

Summary
Defining how self‐antigens are perceived by the immune system is pivotal to understand how tolerance is maintained under homeostatic conditions. Clinically relevant, natural autoantigens targeted by autoantibodies, in e.g. systemic lupus erythematosus (SLE), commonly have an intrinsic ability to engage not only the B cell receptor (BCR), but also a co‐stimulatory pathway in B cells, such as the Toll‐like receptor (TLR)‐7 pathway. Here we developed a novel mouse model displaying inducible expression of a fluorescent epidermal neo‐autoantigen carrying an OT‐II T cell epitope, B cell antigen and associated ribonucleic acids capable of stimulating TLR‐7. The neo‐autoantigen was expressed in skin, but did not drain in intact form into draining lymph nodes, even after ultraviolet B (UVB)‐stimulated induction of apoptosis in the basal layer. Adoptively transferred autoreactive B cells were excluded follicularly and perished at the T–B border in the spleen, preventing their recirculation and encounter with antigen peripherally. This transitional check‐point was bypassed by crossing the reporter to a BCR knock‐in line on a C4‐deficient background. Adoptively transferred OT‐II T cells homed rapidly into cutaneous lymph nodes and up‐regulated CD69. Surprisingly, however, tolerance was not broken, as the T cells subsequently down‐regulated activation markers and contracted. Our results highlight how sequestration of intracellular and peripheral antigen, the transitional B cell tolerance check‐point and T cell regulation co‐operate to maintain immunological tolerance in vivo.
Keywords: autoimmunity, B cell, skin antigen, systemic lupus erythematosus, tolerance
Introduction
Autoimmunity ranks third on the list of most prevalent causes of morbidity and mortality in the Western world. Systemic lupus erythematosus (SLE) is considered a prototypical autoimmune disease. B cells are central players in SLE, both as drivers of disease through autoantibody production and as modulators of disease progression through the regulatory B cell (Breg) subset 1, 2. Pathogenic B cells are responsible for production of the hallmark anti‐nucleic acid autoantibodies which, in turn, lead to immune complex deposition and subsequently organ damage and, in severe cases, organ failure. The central role for B cells is supported by the therapeutic potential of B cell‐depleting treatments reported in recent years 1. Interestingly, this paradigm extends to several other diseases, traditionally not considered B cell‐driven. For example, B cell depletion and reprogramming via anti‐CD22 monoclonal antibody therapy was found to reverse diabetes in non‐obese diabetic (NOD) mice 3.
Understanding how self‐antigens are presented and perceived by the immune system and how tolerance is maintained under homeostatic conditions is pivotal to explain the disease processes underlying break of tolerance and development of autoimmune diseases, such as SLE. According to the danger theory, the context in which (auto)antigens are encountered directs the immunological outcome 4. Clinically relevant, natural autoantigens targeted by autoantibodies in SLE commonly have an intrinsic ability to engage not only the B cell receptor (BCR) but also a co‐stimulatory pathway in B cells, such as the Toll‐like receptor (TLR)‐7 5 or TLR‐9 pathways 6. It has been suggested that autoreactivity focuses first on self‐components which carry TLR ligands because, uniquely, these will be able to activate B cells independently of T cells, and subsequent T–B interactions then activate autoreactive T cells, resulting in chronic autoimmunity 7. TLR‐7 has been found to be particularly relevant in the context of break of tolerance 8.
The skin serves as a barrier to the external environment, and hosts a broad repertoire of immune cells, including Langerhans cells and several subsets of skin‐resident dendritic cells (DCs) 9. For this reason, the skin may be a central tissue in development of autoimmune responses. Application of bacterial products to intact skin can induce and augment organ‐specific autoimmune disease at distant anatomical sites 10. More recently it was found that chronic topical application of the TLR‐7 agonist R848 to skin could induce systemic disease resembling SLE 11. This raised the possibility that peripheral autoantigens in the context of a tonic danger signal can precipitate systemic autoimmunity.
Several transgenic mice expressing epidermal neo‐autoantigens have been generated previously to study T cell‐mediated immune responses to skin‐associated antigens. Despite differences in the promoter used to drive expression of the neo‐autoantigen [e.g. keratin 5 (KRT5) or keratin 14 (KRT14)] or in the structure of the self‐antigen expressed [e.g. membrane‐bound chicken ovalbumin (mOva) or Ova peptide (Ovap)], a number of these mouse models developed inflammatory skin disease upon adoptive transfer of naive Ova peptide‐specific CD8 (OT‐I, H‐2Kb restricted) T cells for KRT5‐mOva 12, KRT14‐mOva 13, KRT14‐Ovap 14, or when crossed to the OT‐I transgenic line for KRT14‐Ovap 15. Conversely, in another study using one of these models, DCs induced deletional tolerance 16 (KRT5‐mOva). This finding was supported by a study reporting that an additional inflammatory signal, in that case provided by tape stripping, was required for development of inflammatory skin disease 17 (KRT14‐mOva). Common to all these models was the focus on CD8 T cell‐mediated mechanisms (OT‐I restricted), the use of constitutive tissue‐specific promotors and ‘foreign’ (Ova) protein neo‐autoantigen.
In a different line of investigation, Kawahata and colleagues employed a nuclear localized version of Ova (nOva) expressed ubiquitously under a major histocompatibility complex (MHC) class I promotor and examined the response of adoptively transferred Ova‐specific DO11.10 CD4 T cells (equivalent to the H2b‐restricted CD4 OT‐II system, but for H2d instead) 18, 19. In that setting, low‐level antigen presentation by DCs was observed to induce tolerance in the cognate CD4 T cells. A more recent study employed a doxycycline‐inducible KRT5 promotor driving expression of a fusion protein linking the transferrin receptor transmembrane domain, green fluorescent protein (GFP) and amino acids 230–359 of Ova 20. This line was crossed onto the DO11.10 T cell receptor (TCR)‐transgenic background. Despite noticeable negative selection of DO11.10 T cells, doxycycline induction of transgene expression caused an acute inflammatory dermatitis. However, this dermatitis resolved spontaneously after prolonged antigen expression, a process associated with the emergence of antigen‐specific T regulatory cells 20.
Here we have generated mice harbouring a floxed‐stop‐cassette encoding Sjögren's syndrome B (SSB) protein (an autoantigen commonly targeted in SLE), coupled to blue fluorescent protein (BFP) through a linker including an OTII epitope (flox‐stop‐flox‐BFP‐OTII‐SSB). This line was crossed to a driver line in which the KRT14 promotor drives expression of Cre under the control of an engineered oestrogen receptor [KRT14‐Cre/oestrogen receptor (ERT)], which is unresponsive to endogenous oestrogen but activated by the synthetic molecular analogue tamoxifen. Thus, this set‐up allows tamoxifen‐induced expression of the neo‐autoantigen in the skin. We have demonstrated previously that SSB ribonuclear complexes are the antigenic targets of knock‐in B cells derived from the 564Igi mouse 21, a well‐characterized murine model of SLE generated by knock‐in of the heavy (H) and kappa light (K) chain of an autoreactive B cell clone 5. We have shown that ribonuclear complexes containing SSB are directly able to stimulate TLR‐7 and induce a type‐I interferon (IFN) response 22. In heterozygous 564Igi mice carrying a single copy of the knock‐in H and K chain, approximately 50% of circulating B cells express the knock‐in B cell receptor and are termed idiotype‐positive (Id+) cells. The remaining half of the naive repertoire is idiotype‐negative (Id–) due to expression of the endogenous IgH allele, receptor editing or allelic inclusion 5, 21, 23. The mice present with high titres of immunoglobulin (Ig)G directed towards nuclear‐associated antigens; however, 564Igi mice do not exhibit overt manifestations of disease until late in life. Combining the KRT14‐Cre/ERT BFP‐OTII‐SSB reporter with the 564Igi model and OTII system thus allows for testing responses of cognate B cells and cognate T cells in the setting of a TLR‐7‐agonistic nuclear antigen localized to the skin.
Materials and methods
Labelled antibodies and dyes
We used commercial antibodies from BioLegend (San Diego, CA, USA): phycoerythrin‐cyanin 7 (PE‐Cy7) anti‐mouse CD69, αB220‐peridinin chlorophyll (PerCP)‐Cy5.5, αB220‐allophycocyanin (APC), αIgMb‐fluorescein isothiocyanate (FITC), αIgMa‐PE, αCD45.2‐FITC, αCD45.2‐APC, αCD45.1‐FITC, αCD45.1‐PE, αIgD‐PB, αCD21/35 (7E9)‐PB, αCD38‐PE‐Cy7, APC anti‐mouse/human CD207 (Langerin), APC anti‐mouse Ly‐6G, PE anti‐mouse CD3ɛ, PE anti‐mouse CD169, PE anti‐mouse IgM, PE‐Cy7 anti‐mouse CD11c, and 7‐aminoactinomycin D (7AAD) viability staining solution; commercial antibodies from eBioscience (San Diego, CA, USA): CD95 (APO‐1/Fas)‐PE (clone 15A7), anti‐mouse/rat Ki‐67 eFluor 660 (clone SolA15) and viability dye fixable live/dead stain eFluor 780; commercial antibodies from Thermo Fisher Scientific (Waltham, MA, USA): rabbit anti‐goat Alexa 488; polyclonal rabbit anti‐Tag red fluorescent protein (RFP) (cross‐reactive with TagBFP) from Evrogen (Richmond, VA, USA); polyclonal rabbit anti‐cleaved caspase‐3 (Asp175) from Cell Signaling Technology (Danvers, MA, USA); Alexa Fluor 568 phalloidin from Life Technologies; propidium iodide from Sigma (St Louis, MO, USA); dextran rhodamine B 70,000 MW neutral from Molecular Probes (Eugene, OR, USA); in‐house‐generated anti‐idiotype antibody, clone 9D11, conjugated to Alexa Fluor 647 or Alexa Fluor 568 from Life Technologies (Camarillo, CA, USA); and in‐house‐generated Lyve‐1 (mab2125), conjugated to Alexa Fluor 405 from Life Technologies.
Mice
All animal experiments were carried out in agreement with the institutional guidelines at Harvard Medical School, following approval of ethical protocols by the local Institutional Animal Care and Use Committee (protocol numbers IS00000095 and IS00000111) and per applicable laws and regulations. C57Bl/6J and OT‐II CD45.1 mice were purchased from Jackson Laboratories (Cambridge, Bar Harbor, MA, USA) and maintained under specific pathogen‐free (SPF) conditions in the animal facility at Harvard Medical School. 564Igi mice were kindly provided by T. Immanishi‐Kari and were similarly maintained locally. Tg(KRT14‐cre/ERT)20Efu/J mice were purchased from Jackson Laboratories and crossed to C57Bl/6J for several generations, before being crossed to BFP‐OTII‐SSB reporters (see below) and back‐crossed to generate KRT14‐Cre+/– BFP‐OTII‐SSB+/+ male breeders, which were mated to C57Bl/6J to generate Cre+ and Cre– BFP‐OTII‐SSB+/– littermates used in experiments. A similar breeding set‐up was used for crosses to 564 H+/+K+/+ to generate Cre+ and Cre– BFP‐OTII‐SSB+/– 564 H+/–K+/– littermates. Furthermore, KRT14‐Cre BFP‐OTII‐SSB reporters were crossed to the C4–/– background, and then set up in a similar cross to C4–/– mice to generate Cre+ and Cre– BFP‐OTII‐SSB+/– 564 H+/–K+/– C4–/– littermates.
Generation of BFP‐OTII‐SSB transgenic reporter line
The cDNA sequence encoding BFP‐OTII‐SSB targeting construct was assembled in Topo cloning vector and then cloned into the ai6 targeting vector 24. The targeting vector includes 5' and 3' flanking sequences homologous with the Rosa26 locus, and expression of the fusion protein is driven by the cytomegalovirus early enhancer/chicken beta actin/rabbit beta‐globin splice acceptor (CAG) promoter. To regulate expression, a STOP cassette flanked by lox P sites is included. The recombinant vector was confirmed by restriction endonuclease mapping and Sanger sequencing of the cDNA. Embryonic stem (ES) cells on a C57Bl/6 background (BRUCE 4 ES cells) 25 were transfected with the ai6 BFP‐OTII‐SSB targeting vector and then plated on feeder cells in neo‐selection medium. Surviving colonies were picked and triplicates prepared (one master and two for screening) and expanded on 96‐well plates, as described previously 26. Genomic DNA was extracted from surviving cells and digested with HindIII restriction enzyme and separated on a 0·7% agarose gel. Subsequently, DNA was transferred to membrane and hybridized with a 1 kb genomic DNA probe representing intron 1 of the Rosa26 locus 24. The expected wild‐type (WT) band is 4·5 and mutant targeted allele 5·6 Kb. Approximately 25% of screened clones were positive for the mutant band. Selected ES cells were transfected subsequently with a PhiC31‐expressing plasmid to delete the phosphoglycerate kinase I (PGK) neo‐marker using the flanking pair of PhiC31 recognition sites, AttB/AttP 24. Knock‐in mice were prepared as described 26. Briefly, embryos microinjected with mutant ES cells were transferred into pseudo‐pregnant females and germline transmission was confirmed by polymerase chain reaction (PCR) of tail DNA from the resulting offspring, which were subsequently bred to homozygosity.
Tamoxifen induction
Mice, 5–8 weeks old, were treated with tamoxifen to recombine the knock‐in cassette and thereby turn on transgene expression. Tamoxifen (Sigma) was dissolved in sunflower oil to a concentration of 10 mg/ml, and 100 µl was injected intraperitoneally (i.p.) on each of four consecutive days, for a cumulative dose of 4 mg tamoxifen.
Genotyping and fluorescence activated cell sorter (FACS) typing
Genotyping was performed on tail‐snips using appropriate primers and reaction conditions. For FACS typing, mice were bled retro‐orbitally using heparinized capillary tubes; approximately 60–100 µl blood was drawn into Eppendorf tubes containing 30 µl acid–citrate–dextrose solution. Following collection, the stabilized blood was briefly spun down, then underlayered with 1 ml of lymphocyte separation medium and spun for 25 min, 400 g at room temperature (RT). The mononuclear cell layer was aspirated and transferred into 1 ml ice‐cold FACS buffer [PBS (phosphate‐buffered saline), 2% FCS, 1 mM ethylenediamine tetraacetic acid (EDTA)], mixed, then pelleted at 200 g for 5 min. Cells were resuspended in FACS buffer and processed for flow cytometric analysis as described further below. FACS typing of 564Igi mice was performed using B220, anti‐IgMa, anti‐IgMb and 9D11.
UVB irradiation protocol
A controlled ultraviolet B (UVB) irradiation protocol was established for localized irradiation of the ear skin. To generate a UV head‐shield, the lower quarter of a 50‐ml conical plastic tube was cut off, and from this piece the tip of the conical bottom was removed to allow breathing. Two slits were cut out on the curved surface, positioned approximately 70 degrees apart, to allow protrusion of the ears, and the device was wrapped in tinfoil to block light from the eyes and the rest of the head. Mice were weighed and anaesthetized by i.p. injection with xylazine–ketamine (90 : 10 mg/kg), then placed with their heads inside the UV head‐shield, and either ear to be irradiated was pulled gently through the appropriate slit to allow exposure. A UVP model UVM‐57 hand‐held UV lamp (6 watts, 302 nm, cat. no. 95–0104‐01) was bridged over the heads of the mice on two glove‐boxes, and a general tool UV513AB, digital UVAB meter, with a metering range of 290–370 nm, calibration point set at 365 nm, with sensitivity of 1 µW/cm2–40 mW/cm2, was placed in the field to allow continuous monitoring and measurement of cumulative dose at the level of the ears. In preliminary experiments a range of doses were tested to arrive at an appropriate dose of 1 J/cm2 (∼two to four times the minimal erythema dose) for induction of skin erythema and robust inflammation both locally and in draining lymph nodes.
Adoptive transfers
Donor mice were euthanized by isofluorane overdose followed by cervical dislocation, and spleens were harvested into ice‐cold digestion buffer [RPMI‐1640 supplemented with 100 µg/ml DNAseI and 50 µg/ml Liberase DH from Roche (Basel, Switzerland)], and dissociated mechanically with forceps. Spleens were then incubated at 37°C for 45 min to 1 h, and passed subsequently through a 70‐µm cell strainer, followed by centrifugation at 200 g for 5 min. The supernatant was decanted and the pellet resuspended in red blood cell (RBC) lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0·1 mM EDTA), incubated for 2–3 min then spun down as before, washed with PBS and resuspended in FACS buffer containing a cocktail of appropriate antibodies. For untouched B cell purification, the following biotinylated antibodies from BD Biosciences was used: CD11b, CD11c, CD69, CD3ɛ, TCRβ and Ter119. For untouched purification of OT‐II T cells, the following biotinylated antibodies were used: CD11b, CD11c, B220, CD8α, CD14, Gr1, NK1.1, MHC class II (IA/IE) and Ter119. Cells were incubated with staining mix on ice for 30 min, then washed, and added to streptavidin magnetic beads (Pierce, Rockford, IL, USA) in FACS buffer. After another 15‐min incubation on ice, cells were again washed, then resuspended in FACS buffer and loaded on a prewetted magnetic activated cell sorting (MACS) LD column. The flow‐through containing untouched purified cells was collected. Cells were either transferred immediately to recipients or labelled by incubation with 5 µM CFDA‐SE [carboxyfluorescein succinimidyl ester (CFSE)] in PBS for 10 min at 37°C, washed, then transferred. Recipients were anaesthetized with isofluorane and cell suspensions were injected intravenously (i.v.) through the retro‐orbital sinus.
Flow cytometry
Mice were euthanized by isoflurane overdose followed by cervical dislocation. Ears were cut off, placed in an Eppendorf tube containing digestion buffer, cut into small pieces, then incubated for ∼45 min at 37°C. After incubation, the tissue was gently dissociated with a pestle, passed through a 70‐µm cell strainer and the cells were recovered by centrifugation. Spleens and lymph nodes (LNs) were harvested into ice‐cold FACS buffer and dissociated mechanically using pestles in 1·5 ml Eppendorf tubes. Samples were filtered through 70‐µm cell strainers (Corning, New York, NY, USA) and spun down at ∼200 g for 5 min. Ear and LN samples were resuspended in FACS buffer. Spleen samples were resuspended in RBC lysis buffer, incubated for 2–3 min then spun down as before and resuspended in FACS buffer. Samples were added to wells of 96‐well round‐bottomed plates, spun down and resuspended in 100 µl staining mix (appropriate antibody and viability dye cocktail in FACS buffer). Staining was performed for 30 min on ice, followed by addition of 150 µl wash buffer. Plates were centrifuged subsequently 200 g for 5 min, and supernatants flicked out of the plates. For two‐step staining procedures secondary staining mix was added (appropriate secondary antibody mix and viability dye in FACS buffer), and the process repeated. Following the last wash, samples were resuspended in 200 µl FACS buffer and transferred to FACS tubes. Flow cytometric analyses were performed on one of three different instruments (BD Biosciences), depending on the experiment: a four‐colour, six‐parameter FACSCalibur equipped with 488 nm and 633 nm lasers; a standard three lasers‐configuration (405 nm, 488 nm and 633 nm) FASCSCanto2, with eight‐colour and 10‐parameter analytical capabilities; and a five‐laser, 20‐parameter FACSAria 2 SORP (lasers: 355 nm, 488 nm, 633 nm, 405 nm, 594 nm) equipped with a photomultiplier (PMT) option for forward scatter (FSC), enhanced optics and digital focusing and a 300 mW 488 nm laser from Coherent (Santa Clara, CA, USA).
Confocal imaging
Lymph nodes were cleaned of fat and placed in freshly thawed 4% paraformaldehyde (PFA) in PBS on ice for 4 h, then rinsed three times in PBS and transferred to 30% sucrose until sinking to the bottom of the tube. The nodes were then placed in optimum cutting temperature (OCT) (TissueTek/Sakura Finetech USA Inc., Torrance, CA, USA) and frozen at −80°C. Freshly harvested ears or spleens were embedded in OCT and frozen immediately at −80°C. Tissue blocks were equilibrated at cutting temperature (–16 to −20°C depending on tissue type) and 10‐µm thick sections were cut on a cryostat. Tissue sections were mounted on SuperFrost+ slides (Fisher Scientific) and fixed with freshly thawed 4% PFA in PBS for 5–10 min. PFA‐fixed slides were incubated with Tris‐buffered saline (TBS) to block residual primary amine reactivity. Slides were then rinsed with PBS and incubated with block/perm buffer (PBS, 2% FBS, 0·1% NaN3 and 0·1% Triton‐X100) for 30 min. This was followed by incubation with primary antibody mixture in staining buffer (PBS, 2% FBS, 0·1% NaN3) overnight at 4°C. For two‐step staining procedures, the slides were washed three times with PBS, 0·01% Tween‐20, then overlayed with secondary antibody mixture in staining buffer and incubated for 2 h at RT. At the end of either one‐ or two‐step staining procedures, slides were washed once with staining buffer for 5 min, then three times for 5 min with PBS, 0·01% Tween‐20. Slides were spot‐dried, then mounted in Fluoro‐Gel (Electron Microscopy Sciences, Hatfield, PA, USA) and coverslipped. Imaging was performed using a Fluoview FV1000 inverted Olympus IX 81 confocal microscope, equipped with six laser lines (405, 457, 488, 515, 559, 635 nms) and four fluorescence + 1 transmission detector (PMTs).
Image analysis
Images were quantified manually using ImageJ 27 or automatically using CellProfiler 28. In CellProfiler, appropriate masks were generated based on signals and the mean pixel intensity was measured within each mask (see Supporting information, Fig. S2 for example).
Two‐photon imaging
For two‐photon imaging in live animals, mice were anaesthetized using an isoflurane vaporizer with 1·5–2% isoflurane for the induction phase, followed by 0·5–1% for maintenance. Adequate surgical plane of anaesthesia was verified frequently throughout the procedure by testing the pedal withdrawal reflex. Rhodamine‐labelled dextran (MW 70 kDa) was injected i.v. 10 min before imaging to label blood vessels. Imaging was performed on an upright Olympus FV1200 MPE multiphoton system microscope fitted with a 20X 0.95NA Plan water‐immersion objective, a MaiTai HP DeepSee Ti‐Sapphire laser (Spectraphysics, Santa Clara, CA, USA) and four non‐descanned detectors (two GaAsP and two regular PMTs). Fluorescence emission was collected in two channels, using 420–460 nm and 575–630 nm filters for BFP and rhodamine detection, respectively.
Statistical analyses
All statistical analyses were performed using GraphPad Prism version 6. Statistical analyses as indicated throughout the text and figure legends; P < 0·05 between groups was considered significant.
Results
Generation of a novel RNA‐associated autoantigen transgenic reporter line
To mimic experimentally the presentation of TLR‐agonistic autoantigen in vivo, mice were generated, which harboured a floxed‐stop‐cassette encoding SSB protein coupled to BFP through a linker including an OTII epitope, i.e. flox‐stop‐flox‐BFP‐OTII‐SSB, knocked into the ROSA locus (Fig. 1). This was performed by first assembling the cDNA sequence encoding mTagBFP 29, the sequence coding for the OTII peptide 18 and the cDNA sequence for murine SSB. This construct was subsequently cloned into the Ai6 targeting vector 24, placing it immediately downstream of the strong, constitutive CAG promotor and a loxP‐flanked stop cassette allowing Cre‐regulated control of expression, and upstream of the woodchuck hepatitis virus post‐transcriptional regulatory element (WPRE) and the bovine growth hormone polyadenylation signal (BGH‐pA), stabilizing the transcript (Fig. 1a). The Ai6 vector additionally included an AttB/AttP‐flanked neo‐cassette allowing selection and subsequent removal, and homology arms targeting the Rosa26 locus, a well‐characterized, ubiquitously expressed locus, which permits high‐level expression from an exogenous promoter. Embryonic stem (ES) cells on a C57Bl/6 background were transfected with the Ai6 BFP‐OTII‐SSB targeting vector, selected for neomycin resistance, and HindIII digested genomic DNA extracted from resulting selectants was analysed by Southern blot (Fig. 1b). Selected ES cells were then transfected with PhiC31‐expressing plasmid to remove the PGK neo‐cassette via site‐specific recombination of the flanking AttB/AttP sites. Embryos were microinjected with mutant ES cells and transferred into pseudo‐pregnant females. Germline transmission was confirmed by PCR analysis of tail DNA from the resulting offspring (Fig. 1c), which were subsequently bred to homozygosity.
Figure 1.
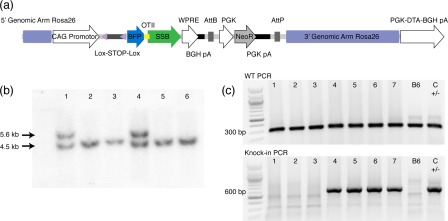
Generation of blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) reporter. (a) Schematic overview of the targeting vector. A CAG promotor is situated upstream of a floxed stop region, followed by the open reading frame for BFP, the OTII peptide sequence and SSB protein. This is followed by a woodchuck hepatitis virus post‐transcriptional regulatory element (WPRE) and a bovine growth hormone polyadenylation signal (BGH‐pA), and finally a phosphoglycerate kinase promotor (PGK) driving expression of the neomycin selection marker. The full cassette is flanked by targeting arms for stable integration into the Rosa26 locus, and a Diphtheria toxin A fragment expression cassette (PGK‐DTA‐BGH pA) drives negative selection of random integration events. (b) Southern blot of HindIII digested genomic DNA extracted from transfected BRUCE 4 embryonic stem (ES) cells after neomycin selection. Results for six different ES clones, labelled 1–6, are shown. Expected wild‐type band is 4·5 kb, whereas the mutant targeted allele is 5·6 kb. As can be seen, two of six ES clones harbour the knock‐in. (c) Example of genotyping results for the knock‐in line. Top gel shows polymerase chain reaction (PCR) for wild‐type, for seven unknown tail DNA samples (1–7), a B6 control and a heterozygous knock‐in control. Bottom gel shows PCR for knock‐in allele for the same samples.
Tamoxifen‐dependent expression of the RNA‐associated autoantigen reporter in keratinized skin
The novel transgenic line was crossed to a KRT14‐Cre/ERT driver line 30, allowing tamoxifen‐regulated expression of Cre localized to the skin, directing specific recombination and subsequent expression of the transgene in the skin (Fig. 2a). As expected, expression of the BFP reporter was tamoxifen‐dependent (Fig. 2b,c), although a rare, sporadic BFP‐positive cell was observed occasionally in untreated mice (Fig. 2b, left, bottom image). The BFP signal localized to nuclei, and more specifically to nucleoli (Fig. 2d), and was restricted to cells in the basal layer of the skin, including those associated with hair follicles (Fig. 2e and Supporting information, Fig. S1). Taken together, this validated the expression pattern of the KRT14 promotor, the stringency of the ERT regulator and the functional integrity of the SSB component allowing the correct subcellular localization. Expression of the BFP‐OTII‐SSB transgene did not give rise to any observable phenotype within 4 weeks of tamoxifen induction.
Figure 2.
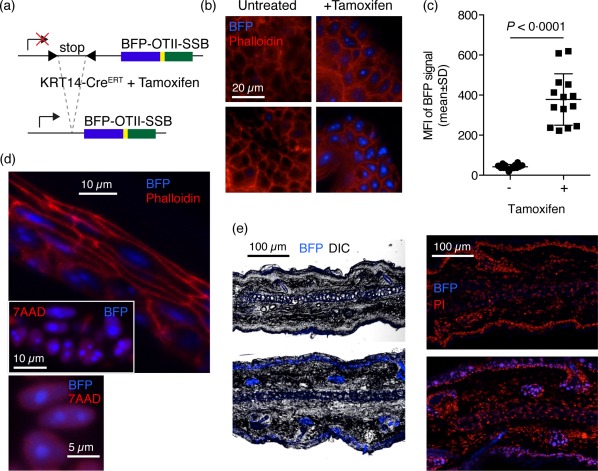
Tamoxifen dependence of blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) expression, subcellular and gross histological localization. (a) Schematic overview of the knock‐in BFP‐OTII‐SSB Rosa locus and the tamoxifen‐induced Cre recombination event permitting its expression. Tamoxifen‐controlled nuclear translocation of Cre expressed under the KRT14 promotor allows Cre‐mediated excision of the stop cassette blocking expression of BFP‐OTII‐SSB fusion protein. (b) Representative images of ear skin either untreated (left panels) or after tamoxifen treatment regimen (right panels). Blue = BFP; red = phalloidin‐A568 counterstain. Scale bar indicated. The two images for the untreated sample are representative of either end of the spectrum observed, i.e. no background (left, top image) and the highest level of observed background (left, bottom image). (c) Quantification of mean fluorescence intensity (MFI) in ear skin of untreated (–) versus tamoxifen‐treated (+) animals. Total image MFI was measured in ImageJ. Individual data points and mean ± standard deviation (s.d.) for two slices per stack for three to four stacks per animal, for two mice in each group, from two independent experiments. Statistical significance given for two‐tailed unpaired Mann–Whitney test. (d) Subcellular localization of BFP signal. Top: transection of epidermis, showing nuclear localization of BFP, counterstained with phalloidin in red. Inset: high magnification view of nuclear and nucleolar localized BFP in the epidermis, with nuclear counterstain [7‐aminoactinomycin D (7AAD)] in red. Bottom: ultra‐high magnification image showing defined nucleolar and diffuse nuclear localization of BFP in relation to nuclear counterstain (7AAD) in red. (e) Left: low magnification overview of BFP signal in transection of the ear, with overlay of the differential interference contrast (DIC) channel for gross morphology. Right: low magnification overview of BFP signal in transection of the ear, with overlay of propidium iodide (PI) for nuclear counterstain. Top panels are from Cre– mice, bottom panels are from Cre+ mice.
UVB‐induced apoptosis in the basal layer inducing local and draining lymph node inflammation
Autoantigens targeted by autoantibodies in SLE have been found to localize to the surface of apoptotic keratinocytes 31. To promote release of labelled autoantigen in vivo, simulating that observed in SLE patients following sun exposure, we developed a UVB‐irradiation protocol (302 nm, 1 J/cm2) allowing timed induction of apoptosis in the basal layer of the ear skin, as evidenced by cleaved caspase 3 positivity (Fig. 3a). The auricular nodes, which are the proximal downstream nodes draining the ear, were found to be inflamed both macro‐ and microscopically, as evidenced by gross swelling and erythema at day 1 post‐irradiation and histologically by influx of neutrophils, defined as Ly6G+ cells, into the medullary areas, expansion of the lymphatic endothelium defined by Lyve‐1, and induction of medullary B cell clusters (Fig. 3a). Based on image quantification using CellProfiler, there was a marked and statistically significant increase in cleaved caspase 3 staining in the basal layer at day 1 following UVB irradiation (Fig. 3b and Supporting information, Fig. S2). A smaller, but significant, decrease in BFP signal was similarly observed (Fig. 3c). We speculate that this could be due either to low‐grade photobleaching or apoptosis‐associated degradation of BFP protein. Concomitant with the increase in cleaved caspase 3 signal, a significant increase in Ly6G expressing cells was observed (Fig. 3d), suggestive of inflammation‐associated neutrophil infiltration 32. Finally, a significant decrease in CD207+ cells was observed (Fig. 3e), in support of inflammation‐associated egress of Langerhans cells 33. Taken together, these physiological responses indicated the potential for increased immune surveillance of the neo‐autoantigen.
Figure 3.
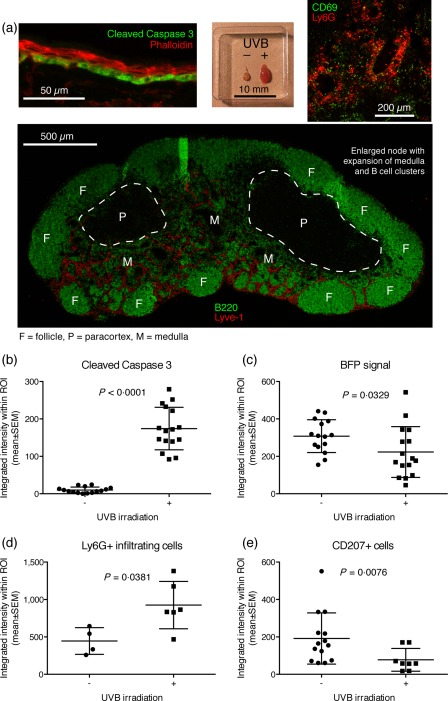
Establishment of ultraviolet B (UVB) irradiation protocol. (a) A UVB irradiation protocol was established allowing timing of induction of apoptosis in the basal layer of ear skin, as evidenced by cleaved caspase 3 positivity in the basal layer at day 1 post‐irradiation (top, left panel), gross enlargement of the draining auricular lymph node compared to the contralateral control (top, middle panel), influx of Ly6G+ neutrophils into the medulla of the node (top, right panel), expansion of the Lyve‐1+ lymphatic network and induction of B cell clusters in the medulla (bottom panel). Images are representative of observations in more than five mice. (b) Quantification of cleaved caspase 3 signal in the basal layer of ear skin. (c) Quantification of BFP signal in the basal layer. For b and c, n = 15 measurements from five mice for the non‐irradiated group and n = 16 measurements from five mice for the irradiated group, pooled from two independent experiments. (d) Quantification of Ly6G+ infiltrating cells in the ear, measured in a subset of the samples presented in b and c. (e) Quantification of CD207+ Langerhans cells in the ear, for a subset of the samples presented in b and c. In b–e, lines represent mean ± standard error of the mean (s.e.m.). The exact P‐values for two‐tailed Mann–Whitney comparison of non‐irradiated and irradiated groups are given for each of the parameters in their respective graph.
Adoptively transferred OT‐II cells home to draining lymph nodes and are activated, but B cells remain follicularly excluded in the spleen
An experiment was designed to mimic in vivo a situation in which naive autoreactive B cells emerge and enter circulation, neo‐autoantigen carrying TLR‐7 agonist is released in peripheral tissues, and cognate T cells are available. This was performed by turning on BFP‐OTII‐SSB expression in the skin, UVB irradiating the ears then co‐transferring OT‐II T cells and 564Igi B cells adoptively (Fig. 4a). Mice that did not receive lymphocytes served as baseline controls. No BFP signal could be detected in draining lymph nodes, neither directly nor by development with a polyclonal antibody directed against the BFP protein (αTagRFP), which was otherwise able to detect BFP in the basal layer of the skin (Supporting information, Fig. S3). However, adoptively transferred OT‐II T cells homed to the auricular lymph nodes (Fig. 4b) [statistically significant higher level of OT‐IIs in AuLN compared to spleen, across days 1 and 3, by regular analysis of variance (anova) with Sidak's post‐test] and up‐regulated CD69 (Fig. 4c), suggesting that they were responding to cognate antigen. Conversely, the adoptively transferred 564Igi B cells were largely follicularly excluded in the spleen (Fig. 4d,e), as has been described previously for B cells which become antigen‐engaged when entering circulation, but fail to receive cognate T help 34, 35. Follicularly excluded B cells have been reported to perish at the T–B border within 2–3 days of their exclusion, if they do not receive T cell help 36. At day 3 following adoptive transfer, OT‐II T cells in the auricular nodes down‐regulated CD69 (Fig. 4c) (statistically significant lower CD69 at day 3 compared to day 1, by regular two‐way anova with Sidak's post‐test), while the number of 564Igi B cells in the spleen decreased (Fig. 4d) (statistically significant lower number of CFSE 564Igi B cells in spleen on day 3 compared to day 1, by regular anova with Sidak's post‐test). Of note, the CFSE+ cells remaining in spleen and auricular lymph nodes by day 3 were predominantly idiotype‐negative, indicating that cells persisting in the spleen or making it into the periphery were probably derived from a minor contaminant of non‐564Igi cells in the adoptive transfer (Supporting information, Fig. S4). Thus, 564Igi cells did not break follicular exclusion, and no productive response was induced.
Figure 4.
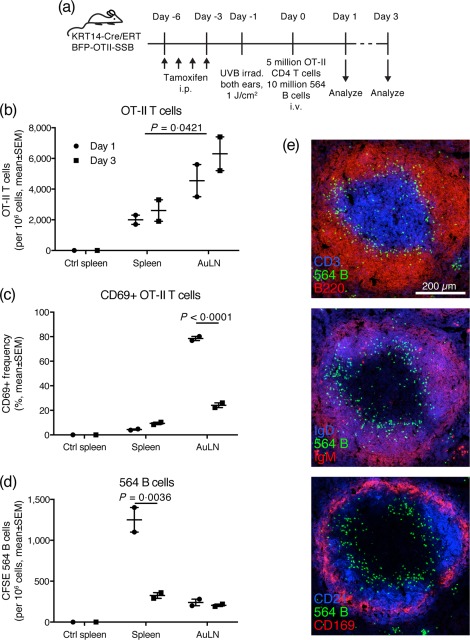
Adoptively transferred OT‐II T cells home to skin‐draining lymph nodes and up‐regulate CD69, whereas the bulk of adoptively transferred 564Igi B cells are follicularly excluded in spleen and eliminated. (a) Overview of experimental layout. Mice were used that carry a KRT14‐driven Cre under the control of a mutated form of the ligand‐binding domain of the oestrogen receptor that only binds tamoxifen (ERT), and a floxed‐stop expression cassette for a fusion construct of blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB). Expression of BFP‐OTII‐SSB was turned on in the skin by intraperitoneal (i.p.) administration of tamoxifen (days −6 to −3), followed by ultraviolet B (UVB) irradiation of both ears to induce apoptosis in the basal layer (day −1). Subsequently, 5 × 106 OT‐II CD4 T cells and 1 × 107 564Igi B cells were transferred adoptively into the mice by intravenous (i.v.) injection (day 0). Spleen and auricular lymph nodes were analysed on day 1 or day 3. (b) Adoptively transferred OT‐II cells home preferentially to skin‐draining lymph nodes. (c) Adoptively transferred OT‐II cells up‐regulate CD69 in skin‐draining lymph nodes at day 1, then down‐regulate it again by day 3. (d) On day 1, adoptively transferred 564Igi B cells remain mainly in spleen due to follicular exclusion, and few escape to peripheral skin‐draining nodes. By day 3, the majority of follicularly excluded 564Igi B cells have perished. For b–d, mean ± standard error of the mean (s.e.m.) is indicated. For control spleen, n = 1, whereas for spleen and AuLN, n = 2, with each point representative of two mice (samples pooled 2 and 2), i.e. n = 4 per time‐point for a total number of eight animals. P‐values given for regular two‐way analysis of variance (anova) with Sidak's post‐test. Representative of two experiments with similar results. (e) Serial sections from a representative spleen sample illustrating follicular exclusion of adoptively transferred carboxyfluorescein succinimidyl ester (CFSE)‐labelled 564Igi B cells, as demonstrated by counterstaining for: CD3 in blue and B220 in red (top); immunoglobulin (Ig)D in blue and IgM in red (middle); CD21 in blue and CD169 in red (bottom).
Constitutive expression of the RNA‐associated neo‐autoantigen does not induce hallmarks of immune activation
The question remained whether constitutive, rather than acute, presentation of the TLR‐agonistic neo‐autoantigen over time would cause a breakdown in tolerance. Therefore, we examined whether there was any indication of an inflammatory response or induction of adaptive response after long‐term expression of BFP‐SSB. To allow presence of 564Igi B cells, the transgenic reporter was crossed to the 564Igi model, resulting in KRT14‐Cre/ERT BFP‐OTII‐SSB 564 H+/– K+/– mice, and mice were examined at 2 and 12 weeks post‐tamoxifen induction (Fig. 5a). BFP expression was robust and consistent between the two time‐points, as ordinary two‐way anova indicated that Cre status was the only significant factor (P = 0·0015), and there was no significant difference in the level of BFP expression in Cre+ animals at 2 and 12 weeks post‐tamoxifen induction, as determined by Sidak's post‐test (Fig. 5b). Locally in the ear skin, a slight increase in Ly6G+ (Fig. 5c) and CD11c+ (Fig. 5d) cells was observed in Cre+ mice compared to Cre– mice at the 12‐week time‐point. However, the only statistically significant difference (ordinary two‐way anova with Sidak's post‐test, P = 0·0006) was an elevated frequency of Ly6G+ cells in the spleens of Cre+ versus Cre– mice (Fig. 5c), potentially indicating increased systemic inflammation. However, the overall frequencies of B cells (Fig. 5e) and germinal centre B cells (Fig. 5f) remained comparable between the two groups. There were no overt signs of inflammation in the skin.
Figure 5.
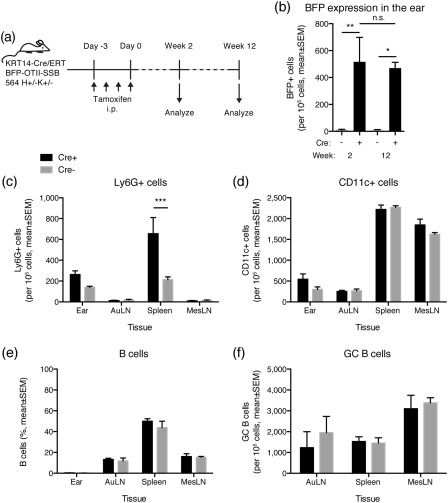
Long‐term consequences of blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) expression. (a) Overview of experimental layout. 564Igi mice were crossed with mice that carry a KRT14‐driven Cre under the control of a mutated form of the ligand‐binding domain of the oestrogen receptor that only binds tamoxifen (ERT), and a floxed‐stop expression cassette for a fusion construct of BFP‐OTII‐SSB, and Cre– and Cre+ littermates were used. Expression of BFP‐OTII‐SSB was turned on in the skin by intraperitoneal (i.p.) administration of tamoxifen (days −3 to 0). The ear skin, auricular lymph nodes, spleen and mesenteric lymph nodes were analysed at 2 or 12 weeks. (b) BFP expression in ears of KRT14 Cre– (grey bars) versus Cre+ (black bars) BFP‐OTII‐SSB 564 H+/– K+/– mice at 2 and 12 weeks after tamoxifen application. Mean ± standard error of the mean (s.e.m.) is shown. For week 2 measurements, n = 6 in each group, for week 12 measurements, n = 4 for Cre+ and n = 3 for Cre–. Cre+ and Cre– were compared by two‐way analysis of variance (anova) with Holm–Sidak's post‐test (*P < 0·05; **P < 0·01). There was no statistically significant difference between the time‐points for either Cre– or Cre+, as assessed by two‐way anova with Holm–Sidak's post‐test (n.s. = not significant). (c) Frequency of Ly6G‐positive cells in ear, auricular lymph nodes, spleen and mesenteric lymph nodes of Cre+ (black bars, mean ± s.e.m. of n = 4 mice) versus Cre– (grey bars, mean ± s.e.m. of n = 3 mice) at 12 weeks post‐tamoxifen treatment. Statistical significance of differences between Cre+ and Cre– assessed by two‐way anova with Sidak's multiple comparisons test (***P < 0·001). (d) As c, but showing CD11c+ cell frequencies. (e) As c, but showing B cell frequencies. (f) As c, but showing GC B cell frequencies.
Tolerance is maintained acutely in the presence of RNA‐associated autoantigen and cognate T and B cells on a permissive C4‐deficient background
Even in 564Igi knock‐in mice with chronic output of autoreactive B cells, the vast majority of emerging B cells are eliminated centrally and at the transitional stage 5. We have demonstrated previously that the transitional check‐point breaks down in the absence of C4 21. Therefore, we asked whether the combination of our neo‐autoantigen reporter, constitutive output of autoreactive 564Igi B cells on a permissive background (C4–/–), and the availability of cognate OT‐II T cells would result in overt break of tolerance. To test this, the KRT14/Cre‐ERT BFP‐OTII‐SSB reporter was crossed to the C4–/– background, and subsequently to 564 H+K+C4–/– mice, generating a quadruple knock‐in and single knock‐out transgenic line. Expression of the BFP‐OTII‐SSB transgene was turned on by tamoxifen administration, and purified naive OT‐II T cells were subsequently transferred in, and the ears of the mice were UVB‐irradiated to stimulate release of autoantigen to the downstream auricular nodes (Fig. 6a). Stable expression of the transgene was confirmed during the time–course of the experiment, as two‐way anova with Sidak's post‐test revealed no significant differences in the level of BFP expression of Cre+ mice between different time‐points (Fig. 6b). In both Cre+ and Cre– littermate controls, an approximately even distribution of adoptively transferred OT‐II cells was seen between spleen, auricular, inguinal and mesenteric lymph nodes at day 1 post‐transfer (Fig. 6c). Two‐way anova with Dunnet's post‐test, comparing each subsequent time‐point to day 1 within each tissue revealed a statistically significant increase in OT‐II cell presence in the auricular and inguinal lymph nodes of Cre+ mice, but not Cre– mice. This could be caused either by redistribution to or preferential expansion in the auricular and inguinal nodes. The similar increase in auricular (downstream, draining irradiated ear) and inguinal (distal, not draining the irradiated ear) lymph nodes indicated that the homing to and/or expansion in these cutaneous draining sites was a direct consequence of constitutive neo‐autoantigen expression, independent of UVB irradiation. By day 8, an apparent contraction occurred. No redistribution or expansion of OT‐II cells was seen in Cre– littermate controls during the time–course of the experiment (Fig. 6c). Already on day 1, a large fraction of the OT‐II T cells localizing to the auricular and inguinal lymph nodes of Cre+ mice were CD69+ (Fig. 6d). This fraction decreased by day 3, and further by day 8, indicating the back end of a response initiated before the day 1 time‐point. In Cre– littermate controls, no CD69+ OT‐II T cells were observed in any of the tissues examined (Fig. 6d), demonstrating that CD69 up‐regulation was a specific induced response in the Cre+ mice, not a pre‐existing phenomenon or an artefact of the purification or transfer. Absolute, non‐normalized frequencies of OT‐II T cells followed the same pattern as when normalizing to the spleen (Fig. 6e). Based on the sum of those frequencies, no change occurred in Cre– mice, whereas in Cre+ mice there may have been a small but statistically non‐significant expansion, followed by a marked and statistically significant contraction by day 8. An early expansion of OT‐II T cells in skin‐draining lymph nodes was supported by imaging data demonstrating massive presence of Ki67‐positive OT‐II T cells in the lymph nodes at day 3 (Fig. 6f). OT‐II T cells were not found locally in the ear at any of the time‐points in either Cre+ or Cre– mice.
Figure 6.
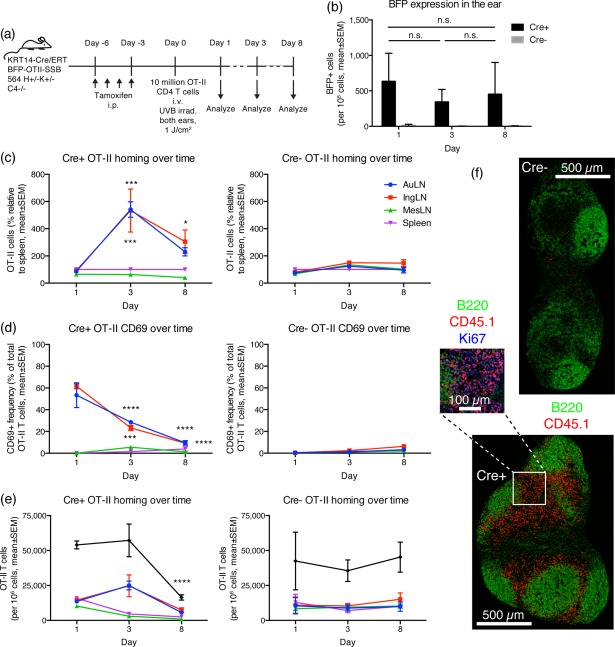
Adoptively transferred OT‐II T cells home to skin‐draining lymph nodes, independent of ultraviolet B (UVB) irradiation, show hallmarks of activation and expand then subsequently contract again. (a) Schematic overview of experimental layout. 564Igi C4–/– mice were crossed with mice that carry a KRT14‐driven Cre under the control of a mutated form of the ligand‐binding domain of the oestrogen receptor that only binds tamoxifen (ERT), and a floxed‐stop expression cassette for a fusion construct of blue fluorescent protein, an OTII epitope, and Sjögren's syndrome B protein (BFP‐OTII‐SSB), on a C4‐deficient background. Cre– and Cre+ littermates were used, and expression of BFP‐OTII‐SSB was turned on in the skin by intraperitoneal (i.p.) administration of tamoxifen (days −6 to −3). This was followed by UVB irradiation of both ears to induce apoptosis in the basal layer, and adoptive transfer of 1 × 107 OT‐II CD4 T cells by intravenous (i.v.) injection (day 0). Ear skin, auricular lymph nodes, inguinal lymph nodes, mesenteric lymph nodes and spleen were analyzed on days 1, 3 or 8. (b) BFP expression in the ears of adoptive transfer recipients. Two‐way analysis of variance (anova) with Sidak's multiple comparisons test (n.s. = not significant). (c) Distribution of OT‐II cells (as percent relative to spleen) following adoptive transfer into UVB irradiated KRT14 Cre+ (left panel) versus Cre– (right panel) BFP‐OTII‐SSB 564Igi H+/–K+/– C4–/– recipients. (d) CD69 expression on adoptively transferred OT‐II cells. (e) Absolute frequencies of OT‐II cells following adoptive transfer. For b–e, mean ± standard error of the mean (s.e.m.) of measurements from two mice are shown per group, per time‐point, for a total of 12 mice. Results are representative of two experiments with similar results. For c–e, statistical significance is indicated for two‐way anova with Dunnett's multiple comparisons test (*P < 0·05; ***P < 0·001; ****P < 0·0001). (f) Stitched whole‐node views of inguinal lymph node from a Cre– (top) or Cre+ (bottom) mouse at day 3. B220 in green, CD45.1+ OT‐II T cells in red. Scale bar indicated next to the top image. The blow‐out from the Cre+ node is at an additional ×2 magnification, and includes Ki67 staining in blue.
Discussion
We have presented here a novel mouse model, KRT14‐Cre/ERT BFP‐OTII‐SSB, with the aim of allowing tracking of peripheral TLR‐7‐agonistic autoantigen and investigation of downstream immunological consequences. Expression of the BFP‐OTII‐SSB reporter was detectable directly in ear skin by confocal microscopy, two‐photon microscopy and flow cytometry, but could not be detected directly nor indirectly by antibody, in downstream nodes or centrally. The inability to detect BFP outside the ear extended even to experiments aimed at promoting release by induction of apoptosis or after extended periods of transgene expression. However, adoptively transferred OT‐II T cells were observed to home to or expand in skin‐draining lymph nodes, specifically in Cre+ mice, but not Cre– littermates, indicating immune surveillance of OT‐II peptide derived from the transgene product. Importantly, this occurred irrespective of the UVB irradiation protocol, indicating that this surveillance was a constitutive, not induced, process. This finding is in line with a prior study reporting constitutive self‐antigen trafficking from skin to regional lymph nodes 37, and another study reporting that KRT14‐driven expression of a single viral protein in skin epithelium was sufficient to induce lymphocyte recruitment 38. In contrast, 564Igi B cells did not appear to see their cognate SSB antigen, due potentially to the absence of intact draining BFP‐OTII‐SSB protein. Although apoptotic keratinocytes have been reported to present SLE‐associated autoantigens on their surface 31, the in‐situ clearance of these cells may preclude direct immune surveillance by B cells.
Tolerance was maintained in Cre+ mice despite UVB irradiation, presence of 564Igi B cells and OT‐II T cells, even on a C4‐deficient background, an otherwise well‐recognized main predisposition for break‐of‐tolerance 21, 39. This finding highlights the power and redundancy of tolerance mechanisms in maintenance of tissue homeostasis, at least in the short term. Perhaps the most striking observation was that adoptively transferred OT‐II T cells, which homed initially to or expanded in skin‐draining nodes and up‐regulated CD69, by day 8 had again down‐regulated CD69 and had contracted substantially. This could be a result of presentation of their cognate antigen in a tolerogenic environment, as it has been found that DCs induce immunity or tolerance depending solely upon their activation status 40. However, in the present study this explanation appears unlikely, given the established general proinflammatory environment in 564Igi mice 5, C4‐deficient mice 41, 42, and 564Igi C4‐deficient mice in particular 21. Consequently, we speculate that it is an active dominant‐negative tolerance mechanism, mediated probably by forkhead box protein 3 (FoxP3)+ regulatory T cells (Tregs). Although we did not investigate this further, we noted the presence of a large population of Ki67+ cells associated with the adoptively transferred OT‐IIs in the lymph nodes at day 3 (see Fig. 6f). The absence of transgene expression in the basal layer of mice which were not tamoxifen‐treated (Fig. 1c) demonstrates the stringent control of Cre by the ERT. Therefore, even if KRT14 has been demonstrated previously to be an Aire‐regulated gene 43, we speculate that BFP is not expressed thymically and hence is not directly subject to central tolerance mechanisms before tamoxifen induction (although we did not evaluate this directly). Furthermore, we employed adoptive transfers of OT‐II transgenic cells, bypassing the thymic tolerance checkpoint. Although thymic bone marrow‐derived cells can cross‐present antigen 44, it is unclear whether extrathymic antigen can enter this pathway. Some studies have demonstrated that self‐antigens from the periphery can enter the thymic medulla via the bloodstream or be imported via immigrating DCs carrying antigen 45, 46. In our setting, where the neo‐autoantigen is not even detectable in the downstream draining lymph node, this seems unlikely. A recent study found that ubiquitously expressed nuclear neo‐self‐antigen Ova was presented efficiently via MHC class II by both medullary thymic epithelial cells (mTECs) and thymic DCs; however, presentation by DCs was highly dependent upon antigen expression by TECs, and haematopoietic cells did not substitute for this antigen source 47. Even so, our data suggest that a dynamic peripheral tolerance induction occurs in the presented setting, either through regulatory T cell activity derived from the endogenous repertoire or by a direct effect on adoptively transferred OT‐II T cells.
Overall, our results are in line with those of Kawahata and colleagues 19, whereby low‐level antigen presentation by DCs induced tolerance of adoptively transferred DO11.10 CD4 T cells. However, it was surprising that expression of a skin‐localized neo‐autoantigen carrying associated RNA in the context of low‐grade inflammation elicited by UVB irradiation, and in the presence of adoptively transferred cognate T cells, in an environment in which cognate B cells are already present, fails to elicit an overt break‐of‐tolerance. Nonetheless, we did not see any skin inflammation, nor hallmarks of a productive response centrally, despite evidence that the T cells were seeing their cognate peptide in the skin‐draining lymph nodes. This functional tolerance induction may be explained as a consequence of the in‐situ clearance of apoptotic keratinocytes and drainage only of heavily degraded antigens derived from this source, as evidenced by absence of both BFP signal and αTagRFP amplified signal. We hypothesize that B cells never encounter their native antigen in the draining lymph nodes, and hence are neither activated by antigen binding nor associated TLR ligands, and never become capable of presenting peptide to OT‐II T cells. Similarly, DCs or Langerhans cells presenting OT‐II peptide in the skin‐draining lymph nodes may have taken up only antigen degraded to a degree where the RNA component is no longer sufficiently intact to drive their maturation, and hence the peptide is presented in a non‐immunogenic context. Thus, the subtle immunostimulatory action of endogenous ribonuclear complexes and mild UVB irradiation appear insufficient to tip the scales in favour of immunogenicity and do not elicit overt autoimmunity in this model.
One aspect, which has not been investigated at this point, is the potential role of B regulatory cells in dampening inflammation in the model. As mentioned in the Introduction, Bregs have emerged in recent years as key players in immunomodulation and suppression of immune responses in connection with autoimmune diseases. This includes both B cell‐centric diseases such as SLE 1 and, perhaps more surprisingly, autoimmune disorders typically considered T cell‐mediated, such as type I diabetes 2. These effects are probably accomplished through the production of IL‐10, transforming growth factor (TGF)‐β and IL‐35, whereby Bregs can both suppress differentiation of proinflammatory lymphocytes and induce differentiation of immunosuppressive T cell subsets 48. Future studies should address this important question in the context of the novel model presented here.
Disclosure
The authors declare that they have no conflicts of interest in relation to the work presented in this paper.
Author contributions
M. C. C. conceived and directed the project. E. A. generated the BFP‐OTII‐SSB line. S. E. D. conducted the work, analysed the data and drafted the paper. All authors provided critical feedback on the paper and approved its final form.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Two‐photon microscopy of hair follicles in skin of KRT14‐Cre/ERT blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) reporters. Two‐photon intravital imaging of BFP+ basal layer cells and vasculature of hair follicles. Z‐projection of stacks from 50‐70 µm depth below the epidermal surface, showing hair follicles with central shaft (blue autofluorescence, indicated by white annulus) surrounded by basal layer cells with BFP‐positive nuclei (blue). The vasculature supplying the hair follicle has been labelled by injection of rhodamine‐dextran (red). In the top image, autofluorescence arising from overlying epidermis can be seen in purple. Scale bars indicated.
Fig. S2. Example of CellProfiler masks applied in image quantification pipeline. Left, non‐irradiated specimen; right, ultraviolet B (UVB)‐irradiated specimen. Top, raw image overlay of BFP and cleaved caspase 3 signals. Middle, BFP outlines defined by CellProfiler. Bottom, cleaved caspase 3 outlines defined by CellProfiler.
Fig. S3. Ability to follow blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) independently of BFP fluorescence via polyclonal anti‐BFP peptide antibody. Left two panels: split channel view of BFP and CD45 signal (top) and anti‐Tag‐red fluorescent protein (RFP) and CD45 signal (bottom), from the same section of ear skin from a Cre+ reporter. Right, top: overlay of the two left panels, showing gross co‐localization of BFP and anti‐TagRFP signals. Right, bottom: overlay as in the above panel, but for a Cre– littermate.
Fig. S4. Idiotype frequencies within adoptively transferred carboxyfluorescein succinimidyl ester (CFSE)‐labelled population recovered on day 3. Idiotype frequencies within the CFSE+ gate in two samples of spleen and auricular lymph nodes on day 3 post‐adoptive transfer.
Acknowledgements
We thank Harry Leung of the Optical Microscopy Core and Natasha Barteneva and Kenneth Ketman of the Flow and Imaging Cytometry Resource at the Program in Cellular and Molecular Medicine for expert technical assistance. We are grateful to Sarah A. Jones for assistance designing the BFP‐OTII‐SSB targeting construct and to Dvora Ghitza for performing the microinjections. This work was supported by grants from the Benzon Foundation and by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme (to SED) and by the NIH (R37 5AI054636) (to MCC).
Contributor Information
S. E. Degn, Email: sdegn@biomed.au.dk.
M. C. Carroll, Email: Michael.Carroll@childrens.harvard.edu
References
- 1. Fiorina P, Sayegh MH. B cell‐targeted therapies in autoimmunity: rationale and progress. F1000 Biol Rep 2009; 1:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kleffel S, Vergani A, Tezza S et al Interleukin‐10+ regulatory B cells arise within antigen‐experienced CD40+ B cells to maintain tolerance to islet autoantigens. Diabetes 2015; 64:158–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fiorina P, Vergani A, Dada S et al Targeting CD22 reprograms B‐cells and reverses autoimmune diabetes. Diabetes 2008; 57:3013–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12:991–1045. [DOI] [PubMed] [Google Scholar]
- 5. Berland R, Fernandez L, Kari E et al Toll‐like receptor 7‐dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 2006; 25:429–40. [DOI] [PubMed] [Google Scholar]
- 6. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak‐Rothstein A. Chromatin‐IgG complexes activate B cells by dual engagement of IgM and Toll‐like receptors. Nature 2002; 416:603–7. [DOI] [PubMed] [Google Scholar]
- 7. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 2002; 297:2066–70. [DOI] [PubMed] [Google Scholar]
- 8. Green NM, Moody K‐S, Debatis M, Marshak‐Rothstein A. Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. J Biol Chem 2012; 287:39789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol 2014; 14:289–301. [DOI] [PubMed] [Google Scholar]
- 10. Riminton DS, Kandasamy R, Dravec D, Basten A, Baxter AG. Dermal enhancement: bacterial products on intact skin induce and augment organ‐specific autoimmune disease. J Immunol 2004; 172:302–9. [DOI] [PubMed] [Google Scholar]
- 11. Yokogawa M, Takaishi M, Nakajima K et al Epicutaneous application of toll‐like receptor 7 agonists leads to systemic autoimmunity in wild‐type mice: a new model of systemic lupus erythematosus. Arthritis Rheumatol 2014; 66:694–706. [DOI] [PubMed] [Google Scholar]
- 12. Azukizawa H, Kosaka H, Sano S et al Induction of T‐cell‐mediated skin disease specific for antigen transgenically expressed in keratinocytes. Eur J Immunol 2003; 33:1879–88. [DOI] [PubMed] [Google Scholar]
- 13. Shibaki A, Sato A, Vogel JC, Miyagawa F, Katz SI. Induction of GVHD‐like skin disease by passively transferred CD8(+) T‐cell receptor transgenic T cells into keratin 14‐ovalbumin transgenic mice. J Invest Dermatol 2004; 123:109–15. [DOI] [PubMed] [Google Scholar]
- 14. Mayerova D, Parke EA, Bursch LS, Odumade OA, Hogquist KA. Langerhans cells activate naive self‐antigen‐specific CD8 T cells in the steady state. Immunity 2004; 21:391–400. [DOI] [PubMed] [Google Scholar]
- 15. McGargill MA, Mayerova D, Stefanski HE et al A spontaneous CD8 T cell‐dependent autoimmune disease to an antigen expressed under the human keratin 14 promoter. J Immunol 2002; 169:2141–7. [DOI] [PubMed] [Google Scholar]
- 16. Waithman J, Allan RS, Kosaka H et al Skin‐derived dendritic cells can mediate deletional tolerance of class I‐restricted self‐reactive T cells. J Immunol 2007; 179:4535–41. [DOI] [PubMed] [Google Scholar]
- 17. Bianchi T, Pincus LB, Wurbel M‐A et al Maintenance of peripheral tolerance through controlled tissue homing of antigen‐specific T cells in K14‐mOVA mice. J Immunol 2009; 182:4665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robertson JM, Jensen PE, Evavold BD. DO11.10 and OT‐II T cells recognize a C‐terminal ovalbumin 323–339 epitope. J Immunol 2000;164:4706–12. [DOI] [PubMed] [Google Scholar]
- 19. Kawahata K, Misaki Y, Yamauchi M et al Peripheral tolerance to a nuclear autoantigen: dendritic cells expressing a nuclear autoantigen lead to persistent anergic state of CD4+ autoreactive T cells after proliferation. J Immunol 2002; 168:1103–12. [DOI] [PubMed] [Google Scholar]
- 20. Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak‐Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature 2012; 480:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatterjee P, Agyemang AF, Alimzhanov MB et al Complement C4 maintains peripheral B‐cell tolerance in a myeloid cell dependent manner. Eur J Immunol 2013; 43:2441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Das A, Heesters BA, Bialas A et al Follicular dendritic cell activation by TLR ligands Promotes autoreactive B cell responses. Immunity 2017; 46:106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luning Prak ET, Monestier M, Eisenberg RA. B cell receptor editing in tolerance and autoimmunity. Ann N Y Acad Sci 2011; 1217:96–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Madisen L, Zwingman TA, Sunkin SM et al A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010; 13:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Köntgen F, Süss G, Stewart C, Steinmetz M, Bluethmann H. Targeted disruption of the MHC class II Aa gene in C57BL/6 mice. Int Immunol 1993; 5:957–64. [DOI] [PubMed] [Google Scholar]
- 26. Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA 1995; 92:11490–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schindelin J, Arganda‐Carreras I, Frise E et al Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012; 9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lamprecht M, Sabatini D, Carpenter A. CellProfiler™: free, versatile software for automated biological image analysis. Biotechniques 2007; 42:71–5. [DOI] [PubMed] [Google Scholar]
- 29. Subach OM, Gundorov IS, Yoshimura M et al Conversion of red fluorescent protein into a bright blue probe. Chem Biol 2008; 15:1116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA 1999; 96:8551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casciola‐Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 1994; 179:1317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee PL, van Weelden H, Bruijnzeel PLB. Neutrophil infiltration in normal human skin after exposure to different ultraviolet radiation sources. Photochem Photobiol 2008; 84:1528–34. [DOI] [PubMed] [Google Scholar]
- 33. Dandie GW, Clydesdale GJ, Radcliff FJ, Muller HK. Migration of Langerhans cells and gammadelta dendritic cells from UV‐B‐irradiated sheep skin. Immunol Cell Biol 2001; 79:41–8. [DOI] [PubMed] [Google Scholar]
- 34. Schmidt KN, Cyster JG. Follicular exclusion and rapid elimination of hen egg lysozyme autoantigen‐binding B cells are dependent on competitor B cells, but not on T cells. J Immunol 1999; 162:284–91. [PubMed] [Google Scholar]
- 35. Cyster JG, Goodnow CC. Antigen‐induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity 1995; 3:691–701. [DOI] [PubMed] [Google Scholar]
- 36. Ekland EH, Forster R, Lipp M, Cyster JG. Requirements for follicular exclusion and competitive elimination of autoantigen‐binding B cells. J Immunol 2004; 172:4700–8. [DOI] [PubMed] [Google Scholar]
- 37. Yoshino M, Yamazaki H, Shultz LD, Hayashi S‐I. Constant rate of steady‐state self‐antigen trafficking from skin to regional lymph nodes. Int Immunol 2006; 18:1541–8. [DOI] [PubMed] [Google Scholar]
- 38. Choyce A, Yong M, Narayan S et al Expression of a single, viral oncoprotein in skin epithelium is sufficient to recruit lymphocytes. PLoS One 2013; 8:e57798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol 2004; 22:(1):431–56. [DOI] [PubMed] [Google Scholar]
- 40. Probst HC, Lagnel J, Kollias G, van den Broek M. Inducible transgenic mice reveal resting dendritic cells as potent inducers of CD8+ T cell tolerance. Immunity 2003; 18:713–20. [DOI] [PubMed] [Google Scholar]
- 41. Paul E, Pozdnyakova OO, Mitchell E, Carroll MC. Anti‐DNA autoreactivity in C4‐deficient mice. Eur J Immunol 2002; 32:2672–9. [DOI] [PubMed] [Google Scholar]
- 42. Finke D, Randers K, Hoerster R et al Elevated levels of endogenous apoptotic DNA and IFN‐α in complement C4‐deficient mice: implications for induction of systemic lupus erythematosus. Eur J Immunol 2007; 37:1702–9. [DOI] [PubMed] [Google Scholar]
- 43. Oftedal BE, Hellesen A, Erichsen MM et al Dominant mutations in the autoimmune regulator AIRE are associated with common organ‐specific autoimmune diseases. Immunity 2015; 42:1185–96. [DOI] [PubMed] [Google Scholar]
- 44. Gallegos AM, Bevan MJ. Central tolerance to tissue‐specific antigens mediated by direct and indirect antigen presentation. J Exp Med 2004; 200:1039–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bonasio R, Scimone ML, Schaerli P, Grabie N, Lichtman AH, Andrian von UH. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol 2006; 7:1092–100. [DOI] [PubMed] [Google Scholar]
- 46. Klein L, Roettinger B, Kyewski B. Sampling of complementing self‐antigen pools by thymic stromal cells maximizes the scope of central T cell tolerance. Eur J Immunol 2001; 31:2476–86. [DOI] [PubMed] [Google Scholar]
- 47. Koble C, Kyewski B. The thymic medulla: a unique microenvironment for intercellular self‐antigen transfer. J Exp Med 2009; 206:1505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42:607–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Two‐photon microscopy of hair follicles in skin of KRT14‐Cre/ERT blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) reporters. Two‐photon intravital imaging of BFP+ basal layer cells and vasculature of hair follicles. Z‐projection of stacks from 50‐70 µm depth below the epidermal surface, showing hair follicles with central shaft (blue autofluorescence, indicated by white annulus) surrounded by basal layer cells with BFP‐positive nuclei (blue). The vasculature supplying the hair follicle has been labelled by injection of rhodamine‐dextran (red). In the top image, autofluorescence arising from overlying epidermis can be seen in purple. Scale bars indicated.
Fig. S2. Example of CellProfiler masks applied in image quantification pipeline. Left, non‐irradiated specimen; right, ultraviolet B (UVB)‐irradiated specimen. Top, raw image overlay of BFP and cleaved caspase 3 signals. Middle, BFP outlines defined by CellProfiler. Bottom, cleaved caspase 3 outlines defined by CellProfiler.
Fig. S3. Ability to follow blue fluorescent protein‐OTII epitope‐Sjögren's syndrome B protein (BFP‐OTII‐SSB) independently of BFP fluorescence via polyclonal anti‐BFP peptide antibody. Left two panels: split channel view of BFP and CD45 signal (top) and anti‐Tag‐red fluorescent protein (RFP) and CD45 signal (bottom), from the same section of ear skin from a Cre+ reporter. Right, top: overlay of the two left panels, showing gross co‐localization of BFP and anti‐TagRFP signals. Right, bottom: overlay as in the above panel, but for a Cre– littermate.
Fig. S4. Idiotype frequencies within adoptively transferred carboxyfluorescein succinimidyl ester (CFSE)‐labelled population recovered on day 3. Idiotype frequencies within the CFSE+ gate in two samples of spleen and auricular lymph nodes on day 3 post‐adoptive transfer.