.r. | n.r. | 36 |
|
CheckMate‐012 Phase 1 110 |
NSCLC | Niv (3 mg/kg) + Ipi versus Niv (3 mg/kg) + Ipi | 77 | Not calculated versus 69 | n.r. | 47 versus 38 | 8·1 versus 3·9 | Yes | 37 versus 33 |
Sq = squamous; non‐sq = non‐squamous; niv = nivolumab; pembro = pembrolizumab; atezo = atezolizumab; ipi = ipilumumab; OS = overall survival; PFS = progression‐free survival; n.r. = not reported.
In the Phase II for patients with atezolizumab versus docetaxel for previously treated non‐small‐cell lung cancer (POPLAR) trial, atezolizumab (Genentech, South San Francisco, CA, USA), an antibody targeting the PD‐L1 rather than PD‐1, showed improved OS for NSCLC patients who had progressed following platinum chemotherapy 107. This study included prospective evaluation of tumour PD‐L1 expression, and used immune gene expression to define and quantify effector T cell activity within the TME. It was clear that efficacy was related closely to both PD‐L1 expression and effector T cell abundance in the tumour. Future studies should consider incorporating this pretreatment analysis of the presence of active tumour‐infiltrating effector T cells that may be a predictive marker of ICI efficacy. The clinical outcomes of POPLAR were reiterated in atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (OAK), a Phase III clinical trial demonstrating improved median OS with atezolizumab versus docetaxel (13·8 versus 9·6 months) in NSCLC patients progressing on platinum chemotherapy, although improvement in OS was seen regardless of tumour PD‐L1 expression 108, 109. As a result of these trials, in 2016 the FDA approved atezolizumab for the treatment of metastatic NSCLC that has progressed on platinum chemotherapy 94.
Significantly, PD‐L1/PD‐1 inhibitors not only improve OS but they are also associated with fewer treatment‐related adverse events (≥ grade 3) than chemotherapy 99, 100, 102, 105, 106, 107, 108. Moreover, they may have broad usage: although strongest responses were seen in patients with high tumour PD‐L1 expression, they also showed similar, if not greater, efficacy than chemotherapy in patients with very low tumour PD‐L1 expression 99, 100, 101, 102, 103, 104, 107, 108. It has not yet been possible to stratify patient responses accurately based on PD‐L1 expression, although this typically involves the analysis of a single pretreatment tumour biopsy, sometimes taken long before starting checkpoint blockade therapy. In the Checkmate‐063 trial, for example, there was a median of 1·3 years between biopsy and commencing nivolumab 101. Thus, more detailed studies are required to explore the prognostic value of tumour PD‐L1 expression, and ideally analysis of effector T cell abundance and phenotype should also be included.
ICIs offer new hope for treating NSCLC which, historically, has poor survival outcomes in patients with metastatic disease, and they have become standard second‐line therapies in NSCLC patients. They are also being used as first‐line treatments, particularly for patients with PD‐L1‐expressing tumours, although optimal ICI treatment strategies, with or without chemotherapy, still need to be defined more precisely in this context 111. Trials using a combination of anti‐CTLA4 and anti‐PD‐L1 antibodies have begun in NSCLC patients, with two Phase I studies demonstrating safety profiles comparable to those seen in melanoma patients (Table 2) 110, 112. The results of ongoing Phase II/III trials are eagerly anticipated 113, 114, 115. Other studies have focused upon including ICIs into treatment strategies either in combination or sequentially after other existing therapies in NSCLC. For example, patients (n = 713) with locally advanced unresectable NSCLC were assigned randomly (2 : 1) to receive consolidation therapy with either the anti‐PDL‐1 antibody durvalumab or placebo every 2 weeks for up to 12 months after definitive chemoradiotherapy (chemotherapy with concurrent radiotherapy). Patients treated with durvalumab had a significantly improved progression‐free survival (16·8 versus 5·6 months; HR = 0·52, P < 0·001) and objective response rates 28·4 versus 16%; P < 0·001) 116.
ICIs show efficacy in other cancers
ICIs have been, or are being, investigated in clinical trials across a broad range of other cancers in the hope that responses seen in patients with melanoma or NSCLC can be reproduced in patients with other tumour types.
PD‐L1 expression has long been known to be associated with tumour aggressiveness and poorer outcomes in patients with renal cell carcinoma (RCC) 62, 117. Nivolumab demonstrated efficacy in a Phase II trial of 168 patients with metastatic RCC 118. They received 0·2, 2 or 10 mg/kg of nivolumab: median OS was 18·2, 25·5 or 24·7 months, respectively. In a subsequent Phase III study, patients that had previously received anti‐angiogenic therapy had a median OS of 25·0 months on nivolumab compared with 19·6 months on everolimus [a mechanistic target of rapamycin (mTOR) inhibitor recommended after failed anti‐angiogenic therapy] 119. Nivolumab was subsequently approved for use in patients with advanced RCC after failing anti‐angiogenic therapy 94. Interestingly, high tumour PD‐L1 expression was not associated with improved responses to therapy in this study: median OS in patients with PD‐L1 expression ≥ 1% was 21·6 and 18·8 months (nivolumab and everolimus, respectively), but 27·4 versus 21·2 months in patients with ≤ 1% tumour PD‐L1 expression. This emphasizes the need to consider carefully the suitability of using PD‐L1 expression alone to predict responses.
Following two clinical trials 120, 121, atezolizumab, and more recently nivolumab, have been granted FDA approval for bladder cancer patients with disease progression within 12 months of treatment with platinum‐containing chemotherapy 94. The Phase II trial using atezolizumab improved objective response rates compared with a historical control 120. Approximately 15% of patients responded and in this case, PD‐L1 expression on tumour‐associated immune cells correlated with objective responses. Ongoing responses in 84% of responders demonstrate excellent treatment durability, and for a small fraction (5%) a complete response was observed. This trial attempted to define features of the TME associated with responses. In addition to PD‐L1 expression, it determined CD8+ T cell infiltration, mutational load and molecular subtype. Several ‘immune cell’ genes were associated with complete and partial responses to atezolizumab, including the IFN‐γ‐inducible genes CXCL9 and CXCL10, which encode chemokines that direct leucocyte and CD8+ T cell homing. Mutational load was also significantly higher in responders than non‐responders (12·4 versus 6·4 mutations per Mb). High mutational load has also been correlated with responses to anti‐CTLA‐4 in malignant melanoma 122, and may predict better survival in melanoma patients treated with anti‐PD1 therapy 123. This is most probably because increased mutation will increase the generation of neoantigens recognizable by T cells. Thus, in addition to characterizing PD‐L1 expression and the immune cell infiltrate, efforts to predict ICI responses should take into account the mutational landscape of the tumour.
Tumours with mismatch‐repair (MMR) defects contain a large number of somatic mutations, so may be particularly sensitive to ICIs. Indeed, in a recent Phase II trial pembrolizumab was administered every 2 weeks to patients with MMR‐deficient colorectal cancers (n = 11), MMR‐proficient colorectal cancers (n = 21) or MMR‐deficient cancers that were not colorectal (n = 9) 124. The objective response rate and progression‐free survival rate at 20 weeks were 40 and 78% for MMR‐deficient colorectal cancers; 71 and 67% for non‐colorectal MMR‐deficient cancers; and 0 and 11% for MMR‐proficient colorectal cancers. This apparent sensitivity of MMR‐deficient tumours supports the rationale of combining DNA repair inhibitors with ICIs in clinical trials, and a Phase I/II study combining the poly ADP ribose polymerase (PARP) inhibitor, olaparib, with durvalumab in patients with selected advanced malignancies is ongoing 125.
PD‐1 blockade has shown positive outcomes in classical Hodgkin's lymphoma. Nivolumab and pembrolizumab are now FDA‐approved for treatment of patients who have relapsed after other therapies. Two clinical trials have reported ORRs of 65 and 87%, with complete response rates in 16 and 17% of patients 126, 127. Nivolumab and pembrolizumab are also approved for use in patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) whose disease has progressed after administration of platinum‐based therapeutics 94. Pembrolizumab treatment gave a clinically significant ORR and durable responses were evident in some patients 128, while nivolumab improved overall survival compared to standard single‐agent therapy (methotrexate, docetaxel or cetuximab) 129. However, in a Phase III study of pembrolizumab versus standard of care in patients with recurrent or metastatic SCCHN, pembrolizumab yielded a 19% reduction in the risk of death compared with standard of care but this did not meet the prespecified efficacy boundary, although it is possible that subsequent immunotherapy use in the standard of care arm may have confounded the overall survival analysis 130. Nonetheless, greater differences in overall survival, progression‐free survival and objective response rates were seen in patients with PD‐1‐expressing tumours 130.
Based on a Phase II clinical trial involving 88 patients, avelumab (Merck–Pfizer, New York, NY, USA), a PD‐L1 blocking antibody, received FDA approval for use in patients with metastatic Merkel cell carcinoma 94, 131. This is the first ever drug approved for this rare skin cancer, in which Merkel cell polyomavirus infection is a major aetiological factor 132. Nearly a third of patients showed objective responses, with eight complete responses reported: five grade 3 treatment‐related adverse events were noted 131. Pembrolizumab has also been trialled on 26 patients with this disease, with an ORR of 56% being reported, including four complete responses 133.
In a large Phase III trial involving 1132 patients with extensive late‐stage small‐cell lung cancer (SCLC), ipilimumab with chemotherapy versus placebo with chemotherapy demonstrated no significant difference in OS 134. A previous Phase I/II trial had also given disappointing results in SCLC, with no survival advantage for patients receiving ipilimumab 135. However, nivolumab, either alone or in combination with ipilimumab, showed anti‐tumour activity with durable responses and manageable safety profiles in previously treated patients with SCLC 136. Patients were administered nivolumab (3 mg/kg); nivolumab (1 mg/kg) plus ipilimumab (3 mg/kg); or nivolumab (3 mg/kg) plus ipilimumab (1 mg/kg). Objective responses were seen in 10, 23 and 19% patients, respectively. Pembrolizumab is currently under investigation in patients with PD‐L1+ (≥ 1% expression) SCLC, who have progressed during or after platinum‐based chemotherapy 137. Initial results indicate good tolerability and, while the data are preliminary, the initial report shows that 25% of SCLC patients developed a partial response.
A Phase III trial compared ipilimumab against placebo, following radiation, in patients with metastatic castration‐resistant prostate cancer that progressed on docetaxel chemotherapy 138. They reported no significant improvement in OS: median OS was 11·2 months for ipilimumab and 10·0 months for placebo. However, subgroup analysis revealed improved OS outcomes for patients with better prognostic factors. This is perhaps unsurprising, as it is now established that immunotherapies often take longer to show measurable efficacy 139. Final results from a completed Phase III trial comparing placebo with ipilimumab in minimally symptomatic, previously untreated patients with metastatic castration‐resistant prostate cancer should reveal whether there is a role for ICIs in this malignancy 140. Finally, promising responses have also been observed with anti‐PD‐1 antibody treatment in patients with refractory gastric cancer 141, 142 or hepatocellular carcinoma 143: results of randomized trials are eagerly awaited.
Thus, in addition to melanoma and NSCLC, ICI monotherapies, particularly those targeting the PD‐1 pathway, are yielding positive outcomes in a range of metastatic cancers. Toxicities are evident and need to be managed, and the long‐term health implications of ICI monotherapy have yet to be determined. This might be significant, given the improvements in patient survival seen with ICIs, although the issue of resistance to ICIs raises the challenge of how to treat cancers which fail multiple therapies.
Future prospects for ICIs in combination therapies
Clinical trial data have justifiably generated considerable excitement concerning the effectiveness of ICIs. In many cases responses are durable, perhaps even curative, possibly because they release systemic personalized memory T cell responses against multiple patient‐specific tumour antigens. However, only subsets of patients respond to ICI monotherapy. Combining ICIs can improve outcomes, and further ICI combination trials, with careful side‐effect monitoring, are certainly merited. There is also a large number of ongoing clinical trials exploring ICIs in combination with other treatment modalities, such as radiotherapy, chemotherapy or molecular targeted therapies 144. The administration of ICIs after radiotherapy attempts to augment the ‘abscopal effect’, where localized radiation‐induced tumour cell death can deliver anti‐tumour immune responses against distant tumour deposits (Fig. 1) 145, 146, 147, 148, 149, 150. ICIs even show activity against tumours viewed classically as poorly immunogenic, such as pancreatic adenocarcinoma and glioblastoma, with efforts aimed at administering ICIs while either modifying the TME, up‐regulating neoantigen abundance 151 or, in glioblastoma, potentiating immune responses using adenovirus 152. Modulating the chemokine composition of the TME 153, or blocking chemokine receptors 154, can also enhance ICI efficacy in animal models, and these approaches may translate to humans. Preliminary data in animal tumour models have shown that depletion of Tregs via modified anti‐CD25 antibody plus anti‐PD‐1 antibody can induce complete regression of established tumours 155. In short, combination therapies have the potential to further improve the activity and scope of ICIs, leading to the induction of durable effector T cell responses in more patients, and throughout a greater range of malignancies.
Integrating ICIs into precision oncology
The future of oncology will undoubtedly involve a precision approach to patient care, with treatment tailored to an individual's disease to maximize efficacy and ameliorate side effects as far as possible. Numerous factors determine a patient's response to ICIs. These include, but are not limited to, the distribution of the target immune checkpoint protein; the immune cell context within the tumour; the availability, immunogenicity and frequency of tumour antigens; the pharmacodynamics and pharmacokinetics of the ICI; and the amenability of the TME for effective drug delivery 156. The analysis of these factors must be incorporated into the design of future clinical trials so that combinations of biomarkers that predict response to ICI immunotherapy can be identified and exploited. It will also be important to clarify further the reciprocal relationship between tumour cells and the TME in the context of ICI therapies, particularly if they are to be implemented across a range of different malignancies. Clinical trials have evolved from when tumours were classified according to histological observations, and comprehensive molecular, cellular and genetic phenotyping is now possible. Genomic and transcriptomic analysis of pretreatment melanoma has identified a transcriptional signature associated with innate resistance to anti‐PD1 therapy, and demonstrated that mutations in the DNA repair gene BRCA2 are enriched in patients that do respond 123. The analysis of serial tumour biopsies taken before, during and after ICI therapy will further enhance understanding of how anti‐tumour responses are induced by ICIs, and how tumours adapt to evade or limit these responses. Interestingly, one study has identified mutations in melanoma cells in patients whose disease progressed after initially responding to PD‐1 blockade 157, and at least one trial is under way that aims to understand the selection pressures and evolutionary changes that ICIs impose upon tumour cells 158.
Concluding remarks
The development and application of ICIs has been a major advance in the treatment of cancer, dramatically rekindling academic, clinical and commercial interest in anti‐cancer immunotherapies. As monotherapies, they show considerable promise and remarkable responses have been seen in some patients. However, it is clear that there is improved efficacy when these ICIs are used as part of a concerted therapeutic approach for management of malignant disease. The key issue to be resolved is generation of sufficient evaluable data from combinatorial trials to define effective parameters for treatment. Results from current trials where ICIs are combined with chemotherapy, radiotherapy or with other immunotherapies are starting to demonstrate which malignancies are particularly susceptible to immune reactivation. These studies are also providing clear evidence that, although ICIs have produced dramatic improvements in some patients, they are not necessarily sufficient to mediate long‐term remission or cure in most. Identification and validation of suitable prognostic markers in patient cohorts will also help to stratify treatment modalities and target therapy to those patients most likely to respond. The corollary to this is that suitable prognostic criteria could also minimize the IAEs associated with ICI treatment and immune reactivation 159. The management of side effects and the evolution of resistance are ongoing concerns, but further advances are anticipated as these new medicines are incorporated into combination therapies and integrated into precision oncology. Progress is likely to be swift, given the current explosion in clinical trials of ICIs and the rapidly expanding number of preclinical studies involving these drugs will no doubt stimulate further clinical applications. The range of new ICIs becoming available, and the novel checkpoint targets being identified and characterized in preclinical studies, means that the resistance or relapse seen in some patients could be reversed by using alternative checkpoint inhibitors. There is also a significant new approach to tumour immunotherapy with the recent FDA approval for autologous gene‐modified chimeric antigen receptor (CAR) T cell therapy. This cell therapy (Kymriah; Novartis, Basel, Switzerland) targets CD19 in B cell acute lymphocytic leukaemia and has shown significant efficacy 160. There are several other CAR‐T cell therapies in development and combination therapy with ICIs is already planned, offering the potential for even greater benefits. ICIs are important new weapons in the oncologist's arsenal, and exciting times lie ahead in the field of cancer immunotherapy.
Disclosure
The authors confirm that they have no competing financial interests or activities.
Acknowledgements
The authors would like to thank Alison Schroeer of Schroeer Scientific Illustration for assistance with the preparation of Fig. 2.
References
- 1. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol 2016; 16:599–611. [DOI] [PubMed] [Google Scholar]
- 3. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004; 21:137–48. [DOI] [PubMed] [Google Scholar]
- 4. Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 2008; 27:3889–900. [DOI] [PubMed] [Google Scholar]
- 5. Igney FH, Krammer PH. Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leukoc Biol 2002; 71:907–20. [PubMed] [Google Scholar]
- 6. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest 2007; 117:1137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte–endothelium interactions: identification of caveolin‐1 and nitric oxide as control points of endothelial cell anergy. J Immunol 2007; 178:1505–11. [DOI] [PubMed] [Google Scholar]
- 8. Buckanovich RJ, Facciabene A, Kim S et al Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med 2008; 14:28–36. [DOI] [PubMed] [Google Scholar]
- 9. Oleinika K, Nibbs RJ, Graham GJ, Fraser AR. Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol 2013; 171:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Facciabene A, Motz GT, Coukos GT. Regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012; 72:2162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dong H, Strome SE, Salomao DR et al Tumor‐associated B7‐H1 promotes T cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8:793–800. [DOI] [PubMed] [Google Scholar]
- 13. Zou W, Chen L. Inhibitory B7‐family molecules in the tumour microenvironment. Nat Rev Immunol 2008; 8:467–77. [DOI] [PubMed] [Google Scholar]
- 14. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28‐mediated signalling co‐stimulates murine T cells and prevents induction of anergy in T cell clones. Nature 1992; 356:607–9. [DOI] [PubMed] [Google Scholar]
- 16. McAdam AJ, Schweitzer AN, Sharpe AH. The role of B7 co‐stimulation in activation and differentiation of CD4+ and CD8+ T cells. Immunol Rev 1998; 165:231–47. [DOI] [PubMed] [Google Scholar]
- 17. Acuto O, Michel F. CD28‐mediated co‐stimulation: a quantitative support for TCR signalling. Nat Rev Immunol 2003; 3:939–51. [DOI] [PubMed] [Google Scholar]
- 18. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013; 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krummel MF, Allison JP. CD28 and CTLA‐4 have opposing effects on the response of T cells to Stimulation. J Exp Med 1995; 182:459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walunas TL, Lenschow DJ, Bakker CY et al CTLA‐4 can function as a negative regulator of T cell activation. Immunity 1994; 1:405–13. [DOI] [PubMed] [Google Scholar]
- 21. Alegre ML, Frauwirth KA, Thompson CB. T cell regulation by CD28 and CTLA‐4. Nat Rev Immunol 2001; 1:220–8. [DOI] [PubMed] [Google Scholar]
- 22. Lee KM, Chuang E, Griffin M et al Molecular basis of T cell inactivation by CTLA‐4. Science 1998; 282:2263–6. [DOI] [PubMed] [Google Scholar]
- 23. Schneider H, Downey J, Smith A et al Reversal of the TCR stop signal by CTLA‐4. Science 2006; 313:1972–5. [DOI] [PubMed] [Google Scholar]
- 24. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell‐extrinsic regulator of T cell responses. Nat Rev Immunol 2011; 11:852–63. [DOI] [PubMed] [Google Scholar]
- 25. Greene JL, Leytze GM, Emswiler J et al Covalent dimerization of CD28/CTLA‐4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J Biol Chem 1996; 271:26762–71. [DOI] [PubMed] [Google Scholar]
- 26. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen‐4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002; 16:23–35. [DOI] [PubMed] [Google Scholar]
- 27. Yokosuka T, Kobayashi W, Takamatsu M et al Spatiotemporal basis of CTLA‐4 costimulatory molecule‐mediated negative regulation of T cell activation. Immunity 2010; 33:326–39. [DOI] [PubMed] [Google Scholar]
- 28. Qureshi OS, Zheng Y, Nakamura K et al Trans‐endocytosis of CD80 and CD86: a molecular basis for the cell‐extrinsic function of CTLA‐4. Science 2011; 332:600–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walker LS, Sansom DM. Confusing signals: recent progress in CTLA‐4 biology. Trends Immunol 2015; 36:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA‐4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA‐4. Immunity 1995; 3:541–7. [DOI] [PubMed] [Google Scholar]
- 31. Waterhouse P, Penninger JM, Timms E et al Lymphoproliferative disorders with early lethality in mice deficient in CTLA‐4. Science 1995; 270:985–8. [DOI] [PubMed] [Google Scholar]
- 32. Kuehn HS, Ouyang W, Lo B et al Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014; 345:1623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schubert D, Bode C, Kenefeck R et al Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20:1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeissig S, Petersen B‐S, Tomczak M et al Early‐onset Crohn's disease and autoimmunity associated with a variant in CTLA‐4. Gut 2015; 64:1889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA‐4 blockade. Science 1996; 271:1734–6. [DOI] [PubMed] [Google Scholar]
- 36. Hurwitz AA, Foster BA, Kwon ED et al Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA‐4 blockade. Cancer Res 2000; 60:2444–8. [PubMed] [Google Scholar]
- 37. Selby MJ, Engelhardt JJ, Quigley M et al Anti‐CTLA‐4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 2013; 1:32–42. [DOI] [PubMed] [Google Scholar]
- 38. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM–CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest 2006; 116:1935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA‐4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti‐CTLA‐4 antibodies. J Exp Med 2009; 206:1717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Simpson TR, Li F, Montalvo‐Ortiz W et al Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti‐CTLA‐4 therapy against melanoma. J Exp Med 2013; 210:1695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bulliard Y, Jolicoeur R, Windman M et al Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor‐targeting antibodies. J Exp Med 2013; 210:1685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol 2014; 92:475–80. [DOI] [PubMed] [Google Scholar]
- 43. Śledzińska A, Menger L, Bergerhoff K, Peggs KS, Quezada SA. Negative immune checkpoints on T lymphocytes and their relevance to cancer immunotherapy. Mol Oncol 2015; 9:1936–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Romano E, Kusio‐Kobialka M, Foukas PG et al Ipilimumab‐dependent cell‐mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA 2015; 112:6140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD‐1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992; 11:3887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Agata Y, Kawasaki A, Nishimura H et al Expression of the PD‐1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996; 8:765–72. [DOI] [PubMed] [Google Scholar]
- 47. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamazaki T, Akiba H, Iwai H et al Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002; 169:5538–45. [DOI] [PubMed] [Google Scholar]
- 49. Eppihimer MJ, Gunn J, Freeman GJ et al Expression and regulation of the PD‐L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 2002; 9:133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McDermott DF, Atkins MB. PD‐1 as a potential target in cancer therapy. Cancer Med 2013; 2:662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Latchman Y, Wood CR, Chernova T et al PD‐L2 is a second ligand for PD‐1 and inhibits T cell activation. Nat Immunol 2001; 2:261–8. [DOI] [PubMed] [Google Scholar]
- 52. Freeman GJ, Long AJ, Iwai Y et al Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hui E, Cheung J, Zhu J et al T cell costimulatory receptor CD28 is a primary target for PD‐1–mediated inhibition. Science 2017; 355:1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fife BT, Pauken KE, Eagar TN et al Interactions between PD‐1 and PD‐L1 promote tolerance by blocking the TCR‐induced stop signal. Nat Immunol 2009; 10:1185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death‐1 ligand 1 interacts specifically with the B7‐1 costimulatory molecule to inhibit T cell responses. Immunity 2007; 27:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus‐like autoimmune diseases by disruption of the PD‐1 gene encoding an ITIM motif‐carrying immunoreceptor. Immunity 1999; 11:141–51. [DOI] [PubMed] [Google Scholar]
- 57. Nishimura H, Okazaki T, Tanaka Y et al Autoimmune dilated cardiomyopathy in PD‐1 receptor deficient mice. Science 2001; 291:319–22. [DOI] [PubMed] [Google Scholar]
- 58. Zajac AJ, Blattman JN, Murali‐Krishna K et al Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 1998; 188:2205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gallimore A, Glithero A, Godkin A et al Induction and exhaustion of lymphocytic choriomeningitis virus‐specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I‐peptide complexes. J Exp Med 1998; 187:1383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barber DL, Wherry EJ, Masopust D et al Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439:682–7. [DOI] [PubMed] [Google Scholar]
- 61. Kamphorst AO, Wieland A, Nasti T et al Rescue of exhausted CD8 T cells by PD‐1–targeted therapies is CD28‐dependent. Science 2017; 355:1423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thompson RH, Gillett MD, Cheville JC et al Costimulatory B7‐H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA 2004; 101:17174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thompson RH, Dong H, Lohse CM et al PD‐1 is expressed by tumor‐infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res 2007; 13:1757–61. [DOI] [PubMed] [Google Scholar]
- 64. Ahmadzadeh M, Johnson LA, Heemskerk B et al Tumor antigen‐specific CD8 T cells infiltrating the tumor express high levels of PD‐1 and are functionally impaired. Blood 2009; 114:1537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Y, Huang S, Gong D, Qin Y, Shen Q. Programmed death‐1 upregulation is correlated with dysfunction of tumor‐infiltrating CD8+ T lymphocytes in human non‐small cell lung cancer. Cell Mol Immunol 2010; 7:289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hino R, Kabashima K, Kato Y et al Tumor cell expression of programmed cell death‐1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010; 116:1757–66. [DOI] [PubMed] [Google Scholar]
- 67. Shi F, Shi M, Zeng Z et al PD‐1 and PD‐L1 upregulation promotes CD8(+) T cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer 2011; 128:887–96. [DOI] [PubMed] [Google Scholar]
- 68. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci USA 2002; 99:12293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Blank C, Brown I, Peterson AC et al PD‐L1/B7H‐1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 2004; 64:1140–5. [DOI] [PubMed] [Google Scholar]
- 70. Kuang DM, Zhao Q, Peng C et al Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD‐L1. J Exp Med 2009; 206:1327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Curiel TJ, Wei S, Dong H et al Blockade of B7‐H1 improves myeloid dendritic cell‐mediated antitumor immunity. Nat Med 2003; 9:562–7. [DOI] [PubMed] [Google Scholar]
- 72. Francisco LM, Salinas VH, Brown KE et al PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009; 206:3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hodi FS, O'Day SJ, McDermott DF et al Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rosenberg SA, Yang JC, Schwartzentruber DJ et al Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 1998; 4:321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Smith FO, Downey SG, Klapper JA et al Treatment of metastatic melanoma using interleukin‐2 alone or in conjunction with vaccines. Clin Cancer Res 2008; 14:5610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weber J, Thompson JA, Hamid O et al A randomized, double‐blind, placebo‐controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res 2009; 15:5591–8. [DOI] [PubMed] [Google Scholar]
- 77. Wolchok JD, Neyns B, Linette G et al Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double‐blind, multicentre, phase 2, dose‐ranging study. Lancet Oncol 2010; 11:155–64. [DOI] [PubMed] [Google Scholar]
- 78. O'Day SJ, Maio M, Chiarion‐Sileni V et al Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single‐arm phase II study. Ann Oncol 2010; 21:1712–7. [DOI] [PubMed] [Google Scholar]
- 79. Schadendorf D, Hodi FS, Robert C et al Pooled analysis of long‐term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015; 33:1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Robert C, Schachter J, Long GV et al Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015; 372:2521–32. [DOI] [PubMed] [Google Scholar]
- 81. Schachter J, Ribas A, Long GV et al Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival analysis of KEYNOTE‐006. J Clin Oncol 2016; 34: [Google Scholar]
- 82. Topalian SL, Hodi FS, Brahmer JR et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Topalian SL, Sznol M, McDermott DF et al Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32:1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Robert C, Long GV, Brady B et al Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–30. [DOI] [PubMed] [Google Scholar]
- 85. Wolchok JD, Kluger H, Callahan MK et al Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sznol M, Kluger HM, Callahan MK et al Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti‐PD‐1, BMS‐936558, ONO‐4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL). J Clin Oncol 2014; 32:9003. [Google Scholar]
- 87. Postow MA, Chesney J, Pavlick AC et al Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015; 372:2006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hodi FS, Chesney J, Pavlick AC et al Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2‐year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2016; 17:1558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Larkin J, Chiarion‐Sileni V, Gonzalez R et al Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373:23–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wolchok JD, Chiarion‐Sileni V, Gonzalez R et al Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017; 377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Weber JS, Gibney G, Sullivan RJ et al Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open‐label, randomised, phase 2 trial. Lancet Oncol 2016; 17:943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Robert C, Ribas A, Wolchok JD et al Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: a randomised dose‐comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–17. [DOI] [PubMed] [Google Scholar]
- 93. Eisenhauer EA, Therasse P, Bogaerts J et al New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45:228–47. [DOI] [PubMed] [Google Scholar]
- 94.FDA.gov. Hematology/oncology (cancer) approvals and safety notifications. Available at: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm (accessed 13 October 2017).
- 95. Weber JS, D'Angelo SP, Minor D et al Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti‐CTLA‐4 treatment (CheckMate 037): a randomised, controlled, open‐label, phase 3 trial. Lancet Oncol 2015; 16:375–84. doi: 10.1016/S1470‐2045(15)70076‐8. [DOI] [PubMed] [Google Scholar]
- 96. Zatloukal P, Heo DS, Park K et al Randomized phase II clinical trial comparing tremelimumab (CP‐675,206) with best supportive care (BSC) following first‐line platinum‐based therapy in patients (pts) with advanced non‐small cell lung cancer (NSCLC). J Clin Oncol 2009; 27:abstr8071. [Google Scholar]
- 97. Lynch TJ, Bondarenko I, Luft A et al Ipilimumab in combination with paclitaxel and carboplatin as first‐line treatment in stage IIIB/IV non‐small‐cell lung cancer: results from a randomized, double‐blind, multicenter phase II study. J Clin Oncol 2012; 30:2046–2054. [DOI] [PubMed] [Google Scholar]
- 98. Govindan R, Szczesna A, Ahn M‐J et al Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non‐small‐cell lung cancer. J Clin Oncol 2017; 35:3449–57. [DOI] [PubMed] [Google Scholar]
- 99. Brahmer J, Reckamp KL, Baas P et al Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med 2015; 373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Borghaei H, Paz‐Ares L, Horn L et al Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med 2015; 373:1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rizvi NA, Mazières J, Planchard D et al Activity and safety of nivolumab, an anti‐PD‐1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non‐small‐cell lung cancer (CheckMate 063): a phase 2, single‐arm trial. Lancet Oncol 2015; 16:257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Herbst RS, Baas P, Kim D‐W et al Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet 2016; 387:1540–50. [DOI] [PubMed] [Google Scholar]
- 103. Herbst RS, Baas P, Kim D‐W et al Pembrolizumab (pembro) vs docetaxel (doce) for previously treated, PD‐L1–expressing NSCLC: updated outcomes of KEYNOTE‐010. Ann Oncol 2016; 387: 1540–50. [DOI] [PubMed] [Google Scholar]
- 104. Garon EB, Rizvi NA, Hui R et al Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med 2015; 372:2018–28. [DOI] [PubMed] [Google Scholar]
- 105. Reck M, Rodríguez‐Abreu D, Robinson AG et al Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med 2016; 375:1823–33. [DOI] [PubMed] [Google Scholar]
- 106. Carbone DP, Reck M, Paz‐Ares L et al First‐line nivolumab in stage IV or recurrent non‐small‐cell lung cancer. New Engl J Med 2017; 376:2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fehrenbacher L, Spira A, Ballinger M et al Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet 2016; 387:1837–46. [DOI] [PubMed] [Google Scholar]
- 108. Rittmeyer A, Barlesi F, Waterkamp D et al Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet 2017; 389:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Barles F, Park K, Ciardiello F et al Primary analysis from OAK, a randomized phase III study comparing atezolizumab with docetaxel in 2L/3L NSCLC. Ann Oncol 2016; 27:vi552–87. [Google Scholar]
- 110. Hellmann MD, Rizvi NA, Goldman JW et al Nivolumab plus ipilimumab as first‐line treatment for advanced non‐small‐cell lung cancer (CheckMate 012): results of an open‐label, phase 1, multicohort study. Lancet Oncol 2017; 18:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Remon J, Pardo N, Martinez‐Martí A et al Immune‐checkpoint inhibition in first‐line treatment of advanced non‐small cell lung cancer patients: current status and future approaches. Lung Cancer 2017; 106:70–75. [DOI] [PubMed] [Google Scholar]
- 112. Antonia S, Goldberg SB, Balmanoukian A et al Safety and antitumour activity of durvalumab plus tremelimumab in non‐small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol 2016; 17:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.A global study to assess the effects of MEDI4736, given as monotherapy or in combination with tremelimumab determined by PD‐L1 expression versus standard of care in patients with locally advanced or metastatic non small cell lung cancer (ARCTIC). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/show/NCT02352948 (accessed 13 October 2017).
- 114.Phase III open label first line therapy study of MEDI 4736 (Durvalumab) with or without tremelimumab versus soc in non small‐cell lung cancer (NSCLC). (MYSTIC). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/show/NCT02453282 (accessed 13 October 2017).
- 115.Study of 1st line therapy study of durvalumab with tremelimumab versus SoC in non small‐cell lung cancer (NSCLC) (NEPTUNE). (NEPTUNE). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/show/NCT02542293 (accessed 13 October 2017).
- 116. Antonia SJ, Villegas A, Daniel D et al Durvalumab after chemoradiotherapy in stage III non‐small‐cell lung cancer. New Engl J Med 2017; 77:1919–29. [DOI] [PubMed] [Google Scholar]
- 117. Thompson RH, Kuntz SM, Leibovich BC et al Tumor B7‐H1 is associated with poor prognosis in renal cell carcinoma patients with long‐term follow‐up. Cancer Res 2006; 66:3381–5. [DOI] [PubMed] [Google Scholar]
- 118. Motzer RJ, Rini BI, McDermott DF et al Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol 2015; 33:1430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Motzer RJ, Escudier B, McDermott DF et al Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med 2015; 373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rosenberg JE, Hoffman‐Censits J, Powles T et al Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet 2016; 387:1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sharma P, Callahan MK, Bono P et al Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open‐label, two‐stage, multi‐arm, phase 1/2 trial. Lancet Oncol 2016; 17:1590–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Snyder A, Makarov V, Merghoub T et al Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014; 371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hugo W, Zaretsky JM, Sun L et al Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2016; 165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Le DT, Uram JN, Wang H et al PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015; 372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Domchek S, Bang Y‐J, Coukos K et al MEDIOLA: a phase I/II, open‐label trial of olaparib in combination with durvalumab (MEDI4736) in patients with advanced solid tumours. Ann Oncol 2016; 27:1103TiP. [Google Scholar]
- 126. Ansell SM, Lesokhin AM, Borrello I et al PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015; 374:311–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Armand P, Shipp MA, Ribrag V et al Programmed death‐1 blockade with pembrolizumab in patients with classical hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol 2016; 34:373–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chow LQ, Haddad R, Gupta S et al Antitumor activity of pembrolizumab in biomarker‐unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE‐012 expansion cohort. J Clin Oncol 2016; 34:3838–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ferris RL, Blumenschein G, Fayette J et al Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med 2016; 375:1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cohen EE, Harrington KJ, Le Tourneau C et al Pembrolizumab versus standard of care for recurrent or metastatic head and neck squamous cell carcinoma: phase 3 KEYNOTE‐040 trial. Ann Oncol 2017; 28: 628LBA45. 28028033 [Google Scholar]
- 131. Kaufman HL, Russell J, Hamid O et al Avelumab in patients with chemotherapy‐refractory metastatic Merkel cell carcinoma: a multicentre, single‐group, open‐label, phase 2 trial. Lancet Oncol 2016; 17:1374–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Liu W, MacDonald M, You J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr Opin Virol 2016; 20:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Nghiem PT, Bhatia S, Lipson EJ et al PD‐1 blockade with pembrolizumab in advanced Merkel‐cell carcinoma. N Engl J Med 2016; 374:2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Reck M, Luft A, Szczesna A et al Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive‐stage small‐cell lung cancer. J Clin Oncol 2016; 34:3740–48. [DOI] [PubMed] [Google Scholar]
- 135. Reck M, Bondarenko I, Luft A et al Ipilimumab in combination with paclitaxel and carboplatin as first‐line therapy in extensive‐disease‐small‐cell lung cancer: results from a randomized, double‐blind, multicenter phase 2 trial. Ann Oncol 2013; 24:75–83. [DOI] [PubMed] [Google Scholar]
- 136. Antonia SJ, López‐Martin JA, Bendell J et al Nivolumab alone and nivolumab plus ipilimumab in recurrent small‐cell lung cancer (CheckMate 032): a multicentre, open‐label, phase 1/2 trial. Lancet Oncol 2016; 17:88395. [DOI] [PubMed] [Google Scholar]
- 137. Ott PA, Fernandez ME, Hiret S et al Pembrolizumab (MK‐3475) in patients (pts) with extensive‐stage small cell lung cancer (SCLC): preliminary safety and efficacy results from KEYNOTE‐028. J Clin Oncol 2015; 33: abstr7502. [Google Scholar]
- 138. Kwon ED, Drake CG, Scher HI et al Ipilimumab versus placebo after radiotherapy in patients with metastatic castration‐resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184‐043): a multicentre, randomised, double‐blind, phase 3 trial. Lancet Oncol 2014; 15:700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wolchok JD, Hoos A, O'Day S et al Guidelines for the evaluation of immune therapy activity in solid tumors: immune‐related response criteria. Clin Cancer Res 2009; 15:7412–20. [DOI] [PubMed] [Google Scholar]
- 140. Phase 3 study of immunotherapy to treat advanced prostate cancer . ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT01057810 (accessed 14 April 2017).
- 141. Muro K, Chung HC, Shankaran V et al Pembrolizumab for patients with PD‐L1‐positive advanced gastric cancer (KEYNOTE‐012): a multicentre, open‐label, phase 1b trial. Lancet Oncol 2016; 17:717–26. [DOI] [PubMed] [Google Scholar]
- 142. Le DT, Bendell JC, Calvo E et al Safety and activity of nivolumab monotherapy in advanced and metastatic (A/M) gastric or gastroesophageal junction cancer (GC/GEC): results from the CheckMate‐032 study. J Clin Oncol 2016; 34:6. 26578608 [Google Scholar]
- 143. El‐Khoueiry AB, Melero I, Crocenzi TS et al Phase I/II safety and antitumour activity of nivolumab in patients with advanced hepatocellular carcinoma (HCC): CA209‐040. J Clin Oncol 2015; 33:LBA101. [Google Scholar]
- 144.ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/results?term=checkpoint+inhibitors&Search=Search (accessed 13 October 2017).
- 145. Twyman‐Saint Victor C, Rech AJ, Maity A et al Radiation and dual checkpoint blockade activate non‐redundant immune mechanisms in cancer. Nature 2015; 520:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Postow MA, Callahan MK, Barker CA et al Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012; 366:925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Schoenfeld JD, Mahadevan A, Floyd SR et al Ipilmumab and cranial radiation in metastatic melanoma patients: a case series and review. J Immunother Cancer 2015; 3:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Simone CB, Burri SH, Heinzerling JH. Novel radiotherapy approaches for lung cancer: combining radiation therapy with targeted and immunotherapies. Transl Lung Cancer Res 2015; 4:545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Palma DA, Louie AV, Rodrigues GB. New strategies in stereotactic radiotherapy for oligometastases. Clin Cancer Res 2015; 21:5198–204. [DOI] [PubMed] [Google Scholar]
- 150. Okwan‐Duodu D, Pollack BP, Lawson D, Khan MK. Role of radiation therapy as immune activator in the era of modern immunotherapy for metastatic malignant melanoma. Am J Clin Oncol 2015; 38:119–25. [DOI] [PubMed] [Google Scholar]
- 151. Immune checkpoint inhibition in combination with radiation therapy in pancreatic cancer patients (CheckPAC) . ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT02866383?term=pancreas+AND+ipilimumab&rank=5 (accessed 13 October 2017).
- 152. Combination Adenovirus + Pembrolizumab to Trigger Immune Virus Effects (CAPTIVE) . ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT02798406?term=checkpoint+inhibitors&rank=5 (accessed 20 August 2016).
- 153. Peng D, Kryczek I, Nagarsheth N et al Epigenetic silencing of TH1‐type chemokines shapes tumour immunity and immunotherapy. Nature 2015; 527:249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Steele CW, Karim SA, Leach JDG et al CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016; 29:832–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Arce Vargas F, Furness AJS, Solomon I et al Fc‐optimized anti‐CD25 depletes tumor‐infiltrating regulatory T cells and synergizes with PD‐1 blockade to eradicate established tumors. Immunity 2017; 46:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 348: 56–61. [DOI] [PubMed] [Google Scholar]
- 157. Zaretsky JM, Garcia‐Diaz A, Shin DS et al Mutations Associated with Acquired Resistance to PD‐1 Blockade in Melanoma. N Engl J Med 2016; 375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Selection Pressure and Evolution Induced by Immune Checkpoint Inhibitors and Other Immunologic Therapies (SPECIAL) . ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT02724488?term=checkpoint+inhibitors&rank=1 (accessed 20 August 2016).
- 159. Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD‐1/PD‐L1 immune checkpoint blockade therapy. J Hematol Oncol 2016; 9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Khalil D, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nature Rev Clin Oncol 2016; 13:273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]