

Abstract
Background and Purpose
Dexpramipexole, a drug recently tested in patients with amyotrophic lateral sclerosis (ALS,) is able to bind F1Fo ATP synthase and increase mitochondrial ATP production. Here, we have investigated its effects on experimental ischaemic brain injury.
Experimental Approach
The effects of dexpramipexole on bioenergetics, Ca2+ fluxes, electrophysiological functions and death were evaluated in primary neural cultures and hippocampal slices exposed to oxygen‐glucose deprivation (OGD). Effects on infarct volumes and neurological functions were also evaluated in mice following proximal or distal middle cerebral artery occlusion (MCAo). Distribution of dexpramipexole within the ischaemic brain was evaluated by means of mass spectrometry imaging.
Key Results
Dexpramipexole increased mitochondrial ATP production in cultured neurons or glia and reduces energy failure, prevents intracellular Ca2+ overload and affords cytoprotection when cultures are exposed to OGD. This compound also counteracted ATP depletion, mitochondrial swelling, anoxic depolarization, loss of synaptic activity and neuronal death in hippocampal slices subjected to OGD. Post‐ischaemic treatment with dexpramipexole, at doses consistent with those already used in ALS patients, reduced brain infarct size and ameliorated neuroscore in mice subjected to transient or permanent MCAo. Notably, the concentrations of dexpramipexole reached within the ischaemic penumbra equalled those found neuroprotective in vitro.
Conclusion and Implications
Dexpramipexole, a compound able to increase mitochondrial F1Fo ATP‐synthase activity reduced ischaemic brain injury. These findings, together with the excellent brain penetration and favourable safety profile in humans, make dexpramipexole a drug with realistic translational potential for the treatment of stroke.
Linked Articles
This article is part of a themed section on Inventing New Therapies Without Reinventing the Wheel: The Power of Drug Repurposing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.2/issuetoc
Abbreviations
- AD
anoxic depolarization
- ALS
amyotrophic Lateral Sclerosis
- DCD
delayed Ca2+ deregulation
- DEX
dexpramipexole
- MCAo
middle cerebral artery occlusion
- OGD
oxygen‐glucose deprivation
- PI
propidium iodide
Introduction
Stroke is among the most common causes of death worldwide (Mozaffarian et al., 2016), and there is urgent need to identify treatments to protect the ischaemic brain (Moskowitz et al., 2010). Although all clinical trials in stroke have failed, there are no obvious pathophysiological reasons that post‐ischaemic neuroprotection in stroke victims cannot be achieved by pharmacological interventions (Moskowitz, 2010). However, it is now acknowledged that drugs with realistic neuroprotective potential must target pathophysiological events occurring early after the ischaemic insult and shared by all the components of neurovascular unit (del Zoppo, 2006; Moskowitz et al., 2010). Inevitably, even chemicals endowed with these features may fail when tested in stroke patients for safety reasons. On this basis, attention is now moving to repositioning, for stroke treatment, drugs that have already been used clinically.
Compounds able to sustain energy dynamics within the ischaemic penumbra may represent attractive therapeutics for stroke therapy (George and Steinberg, 2015). Therefore, we focused on compounds able to counteract ischaemic energy failure, the earliest event within all the components of the ischaemic neurovascular unit, and upstream to the pathophysiological processes accompanying stroke injury. Among chemicals affecting cellular bioenergetics, we directed our attention to molecules able to support mitochondrial energy production. This is because mitochondria produce most of brain energy (Harris et al., 2012) and also prompt the necrotic and apoptotic programs when dysfunctional (Dirnagl et al., 1999).
This search led us to the identification of dexpramipexole (DEX). DEX is the R‐isomer of the antiparkinson drug pramipexole but with very low affinity for dopamine receptors. Interestingly, DEX has the unique ability to bind mitochondrial F1Fo‐ATP synthase and improve mitochondrial efficiency by concomitant increase of ATP synthesis and reduction of O2 consumption (Alavian et al., 2012; Alavian et al., 2015). Consistent with this, and with recent evidence that F1Fo‐ATP synthase participates in the supramolecular complex organization of the mitochondrial transition pore complex (Bonora et al., 2015; Jonas et al., 2015), DEX inhibits depolarizing membrane currents in dysfunctional mitochondria and prevents organelle swelling in conditions predisposing to pore opening (Cassarino et al., 1998; Sayeed et al., 2006). Remarkably, DEX also detoxifies mitochondrial reactive oxygen species and reduces cell death in in vitro models of neurotoxicity (Cassarino et al., 1998; Danzeisen et al., 2006; Ferrari‐Toninelli et al., 2010). Because of these cytoprotective properties, the drug has been recently tested in patients affected by amyotrophic lateral sclerosis (ALS). Unfortunately, however, the positive results of aPhase II study were not confirmed by a large Phase III trial. Still, the drug showed a very favourable safety profile in patients treated with daily doses of 300 mg for almost one year (Cudkowicz et al., 2011; Cudkowicz et al., 2013). Crucially, DEX readily crosses the blood brain barrier and accumulates within the brain (brain/plasma ratio > 15) (Bozik et al., 2011; Cheah and Kiernan, 2010).
In spite of these favourable mitochondrial actions and cytoprotective properties, as well as pharmacokinetics and safety profiles, there is no information on whether DEX affects bioenergetics derangement and cell fate of neurons exposed to hypoxia/ischaemia. Thus, to understand whether DEX might be of relevance to stroke treatment, in the present study, we tested its effects in different models of ischaemic neurodegeneration.
Methods
Animals
All animal care and experimental procedures were conducted according to the European Community Guidelines for Animal Care, DL 116/92, and application of the European Communities Council Directive (86/609/EEC) and approved by the Committee for Animal Care and Experimental Use of the University of Florence. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). C57/Bl6 male mice (20–25 g) or SD male rats (250–300 g) from Charles River (Calco, Italy) were used for all experiments. Animals had free access to food and water and were kept under a 12 h light/dark cycle in a well‐ventilated room at an approximate temperature of 22°C.
Primary cultures and OGD exposure
Neuronal/astrocytes cultures were prepared from rat embryos (E‐17/E‐19) or pups (P‐1/P‐2), as reported (Chiarugi et al., 2003). Briefly, the cerebral cortex was minced using medium stock (MS) (Eagle's minimal essential medium with Earle's salts, glutamine‐ and NaHCO3‐free, NaHCO3 38 mM, glucose 22 mM, penicillin 100 U·mL−1 and streptomycin 100 µg·mL−1) and then incubated for 10 (neurons) and 45 min (astrocytes) at 37°C in MS supplemented with 0.25% trypsin and 0.05% DNase. Enzymic digestion was terminated by incubation (10 min at 37°C) in MS supplemented with 10% heat‐inactivated horse serum (HIHS) and 10% FBS. Following tissue mechanical disruption, cells were counted and plated. For mixed cortical cell cultures, neurons were re‐suspended at a density of 4 × 105 cells·mL−1 and plated in 15 mm multiwell on a layer of confluent astrocytes using MS supplemented with 10% HIHS, 10% FBS and 2 mM glutamine. After 4–5 days in vitro, non‐neuronal cell division was halted by the application of 3 µM cytosine arabinoside for 24 h. Cell cultures were subjected to oxygen‐glucose deprivation (OGD) in the presence or absence of DEX in a serum‐ and glucose‐free medium saturated with 95% N2 and 5% CO2. Following 2 h of incubation at 37°C in an anoxic chamber, the cultures were transferred to oxygenated serum‐free medium (75% Eagle's minimal essential medium; 25% Hank's balanced salt solution; 2 mM l‐glutamine; 3.75 µg·mL−1 amphotericin B; and 5 mg·mL−1 glucose) and returned to normoxic conditions in the presence or absence of DEX. Propidium iodide (PI) fluorescence was evaluated 24 h later.
Evaluation of ATP
ATP contents in cell cultures and organotypic hippocampal slices have been quantified by means of the ATPlite Kit (Perkin‐Elmer, Milan, Italy) as previously described (Cipriani et al., 2005). The ATP contents within the ischaemic brain tissue have been evaluated by means of the above mentioned kit in brain cortex extracts obtained by tissue sonication in lysis buffer of the kit. Mitochondrial ATP production was evaluated by means of mitochondria‐targeted luciferase as described (Cipriani et al., 2005). Light emission was evaluated 48 h after transfection by 5 min incubation with 100 µM luciferin dissolved in growth medium, followed by 1 min luminometric analysis. Mit‐luciferase‐transfected cells were exposed or not to DEX 10 µM for 6 h prior to luciferin exposure.
Ca2+ imaging in cultured hippocampal neurons
Cultured hippocampal neurons obtained as described (Chiarugi, 2002) were incubated in a solution containing (mM) the following: 140 NaCl, 2 CaCl2, 1 MgCl2, 3 KCl, 10 HEPES, 10 D(+)glucose and pH 7.3 at room temperature for 30 min with fluo‐3 acetoxymethyl (AM) ester (fluo‐3 AM, 10 µg·mL−1; Molecular Probes, Milano, Italy). Coverslips were transferred to a perfused (1 mL·min−1) microscope chamber. Images were visualized with a 20X/0.5W Fluor objective (Nikon) and acquired every 3 s. Exposure time was set to 50 ms (ex 450–490 nm, em 510–560 nm) using a Photometrics Coolsnap HP Camera. Fluorescence intensity was measured using Imaging Workbench 6 software (Indec BioSystem, Los Altos, CA, USA) and expressed as the ratio of (F‐F0)/F0, where F0 is the fluorescence intensity before OGD.
OGD and glutamate excitotoxicity in organotypic hippocampal slices
Organotypic hippocampal slice cultures were prepared as previously reported (Pellegrini‐Giampietro et al., 1999a , b). Briefly, hippocampi were removed from the brains of C57Bl mice or SD rat pups, sliced (420 µm) and transferred onto 30‐mm‐diameter semiporous membranes inserts (Millicell‐CM PICM03050; Millipore, Italy), placed in 6‐well tissue culture plates containing 1.2 mL medium per well. Slices were subjected to OGD as previously described (Pellegrini‐Giampietro et al., 1999a , b). After OGD, hippocampal slices were cultured for an additional 24 h in a fresh serum‐free medium (with or without DEX) and then evaluated for CA1 pyramidal cell injury. To induce glutamate excitotoxicity, slices were exposed to glutamate (1 mM per·6 h), then washed and exposed or not to DEX. Neuronal injury was evaluated 18 h later by PI fluorescence analysis.
Electron microscopy
Electron microscopy was conducted as described (Moroni et al., 2001). Briefly, slices were washed with cold 0.01 M phosphate‐buffered saline, pH 7.4, and were directly fixed in cold 4% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) overnight at 4°C and post‐fixed in cold 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 1 h at RT. The samples were dehydrated in graded acetone, passed through propylene oxide and embedded in Epon 812. Ultrathin sections were stained with uranyl acetate and alkaline bismuth subnitrate and examined under a JEM 1010 electron microscope (Jeol, Tokyo, Japan) at 80 kV.
Acute hippocampal slice preparation and OGD exposure
Acute hippocampal slices were prepared from male SD rats (Charles River, Calco, Italy, 150–200 g) as described (Pugliese et al., 2009). Hippocampi were removed and placed in ice‐cold oxygenated artificial CSF of the following composition (mM): NaCl 125, KCl 3, NaH2PO4 1.25, MgSO4 1, CaCl2 2, NaHCO3 25 and D‐glucose 10. Slices of 400 mm were prepared and kept in oxygenated aCSF for at least 1 h at RT. A single slice was then placed on a nylon mesh, completely submerged in a small chamber (0.8 mL) and superfused with oxygenated aCSF (31–32°C) at a constant flow rate of 1.5–1.8 mL·min−1. Under OGD condition, the slice was superfused with aCSF without glucose and gassed with 95%N2–5% CO2. This caused a drop in pO2 in the recording chamber from ~500 mmHg (normoxia) to a range of 35–75 mmHg (after 7 min OGD). (Pugliese et al., 2003) At the end of the ischaemic period, the slice was again superfused with normal, glucose‐containing, oxygenated aCSF. Control slices were not subjected to OGD or drug treatment but were incubated in oxygenated aCSF for identical time intervals. Hippocampal slices were (i) incubated for at least 1 h before electrophysiological recordings in the presence of DEX, which was maintained throughout the experiments or (ii) superfused in the presence of DEX at least 30 min before and after OGD application.
Extracellular recording
Test pulses (80 ms, 0.066 Hz) were delivered through a bipolar nichrome electrode positioned in the stratum radiatum. Evoked extracellular potentials were recorded with glass microelectrodes (2–10 MΩ, Harvard Apparatus LTD, Edenbridge, UK) filled with 150 mM NaCl and placed in the CA1 region of the stratum radiatum. Responses were amplified (BM 622, Mangoni, Pisa, Italy), digitized (sample rate, 33.33 kHz) and stored for later analysis with LTP (version 2.30D) program. Stimulus–response curves were obtained by gradual increases in stimulus strength at the beginning of each experiment, when a stable baseline of evoked response was reached. The test stimulus pulse was then adjusted to produce a field EPSP whose slope and amplitude was 40–50% of the maximum and was kept constant throughout the experiment. The fEPSP amplitude was routinely measured and expressed as the percentage of the average amplitude of the potentials measured during the 5 min preceding exposure of the hippocampal slices to OGD. Anoxic depolarization (AD) was recorded as negative extracellular direct current (d.c.) shifts induced by 7 or 30 min of OGD. The d.c. potential is an extracellular recording considered to provide an index of the polarization of cells surrounding the tip of the glass electrode (Pugliese et al., 2009).
Transient and permanent MCAo induction and neuroscore evaluation
A total of 207 C57/Bl6 male mice (20–25 g) or a total of 22 SD male rats (250–300 g) from Charles River (Calco, Italy) were randomized and subjected to transient or permanent middle cerebral artery occlusion (MCAo). Two different species of rodents were used in order to rule out the possibility of species‐specific pharmacological effects. Rodents were anaesthetized (1.5% isoflurane in 70% nitrous oxide and 30% oxygen), and regional cerebral blood flow was measured by Laser‐Doppler (PF2B; Perimed, Stockholm, Sweden). In randomly selected animals, the left femoral artery was cannulated with a PE‐10 polyethylene tube for blood gas determination. Rectal temperature was maintained between 36.5 and 37.5°C with a homeothermic blanket. Middle cerebral artery was occluded proximally for 1 h using the intraluminal filament technique (tMCAo) (Muzzi et al., 2012) or distally by cauterization (dMACo) (Faraco et al., 2007). The number of mice per group was for tMCAo n = 8 (saline) and n = 9 (DEX); for ATP content within ischaemic brain n = 8 per group; for neuroscore and weight evaluation n = 14 per group; for survival analysis n = 30 (saline) and n = 28 (DEX); for dMCAo n = 21 per group (DEX at reperfusion); and n = 23 per group (DEX post‐treatment). The number of tMCAo rats was 11 per group. DEX was i.p. injected bid after reperfusion or cauterization, randomly, in a double‐blinded manner. In post‐treatment experiments, DEX was injected starting 1 h after artery cauterization. Animals were killed 48 h later and the brains snap‐frozen in N2 vapour for cryostat sectioning. Infarction areas were quantitated, without knowledge of the treatments, by means of MCID M4 image analysis software on toluidine blue‐stained sections. Infarction volumes were calculated as reported (Muzzi et al., 2012). Neuroscore evaluation was performed, blindly, by examining different sensorimotor functions, as reported (Garcia et al., 1995).
Imaging mass spectrometry
Imaging mass spectrometry (MS) analysis was conducted as described (Reyzer et al., 2003). Brains were rapidly snap frozen into 80°C isopentane, cut (6 µm) and placed on glass slides. Brain sections were placed into a vacuum desiccator (1 h) and then coated with MALDI matrix using an automated device (ImagePrep, Bruker Daltonics, Bremen, Germany). To increase analyte extraction from tissues and uniformity of coating, the previously described (Gimenez et al., 2010) ionic liquid matrix mixture was used. ImagePrep matrix application method was in‐house edited and tested in order to ensure a uniform coating and reduce analyte delocalization on the tissue. MS experiments were performed in a MALDI LTQ‐Orbitrap XL (Thermo Scientific, San Jose, CA, USA) equipped with a 337 nm nitrogen laser operating at 60 Hz. Optimal MS/MS conditions for DEX quasi‐molecular ions were achieved using the linear ion trap for ion isolation and collision‐induced dissociation (CID) and the Orbitrap analyzer for detecting product ions, working in positive ion mode at 60 000 resolution. CID energy was set at 35%, and the precursor ion isolation window was 2 m/z. For all the imaging experiments, the MALDI source worked at 10 µJ laser power in raster mode (spatial resolution 50 µm), accumulating 30 laser shots for the MS/MS spectrum in every position of the tissue. Tissue drug concentrations were calculated as described (Porta et al., 2015). Briefly, two sections from an untreated brain were placed onto the same glass slide and spotted with different content of DEX in 0,5 µL, to obtain a standard curve. The software Thermo Image Quest 1.0 (Thermo Scientific) was used for data processing. Comparison of treated and untreated samples was conducted by rescaling images to the same raw intensity value.
Synthesis of Fluo‐DEX
Fluo‐DEX [(±)‐5‐dimethylamino‐naphthalene‐1‐(2‐amino‐4,5,6,7‐tetrahydro‐benzothiazol‐6‐yl)‐sulfonamide] was synthesized by suspending the compound [(±)‐2,6‐diamino‐4,5,6,7‐tetrahydrobenzothiazole hydrochloride] in dichloromethane together with dansyl chloride using triethylamine as base. The reaction mixture was stirred at room temperature for 2 h, and the reaction progress was monitored with TLC. After purification, with flash chromatography and a further trituration with a mixture of diethyl ether and n‐hexane, Fluo‐DEX was obtained as pale yellow pure solid, in about 40% yield. Molecular structure and purity was checked by recording 1H NMR and 13C NMR spectra at 400 and 100.6 MHz respectively. All reagents and solvents were from commercial sources.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All numerical data are expressed as mean ± SEM. Data were analysed using WinLTP 1.11 reanalysis program and the software package GRAPHPAD PRISM (version 4.0; GraphPad Software, San Diego, CA, USA). Statistical significance was evaluated using paired two‐tailed Student's t‐test or one‐way ANOVA plus Tukey's post hoc test, if F achieved the necessary level of statistical significance (P < 0.05) and there was no significant variance inhomogeneity, Differences between means were considered significant when P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/2016 (Alexander et al., 2015a,b).
Results
DEX increases mitochondrial ATP production and resistance to in vitro ischaemia of primary cultures of neurons or glia
In contrast with the notion that DEX interacts with mitochondrial F1Fo ATP synthase and with its use in large clinical trials with hundreds of ALS patients, there is little information on the effects of this compound on neural cell bioenergetics. We therefore first asked whether DEX improves energy dynamics of primary cultures of neural cells. Cultures of pure cortical neurons or glia from mice were therefore exposed to the drug and intracellular ATP concentrations measured by different means. We found that DEX increased ATP content in both types of cultures (Figure 1A, B). Of note, the ATP increase was blunted in the presence of the mitochondrial ATP synthase inhibitor oligomycin but still present when glycolysis was suppressed by glucose starvation in the presence of the mitochondrial substrates pyruvate and glutamine in glial cells (Figure 1C). These findings suggested that DEX increases energy production in neural cells by promoting mitochondrial ATP production. To confirm this hypothesis, we next monitored ATP production within mitochondria of living neurons or astrocytes by means of a mitochondrially targeted luciferase as sensor of ongoing mitochondrial ATP synthesis. Again, we found that preincubation with DEX increased photon emission of transfected neurons or astrocytes (Supporting Information Figure S1). Next, to further corroborate evidence that the drug improves cellular bioenergetics by direct interaction with mitochondria, we synthesized a fluorescent DEX analogue (see Methods and Supporting Information Figure S2) and followed its intracellular distribution. Notably, the intraneuronal distribution pattern of the DEX derivative perfectly matched that of tetramethylrhodamine, ethyl ester, a prototypical mitochondrial marker, and the intensity of the fluorescent signal, as well as mitochondrial staining, was decreased by the concomitant presence of unlabelled DEX (Figure 1D). This latter finding indicates that the fluorescent moiety does not affect the cellular distribution of DEX. These findings taken together are in good agreement with the ability of DEX to bind F1Fo ATP synthase (Alavian et al., 2015). Prompted by the ability of DEX to increase mitochondrial ATP production, we then wondered whether the drug could counteract energy derangement during an ischaemic insult. We found that DEX reduced ATP loss in mixed cortical cell cultures exposed to in vitro ischaemia. The drug also improved energy recovery upon re‐exposure of cultures to nutrients and normoxia (Figure 1E). To understand whether the improved bioenergetics of cultured neurons during OGD had a functional correlate, we monitored their Ca2+ homeostasis. It is well known, indeed, that delayed Ca2+ deregulation (DCD) occurs downstream of energy failure in ischaemic neurons and is mainly due to ATP loss‐dependent massive extracellular Ca2+ influx and exhaustion of mitochondrial Ca2+ buffering capacity (Nicholls et al., 2007). Consistent with the effects of DEX on mitochondrial bioenergetics, we found that the drug reduced both the extent of intracellular Ca2+ increase and the number of cells undergoing DCD in cultured primary neurons exposed to OGD (Figure 1F–H). Importantly, in keeping with the causative role of DCD in ischaemic neurodegeneration, DEX reduced cell death of both primary neuronal or glial cultures exposed to OGD (Supporting Information Figure S3).
Figure 1.
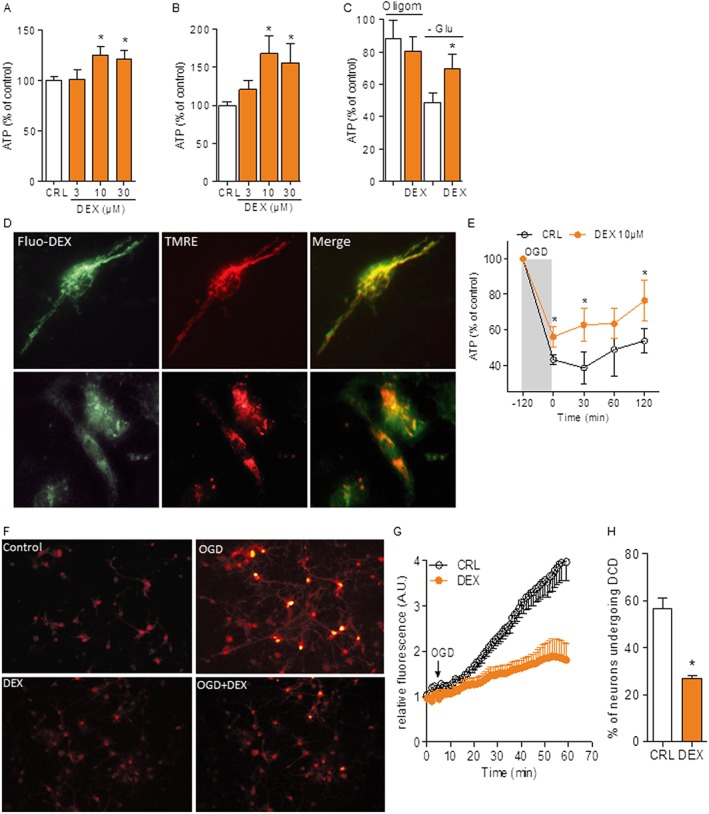
DEX improves neural cell bioenergetics and resistance to OGD. ATP contents in resting mouse cultures of pure cortical neurons (A) or glial cells (B) after a 6 h exposure to different DEX concentrations. Values of ATP under control condition were 8.3 ± 2.21 and 1.1 ± 0.54 nmol·mg protein−1 for neurons and glia respectively. (C) Effects of the F1Fo ATP synthase inhibitor oligomycin (10 µM) or glucose starvation in the presence of pyruvate and glutamine on ATP content in glial cells exposed to DEX (10 µM). (D) Representative images of the intracellular distribution of 10 µM Fluo‐DEX (green) in the absence or presence of unlabelled 100 µM DEX and the potentiometric mitochondrial dye TMRE (red) in hippocampal neurons in culture. (E) ATP contents in cultures of mixed cortical cells during OGD and after returning to normoxia and normal growth medium. Value of ATP under control condition was 10.5 ± 1.82 nmol·mg protein−1. (F) Visualization of intracellular Ca2+ contents in primary cultures of hippocampal neurons exposed to OGD. DEX was added to the culture 10 min before OGD. The temporal pattern of Ca2+ increase (G) and percentage of neurons undergoing DCD (H) are also shown. Data represent the mean ± SEM of at least three experiments. *P < 0.05, significantly different from corresponding control (CRL); ANOVA plus Tukey's post hoc test.
DEX prevents mitochondrial swelling and death in organotypic hippocampal slices exposed to in vitro ischaemia or excitotoxicity
We then investigated whether the effects of DEX obtained in dissociated cell cultures were also reproducible in a more integrated neural network. To this end, we exposed organotypic hippocampal slices to DEX and found that it increased ATP content under basal conditions (Figure 2A) and prevented OGD‐dependent ATP depletion (Figure 2B). Again, this early effects on ATP content correlated with the drug's ability to confer delayed cytoprotection of CA1 neurons in hippocampal slices exposed to OGD (Figure 2C, D). In light of the original mechanism of action of the drug, a key point was to understand whether DEX‐dependent neuroprotection must be ascribed to an increased ability of neurons to withstand the ischaemic insult or, alternatively, to counteract delayed neurodegenerative processes prompted by the OGD challenge. To answer this question, we analysed ultrastructural integrity of CA1 neurons immediately after OGD. Strikingly, we found that the prototypical swelling of both somata and mitochondria of CA1 neurons at the end of the OGD insult was abolished by DEX (Figure 2E). Maintenance of organelle integrity is in keeping with the ability of DEX to inhibit depolarizing membrane currents and swelling of dysfunctional mitochondria (Cassarino et al., 1998; Sayeed et al., 2006). Ischaemic neuroprotection occurred even when a post‐treatment window of 1 h was adopted (Figure 2F). Notably, post‐treatment with DEX also protected CA1 neurons from glutamate‐induced cell death (Supporting Information Figure S4), consistent with the causal role of mitochondrial energy failure and deregulation of Ca2+ homeostasis in excitotoxicity (Nicholls et al., 2007). Overall, data indicate that DEX improved bioenergetics and survival of ischaemic neural cells.
Figure 2.
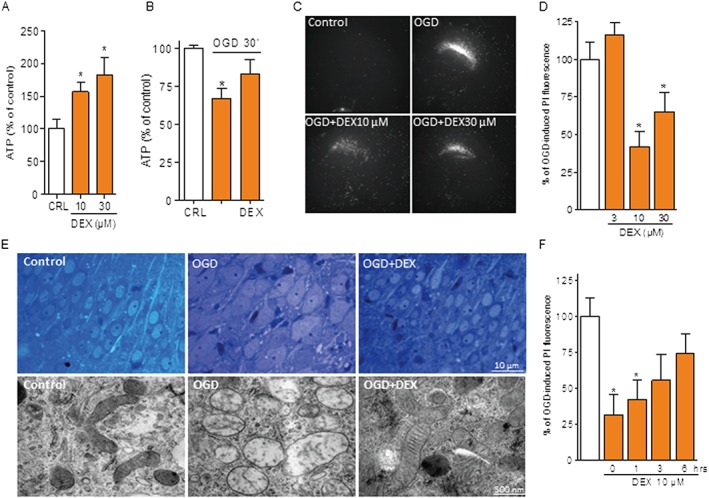
DEX protects hippocampal slices from OGD‐dependent neurodegeneration. ATP content of organotypic hippocampal slices 6 h after different concentrations of DEX (A) or immediately after OGD (30 min) in the absence or presence of 10 µM DEX added 10 min before OGD (B). Value of ATP under control condition was 14.8 ± 3.51 nmol·mg protein−1. Representative images (C) and quantification (D) of the effect of different concentrations of DEX (added at the end of the OGD insult) on cell death (revealed by PI staining) of CA1 neurons of organotypic hippocampal slices exposed to 30 min OGD/24 h reoxygenation. (E) DEX (10 µM 10 min before OGD) prevents swelling of somata of hippocampal CA1 neurons and their mitochondria in organotypic hippocampal slices exposed to 30 min OGD. (F) Effect of different post‐treatment windows with 10 µM DEX on cell death (revealed by PI staining) of CA1 neurons of organotypic hippocampal slices exposed to 30 min OGD/24 h reoxygenation. Data represent the mean ± SEM of at least three experiments. *P < 0.05, significantly different from corresponding control (CRL); ANOVA plus Tukey's post hoc test.
DEX improves resistance to anoxic depolarization and preserves synaptic transmission during OGD
Because of the ability of DEX to sustain neuronal energy dynamics and ion homeostasis during the ischaemic insult, we next investigated whether the drug also affects OGD‐dependent AD and loss of synaptic activity, two typical events within the ischaemic brain tissue almost concomitant to energy failure (Somjen, 2001). Remarkably, DEX fully prevented AD in the CA1 region of hippocampal slices exposed to 7 min OGD (Figure 3A). When slices were exposed to a stronger insult by extending OGD to 30 min, DEX still delayed and reduced amplitude of AD even though it was unable to prevent it (Figure 3B). We then tested DEX in the 30 min OGD protocol under hypothermic conditions (30°C) in order to cause a reduced metabolic demand of the ischaemic tissue. Interestingly, we found that DEX prevented AD in about half (three out of seven) of the hypothermic hippocampal slices exposed to the 30 min OGD insult (Figure 3C, D). Of note, we also found that DEX fully prevented loss of neurotransmission that typically occurs in slices reperfused after 7 min OGD (Figure 3E). When tested in the 30 min OGD model the drug did not restore synaptic activity (Figure 3F) but prompted partial, transient recovery under hypothermic conditions (Figure 3G). Taken together, electrophysiological data indicate that DEX counteracted depolarization of ischaemic brain tissue and ensuing loss of neurotransmission, in good agreement with its ability to sustain bioenergetics and reduce neurodegeneration in cultured neural cells and hippocampal slices exposed to OGD.
Figure 3.
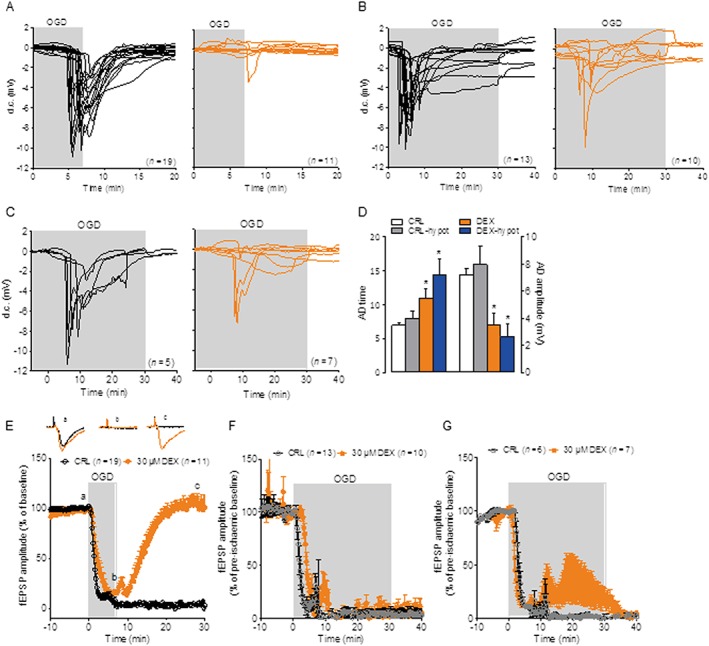
DEX protects hippocampal slices from OGD‐dependent anoxic depolarization. (A) AD (expressed as d.c. shift) occurring in acute hippocampal slices exposed to 7 min OGD is prevented by co‐incubation with DEX (30 µM, red traces). (B) AD occurring in acute hippocampal slices exposed to 30 min OGD is not prevented by co‐incubation with DEX (30 µM). (C) DEX suffices to prevent AD in three out of seven slices exposed to 30 min OGD under hypothermic conditions (30°C bath temperature). (D) Effect of 30 µM DEX on AD time and amplitude in slices exposed to 30 min OGD under normothermic or hypothermic (30°C) conditions. (E) Neurotransmission (measured as filed EPSP) in the CA1 region of acute hippocampal slices exposed to 7 min OGD is irreversibly lost in control slices but fully restored upon reoxygenation in those incubated with DEX 30 µM. DEX does not prevent loss of neurotransmission in slices exposed to 30 min OGD (F) but leads to partial and transient recovery under hypothermic conditions (30°C) (G). Data represent the mean ± SEM of at least three experiments. *P < 0.05, significantly different from corresponding control (CRL); ANOVA plus Tukey's post hoc test.
Post‐ischaemic treatment with DEX reduces brain infarcts in mice exposed to transient or permanent MCAo
The original pharmacodynamic properties shown by DEX in in vitro models of cerebral ischaemia prompted us to test whether it also provides ischaemic neuroprotection in vivo. To this end, we first adopted a model of transient ischaemia (tMCAo) and doses of DEX (3 mg·kg−1, i.p. twice daily) consistent with those already used and well tolerated by ALS patients (150 mg bid) (Cudkowicz et al., 2011; Cudkowicz et al., 2013). As shown in Figure 4A–C, DEX administered twice daily starting at reperfusion reduced ischaemic brain injury in mice subjected to tMCAo. To confirm the ability of DEX to support mitochondrial bioenergetics, we analysed ATP content within the ischaemic penumbra early (3 h) after reperfusion. Data shown in Figure 4D indicate that energy content of the ischaemic brain tissue was higher in mice treated with DEX. We then prolonged the same dosing up to 7 days and evaluated neuroscore of tMCAo mice for 1 month. Remarkably, DEX provided functional neuroprotection, leading to significant recovery of sensorimotor functions (Figure 4E, F). The drug, however, only marginally improved post‐stroke weight recovery (Figure 4G) and showed a tendency to reduce mortality that did not reach statistical significance (Figure 4H). In order to corroborate the therapeutic potential of DEX for stroke treatment, we then tested the drug in a mouse model of permanent brain ischaemia that, compared with a transient ischaemic insult, more closely reproduces pathophysiology and clinical outcomes of human stroke (Macrae, 2011). To this end, we adopted the treatment protocol used in the transient model (3 mg·kg−1 i.p. twice daily) and subjected mice to distal MCAo (Figure 4I–K). We found that DEX reduced injury of permanent brain ischaemia when treatment started either immediately or even 1 h after artery cauterization (Figure 4L, M). Remarkably, the drug afforded ischaemic neuroprotection even in rats subjected to permanent brain ischaemia (Figure 4N–P), thereby indicating that DEX‐dependent ischaemic neuroprotection in vivo is not species specific.
Figure 4.
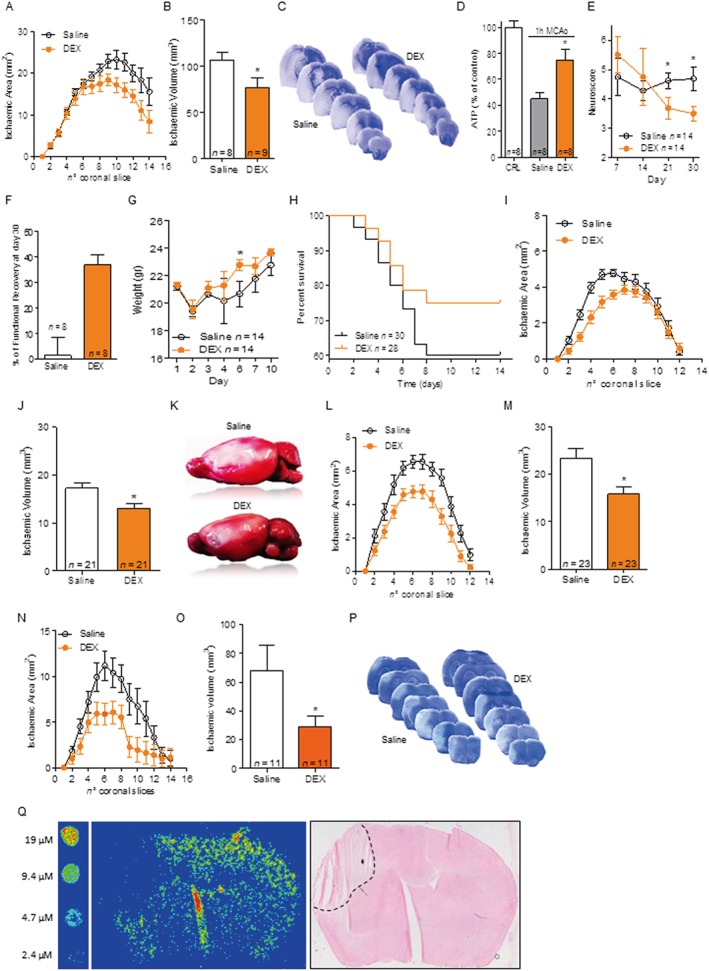
DEX is functionally neuroprotective in experimental stroke models. DEX (3 mg·kg−1 i.p.) administered at reperfusion and then every 12 h reduces infarct areas (A) and volumes (B) of mice subjected to 1 h MCAo/48 h reperfusion. (C) Visualization of infarct distribution (pale areas) in toluidine blue‐stained coronal brain slices of representative saline‐ or DEX‐treated mice. (D) Content of ATP in the brain of control mice (10.3 ± 4.06 nmol·mg protein−1) and in the penumbra of ischaemic (1 h MCAo) brain 3 h after reperfusion in mice treated or not with DEX 3 mg·kg−1 i.p. Effect of a 7 day treatment with DEX (3 mg·kg−1 i.p. bid) on neuroscore (E) and functional recovery (F) over 1 month in mice subjected to 1 h MCAo. Effect of DEX on weight recovery (G) and survival (H) compared with saline treatment in mice subjected to 1 h MCAo. Effect of DEX (3 mg·kg−1 i.p. bid, first dose immediately after artery occlusion) on infarct areas (I) and volumes (J) of mice subjected to permanent MCAo (distal MCA cauterization). (K) Visualization by TTC staining of the extent of cortical infarct (pale area) in representative brains of saline‐ or DEX‐treated mice. Effect of a 1 h post‐treatment with DEX (3 mg·kg−1 bid) on cortical infarct areas (L) or volumes (M) of mice subjected to permanent MCAo. Effect of DEX (3 mg·kg−1 i.p. bid, first dose immediately after artery occlusion) on infarct areas (N) and volumes (O) of rats subjected to permanent MCAo. (P) Visualization of infarct distribution (pale areas) in toluidine blue‐stained coronal brain slices of representative saline‐ or DEX‐treated rats. (Q) Pseudocolour visualization by means of imaging mass spectrometry of DEX distribution in a mouse brain subjected to 48 h distal MCAo cauterization and 1 h treatment with 3 mg·kg−1 DEX i.p. The pseudocolour intensity of standard concentrations spotted on a control brain slice is shown on the left. Note that the increased signal in the slice fracture is an artefact due to analyte delocalization (see Methods). Haematoxylin and eosin staining of the slice analysed by mass spectrometry is shown on the right. The dotted line delimits the infarct boundary. Data represent the mean ± SEM of at least three experiments. *P < 0.05, significantly different from saline; Student's t‐test or ANOVA plus Tukey's post hoc test.
It is known that DEX readily crosses the blood brain barrier and accumulates within the brain compartment (Danzeisen et al., 2006; Bozik et al., 2011). Thus, to understand whether DEX reaches mouse brain concentrations consistent with those found able to provide neuroprotection in vitro, we took advantage of imaging mass spectrometry that allows determining tissue drug contents and distribution. We found that when injected at doses providing ischaemic neuroprotection in vivo (3 mg·kg−1), DEX was evenly distributed in the brain and penumbra of ischaemic mice, reaching brain concentrations in the range of 10–20 µM (Figure 4Q), which equal those we found neuroprotective in OGD experiments.
Discussion
We report here that DEX shows a selective mitochondrial localization within the cell and improves mitochondrial ATP production, Ca2+‐handling capacity, energy recovery and survival of cultured neural cells exposed to OGD. Remarkably, we also show that DEX prompts neuroprotection in different models of experimental stroke. To our knowledge, this is the first evidence that DEX, which significantly advanced in clinical development, may be of therapeutic relevance to stroke treatment. It is also worth noting that the present study originally identifies F1Fo ATP synthase as a therapeutic target to reduce ischaemic brain injury. Although mechanisms in addition to increased F1Fo ATP synthase efficiency might have contributed to ischaemic neuroprotection by DEX, current evidence does not indicate any additional target of potential significance to neuroprotection. Even the very low affinity for dopamine D3 receptors (1000/10.000‐fold lower than that of pramipexole) (Cheah and Kiernan, 2010; Bozik et al., 2011) rules out a possible participation of these receptors, if any, to ischaemic neuroprotection. This is in keeping with the notion that DEX reduces neuronal cell death in vitro through mechanisms unrelated to dopamine receptor activation (Bozik et al., 2011). As for the bioenergetic effects of DEX, our findings with mitochondrial‐targeted luciferase represent an unequivocal demonstration of the drug's ability to increase mitochondrial ATP production. Data are in keeping with a recent report showing the binding of DEX to specific subunits of F1Fo ATP synthase (Alavian et al., 2015) and with our data showing the ability of the fluorescent DEX analogue to selectively localize into mitochondria. Further evidence that DEX improves mitochondrial bioenergetics comes from data of Figure 2E showing that mitochondrial integrity of CA1 neurons is preserved by DEX immediately after the OGD insult. This indicates the ability of the compound to target an early, detrimental event during the ischaemic insult, in keeping with its ability to improve activity of F1Fo ATP synthase and preserve ATP content of hippocampal slices exposed to OGD. Our interpretation is further strengthened by electrophysiological data showing that DEX prevents or reduces (according to insult duration) AD of hippocampal slices exposed to OGD. Indeed, these findings are a clear demonstration that DEX acts during the ischaemic insult conferring on the brain tissue the ability to maintain cellular membrane potentials in spite of reduced oxygen/nutrient supply. Given that loss of membrane potential is an early event immediately following energy depletion, our findings point to the ability of DEX to increase efficiency of mitochondrial ATP production as the pharmacodynamic mechanism underlying ischaemic neuroprotection. It is therefore tempting to speculate that within the ischaemic penumbra, DEX increases the efficiency of F1Fo ATP synthase (i.e. the capability of producing a higher amount of ATP at a certain degree of oxygen supply) above a given threshold sufficient to preserve membrane ion homeostasis, overall cellular bioenergetics and survival. To our knowledge, this is an unprecedented mechanism of ischaemic neuroprotection that may open new avenues for stroke and other ischaemic disorders. Also, our study may pave the way to development of compounds that more potently increase activity of F1Fo ATP synthase. In this regard, it is worth noting that drugs such as cyclosporin, which is also able to confer ischaemic neuroprotection by preventing mitochondrial derangement (Uchino et al., 2002), target mechanisms leading to permeability transition pore opening, rather than F1Fo ATP synthase activity. Even though we now know that different subunits of mitochondrial ATP synthase participate in the organization of the permeability transition pore complex (Bernardi et al., 2015; Bonora et al., 2015; Jonas et al., 2015) the mode of action of cyclosporin and DEX are substantially different with respect to the mechanisms operating during ischaemic neurodegeneration. Indeed, whereas a potentiation of mitochondrial ATP production by DEX can prevent almost all of the detrimental events prompted by the ischaemic insults, inhibition of mitochondrial permeability transition by cyclosporin is typically downstream from both energy failure and early necrotic or apoptotic signalling within the neurovascular unit. In this context, the ability of DEX to improve ATP production and survival not only of neurons but also of astrocytes (see Figure 1) may have well contributed to protect the ischaemic brain tissue and further underscores the drug's therapeutic potential as a neuroprotectant in stroke. Admittedly, DEX might improve neural cell bioenergetics by interacting with mitochondrial targets in addition to F1Fo ATP synthase such as the adenylate transporter or by inhibiting detrimental ATP‐consuming enzymes. Alongside the novel mechanism of ischaemic neuroprotection we report here, our study might have remarkable translational implications, although confirmation in gyrencephalic animals is needed. Indeed, DEX protects the brain from permanent ischaemia adopting a post‐treatment paradigm, two features that closely resemble the clinical setting of stroke treatment. Also, evidence that DEX reduces ischaemic brain injury at doses consistent with those very well tolerated in humans and reaches neuroactive concentrations within the ischaemic penumbra (i.e. pharmacokinetics/pharmacodynamics relationship) corroborates its translational potential to stroke treatment.
Author contributions
M.M., E.G., D.B., E.C., F.R., L.F., R.Z., L.T., D.G., M.F., E.C., A.M., D.E.P.‐G., G.M., D.B., A.M.P. and A.C. participated in the design of experiments and interpretation of results. M.M., E.G., D.B., E.C., F.R., R.Z., L.T., D.G., M.F. and A.M.P participated in the acquisition and analysis of data. M.M. and A.C. supervised the research and wrote the manuscript.
Conflict of interest
Authors have filed a patent application on the use of dexpramipexole for the treatment of cerebral ischaemia.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of DEX on mitochondrial ATP synthesis. Dex increases photon emission by primary cultures of neuronal (A) or glial (B) cells transfected with a mitochondria‐targeted luciferase. The effect of DEX (10 µM) was evaluated by luminometric detection. Light emission was evaluated 48 h after transfection by 5 min incubation with 100 µM luciferin dissolved in growth medium, followed by 1 min luminometric analysis. Each point represents the mean ± SEM of 2 (A) and 4 (B) experiments conducted in triplicate. *P < 0.05, **P < 0.01, versus CTRL. ANOVA plus Tukey's post hoc test.
Figure S2 Chemical structures of DEX and Fluo‐DEX. Chemical structures of Fluo‐DEX is shown. DEX structure is shown for comparison. See methods for chemical synthesis of Fluo‐DEX.
Figure S3 Effect of DEX on OGD‐dependent cell death in cultured neurons and glia. The effect of DEX (added 10 min before and during OGD) on cell death of cultured cortical neurons or glia exposed to 2 h OGD is shown. Cell death has been evaluated 24 h after OGD by means of PI staining and representative images are shown in (A). Fluorescence was quantified and expressed as arbitrary units for glial (B) and neuronal (C) cell cultures. Each bar represents the mean ± SEM of 4 experiments conducted in duplicate. ***P < 0.001, versus CTRL. ANOVA plus Tukey's post hoc test.
Figure S4 Effect of DEX on glutamate‐dependent excitotoxic cell death in organotypic hippocampal slices. The effect of DEX dissolved in growth medium on glutamate‐dependent cell death of CA1 neurons of organotypic hippocampal slices is shown. Cell death was evaluated 18 h after exposure to glutamate (1 mM per 6 h). DEX was added at the end of glutamate exposure at the indicated concentrations. CA1 excitotoxic cell death was quantified by means of PI staining. Representative images are shown in (A). Bars represent the mean ± SEM of 3 experiments conducted in quadruplicate. *P < 0.05, **P < 0.01 versus CTRL. ANOVA plus Tukey's post hoc test.
Acknowledgements
We gratefully acknowledge Pietro Franceschi (Biostatistics and Data Handling Research Group, Edmund Mach Foundation, Trento, Italy) for his advice and support on Imaging Mass Spectrometry. This study has been supported by grants from Italian Foundation for the Study of Amyotrophic Lateral Sclerosis (ARISLA) (HDACALS‐2), Regione Toscana Mitochondrial Health Projects 2009 and 2012 and Italian Foundation for Multiple Sclerosis 2014/R/6.
Muzzi, M. , Gerace, E. , Buonvicino, D. , Coppi, E. , Resta, F. , Formentini, L. , Zecchi, R. , Tigli, L. , Guasti, D. , Ferri, M. , Camaioni, E. , Masi, A. , Pellegrini‐Giampietro, D. E. , Mannaioni, G. , Bani, D. , Pugliese, A. M. , and Chiarugi, A. (2018) Dexpramipexole improves bioenergetics and outcome in experimental stroke. British Journal of Pharmacology, 175: 272–283. doi: 10.1111/bph.13790.
References
- Alavian KN, Dworetzky SI, Bonanni L, Zhang P, Sacchetti S, Mariggio MA et al. (2012). Effects of dexpramipexole on brain mitochondrial conductances and cellular bioenergetic efficiency. Brain Res 1446: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Dworetzky SI, Bonanni L, Zhang P, Sacchetti S, Li H et al. (2015). The mitochondrial complex V‐associated large‐conductance inner membrane current is regulated by cyclosporine and dexpramipexole. Mol Pharmacol 87: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Rasola A, Forte M, Lippe G (2015). The mitochondrial permeability transition pore: channel formation by F‐ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 95: 1111–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L et al. (2015). Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34: 1475–1486. [DOI] [PubMed] [Google Scholar]
- Bozik ME, Mather JL, Kramer WG, Gribkoff VK, Ingersoll EW (2011). Safety, tolerability, and pharmacokinetics of KNS‐760704 (dexpramipexole) in healthy adult subjects. J Clin Pharmacol 51: 1177–1185. [DOI] [PubMed] [Google Scholar]
- Cassarino DS, Fall CP, Smith TS, Bennett JP Jr (1998). Pramipexole reduces reactive oxygen species production in vivo and in vitro and inhibits the mitochondrial permeability transition produced by the parkinsonian neurotoxin methylpyridinium ion. J Neurochem 71: 295–301. [DOI] [PubMed] [Google Scholar]
- Cheah BC, Kiernan MC (2010). Dexpramipexole, the R(+) enantiomer of pramipexole, for the potential treatment of amyotrophic lateral sclerosis. IDrugs 13: 911–920. [PubMed] [Google Scholar]
- Chiarugi A (2002). Characterization of the molecular events following impairment of NF‐kappaB‐driven transcription in neurons. Brain Res Mol Brain Res 109: 179–188. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Meli E, Calvani M, Picca R, Baronti R, Camaioni E et al. (2003). Novel isoquinolinone‐derived inhibitors of poly(ADP‐ribose) polymerase‐1: pharmacological characterization and neuroprotective effects in an in vitro model of cerebral ischemia. J Pharmacol Exp Ther 305: 943–949. [DOI] [PubMed] [Google Scholar]
- Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, Chiarugi A (2005). Nuclear poly(ADP‐ribose) polymerase‐1 rapidly triggers mitochondrial dysfunction. J Biol Chem 280: 17227–17234. [DOI] [PubMed] [Google Scholar]
- Cudkowicz M, Bozik ME, Ingersoll EW, Miller R, Mitsumoto H, Shefner J et al. (2011). The effects of dexpramipexole (KNS‐760704) in individuals with amyotrophic lateral sclerosis. Nat Med 17: 1652–1656. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A et al. (2013). Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double‐blind, phase 3 trial. Lancet Neurol 12: 1059–1067. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzeisen R, Schwalenstoecker B, Gillardon F, Buerger E, Krzykalla V, Klinder K et al. (2006). Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2‐amino‐4,5,6,7‐tetrahydro‐6‐Lpropylamino‐benzathiazole dihydrochloride]. J Pharmacol Exp Ther 316: 189–199. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22: 391–397. [DOI] [PubMed] [Google Scholar]
- Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B et al. (2007). High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem 103: 590–603. [DOI] [PubMed] [Google Scholar]
- Ferrari‐Toninelli G, Maccarinelli G, Uberti D, Buerger E, Memo M (2010). Mitochondria‐targeted antioxidant effects of S(−) and R(+) pramipexole. BMC Pharmacol 10: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ (1995). Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26: 627–634. [DOI] [PubMed] [Google Scholar]
- George PM, Steinberg GK (2015). Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 87: 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez E, Benavente F, Barbosa J, Sanz‐Nebot V (2010). Ionic liquid matrices for MALDI‐TOF‐MS analysis of intact glycoproteins. Anal Bioanal Chem 398: 357–365. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D (2012). Synaptic energy use and supply. Neuron 75: 762–777. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Porter GA Jr, Beutner G, Mnatsakanyan N, Alavian KN (2015). Cell death disguised: The mitochondrial permeability transition pore as the c‐subunit of the F(1)F(O) ATP synthase. Pharmacol Res 99: 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae IM (2011). Preclinical stroke research – advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol 164: 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R et al. (2001). Poly(ADP‐ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ 8: 921–932. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA (2010). Brain protection: maybe yes, maybe no. Stroke 41: S85–S86. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C (2010). The science of stroke: mechanisms in search of treatments. Neuron 67: 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al. (2016). Executive summary: heart disease and stroke statistics – 2016 Update: a report from the American Heart Association. Circulation 133: 447–454. [DOI] [PubMed] [Google Scholar]
- Muzzi M, Felici R, Cavone L, Gerace E, Minassi A, Appendino G et al. (2012). Ischemic neuroprotection by TRPV1 receptor‐induced hypothermia. J Cereb Blood Flow Metab 32: 978–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Johnson‐Cadwell L, Vesce S, Jekabsons M, Yadava N (2007). Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res 85: 3206–3212. [DOI] [PubMed] [Google Scholar]
- Pellegrini‐Giampietro DE, Cozzi A, Peruginelli F, Leonardi P, Meli E, Pellicciari R et al. (1999a). 1‐Aminoindan‐1,5‐dicarboxylic acid and (S)‐(+)‐2‐(3′‐carboxybicyclo[1.1.1]pentyl)‐glycine, two mGlu1 receptor‐preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischemia. Eur J Neurosci 11: 3637–3647. [DOI] [PubMed] [Google Scholar]
- Pellegrini‐Giampietro DE, Peruginelli F, Meli E, Cozzi A, Albani Torregrossa S, Pellicciari R et al. (1999b). Protection with metabotropic glutamate 1 receptor antagonists in models of ischemic neuronal death: time‐course and mechanisms. Neuropharmacology 38: 1607–1619. [DOI] [PubMed] [Google Scholar]
- Porta T, Lesur A, Varesio E, Hopfgartner G (2015). Quantification in MALDI‐MS imaging: what can we learn from MALDI‐selected reaction monitoring and what can we expect for imaging? Anal Bioanal Chem 407: 2177–2187. [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Latini S, Corradetti R, Pedata F (2003). Brief, repeated, oxygen‐glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: role of adenosine receptors. Br J Pharmacol 140: 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese AM, Traini C, Cipriani S, Gianfriddo M, Mello T, Giovannini MG et al. (2009). The adenosine A2A receptor antagonist ZM241385 enhances neuronal survival after oxygen‐glucose deprivation in rat CA1 hippocampal slices. Br J Pharmacol 157: 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyzer ML, Hsieh Y, Ng K, Korfmacher WA, Caprioli RM (2003). Direct analysis of drug candidates in tissue by matrix‐assisted laser desorption/ionization mass spectrometry. J Mass Spectrom 38: 1081–1092. [DOI] [PubMed] [Google Scholar]
- Sayeed I, Parvez S, Winkler‐Stuck K, Seitz G, Trieu I, Wallesch CW et al. (2006). Patch clamp reveals powerful blockade of the mitochondrial permeability transition pore by the D2‐receptor agonist pramipexole. FASEB J 20: 556–558. [DOI] [PubMed] [Google Scholar]
- Somjen GG (2001). Mechanisms of spreading depression and hypoxic spreading depression‐like depolarization. Physiol Rev 81: 1065–1096. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino H, Minamikawa‐Tachino R, Kristian T, Perkins G, Narazaki M, Siesjo BK et al. (2002). Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol Dis 10: 219–233. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ (2006). Stroke and neurovascular protection. N Engl J Med 354: 553–555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of DEX on mitochondrial ATP synthesis. Dex increases photon emission by primary cultures of neuronal (A) or glial (B) cells transfected with a mitochondria‐targeted luciferase. The effect of DEX (10 µM) was evaluated by luminometric detection. Light emission was evaluated 48 h after transfection by 5 min incubation with 100 µM luciferin dissolved in growth medium, followed by 1 min luminometric analysis. Each point represents the mean ± SEM of 2 (A) and 4 (B) experiments conducted in triplicate. *P < 0.05, **P < 0.01, versus CTRL. ANOVA plus Tukey's post hoc test.
Figure S2 Chemical structures of DEX and Fluo‐DEX. Chemical structures of Fluo‐DEX is shown. DEX structure is shown for comparison. See methods for chemical synthesis of Fluo‐DEX.
Figure S3 Effect of DEX on OGD‐dependent cell death in cultured neurons and glia. The effect of DEX (added 10 min before and during OGD) on cell death of cultured cortical neurons or glia exposed to 2 h OGD is shown. Cell death has been evaluated 24 h after OGD by means of PI staining and representative images are shown in (A). Fluorescence was quantified and expressed as arbitrary units for glial (B) and neuronal (C) cell cultures. Each bar represents the mean ± SEM of 4 experiments conducted in duplicate. ***P < 0.001, versus CTRL. ANOVA plus Tukey's post hoc test.
Figure S4 Effect of DEX on glutamate‐dependent excitotoxic cell death in organotypic hippocampal slices. The effect of DEX dissolved in growth medium on glutamate‐dependent cell death of CA1 neurons of organotypic hippocampal slices is shown. Cell death was evaluated 18 h after exposure to glutamate (1 mM per 6 h). DEX was added at the end of glutamate exposure at the indicated concentrations. CA1 excitotoxic cell death was quantified by means of PI staining. Representative images are shown in (A). Bars represent the mean ± SEM of 3 experiments conducted in quadruplicate. *P < 0.05, **P < 0.01 versus CTRL. ANOVA plus Tukey's post hoc test.