

Abstract
Despite substantial advances in the research exploring the pathogenesis of Type 1 Diabetes (T1D), the pathophysiological mechanisms involved remain unknown. We hypothesized in this study that IFNα participates in the early stages of T1D development by triggering endoplasmic reticulum (ER) stress. To test our hypothesis, human islets and human EndoC-βH1 cells were exposed to IFNα and tested for ER stress markers, glucose stimulated insulin secretion (GSIS) and insulin content. IFNα treatment induced upregulation of ER stress markers including Binding immunoglobulin Protein, phospho-eukaryotic translation initiation factor 2α, spliced- X-box binding protein-1, C/EBP homologous protein and activating transcription factor 4. Intriguingly, IFNα treatment did not impair GSIS but significantly decreased insulin production in both human islets and EndoC-βH1 cells. Furthermore, IFNα decreased the expression of both proinsulin convertase 1 and proinsulin convertase 2, suggesting an altered functional state of the beta cells characterized by a slower proinsulin-insulin conversion. Pretreatment of both human islets and EndoC-βH1 cells with chemical chaperones 4-phenylbutyric acid and tauroursodeoxycholic acid completely prevented IFNα effects, indicating an ER stress-mediated impairment of insulin production. We demonstrated for the first time that IFNα elicits ER stress in human beta cells providing a novel mechanistic role for this virus-induced cytokine in the development of T1D. Compounds targeting molecular processes altered in ER-stressed beta cells could represent a potential therapeutic strategy to prevent IFNα-induced beta cell dysfunction in the early onset of T1D.
Keywords: Interferon alpha, endoplasmic reticulum stress, insulin, proinsulin, Type 1 Diabetes
Graphical Abstract
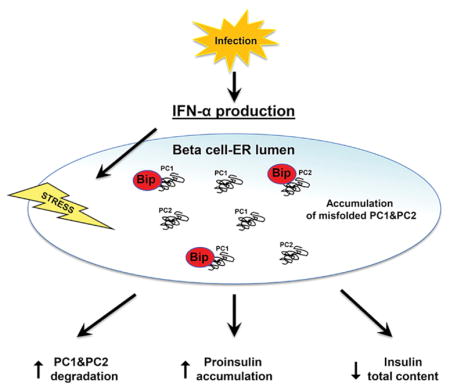
1. Introduction
Type 1 Diabetes (T1D) is an autoimmune disease characterized by pancreatic β-cell destruction and its incidence has been rapidly rising worldwide in the past several decades. Environmental factors such as viruses have been implicated as possible causes of this rise [1–3]. Moreover, growing evidence suggests that the virally-induced cytokine interferon alpha (IFNα) is a key trigger of T1D. Indeed, therapies that neutralize IFNα are able to suppress the beta cell dysfunction that precedes the disease, although the underlying molecular mechanisms are still unclear [4].
All the major beta cell autoantigens, including insulin, traffic through the endoplasmic reticulum (ER), a key organelle specialized in a number of important cellular tasks including protein folding, calcium and redox regulation, and the activation of survival/death pathways. Evidence is accumulating that defective folding and rapid degradation of mutant proteins is one of the causes of various diseases including metabolic disease, neurodegenerative disease, inflammatory disease, and cancer. In all cases the disorders are affecting the folding of exportable proteins that tend to accumulate within the ER [5–8]. In professional secretory cells like beta cells, the rapidly changing demand for insulin production and secretion in response to serum glucose levels relies greatly on ER stability to ensure proper synthesis and folding of proinsulin [9]. Recently, ER stress has been proposed to play a critical role in autoimmune-mediated beta cell destruction. Indeed ER-stressed beta cells in early T1D could eventually die or produce neo-autoantigens which could be targets of the autoimmune response in T1D, initiating a self-perpetuating vicious cycle of failing beta cells and autoimmune attack in genetically susceptible individuals [10–12].
Interestingly, high levels of IFNα have been linked with ER stress associated apoptosis in human urothelial cancer cells [13] connecting IFNα to ER stress in non-pancreatic cells. Moreover, the α subunit of eukaryotic translation initiation factor 2α (eIF2α) connects ER stress and interferon responses, indeed the production of IFNα triggered by viral infection upregulates double-stranded RNA-dependent protein kinase expression which phosphorylates and activates eIF2α [14].
The key role of IFNα in the etiology of T1D and the link between ER stress and beta cell autoimmunity let us to hypothesize that IFNα could trigger T1D by inducing ER stress in beta cells. Here we show strong evidence to support this hypothesis.
2. Materials & methods
2.1 Cell cultures and reagents
Human islets were isolated from deidentified donors by the Integrated Islet Distribution Program (IIDP) as per IIDP protocols (iidp.coh.org/), and transported to the principal investigator within 24 hours. Human pancreata were obtained from 15 non-diabetic organ donors (45 ± 10 years, range 28–60 years). There were 7 female and 7 male donors. (For one donor the gender was not documented). Characteristics of human islet donors are also shown in Supplementary Table 1. Upon receipt, human islets were cultured in RPMI 1640 medium (Thermo Scientific) containing 5.5 mM glucose, 10% FBS (Sigma) and 1% antibiotic antimycotic solution (HyClone) and maintained at 37°C in a humidified atmosphere with 5% CO2 for 24 h before starting experiments. The protocol was approved as exempt by Albert Einstein College of Medicine institutional review board. Insulin-producing EndoC-βH1 cells, a human beta cell line, were cultured as previously described [15]. For different experiments cells were seeded on 10-cm dishes, 6-cm dishes or on 12 mm diameter glass coverslips in 24-well plates. 24 hours later, cells were vehicle treated or treated with the indicated drugs at the indicated experimental settings. Thapsigargin and Tunicamycin were purchased from Sigma, IFNα, 4-phenylbutyric acid (PBA) and tauroursodeoxycholic acid (TUDCA) were purchased respectively from Millipore, Sigma and Calbiochem.
2.2 Immunofluorescence
1.5×105 EndoC-βH1 cells were seeded on Matrigel and fibronectin coated 12-mm glass coverslips in 24-well plates and allowed to attach for 5 days at 37 °C. Cells were then vehicle treated or treated with the indicated drugs at the indicated experimental settings and fixed in 4% PFA (Polysciences, Inc.) in PBS for 15 minutes at room temperature. After being permeabilized by 0.5% Triton X-100 (Bio-Rad) in PBS for 10 minutes, cells were blocked for 1 hour with 1% normal goat serum (Cell Signaling) in PBS (both steps were done at room temperature). Cells were subsequently immunostained overnight at 4°C with the primary antibodies diluted in blocking buffer. Indirect immunofluorescence was performed using guinea pig polyclonal anti insulin, rabbit polyclonal anti C-peptide antibody, and rabbit monoclonal anti PDX-1 as described in Supplementary Table 2. Secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 (Life Technologies, 1:500) were used for 1 hour at room temperature. After final washes with PBS, the coverslips were mounted on microscope slides with ProLong Gold Antifade Reagent containing DAPI to counterstain the nuclei (Cell Signaling). Samples were examined with a Leica DM6000 microscope.
2.3 RNA isolation, reverse transcription, semi-quantitative and real-time RT-PCR
RNA isolation, reverse transcription and real-time reverse transcription polymerase chain reaction (RT-PCR) were performed as previously described [16]. In brief, total RNA from human islets or EndoC-βH1 cells was isolated using TRIzol reagent (Thermo Scientific) in combination with the RNeasy Mini kit (Qiagen) followed by DNase treatment. Five hundred nanograms of total RNA were retrotranscribed using the Superscript III kit (Thermo Scientific) following the manufacturer’s instructions. The cDNAs obtained after retrotranscription were used as templates for quantitative PCR, run on an AbiPRISM 7300 fast real-time cycler using the power SYBR Green real-time PCR master mix kit and quantified by built-in SYBR Green Analysis (all from Applied Biosystems). The relative amount of specific mRNA was normalized to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH). To amplify spliced and unspliced X-box binding protein-1 (Xbp1) mRNA, PCR products were electrophoresed on 2.5% agarose gel and GAPDH was used as a loading control. Primer sequences (Integrated DNA Technologies) used for PCR are shown in Supplementary Table 3.
2.4 Western Blot Assay
Total proteins from adult human islets and EndoC-βH1 cells were extracted as previously described [17, 18]. Supernatants were collected and equal amount of proteins (50 μg per sample) were subjected to electrophoresis on SDS- polyacrylamide gel and transferred to Immobilon-P membranes (Millipore) for 2 hours at 100V on ice. Membranes were incubated in Odyssey Blocking Buffer (LI-COR) for 1 hour at room temperature and then reacted overnight at 4 °C with specific primary antibodies for the protein of interest. The antibodies used in this study are described in Supplementary Table 2. All immunoblots were developed with the LI-COR Odyssey system, usinginfrared-labeled anti-rabbit and anti-mouse IgG (LI-COR 1:5000) secondary antibodies.
2.5 Insulin secretion and insulin/proinsulin contents
For insulin secretion analysis, insulin content, and proinsulin:insulin ratio we used 50 size-matched islets from each individual for each experimental condition. All experiments were repeated at least 3 times using islets from different donors (at least 3 donors). Human islets with or without IFNα were left overnight in glucose starving medium (RPMI without glucose, supplemented with 10% heat-inactivated FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin) and subsequently incubated for 1 h at 37°C with 1 ml Krebs-Ringer bicarbonate buffer solution supplemented with 2 mM glucose or 20 mM glucose. (Krebs-Ringer bicarbonate buffer solution: 120 mmol/L NaCl, 4.7 mmol/LKCl, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 2.4 mmol/L CaCl2, and 20 mmol/LNaHCO3, supplemented with 10 mmol/L HEPES and 0.2% BSA and gassed with a mixture of 95% O2 and 5% CO2). Islets were then pelleted by centrifugation at 1500 rpm for 5 minute at 4°C and supernatants were collected and stored at −20°C for insulin secretion. Insulin secretion in EndoC-βH1 cells was performed as previously described [4]. For insulin and proinsulin content measurements, human islets and EndoC-βH1 cells were lysed in ice-cold acid ethanol (75% ethanol, 1.5% HCl) and incubated overnight in this extracting solution at 4°C. Lysates were then sonicated and centrifuged for 2 minutes at 20,000 g. Samples were kept frozen at −20°C before use. In both systems insulin intracellular content was measured using the Human Insulin kit (Mercodia) and proinsulin was measured using the Human Proinsulin kit (Millipore).
2.6 Statistics
All experiments were performed at least in triplicate. Data are presented as means ± SEM. Statistical differences were determined using a two-tailed Student’s t test. Statistical analysis was performed using GraphPad Prism (version 5.01; GraphPad Software Inc.). A p-value < 0.05 was considered statistically significant.
3. Results
3.1 IFNα induces BiP upregulation in human islets and EndoC-βH1 cells
To investigate if IFNα was able to activate ER stress in human beta cells we exposed human islets and the insulin-producing human beta cell line EndoC-βH1 to increasing concentrations of IFNα. To demonstrate that they are functional human beta cells EndoC-βH1 cells were tested for baseline insulin, C-peptide, pancreatic and duodenal homeobox 1 (PDX1) expression, and for glucose-stimulated insulin secretion (Fig. 1A–D). Treatment of islets and EndoC-βH1 cells with 1000 U/ml IFNα for 48 hours induced upregulation of the diagnostic ER stress marker Binding immunoglobulin Protein (BiP), a molecular chaperone that resides in the ER [19] (Fig. 2A). Such induction was comparable to classic ER stress-inducing agents, Thapsigargin and Tunicamicin, and was dose dependent (Supplementary Fig. 1A). In addition, kinetic analysis done in human islets and EndoC-βH1 cells with 1000 U/ml IFNα showed that BiP mRNA was significantly increased as early as 24 hours after treatment and remained elevated for 72 hours (Supplementary Fig. 1B). To corroborate these findings, we tested the ability of classical ER stress modulators, the chemical chaperones PBA and TUDCA [20], to prevent IFNα-induced BiP mRNA upregulation. PBA is a low molecular weight non-specific chemical chaperone known to stabilize protein conformation, improving ER folding capacity and facilitating the traffic of mutant proteins. Likewise, TUDCA, a taurine-conjugated derivative of bile acid, mitigates ER stress by acting as a chemical chaperone. Both PBA and TUDCA almost completely prevented the effect of IFNα on BiP mRNA upregulation (Fig. 2A) confirming that IFNα upregulates BiP in beta cells by inducing ER stress. The IFNα-induced increase of BiP mRNA and the effect of PBA and TUDCA were also confirmed at the protein levels in both human islets and EndoC-βH1 cells (Fig. 2BD).
Fig. 1. Immunofluorescence analysis of human EndoC-βH1 cells.

(A–C) EndoC-βH1 cells were cultured on 12-mm glass cover slips precoated with Matrigel and fibronectin. After 5 days, the cells were fixed in 4% parafolmaldehyde for 1 hour. Immunofluorescence staining was performed using insulin antibody (green), C-peptide antibody (red) or PDX-1 antibody (red) described in Supplementary Table 2. The secondary antibodies were: Alexa Fluor 488 goat anti-guinea pig antibody (1:500; Life Technologies) for insulin, and Alexa Fluor 568 goat anti-rabbit antibody (1:500; Life Technologies) for C-peptide and PDX-1. Nuclei were counterstained with DAPI (blue). Samples were examined with a Leica DM6000 microscope. (D) Glucose-induced insulin secretion in EndoC-βH1 cells (***p < 0.001 compared to unstimulated cells).
Fig. 2. Effect of IFNα on BiP induction in human islets and EndoC-βH1 cells.

(A–D) Human islets and EndoC-βH1 cells were pretreated overnight with 2.5 mM PBA or with 1 mM TUDCA or with medium only (negative control) and then cultured in presence of IFNα 1000 U/ml for 48 hours. (A) The expression levels of mRNAs for BiP were determined by real-time RT-PCR analysis of total RNA from three different preparations of human islets or EndoC-βH1 cells treated as above. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). Bars represent means ± SEM from three independent experiments. ***p < 0.001 compared to CTRL cells. (B–D) Human islets and EndoC-βH1 cells (from five different preparations) were solubilized and equal amount of proteins (50 μg per sample) were analyzed by immunoblotting. Filters were probed with antibodies against BiP. Representative images are shown. Band intensities were quantified by densitometry using ImageJ software.
3.2 IFNα induces unfolded protein response in human islets and EndoC-βH1 cells
The ability of beta cells to compensate for IFNα-driven increases in ER stress requires an intracellular signaling pathway, the unfolded protein response (UPR), that monitors conditions in the ER, sensing an insufficiency in the ER’s protein folding capacity and communicates this information regarding the status of the ER lumen to gene expression programs of beta cells. Three principal branches of the UPR have been identified. Each branch is defined by a class of transmembrane ER-resident signaling components: activating transcription factor 6 (ATF6), double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK) and inositol requiring enzyme 1 (IRE1) [19]. Among ATF’s targets are prominent ER-resident proteins involved in protein folding, such as BiP (Fig. 2A–D). The second branch of the UPR is mediated by PERK, a kinase that upon sensing ER stress phosphorylates eukaryotic translation initiation factor 2α (p-eIF2α) that induces the transcription of activating transcription factor 4 (ATF4), and C/EBP homologous protein (CHOP) [19]. As shown in Figure 3A–D, treatment with IFNα increased UPR markers p-eIF2α, ATF4, and CHOP in human islets and EndoC-βH1 cells. Moreover, pretreatment with PBA or TUDCA prevented these effects. IRE1 defines the third and best studied branch of the UPR. When activated, IRE1 cleaves the mRNA encoding a specific transcription factor called Xbp1, giving rise to an active spliced mRNA (Xbp1s) that regulates the transcription of genes involved in protein folding [21]. RT-PCR analysis done in human islets and EndoC-βH1 cells showed an increase in both the total (unspliced) and spliced (active) form of Xbp1 mRNA levels following IFNα treatment, this effect was also suppressed by PBA or TUDCA pretreatment (Fig. 3E–G). These data suggest that IFNα causes UPR activation in human beta cells.
Fig. 3. IFNα induces UPR activation in human islets and EndoC-βH1 cells.

(A–G) Human islets and EndoC-βH1 cells were pretreated overnight with 2.5 mM PBA or with 1 mM TUDCA or with medium only (negative control) and then cultured in presence of IFNα (1000 units/ml) for 48 hours. (A–C) Human islets and EndoC-βH1 cells were solubilized and equal amount of proteins (50 μg per sample) were analyzed by immunoblotting. Filters were probed with antibodies against p-eIF2α and β-Actin. Representative images are shown. Band intensities were quantified by densitometry using ImageJ software. (D–E) The expression levels of mRNAs for ATF4, CHOP and Xbp1 were determined by real-time RT-PCR analysis of total RNA from human islets and EndoC-βH1 cells treated as above. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). Bars represent means ± SEM from four to five independent experiments. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells. (F–G) Semi-quantitative RT-PCR analysis for Xbp1 and GAPDH of total RNA extracted from human islets and EndoC-βH1 cells vehicle treated (CTRL) or IFNα treated, pretreated or not with PBA and TUDCA. These results have been replicated three times.
3.3 IFNα-induced ER stress causes a decrease in insulin content and augments proinsulin:insulin ratio
To investigate whether IFNα-mediated ER stress affected beta cells functionality we measured glucose-stimulated insulin secretion (GSIS) in control and IFNα-treated human islets and EndoC-βH1 cells. IFNα exposure did not alter GSIS but impaired basal insulin release in both systems suggesting an effect on insulin content (Fig. 4A). To test this hypothesis, we measured insulin content in human islets and EndoC-βH1 cells untreated and treated with 1000 U/ml IFNα for 48 hours; in both systems the exposure to IFNα caused a significant decrease in insulin content and an increase in proinsulin/insulin ratio. This finding together with the observation that these effects were prevented by PBA and TUDCA (Fig. 4B and C) are suggestive of an ER folding/processing defect. Interestingly, when IFNα was removed from both systems BiP mRNA levels significantly decreased already after 12 hours and insulin production was restored within 24 hours both in human islets and EndoC-βH1 cells (Supplementary Fig. 2A–B); this results can be explained by the short half-life of the cytokine in tissue culture medium.
Fig. 4. IFNα-induced ER stress impairs insulin content and proinsulin:insulin ratio in human islets and EndoC-βH1 cells.

(A) Insulin secretion, (B) insulin content, and (C) proinsulin:insulin ratio in human islets and EndoC-βH1 pretreated or not overnight with 2.5 mM PBA or with 1 mM TUDCA and cultured in presence of IFNα (1000 units/ml) for 48 hours. Values are mean ± SEM of three independent experiments. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells.
3.3 IFNα-induced ER stress impairs PC1 and PC2 expression in human islets and EndoC-βH1 cells
To gain insights into the mechanism of impaired insulin content, we analyzed the contribution of proinsulin convertase (PC) to the proinsulin conversion delay, we performed real-time RT-PCR and western blot analyses of proinsulin convertase 1 (PC1) and proinsulin convertase 2 (PC2) mRNA and protein levels, respectively. As shown in Fig. 5A–E, human islets and EndoC-βH1 cells treated with IFNα exhibited a decreased expression of both PC1 and PC2 mRNA and protein levels suggesting that both suppression of transcription and protein degradation can be involved in such down-regulation. However, pretreatment with PBA or TUDCA prevented these alterations (Fig. 5A–E) demonstrating that not only IFNα causes impairment in insulin content and proinsulin conversion via ER-stress mediated mechanisms, but also that reversing ER stress may promote correct folding of PC1 and PC2 in human beta cells.
Fig. 5. IFNα-induced ER stress impairs PC1–2 levels in human islets and EndoC-βH1 cells.

(A–E) human islets and EndoC-βH1 were pretreated or not overnight with 2.5 mM PBA or with 1 mM TUDCA and cultured in presence of IFNα (1000 units/ml) for 48 hours. (A) Real-time RT-PCR analysis of PC1 and PC2 mRNA levels using the total RNAs from four different preparations of human islets or EndoC-βH1 cells treated as indicated, with GAPDH as internal standard. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). Each bar represents the mean ± SEM of four independent experiments, each performed in triplicate. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells. (B–E) Human islets and EndoC-βH1 cells, both from three different preparations, were solubilized and cell lysates (50 μg per sample) were analyzed by western blotting with anti PC1 and PC2 antibodies using β-Actin as a loading control.
4. Discussion
Currently, there is no cure or preventive regimen for T1D and the early stages of the disease remain poorly defined. Viral infection has been hypothesized to be one of the environmental triggers for the development of T1D and the mechanism of viral infection leading to beta cell destruction involves multiple pathways including IFNα. This cytokine has been recently proposed as a key molecule in the development of T1D creating a favorable inflammatory milieu for the diabetogenic adaptive response. A growing body of evidence associates IFNα with the development of T1D in humans and mice but the mechanisms underlying IFNα-induced beta cell dysfunction are still not known. Almost 25 years ago Timothy Stewart and his group provided important data on the impact of type 1 interferons on beta cell function. They discovered that transgenic non-autoimmune-prone mice expressing IFNα in beta cells develop inflammatory infiltrates of the islets and T1D but the severity depends on the genetic background, suggesting that IFNα may be required for the initiation of T1D in genetically susceptible hosts [22–24]. Moreover, recent additional studies by Qing Li et al. demonstrated that IFNα is an essential initiator in the pathogenesis of T1D in NOD mice [25, 26]. In humans, a pathogenic role for IFNα has been proposed as IFNα treatment of patients with viral infections or with leukemia has been shown to be associated with increased incidence of T1D [27–32] Of interest, transcriptional profiling of NOD islets and pancreatic lymph nodes before T-cell infiltration of islets identified an IFNα-induced gene signature, showing that islet exposure to IFNα is a precipitating event in T1D pathogenesis [33]. Intriguingly, elevated IFNα mRNA transcripts were observed in the pancreata and islets of deceased diabetic patients [34] and beta cells from type 1 diabetic individuals contained immunoreactive IFNα [35]. Finally, IFNα-producing plasmacytoid dendridic cells have been detected in the blood of patients with T1D [36] and recent genetic analysis supports a diabetogenic role for IFNα-induced genes in prediabetic children [37]. Taken together these data pinpoint a key role for local islet IFNα production in starting the autoimmune process in T1D.
Since no clear mechanism linking beta cell dysfunction and IFNα has been identified to date, in the present study we hypothesized that local production of IFNα in the pancreas (e.g. during viral infections) could trigger ER stress leading to beta cell failure. Beta cells have the highest protein secretion burden in the body and secreting functional insulin is a complex process very easy to perturb. For example, mutations in the insulin gene that cause misfolding of the encoded protein result in chronic ER stress triggering beta cell apoptosis [38]. Given the essential role of ER in insulin production and secretion, beta cells, which are professional secretory cells, are very sensitive to ER perturbations. Interestingly, it has been shown very recently that IFNα-induced apoptosis is linked to ER stress also in non-pancreatic cells [39], however, the effect of IFNα on islet cells different from beta cells is still under investigation. Our study showed that IFNα can induce ER stress in human islets and human EndoC-βH1 cells, identifying for the first time ER stress as a molecular mechanism underlying IFNα and beta cell dysfunction that precedes the clinical onset of T1D. IFNα treatment did not alter glucose induced insulin secretion in human beta cells but insulin content was significantly reduced, with a significantly increased proinsulin:insulin ratio. Moreover, exposure of human beta cells to IFNα decreased the expression of both proinsulin convertases PC1 and PC2. Of note, similar observations have been made in beta cells for other proinflammatory cytokines (e.g. IL-1β or combinations of IL-1β and IFN-γ) consistent with our findings and indicating that increased proinsulin blood concentrations in prediabetic conditions can result from exposure of beta cells to proinflammatory cytokines [40, 41]. Indeed, increased blood concentrations of proinsulin as compared to insulin are a recognized marker of beta cell dysfunction in T1D [42, 43]. Interestingly, we found that the deleterious consequences of IFNα treatment were almost completely prevented by the chemical chaperones PBA or TUDCA, suggesting that IFNα causes PC1 and PC2 misfolding in human beta cells. Such misfolding could alter the functional state of the beta cells leading to a delay in proinsulin conversion and an increased ratio of cellular proinsulin over insulin content.
In conclusion our findings have significant translational implications given the potential to use PBA and TUDCA in the pre-diabetic stages of T1D and their excellent safety profiles. Preclinical and clinical studies have recently demonstrated the therapeutic potential of PBA and TUDCA in several metabolic diseases caused by ER stress. The beneficial properties of PBA and TUDCA were already discovered and explored in cystic fibrosis [44], metabolic syndrome [45], obesity [46], and atherosclerosis [47]. Moreover, TUDCA is currently being tested in a clinical trial in recent onset T1D. Indeed, it is possible that these compounds may be used in the future for their ability to restore insulin homeostasis in the early stages of T1D.
Supplementary Material
Supplementary Fig. 1. Effect of IFNα on BiP mRNA in human islets and EndoC-βH1 cells. (A) Human islets and EndoC-βH1 cells were cultured in presence of 0.5 μM thapsigargin or 0.5 μg/ml tunicamycin or with the indicated concentrations of IFNα for 24 hours. (B) Human islets and EndoC-βH1 cells were cultured with 1000 U/ml IFNα (added in the medium every 24 hours) for the indicated times. The expression levels of mRNAs for BiP were determined by real-time RT-PCR analysis of total RNA from three different preparations of human islets or EndoC-βH1 cells treated as above. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). Bars represent means ± SEM from three independent experiments. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells.
Supplementary Fig. 2. Effect of IFNα-removal on bip mRNA and insulin production in human islets and EndoC-βH1 cells. (A–B) Human islets and EndoC-βH1 cells were vehicle treated (CTRL) or treated with 1000 U/ml IFNα for 48 hours followed by 6, 12, 24, and 48 hours in medium without IFNα. (A) BiP mRNA levels were determined by real-time RT-PCR analysis of total RNA from three different preparations of human islets or EndoC-βH1 cells treated as above. (B) Insulin content in human islets and EndoC-βH1 treated as above. Values shown represent the mean ± SEM of three independent experiments. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells.
Acknowledgments
Sources of funding
None of the sources of funding have an interest in the subject matter or materials discussed in the submitted manuscript.
This work was supported in part by grant DK067555 from NIDDK. In addition this material is based upon work supported in part by the Department of Veterans Affairs.
Footnotes
Conflict of interest statement
The authors declare that there is no duality of interest associated with their contribution to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.von Herrath M. Diabetes: A virus-gene collaboration. Nature. 2009;459:518–9. doi: 10.1038/459518a. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez-Calvo T, Sabouri S, Anquetil F, von Herrath MG. The viral paradigm in type 1 diabetes: Who are the main suspects? Autoimmunity reviews. 2016;15:964–9. doi: 10.1016/j.autrev.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 3.de Beeck AO, Eizirik DL. Viral infections in type 1 diabetes mellitus--why the beta cells? Nature reviews Endocrinology. 2016;12:263–73. doi: 10.1038/nrendo.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devendra D, Eisenbarth GS. Interferon alpha--a potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clinical immunology. 2004;111:225–33. doi: 10.1016/j.clim.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiological reviews. 2007;87:1377–408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 6.Cao SS, Kaufman RJ. Targeting endoplasmic reticulum stress in metabolic disease. Expert opinion on therapeutic targets. 2013;17:437–48. doi: 10.1517/14728222.2013.756471. [DOI] [PubMed] [Google Scholar]
- 7.Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. The FEBS journal. 2016;283:2640–52. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. The Journal of biological chemistry. 2014;289:1203–11. doi: 10.1074/jbc.R113.534743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim MK, Kim HS, Lee IK, Park KG. Endoplasmic reticulum stress and insulin biosynthesis: a review. Experimental diabetes research. 2012;2012:509437. doi: 10.1155/2012/509437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–27. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oslowski CM, Urano F. The binary switch that controls the life and death decisions of ER stressed beta cells. Current opinion in cell biology. 2011;23:207–15. doi: 10.1016/j.ceb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. 2016;351:711–4. doi: 10.1126/science.aad2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Zhang XQ, Dinney CN, Benedict WF. Direct cytotoxicity produced by adenoviral-mediated interferon alpha gene transfer in interferon-resistant cancer cells involves ER stress and caspase 4 activation. Cancer gene therapy. 2011;18:609–16. doi: 10.1038/cgt.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell death and differentiation. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 15.Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–97. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lombardi A, Inabnet WB, 3rd, Owen R, Farenholtz KE, Tomer Y. Endoplasmic reticulum stress as a novel mechanism in amiodarone-induced destructive thyroiditis. J Clin Endocrinol Metab. 2015;100:E1–10. doi: 10.1210/jc.2014-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Vitis S, Sonia Treglia A, Ulianich L, Turco S, Terrazzano G, Lombardi A, et al. Tyr phosphatase-mediated P-ERK inhibition suppresses senescence in EIA + v-raf transformed cells, which, paradoxically, are apoptosis-protected in a MEK-dependent manner. Neoplasia. 2011;13:120–30. doi: 10.1593/neo.101152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HJ, Lombardi A, Stefan M, Li CW, Inabnet WB, 3rd, Owen RP, et al. CD40 SIGNALING IN GRAVES’ DISEASE IS MEDIATED THROUGH CANONICAL AND NON-CANONICAL THYROIDAL NF-kappaB ACTIVATION. Endocrinology. 2016:en20161609. doi: 10.1210/en.2016-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 20.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 22.Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon-alpha in transgenic mice. Science. 1993;260:1942–6. doi: 10.1126/science.8100367. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Hultgren B, Dybdal N, Stewart TA. Islet expression of interferon-alpha precedes diabetes in both the BB rat and streptozotocin-treated mice. Immunity. 1994;1:469–78. doi: 10.1016/1074-7613(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 24.Chakrabarti D, Hultgren B, Stewart TA. IFN-alpha induces autoimmune T cells through the induction of intracellular adhesion molecule-1 and B7. 2. Journal of immunology. 1996;157:522–8. [PubMed] [Google Scholar]
- 25.Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12439–44. doi: 10.1073/pnas.0806439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, McDevitt HO. The role of interferon alpha in initiation of type I diabetes in the NOD mouse. Clinical immunology. 2011;140:3–7. doi: 10.1016/j.clim.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Fabris P, Betterle C, Floreani A, Greggio NA, de Lazzari F, Naccarato R, et al. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis. Lancet. 1992;340:548. doi: 10.1016/0140-6736(92)91744-s. [DOI] [PubMed] [Google Scholar]
- 28.Bosi E, Minelli R, Bazzigaluppi E, Salvi M. Fulminant autoimmune Type 1 diabetes during interferon-alpha therapy: a case of Th1-mediated disease? Diabetic medicine : a journal of the British Diabetic Association. 2001;18:329–32. doi: 10.1046/j.1464-5491.2001.00492.x. [DOI] [PubMed] [Google Scholar]
- 29.Schreuder TC, Gelderblom HC, Weegink CJ, Hamann D, Reesink HW, Devries JH, et al. High incidence of type 1 diabetes mellitus during or shortly after treatment with pegylated interferon alpha for chronic hepatitis C virus infection. Liver international : official journal of the International Association for the Study of the Liver. 2008;28:39–46. doi: 10.1111/j.1478-3231.2007.01610.x. [DOI] [PubMed] [Google Scholar]
- 30.Eibl N, Gschwantler M, Ferenci P, Eibl MM, Weiss W, Schernthaner G. Development of insulin-dependent diabetes mellitus in a patient with chronic hepatitis C during therapy with interferon-alpha. European journal of gastroenterology & hepatology. 2001;13:295–8. doi: 10.1097/00042737-200103000-00015. [DOI] [PubMed] [Google Scholar]
- 31.Kose S, Gozaydin A, Akkoclu G, Ece G. Chronic hepatitis B with type I diabetes mellitus and autoimmune thyroiditis development during interferon alpha therapy. Journal of infection in developing countries. 2012;6:364–8. doi: 10.3855/jidc.1632. [DOI] [PubMed] [Google Scholar]
- 32.Guerci AP, Guerci B, Levy-Marchal C, Ongagna J, Ziegler O, Candiloros H, et al. Onset of insulin-dependent diabetes mellitus after interferon-alfa therapy for hairy cell leukaemia. Lancet. 1994;343:1167–8. doi: 10.1016/s0140-6736(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 33.Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nature medicine. 2013;19:65–73. doi: 10.1038/nm.3042. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, Yuang J, Goddard A, Foulis A, James RF, Lernmark A, et al. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. 1995;44:658–64. doi: 10.2337/diab.44.6.658. [DOI] [PubMed] [Google Scholar]
- 35.Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet. 1987;2:1423–7. doi: 10.1016/s0140-6736(87)91128-7. [DOI] [PubMed] [Google Scholar]
- 36.Allen JS, Pang K, Skowera A, Ellis R, Rackham C, Lozanoska-Ochser B, et al. Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes. 2009;58:138–45. doi: 10.2337/db08-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkler C, Lauber C, Adler K, Grallert H, Illig T, Ziegler AG, et al. An interferon-induced helicase (IFIH1) gene polymorphism associates with different rates of progression from autoimmunity to type 1 diabetes. Diabetes. 2011;60:685–90. doi: 10.2337/db10-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15040–4. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi WY, Cao C, Liu L. Interferon alpha Induces the Apoptosis of Cervical Cancer HeLa Cells by Activating both the Intrinsic Mitochondrial Pathway and Endoplasmic Reticulum Stress-Induced Pathway. International journal of molecular sciences. 2016:17. doi: 10.3390/ijms17111832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hostens K, Pavlovic D, Zambre Y, Ling Z, Van Schravendijk C, Eizirik DL, et al. Exposure of human islets to cytokines can result in disproportionately elevated proinsulin release. The Journal of clinical investigation. 1999;104:67–72. doi: 10.1172/JCI6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borjesson A, Carlsson C. Altered proinsulin conversion in rat pancreatic islets exposed long-term to various glucose concentrations or interleukin-1beta. The Journal of endocrinology. 2007;192:381–7. doi: 10.1677/joe.1.06676. [DOI] [PubMed] [Google Scholar]
- 42.Sun J, Cui J, He Q, Chen Z, Arvan P, Liu M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Molecular aspects of medicine. 2015;42:105–18. doi: 10.1016/j.mam.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Truyen I, De Pauw P, Jorgensen PN, Van Schravendijk C, Ubani O, Decochez K, et al. Proinsulin levels and the proinsulin:c-peptide ratio complement autoantibody measurement for predicting type 1 diabetes. Diabetologia. 2005;48:2322–9. doi: 10.1007/s00125-005-1959-0. [DOI] [PubMed] [Google Scholar]
- 44.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1796–801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell metabolism. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 46.Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes. 2011;60:918–24. doi: 10.2337/db10-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nature medicine. 2009;15:1383–91. doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Effect of IFNα on BiP mRNA in human islets and EndoC-βH1 cells. (A) Human islets and EndoC-βH1 cells were cultured in presence of 0.5 μM thapsigargin or 0.5 μg/ml tunicamycin or with the indicated concentrations of IFNα for 24 hours. (B) Human islets and EndoC-βH1 cells were cultured with 1000 U/ml IFNα (added in the medium every 24 hours) for the indicated times. The expression levels of mRNAs for BiP were determined by real-time RT-PCR analysis of total RNA from three different preparations of human islets or EndoC-βH1 cells treated as above. mRNA levels in treated cells are relative to those in vehicle-treated cells (CTRL). Bars represent means ± SEM from three independent experiments. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells.
Supplementary Fig. 2. Effect of IFNα-removal on bip mRNA and insulin production in human islets and EndoC-βH1 cells. (A–B) Human islets and EndoC-βH1 cells were vehicle treated (CTRL) or treated with 1000 U/ml IFNα for 48 hours followed by 6, 12, 24, and 48 hours in medium without IFNα. (A) BiP mRNA levels were determined by real-time RT-PCR analysis of total RNA from three different preparations of human islets or EndoC-βH1 cells treated as above. (B) Insulin content in human islets and EndoC-βH1 treated as above. Values shown represent the mean ± SEM of three independent experiments. *p < 0.001; **p < 0.01; ***p < 0.001 compared to CTRL cells.