

Abstract
Background and Purpose
The PARP inhibitor olaparib has recently been approved for human use for the therapy of cancer. Considering the role of PARP in critical illness, we tested the effect of olaparib in a murine model of burn injury, in order to begin exploring the feasibility of repurposing olaparib for the therapy of burn patients.
Experimental Approach
Mice were subjected to scald burn injury and randomized into vehicle or olaparib (10 mg·kg−1·day−1 i.p.) groups. Outcome variables included indices of organ injury, clinical chemistry parameters, plasma levels of inflammatory mediators (at 24 h, 7 and 21 days) and burn wound size (at 21 days).
Key Results
Olaparib reduced myeloperoxidase levels in heart and lung homogenates and reduced malondialdehyde levels in all tissues 24 h post‐burn. Olaparib also reduced circulating alkaline aminotransferase, amylase and blood urea nitrogen and creatinine levels, indicative of protection against hepatic, pancreatic and renal dysfunction. Pro‐inflammatory mediator (TNF‐α, IL‐1β, IFN‐γ, GCSF, GM‐CSF, eotaxin, KC, MIP‐1‐α and IL‐3, 6 and 12) levels as well as the levels of several mediators that are generally considered anti‐inflammatory (IL‐4, 10 and 13) were reduced by olaparib. Plasma troponin‐I levels (an indicator of skeletal muscle damage) was also attenuated by olaparib. Finally, olaparib stimulated wound healing.
Conclusions and Implications
The clinically approved PARP inhibitor olaparib improves organ function, suppresses inflammatory responses and accelerates wound healing in murine burn injury. The data raise the potential utility of olaparib for severe burn injury.
Linked Articles
This article is part of a themed section on Inventing New Therapies Without Reinventing the Wheel: The Power of Drug Repurposing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.2/issuetoc
Abbreviations
- MDA
malondialdehyde
- MPO
myeloperoxidase
Tables of Links
| LIGANDS | |||
|---|---|---|---|
| Eotaxin (CCL11) | IL‐2 | IL‐12 p70 | MIP‐1α (CCL3) |
| G‐CSF | IL‐3 | IL‐13 | MIP‐1β (CCL4) |
| GM‐CSF | IL‐4 | IL‐15 | Olaparib |
| IFN‐γ | IL‐6 | IP‐10 (CXCL10) | TNF‐α |
| IL‐1α | IL‐12 p40 | KC (CXCL1) | VEGF |
| IL‐1β |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
The enzyme poly(ADP‐ribose) polymerase (PARP) was originally discovered by Chambon and colleagues and was classified as a DNA damage response effector (Chambon et al., 1963). Activation of PARP, in response to DNA damage to alkylating agents, catalyses the cleavage of NAD+ into nicotinamide and ADP‐ribose, as shown in cells challenged with alkylating agents. Recognizing the role of PARP in DNA repair led to the development of PARP inhibitors for the therapy of cancer (reviewed in Shall, 1983; Berger et al., 1987; Griffin et al., 1995; Jagtap and Szabo, 2005). By using PARP inhibitors to suppress DNA repair, and thereby enhancing the anticancer efficacy of DNA‐damaging anticancer therapeutic agents, PARP inhibitors emerged as a new class of targeted anticancer therapeutics. Their efficacy is especially significant in cells deficient in BRCA1 or BRCA2. Such cells are extremely sensitive to the cytotoxic effects of PARP inhibition, due to their defective homologous recombination system (Bryant et al., 2005; Farmer et al., 2005). In BRCA‐deficient cells, PARP inhibition causes synthetic lethality. In wild‐type cells, single strand breakage of the DNA is repaired by the base excision repair system, the activation and recruitment of which is dependent on PARP activation. When base excision repair is impaired, in BRCA‐competent cells, single strand breaks are repaired by a secondary mechanism, the homologous recombination pathway. In contrast, in BRCA‐deficient cells, the homologous recombination pathway is genetically impaired, and therefore the unrepaired broken DNA remains unrepaired: This mechanism forms the scientific basis of PARP‐inhibitor‐based therapy of BRCA‐mutant cancers (reviewed in: Curtin and Szabo, 2013; Drew, 2015). As a result of many clinical trials conducted over the last decade, the first PARP inhibitor (olaparib, 4‐[[3‐[4‐(cyclopropanecarbonyl)piperazine‐1‐carbonyl]‐4‐fluoro‐phenyl]methyl]‐2H‐phthalazin‐1‐one, now marketed as Lynparza, and previously known as AZD2281 and KU59436) was clinically approved for the therapy of patients with ovarian cancer (Deeks, 2015).
The approval and clinical availability of olaparib may also open the door for repurposing of this compound (as well as other PARP inhibitors that are expected to be approved in the future) for other, non‐oncological indications. The theoretical basis of such repurposing efforts are based on the well‐established role of PARP in promoting cell death and enhancing pro‐inflammatory mediator production. As reviewed elsewhere (Jagtap and Szabo, 2005; Curtin and Szabo, 2013) extensive cellular depletion of NAD+, and, secondarily, ATP, in cells exposed to DNA‐damaging agents, can be deleterious to the cell's viability. PARP inhibitors, by preventing the PARP‐activation induced deleterious cellular energetic cycles, have been shown to maintain vital cellular functions in cells treated with alkylating agents (Sims et al., 1983), ROS (Schraufstatter et al., 1986), nitric oxide (Heller et al., 1994; Zhang et al., 1994), peroxynitrite (Szabo et al., 1996) or various pathophysiologically relevant triggers, such as NMDA receptor activation in neurons (Zhang et al., 1994), hypoxia/reoxygenation in myocytes (Gilad et al., 1997), bacterial lipopolysaccharide in macrophages (Zingarelli et al., 1996) and high glucose concentration (simulates hyperglycaemia) in vascular endothelial cells (Soriano et al., 2001). The mode of cell death triggered by PARP overactivation involves mitochondrial dysfunction, which culminates in a regulated (active) form of necrosis (Virág et al., 1998; Ha and Snyder, 1999). The exact PARP‐mediated cell death effector mechanisms depend on the cell type, stimulus and experimental context (Andrabi et al., 2006; Alano et al., 2010; Fatokun et al., 2014).
In addition to mediating cell dysfunction and death, PARP has also been shown to contribute to the production of a variety of pro‐inflammatory mediators (reviewed in Jagtap and Szabo, 2005; Luo and Kraus, 2012). The underlying mechanisms are numerous. Firstly, poly(ADP‐ribosylation) of histones confers negative charge to histones leading to electrostatic repulsion from the DNA. Poly(ADP‐ribosylation) loosens up the chromatin and thereby renders various genes accessible for the transcriptional machinery. Secondly, polyanionic PAR, attached to protein substrates (or as free polymer), serves as a local matrix for core histones released from destabilized nucleosomes. Additionally, PARP‐1 binds to DNA methyltransferase‐1, a key enzyme in the DNA methylation (and a global regulator of gene expression), thereby inhibiting its catalytic function. Moreover, PARP‐1 regulates a variety of pro‐inflammatory signal transduction factors. Importantly, PARP acts as a co‐activator in NF‐κB‐mediated transcription, acts as a stimulator of the Akt (PKB pathway) and can contribute to the activation of a variety of inflammation‐related transcription factors including AP1, AP2, TEF‐1, SP1, Oct‐1, YY1 and STAT1. The functional consequence of these actions is that pharmacological inhibition of PARP can attenuate the production of various pro‐inflammatory mediators [including IL‐6, pro‐IL‐1, ICAM‐1, TNF‐α, COX2, inducible NOS, MIP‐1α (also known as CCL3) and MIP‐2 (also known as CXCL2)], as shown in various in vitro and in vivo models including endotoxin‐induced systemic inflammation, various local inflammatory diseases (e.g. arthritis and colitis), ischaemia–reperfusion and high glucose‐induced endothelial dysfunction (reviewed in Jagtap and Szabo, 2005; Bai and Virág, 2012; García and Conde, 2015).
PARP inhibitors, by protecting against cell death, suppressing the overproduction of multiple pro‐inflammatory mediators and interrupting various deleterious positive feedback cycles of cell injury, exert protective effects in a variety of pathophysiological conditions, ranging from ischaemia–reperfusion to neuroinflammation, neurodegeneration and various forms of critical illness (reviewed in Virág and Szabo, 2002; Jagtap and Szabo, 2005; Pacher and Szabo, 2008; Ba and Garg, 2011; Curtin and Szabo, 2013).
Burn injury is associated with a characteristic systemic cardiovascular, neurohumoral, metabolic and immune response, which is pronounced and long‐lasting (reviewed in Herndon, 2012). The systemic response to burn includes the production of multiple pro‐ and anti‐inflammatory mediators, the generation of various oxygen‐ and nitrogen‐derived oxidants and free radicals, and culminates in systemic inflammation and/or immunosuppression as well as multiple organ failure. There are several studies implicating PARP activation in burn injury. A rapid and sustained activation of PARP – evidenced by increased cell and tissue accumulation of its product, poly(ADP‐ribose) (PAR) – was demonstrated in various parenchymal organs as well as circulating leukocytes following burn injury (Asmussen et al., 2011; Bartha et al., 2011; Lange et al., 2011; Szczesny et al., 2015). PARP inhibitors of various structural classes were found to exert protective effects in terms of inflammatory mediator production, tissue oxidative stress and neutrophil infiltration, intestinal barrier dysfunction and pulmonary dysfunction (Shimoda et al., 2003; Avlan et al., 2004; 2005; Hamahata et al., 2012). Increased poly(ADP‐ribosylation) has also been demonstrated in endothelial cells and leukocytes of skeletal muscle tissue biopsies of paediatric patients with severe burn injury (Oláh et al., 2011). Considering the pathogenetic role of PARP in various forms of critical illness, including burns, in the current study, we have tested the effect of the clinically approved PARP inhibitor, olaparib, in a murine model of burn injury, in order to begin exploring the feasibility of repurposing olaparib for the experimental therapy of patients with severe burn injury.
Methods
Animals and experimental design
Male BALB/c mice (10–12 weeks old) were housed at 24–26°C on a 12:12 light : dark cycle and subjected to burn injury as described previously (Szczesny et al., 2015). Sham and burned mice underwent identical experimental procedures, with the exception of injury. Following an i.p. injection of 0.1 mg·kg−1 buprenorphine, mice were anaesthetised by inhalation of 3–5% isoflurane. Next, ~40% of the dorsum was shaved with electrical clippers and ~1 mL of lactated Ringer's solution was injected under the skin along the spinal column. The dorsa of burn treated animals were then exposed to ~95°C water for 10 s to produce a full thickness scald wound covering ~30% of the total body surface area. Mice were then resuscitated with 2 mL of lactated Ringer's solution. Burn and sham treated mice were individually housed throughout the experimental period. To minimize animal suffering, pain or distress, animals were scored twice daily throughout the post burn period using an IACUC‐approved Rodent Intervention Score Sheet to assess their well‐being and clinical status by certified veterinarian. To minimize suffering of the animals the analgesic buprenorphine was administered when indicated to reduce pain and distress. The dose of olaparib (10 mg·kg−1·day−1; i.p., once a day) was based on recent studies utilizing olaparib in various rodent models of non‐oncological disease (Ghonim et al., 2015a,b; Kapoor et al., 2015; Teng et al., 2016).
The design of the in vivo studies followed the recommendations of Curtis and colleagues (Curtis et al., 2015). In order to ensure that the order of treatment should be randomized at the level of the experimental subject (i.e. all placebo‐treated animals should not be treated systematically before all drug‐treated animals even if animals were previously randomized into these two groups), a randomized block design was used. Animals subjected to burn were randomized into either vehicle or olaparib groups. The experimental design included n = 30 animals burn treated with vehicle and n = 30 burn animals treated with olaparib; 10 animals from each group were killed at three different time points (24 h, 7 days and 21 days) post‐injury. In addition, 10 additional animals (without burn injury) were anaesthetised and killed to provide baseline control values for the various measurements. Blood, heart, lung, liver and kidney tissues were harvested for analysis. In addition, at 21 days, burn wound size was determined as described below. Biochemical measurements were conducted in such a way that the person conducting the analysis was not aware of the identity of the samples until the completion of the assays. No samples were excluded from the statistical analysis.
All investigations conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Eight Edition, 2011) and were performed in accordance with the IACUC, University of Texas Medical Branch, Galveston, TX, USA. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Myeloperoxidase assay
Myeloperoxidase (MPO) activity was measured in heart, lung, liver and kidney samples using a commercially available MPO fluorometric detection kit (Enzo Life Sciences) as described (Ahmad et al., 2016). The assay utilizes a non‐fluorescent detection reagent, which is oxidized in the presence of hydrogen peroxide and MPO to produce its fluorescent analogue. The fluorescence is measured at excitation wavelength of 530 to 571 nm and emission wavelength of 590 to 600 nm.
Malondialdehyde assay
Tissue malondialdehyde (MDA) levels, an index of cellular injury/oxidative stress, were quantified in heart, lung, liver and kidney samples using a fluorimetric MDA‐specific lipid peroxidation assay kit (Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturer's instructions and as described previously (Ahmad et al., 2016). The assay is based on the BML‐AK171 method in which two molecules of the chromogenic reagent N‐methyl‐2‐phenylindole react with one molecule of MDA at 45°C to yield a stable carbocyanine dye with a maximum absorption at 586 nm.
Detection of troponin‐I
Measurement of plasma levels of troponin‐I (a regulatory protein of the striated muscle's actomyosin contractile apparatus) was used as a biomarker of skeletal muscle injury (Vassallo et al., 2009; Burch et al., 2016). A commercial available elisa kit (Life Diagnostics, Inc.,) specific for troponin‐I was used according to the manufacturers instructions.
Measurement of biochemical parameters of organ dysfunction
Blood samples were collected via cardiac puncture and were analysed by using a Vetscan analyser as described (Coletta et al., 2014) for various biochemical parameters within 1 h of collection.
Quantification of plasma cytokine levels
Blood from all groups was collected in heparin‐treated blood collection tubes and centrifuged at 4°C for 10 min at 1000 × g within 30 min of collection. Plasma was isolated, aliquoted and stored at −80°C until used. The EMD Millipore's MILLIPLEX™ MAP Mouse cytokine Magnetic Bead Panel 1 kit was used, as described previously (Ahmad and Szabo, 2016) for the simultaneous quantification of the following analytes: G‐CSF, GM‐CSF, eotaxin (CCL11), INF‐γ, IL‐1α, IL‐1β, IL‐2, IL‐3, IL‐4, IL‐6, IL‐10, IL‐12 (p40), IL‐12(p70), Il‐13, IL‐15, IP‐10 (CXCL10), TNF‐α, VEGF, KC (CXCL1), MIP‐1α (CCL3) and MIP‐1β (CCL4) (Merck Millipore, Darmstadt, Germany). The Luminex instrument acquires and analyses data using the LuminexxMAP fluorescent detection method and the LuminexxPONENT™ acquisition software (Thermo Fisher Scientific, Waltham, MA, USA).
Quantification of burn wound area
Wound areas at 24 h after burn (to obtain a baseline value) and at 21 days were quantified as previously described (Coletta et al., 2015), using a transparent sheet and the NIS Elements Imaging Software (Nikon).
Statistical analysis
All values presented in the text and figures are expressed as means ± SEM for n observations. Student's t‐test, one‐way and two‐way ANOVA with Tukey's post hoc test were used to detect differences between groups. Prism version 5 for Windows (Graph Pad Software) was used. P < 0.05 was considered statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
Olaparib was obtained from LC Laboratories (Woburn, MA). Unless indicated otherwise, all other all chemicals were obtained from Sigma‐Aldrich (St. Louis, MO, USA).
Results
Burn injury resulted in an increase in lung, liver, heart and kidney MPO and MDA levels by 24 h, which, in most tissues, was followed by a further elevation by 7 days, and a return to baseline by 21 days (Figures 1 and 2). One exception was the kidney, where MPO levels remained elevated throughout the 21 days post‐burn time period (Figure 1D); another exception was liver MDA levels, which showed the maximal increase 24 h post‐burn, and tended to decrease by 7 days and further decreased by 21 days (Figure 2C). Heart MPO levels (Figure 1A) and lung MPO levels (Figure 1B) was attenuated by olaparib treatment at 7 days. MDA levels in all tissues were reduced at 24 h by olaparib.
Figure 1.
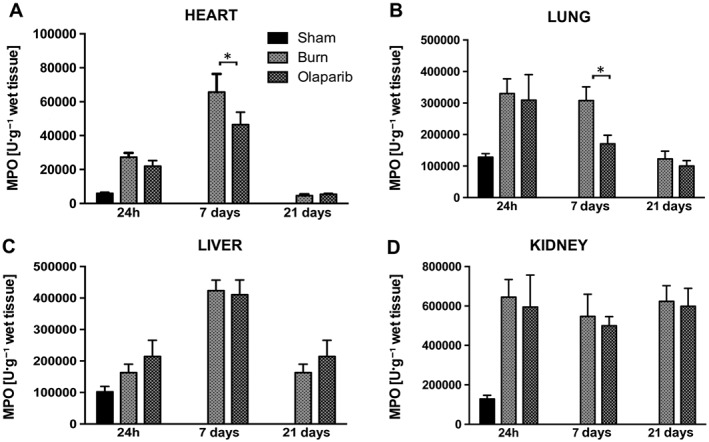
Effect of olaparib treatment on burn‐induced increases in heart, lung, liver and kidney MPO levels. (A) Heart, (B) lung, (C) liver and (D) kidney MPO levels are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in MPO levels in the heart and lung at 24 h and 7 days, in the liver at 7 days and in the kidney at 24 h, 7 and 21 days. Significant effect of olaparib during burn injury in the heart and lung is shown at 7 days (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Figure 2.
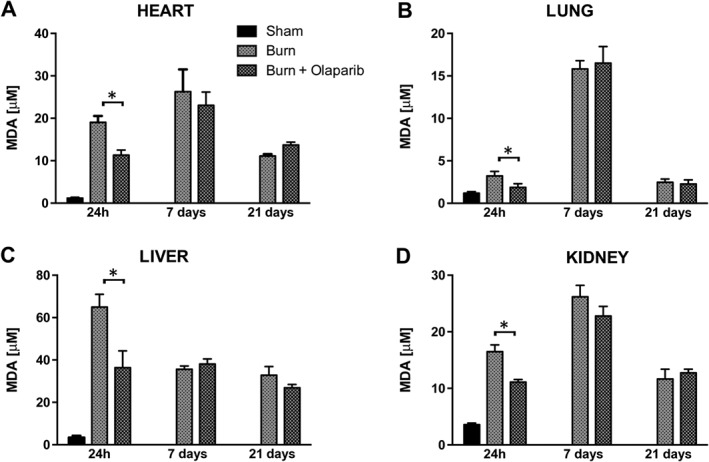
Effect of olaparib treatment on burn‐induced increases in heart, lung, liver and kidney MDA levels. (A) Heart, (B) lung, (C) liver and (D) kidney MDA levels are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in MDA levels in all tissues at all three time points studied. Significant effect of olaparib during burn injury in all four tissues studied is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Burn injury also induced marked changes in the clinical chemistry parameters and organ injury markers of the animals. Notably, there was an increase in the plasma levels of hepatic injury marker alanine aminotransferase (ALT), the hepatic/bone injury marker alkaline phosphatase, the pancreatic injury marker amylase and the renal dysfunction markers creatinine and blood urea nitrogen (Figures 3 and 4). Many of these parameters peaked at 24 h. Olaparib attenuated plasma ALT levels at 24 h (Figure 3C), plasma amylase levels at 24 h (Figure 3D) and plasma blood urea nitrogen (BUN) and creatinine levels at 24 h (Figures 3F and 4C). The electrolyte changes (hypercalcaemia, hypernatraemia, hyperkalaemia) induced by burn remained unaffected by olaparib (Figure 4). There was also a hyperalbuminaemic response to burn, which was unaffected by olaparib (Figure 3A). The early (24 h) but not the later (7 days and 21 days) hyperglycaemic response was attenuated by olaparib (Figure 4D).
Figure 3.
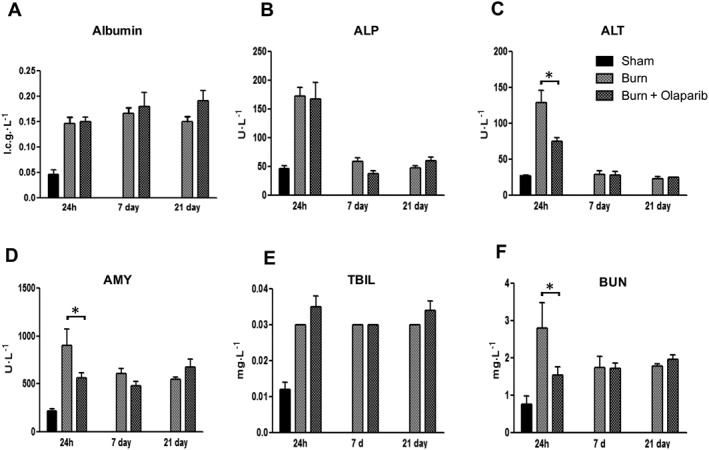
Effect of olaparib treatment in burn‐induced increases in selected parameters of organ injury. Various physiological and organ injury marker levels, albumin, alkaline phosphatase (ALP), alanine aminotransferase (ALT), amylase (AMY), total bilirubin (TBIL), and blood urea nitrogen (BUN) are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in albumin, AMY, bilirubin and BUN levels at all three time points studied and in ALT and ALP levels at 24 h. Significant effect of olaparib on ALT, ALT and BUN levels during burn injury is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Figure 4.
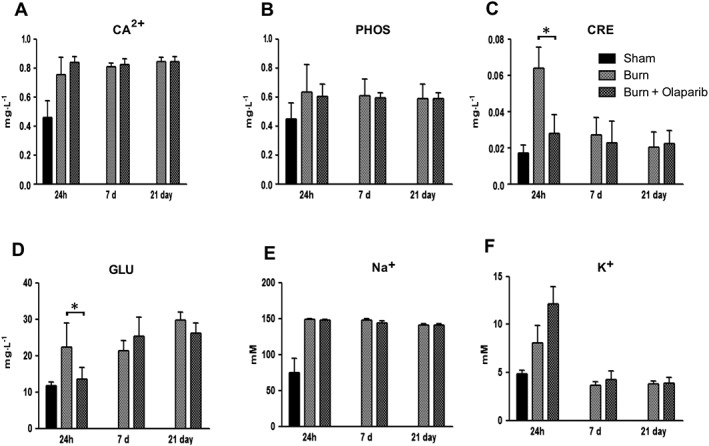
Effect of olaparib treatment on burn‐induced increases in selected parameters of organ injury. Various physiological and organ injury marker levels (plasma calcium [Ca2+], plasma phosphate [PHOS], plasma creatinine [CRE], plasma glucose [GLU], plasma sodium [Na+] and plasma potassium [K+]) are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1 day, 6 days and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in calcium and sodium levels at all three time points studied and in creatinine, glucose and potassium levels at 24 h. Significant effect of olaparib on creatinine and glucose levels during burn injury is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Consistent with the systemic inflammatory response syndrome associated with burns (Dahiya, 2009; Rae et al., 2016), burn markedly increased plasma G‐CSF, GM‐CSF, eotaxin, INF‐γ, IL‐α, IL‐β, IL‐2, IL‐3, IL‐6, IL‐10, IL‐12, IL‐12(p40), IL‐12(p70), IL‐13, IL‐15, IP‐10, TNF‐α, VEGF, KC, MIP‐1α and MIP‐1β levels at 24 h and slightly increased INF‐γ levels at the 21st day; many of mediators were reduced by olaparib treatment (Figures 5, 6, 7, 8). It is important to note that not only pro‐inflammatory mediators, but also mediators that are typically considered anti‐inflammatory (e.g. IL‐4, IL‐10 and IL‐13) were reduced by olaparib.
Figure 5.
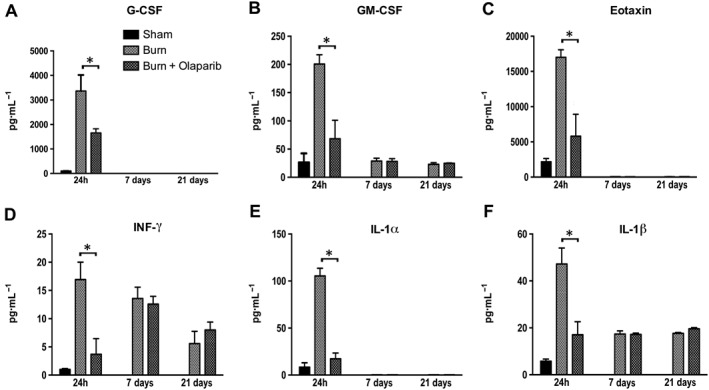
Effect of olaparib treatment on burn‐induced increases in selected plasma inflammatory mediator levels. Plasma levels of G‐CSF, GM‐CSF, eotaxin, IFN‐γ, IL‐1α and IL‐1β are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in GM‐CSF, eotaxin, IFN‐γ, IL‐1α and IL‐1β levels of vehicle‐treated (but not olaparib‐treated) mice at 24 h, and in IFN‐γ and IL‐1β levels in both vehicle‐ and olaparib‐treated mice at 7 and 21 days. Significant effect of olaparib on all six mediators studied during burn injury is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Figure 6.

Effect of olaparib treatment on burn‐induced increases in selected plasma inflammatory mediator levels. Plasma levels of IL‐2, IL‐3, IL‐4, IL‐6, IL‐10 and IL‐12(p40) are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in IL‐3 levels at 24 h and in all other mediator levels at all time points studied. Significant effect of olaparib on all six mediators studied during burn injury is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Figure 7.
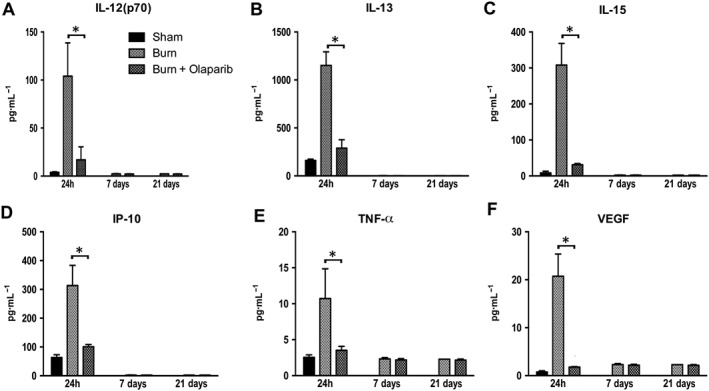
Effect of olaparib treatment on burn‐induced increases in selected plasma inflammatory mediator levels. Plasma levels of IL‐12 (p70), Il‐13, IL‐15, IP‐10, TNF‐α and VEGF are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in plasma levels of all six mediators studied in the vehicle‐treated (but not olaparib‐treated) mice at 24 h. Significant effect of olaparib on all six mediators studied during burn injury is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Figure 8.
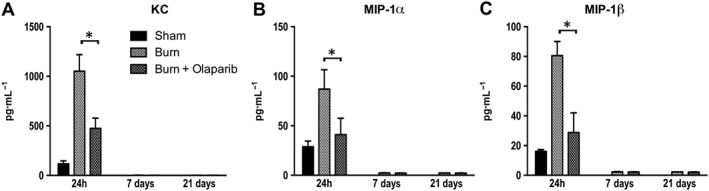
Effect of olaparib treatment on burn‐induced increases in selected plasma chemokine levels. Plasma levels of KC, MIP‐1α and MIP‐1β are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p.) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in plasma levels of KC at 24 h in both vehicle‐treated and olaparib‐treated and in the plasma levels of MIP‐1α and MIP‐1β at 24 h in vehicle‐treated (but not olaparib‐treated) mice. Significant effect of olaparib on all three mediators studied during burn injury is shown at 24 h (*P < 0.05). Data are shown as mean ± SEM of 10 animals for each group.
Burn markedly increased plasma troponin‐I levels at 24 h; this was slightly, but statistically significantly reduced by olaparib treatment, indicative of reduced skeletal muscle injury in PARP inhibitor treated animals (Figure 9A).
Figure 9.
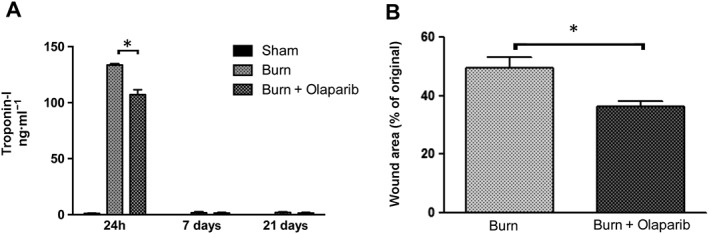
Olaparib reduces the burn‐induced increases in plasma troponin‐I levels and reduces burn wound area. (A) Plasma troponin‐I levels are shown in sham‐control mice and in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p) once a day for 1, 6 and 20 days. Compared to non‐burn sham control, burn produced a significant increase (P < 0.05) in plasma troponin‐I levels at 24 h; olaparib exerted a significant inhibitory effect (*P < 0.05). Data are shown as mean ± SEM of 10 animals per group. (B) % wound area (as percentage of the original burn wound area) is shown on Day 21 in mice subjected to burn injury and in burn mice treated with olaparib (10 mg·kg−1, i.p) once a day for 20 days. *P < 0.05 shows a significant reduction in the wound area in olaparib‐treated animals. Data are shown as mean ± SEM of 10 animals per group.
The original burn wound area underwent an approximately 50% healing response in vehicle‐treated animals by Day 21; treatment with olaparib significantly reduced the % wound area, suggesting that the PARP inhibitor stimulated would healing (Figure 9B).
Discussion
The key findings of the study are the following: (i) olaparib‐treated animals exhibit a partial, but statistically significant reduction in MPO levels in heart and lung homogenates (indicative of reduced polymorphonuclear cell infiltration), and reduced malondialdehyde (MDA) levels in liver homogenates (indicative of reduced intraorgan oxidative stress) 24 h post‐burn. (ii) At 24 h post‐burn, olaparib treatment also reduced circulating alkaline aminotransferase, amylase and blood urea nitrogen/creatinine levels, indicative of protection against the burn‐induced hepatic, pancreatic and renal dysfunction. (iii) Several of the plasma inflammatory mediators measured (TNF‐α, IL‐1β, IFN‐γ, GCSF, GM‐CSF, eotaxin, KC, MIP‐1‐α and IL‐3, 4, 6, 10 and 12) were suppressed by olaparib in burn animals, indicative of a global suppression of inflammatory mediator production by the PARP inhibitor. (iv) Muscle‐specific creatinine kinase (an indicator of skeletal muscle damage) was also attenuated by olaparib treatment. (v) Burn wound size was smaller after 21 days in olaparib‐treated mice compared to the sham controls, suggesting that olaparib enhances burn wound healing responses. (vi) Olaparib, at the 10 mg·kg−1·day−1 dose used, did not affect the clinical chemistry, organ function or inflammatory parameters measured in the animals subjected to burn.
While increased temperature itself has been shown to increase cellular PAR polymer formation in cultured cells in vitro (Yamashita et al., 2016) and in the liver tissue after whole‐body hyperthermia in rats (Zaalishvili et al., 2012), in the current study, the hyperthermic response was local (creating the burn wound, but not elevating the temperature of the animal elsewhere), while the resulting organ dysfunction and inflammatory response observed was global (systemic). This is consistent with the well‐known, and both preclinically and clinically well‐documented systemic inflammatory response associated with burns (Finnerty et al., 2009; Herndon, 2012; Porter et al., 2016). The complex physiological response associated with burns is sometimes also called PICS (persistent inflammatory, immunosuppressed, catabolic syndrome) (Rosenthal and Moore, 2015). Olaparib had marked inhibitory effects on plasma levels of multiple cytokines and chemokines. It is important to mention, however, that not only pro‐inflammatory mediators but also mediators that are typically considered anti‐inflammatory (e.g. IL‐4, IL‐10 and IL‐13) were reduced by olaparib. This is in contrast to the pattern elicited by some of the broad‐spectrum anti‐inflammatory agents (e.g. glucocorticoids, adenosine receptor ligands or cAMP elevators), which reduce pro‐inflammatory mediator synthesis, while boosting anti‐inflammatory (e.g. IL‐10) mediator production (Haskó et al., 1996, 2002; Szabo et al., 1997; Elenkov, 2004). Many (Yang et al., 2000; Soriano et al., 2002; Oumouna et al., 2006; Ahmad et al., 2015) but not all (Gonzalez‐Rey et al., 2007; Ghonim et al., 2015a,b) previous studies have also reported an inhibitory effect of PARP inhibition or PARP1 genetic deficiency on IL‐10 production. The exact molecular mechanism through which olaparib inhibits anti‐inflammatory mediator production remains to be further elucidated; a working hypothesis is that the anti‐inflammatory (counter‐regulatory) response is lower in the olaparib‐treated mice, because the preceding inflammatory response has been markedly reduced by the PARP inhibitor.
The improvement in organ function, the suppression of inflammatory mediator production, and the reduction in tissue neutrophil infiltration and suppression of tissue oxidative stress by olaparib is consistent with the mode of PARP inhibitor's action (a combined effect on protection against oxidative/nitrative stress induced cell necrosis and inhibition of pro‐inflammatory signal transduction, as well as an interruption of positive feedback cycles of injury) (reviewed in Jagtap and Szabo, 2005) and is comparable to the effect of other, earlier‐generation PARP inhibitors, as well as genetic PARP1 deficiency in various models of critical illness and systemic inflammation (Jagtap et al., 2002; Liaudet et al., 2002; Soriano et al., 2002, 2006; Shimoda et al., 2003; Farivar et al., 2004; Szabo, 2007; Wang et al., 2013; Liu et al., 2015; Walko et al., 2015; Zhang et al., 2015). Thus, olaparib – although originally developed and optimized for the purposes of cancer therapy – performs as expected as a PARP inhibitor in the context of inflammation, organ injury and critical illness. It must be noted, nevertheless, that the effects of olaparib, on many of the parameters studied, were only partial. For example, olaparib only reduced MPO and MDA levels in some time points in some organs, and not in others. Interestingly, even in those organs where MPO levels were not reduced by olaparib (e.g. the kidney), olaparib produced significant functional improvements (e.g. improved plasma creatinine and BUN levels). These data indicate that a reduction in inflammatory leukocytes is not an absolute requirement to reduce intraorgan oxidative stress; moreover, a reduction in MPO levels is not an absolute requirement for the organ protective effects of olaparib. We hypothesize that the well‐documented direct protective effect against oxidant‐induced cell necrosis by PARP inhibitors (see Methods) contributes to the organ protection mediated by the PARP inhibitor.
In light of the fact that hyperglycaemia is a common pathophysiological event in burns, which precedes and predicts many of the subsequent adverse events (Gauglitz et al., 2008; Mecott et al., 2010; Finnerty et al., 2014; Jeschke et al., 2016; Ray et al., 2016), it was interesting to note that olaparib attenuated the burn‐associated early hyperglycaemia in our current model. We interpret this finding as an indication that the overall stress response and/or the acute insulin resistance that develops in burns may be, at least in part, dependent on PARP. In this context, it is interesting to note that recent studies have implicated the pathogenetic role of PARP activation in the development of insulin resistance (Pang et al., 2015).
Olaparib also exerted a partial protective effect against skeletal muscle damage (as evidenced by measurement of plasma troponin‐I levels). Skeletal muscle dysfunction is another complication of burn injury (Alloju et al., 2008; Ebid et al., 2012; Ogunbileje et al., 2016), and recent studies have started to investigate the role of PARP in skeletal muscle. While in undifferentiated muscle cells PARP expression is high, its expression decreases with differentiation, and this confers oxidative stress resistance to the myocytes (Oláh et al., 2015). The current results are consistent with the hypothesis that burn (similar to many other organs) also produces an oxidative and/or nitrative stress to the skeletal muscle, and the subsequent PARP activation contributes to muscle damage. Follow‐up studies, investigating functional parameters (e.g. skeletal muscle weakness) are needed to expand on these observations.
The current results also show that olaparib enhances wound healing in burned mice. Although there are conflicting data on the role of PARP in angiogenesis (Rajesh et al., 2006; Pyriochou et al., 2008; Caldini et al., 2011; Binu et al., 2012; Guzyk et al., 2016), multiple lines of in vivo studies indicate that PARP inhibitors of various classes enhance wound healing (El‐Hamoly et al., 2014; Sarras et al., 2014; Byun et al., 2015; El‐Hamoly et al., 2015; Zhou et al., 2016). The current study adds burn injury to the list of conditions, where PARP inhibition may accelerate wound healing. The precise mechanism of olaparib's action, however, remains to be characterized in future studies. Naturally, the wound healing process is drastically different in rodents and in large animals and in humans; for instance, the burn‐associated scarring is poorly recapitulated in animal models. Clearly, additional studies, in more translationally relevant models are needed to further evaluate the potential benefit of PARP inhibitors in the context of burn wound healing.
Several groups have started conducting preclinical studies with olaparib in the context of various non‐oncological applications, with an eye on future therapeutic repurposing. These efforts, so far, produced the following results: (i) In vitro studies with olaparib, in a novel differentiated human neuron system showed that olaparib (2 μM) protects against NMDA receptor‐stimulation induced and oxygen–glucose deprivation‐induced neuronal cell death in vitro (Xu et al., 2016). (ii) In vivo stroke studies, conducted in a mouse model of transient middle cerebral artery occlusion (2 h ischaemia +24 h reperfusion) show that olaparib (3 and 5 mg·kg−1, but not doses higher or lower) produces a reduction in infarct size, reduces IgG extravasation, and improves neurological scores in some (but not all) tests used (Teng et al., 2016). (iii) In a model of acute lung injury and acute kidney injury induced by intratracheal administration of endotoxin in mice, olaparib (5 mg·kg−1) reduces inflammatory cell (in particular neutrophil) infiltration into the lungs and attenuates the development of pulmonary oedema. It also protects against the endotoxin‐induced secondary kidney injury, as shown by improvements in serum urea and creatinine levels. All of these effects are also associated with reduced oxidative stress markers and reduced production of various pro‐inflammatory factors (TNF‐α, IL‐1β and VCAM‐1) (Kapoor et al., 2015). (iv) In an ovalbumin‐induced asthma model in mice, olaparib (1, 5 and 10 mg·kg−1) dose‐dependently reduced ovalbumin‐specific IgE production in the bronchoalveolar lavage fluid, reduced inflammatory cell numbers in the bronchoalveolar lavage and reduced mucus production in the lungs; it also improved the AHR response to metacholine. These effects were associated with the suppression of multiple inflammatory mediators including eotaxin, IL‐2, IL‐4, IL‐5, IL‐6, IL‐13 and M‐CSF (Ghonim et al., 2015a). Finally, (v) protective effects of olaparib (5 mg·kg−1) have also been demonstrated in a house dust mite model of asthma (Ghonim et al., 2015b). The above results, although conducted in markedly different models than our current burn model, are consistent with the overall notion that olaparib exerts cytoprotective and anti‐inflammatory effects in various non‐oncological disease indications.
Although the dose of olaparib in the current study (10 mg·kg−1·day−1) was selected based on prior rodent studies with this compound in non‐oncological indications (Kapoor et al., 2015; Ghonim et al., 2015a,b; Teng et al., 2016), we cannot exclude the possibility that different (perhaps lower) doses of the compound may have produced a more pronounced effect. Indeed, olaparib can exert bell‐shaped dose‐responses – as demonstrated in a stroke model by Teng et al. (2016) where maximal efficacy was seen at the dose of 5 mg·kg−1, and the dose of 10 mg·kg−1 was already associated with some signs of toxicity (e.g. impairment of the blood–brain barrier). It should be noted that the dose used in various studies focusing on non‐oncological indications (including the current study) is lower (in the range of 1–10 mg·kg−1) than the doses of olaparib (typically in the range of 30–200 mg·kg−1) that have previously been used in the preclinical anticancer studies in mice (To et al., 2014; Sui et al., 2015), perhaps predicting that the optimal dose of olaparib for non‐oncological indications may be lower than the doses currently used in oncology.
It is worth mentioning that most of the effects of olaparib, on many of the biochemical and inflammatory parameters, were most evident at the earlier time points studied and less pronounced at Day 21, even though the administration of the PARP inhibitor was continuous throughout the study. This may be explained by the combination of two factors. First, some of the parameters tend to return to near‐baseline levels by 21 days, at which point all of the groups (21 day control and 21 day olaparib) are all close to the non‐burned (control) values. Second, the data may also indicate that in the current experimental model PARP activation (which typically occurs in response to oxidants and free radicals) may be most pronounced at the earlier time points; the inflammatory and organ injury response seen at later time points may, therefore, be triggered by pathophysiological factors other than PARP.
It should be noted that the current study was conducted in burn animals without any concomitant therapies, which is different from the clinical situation where patients with burn receive a variety of drugs targeting various pathophysiological processes. It should also be noted that several studies suggest that the cytoprotective and anti‐inflammatory effect of PARP inhibitors can be dependent on the sex of the animals, with male animals being more responsive than females (Mabley et al., 2005; McCullough et al., 2005). The potential sex‐dependency of olaparib has not been taken into account in the current study, as the study used male animals only.
The design of follow‐on studies should take some of the above factors (concomitant therapies and gender differences) into account, as well as the relevant therapeutic window of intervention (in order to mimic the clinical situation with the time window between the burn injury event and the time of definitive in‐hospital care). Another key step in the process of therapeutic repurposing would be to ensure to confirm target engagement, that is, to ensure that PARP is sufficiently inhibited at the dose used. Studies in oncology have used measurements of PARP activity in peripheral blood leukocytes for this purpose (e.g. Zaremba et al., 2011); similar approaches may also be feasible in the context of burns, because burn is associated with a significant increase in PARP activity in circulating leukocytes (Asmussen et al., 2011; Bartha et al., 2011).
Because of the inhibitory effect of olaparib on DNA repair (Ito et al., 2016; Lee et al., 2016), further studies will be necessary to test whether olaparib affects DNA integrity and chromosomal stability in animal models of those selected non‐oncological pathophysiological conditions that may be candidates for therapeutic repurposing. The risk–benefit analysis of using PARP inhibitors for non‐oncological indications has been previously discussed (e.g. Jagtap and Szabo, 2005; Curtin and Szabo, 2013). At least initially, repurposing of olaparib (or of other clinically used PARP inhibitors) should focus on those non‐oncological indications where the disease is associated with a high risk of mortality, the alternative therapeutic options are limited, and the expected duration of drug administration is relatively short.
Author contributions
A.A: conduction of experiments, analysis of data and preparation of manuscript; G.O.: conduction of experiments; D.H.: experimental design and preparation of manuscript; C.S.: experimental design, data analysis and preparation of manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by a grant from the Shriners of North America (grant #85800) to C.S. and a grant from the National Institutes of Health (2P50GM060338) to D.H. and C.S.
Ahmad, A. , Olah, G. , Herndon, D. N. , and Szabo, C. (2018) The clinically used PARP inhibitor olaparib improves organ function, suppresses inflammatory responses and accelerates wound healing in a murine model of third‐degree burn injury. British Journal of Pharmacology, 175: 232–245. doi: 10.1111/bph.13735.
References
- Ahmad A, Druzhyna N, Szabo C (2016). Delayed treatment with sodium hydrosulfide improves regional blood flow and alleviates cecal ligation and puncture (CLP)‐induced septic shock. Shock 46: 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad SF, Zoheir KM, Ansari MA, Korashy HM, Bakheet SA, Ashour AE (2015). The role of poly(ADP‐ribose) polymerase‐1 inhibitor in carrageenan‐induced lung inflammation in mice. Mol Immunol 63: 394–405. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Szabo C (2016). Both the H2S biosynthesis inhibitor aminooxyacetic acid and the mitochondrially targeted H2S donor AP39 exert protective effects in a mouse model of burn injury. Pharmacol Res 113: 348–355. [DOI] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010). NAD+ depletion is necessary and sufficient for poly(ADP‐ribose) polymerase‐1‐mediated neuronal death. J Neurosci 30: 2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Brit J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloju SM, Herndon DN, McEntire SJ, Suman OE (2008). Assessment of muscle function in severely burned children. Burns 34: 452–459. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M et al. (2006). Poly(ADP‐ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103: 18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmussen S, Bartha E, Olah G, Sbrana E, Rehberg SW, Yamamoto Y et al. (2011). The angiotensin‐converting enzyme inhibitor captopril inhibits poly(adp‐ribose) polymerase activation and exerts beneficial effects in an ovine model of burn and smoke injury. Shock 36: 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avlan D, Taşkinlar H, Unlü A, Oztürk C, Cinel L, Nayci A et al. (2004). The role of poly(ADP‐ribose) synthetase inhibition on the intestinal mucosal barrier after thermal injury. Burns 30: 785–792. [DOI] [PubMed] [Google Scholar]
- Avlan D, Unlü A, Ayaz L, Camdeviren H, Nayci A, Aksöyek S (2005). Poly (adp‐ribose) synthetase inhibition reduces oxidative and nitrosative organ damage after thermal injury. Pediatr Surg Int 21: 449–455. [DOI] [PubMed] [Google Scholar]
- Ba X, Garg NJ (2011). Signaling mechanism of poly(ADP‐ribose) polymerase‐1 (PARP‐1) in inflammatory diseases. Am J Pathol 178: 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Virág L (2012). Role of poly(ADP‐ribose) polymerases in the regulation of inflammatory processes. FEBS Lett 586: 3771–3777. [DOI] [PubMed] [Google Scholar]
- Bartha E, Asmussen S, Olah G, Rehberg SW, Yamamoto Y, Traber DL et al. (2011). Burn and smoke injury activates poly(ADP‐ribose)polymerase in circulating leukocytes. Shock 36: 144–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger NA, Berger SJ, Gerson SL (1987). DNA repair, ADP‐ribosylation and pyridine nucleotide metabolism as targets for cancer chemotherapy. Anticancer Drug Des 2: 203–209. [PubMed] [Google Scholar]
- Binu S, Soumya SJ, Kumar VB, Sudhakaran PR (2012). Poly‐ADP‐ribosylation of vascular endothelial growth factor and its implications on angiogenesis. Adv Exp Med Biol 749: 269–278. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E et al. (2005). Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 434: 913–917. [DOI] [PubMed] [Google Scholar]
- Burch PM, Greg Hall D, Walker EG, Bracken W, Giovanelli R, Goldstein R et al. (2016). Evaluation of the relative performance of drug‐induced skeletal muscle injury biomarkers in rats. Toxicol Sci 150: 247–256. [DOI] [PubMed] [Google Scholar]
- Byun YS, Kang B, Yoo YS, Joo CK (2015). Poly(ADP‐ribose) polymerase inhibition improves corneal epithelial innervation and wound healing in diabetic rats. Invest Ophthalmol Vis Sci 56: 1948–1955. [DOI] [PubMed] [Google Scholar]
- Caldini R, Fanti E, Magnelli L, Barletta E, Tanganelli E, Zampieri M et al. (2011). Low doses of 3‐aminobenzamide, a poly(ADP‐ribose) polymerase inhibitor, stimulate angiogenesis by regulating expression of urokinase type plasminogen activator and matrix metalloprotease 2. Vasc Cell 3: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P, Weill JD, Mandel P (1963). Nicotinamide mononucleotide activation of new DNA‐dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun 11: 39–43. [DOI] [PubMed] [Google Scholar]
- Coletta C, Módis K, Oláh G, Brunyánszki A, Herzig DS, Sherwood ER et al. (2014). Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care 18: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta C, Módis K, Szczesny B, Brunyánszki A, Oláh G, Rios EC et al. (2015). Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3‐mercaptopyruvate sulfurtransferase/H2S pathway: functional impairment by hyperglycemia and restoration by DL‐α‐lipoic acid. Mol Med 21: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, Szabo C (2013). Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med 34: 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiya P (2009). Burns as a model of SIRS. Front Biosci (Landmark Ed) 14: 4962–4967. [DOI] [PubMed] [Google Scholar]
- Deeks ED (2015). Olaparib: first global approval. Drugs 75: 231–240. [DOI] [PubMed] [Google Scholar]
- Drew Y (2015). The development of PARP inhibitors in ovarian cancer: from bench to bedside. Br J Cancer 113: S3–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebid AA, Omar MT, Abd El Baky AM (2012). Effect of 12‐week isokinetic training on muscle strength in adult with healed thermal burn. Burns 38: 61–68. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ (2004). Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci 1024: 138–146. [DOI] [PubMed] [Google Scholar]
- El‐Hamoly T, El‐Denshary ES, Saad SM, El‐Ghazaly MA (2015). 3‐aminobenzamide, a poly (ADP ribose) polymerase inhibitor, enhances wound healing in whole body gamma irradiated model. Wound Repair Regen 23: 672–684. [DOI] [PubMed] [Google Scholar]
- El‐Hamoly T, Hegedűs C, Lakatos P, Kovács K, Bai P, El‐Ghazaly MA et al. (2014). Activation of poly(ADP‐ribose) polymerase‐1 delays wound healing by regulating keratinocyte migration and production of inflammatory mediators. Mol Med 20: 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farivar AS, Woolley SM, Fraga CH, Thomas R, Salzman AL, Szabo C et al. (2004). Intratracheal poly (ADP) ribose synthetase inhibition ameliorates lung ischemia reperfusion injury. Ann Thorac Surg 77: 1938–1943. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, Dawson TM (2014). Parthanatos: mitochondrial‐linked mechanisms and therapeutic opportunities. Br J Pharmacol 171: 2000–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerty CC, Ali A, McLean J, Benjamin N, Clayton RP, Andersen CR et al. (2014). Impact of stress‐induced diabetes on outcomes in severely burned children. J Am Coll Surg 218: 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerty CC, Przkora R, Herndon DN, Jeschke MG (2009). Cytokine expression profile over time in burned mice. Cytokine 45: 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García S, Conde C (2015). The role of poly(ADP‐ribose) polymerase‐1 in rheumatoid arthritis. Mediators Inflamm 2015: 837250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauglitz GG, Herndon DN, Jeschke MG (2008). Insulin resistance postburn: underlying mechanisms and current therapeutic strategies. J Burn Care Res 29: 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonim MA, Pyakurel K, Ibba SV, Wang J, Rodriguez P, Al‐Khami AA et al. (2015a). PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite‐induced disease in mice. Clin Sci (Lond) 129: 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonim MA, Pyakurel K, Ibba SV, Wang J, Rodriguez P, Al‐Khami AA et al. (2015b). PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite‐induced disease in mice. Clin Sci (Lond) 129: 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad E, Zingarelli B, Salzman AL, Szabo C (1997). Protection by inhibition of poly (ADP‐ribose) synthetase against oxidant injury in cardiac myoblasts in vitro. J Mol Cell Cardiol 29: 2585–2597. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Rey E, Martínez‐Romero R, O'Valle F, Aguilar‐Quesada R, Conde C, Delgado M et al. (2007). Therapeutic effect of a poly(ADP‐ribose) polymerase‐1 inhibitor on experimental arthritis by downregulating inflammation and Th1 response. PLoS One 2: e1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RJ, Curtin NJ, Newell DR, Golding BT, Durkacz BW, Calvert AH (1995). The role of inhibitors of poly(ADP‐ribose) polymerase as resistance‐modifying agents in cancer therapy. Biochimie 77: 408–422. [DOI] [PubMed] [Google Scholar]
- Guzyk MM, Tykhomyrov AA, Nedzvetsky VS, Prischepa IV, Grinenko TV, Yanitska LV et al. (2016). Poly(ADP‐ribose) polymerase‐1 (PARP‐1) inhibitors reduce reactive gliosis and improve angiostatin levels in retina of diabetic rats. Neurochem Res 41: 2526–2537. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH (1999). Poly(ADP‐ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 96: 13978–13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamahata A, Enkhbaatar P, Lange M, Yamaki T, Sakurai H, Shimoda K et al. (2012). Administration of poly(ADP‐ribose) polymerase inhibitor into bronchial artery attenuates pulmonary pathophysiology after smoke inhalation and burn in an ovine model. Burns 38: 1210–1215. [DOI] [PubMed] [Google Scholar]
- Haskó G, Szabo C, Németh ZH, Kvetan V, Pastores SM, Vizi ES (1996). Adenosine receptor agonists differentially regulate IL‐10, TNF‐alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol 157: 4634–4640. [PubMed] [Google Scholar]
- Haskó G, Deitch EA, Szabo C, Németh ZH, Vizi ES (2002). Adenosine: a potential mediator of immunosuppression in multiple organ failure. Curr Opin Pharmacol 2: 440–444. [DOI] [PubMed] [Google Scholar]
- Heller B, Bürkle A, Radons J, Fengler E, Jalowy A, Müller M et al. (1994). Analysis of oxygen radical toxicity in pancreatic islets at the single cell level. Biol Chem Hoppe Seyler 375: 597–602. [DOI] [PubMed] [Google Scholar]
- Herndon D. (ed) (2012). Total burn care, 4th edn. Elsevier Health Sciences: Amsterdam, The Netherlands. [Google Scholar]
- Ito S, Murphy CG, Doubrovina E, Jasin M, Moynahan ME (2016). PARP inhibitors in clinical use induce genomic instability in normal human cells. PLoS One 11: e0159341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P, Soriano FG, Virág L, Liaudet L, Mabley J, Szabó E et al. (2002). Novel phenanthridinone inhibitors of poly (adenosine 5'‐diphosphate‐ribose) synthetase: potent cytoprotective and antishock agents. Crit Care Med 30: 1071–1082. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C (2005). Poly(ADP‐ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4: 421–440. [DOI] [PubMed] [Google Scholar]
- Jeschke MG, Abdullahi A, Burnett M, Rehou S, Stanojcic M (2016). Glucose control in severely burned patients using metformin: an interim safety and efficacy analysis of a phase II randomized controlled trial. Ann Surg 264: 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor K, Singla E, Sahu B, Naura AS (2015). PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol Cell Biochem 400: 153–162. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange M, Szabo C, Enkhbaatar P, Connelly R, Horvath E, Hamahata A et al. (2011). Beneficial pulmonary effects of a metalloporphyrinic peroxynitrite decomposition catalyst in burn and smoke inhalation injury. Am J Physiol Lung Cell Mol Physiol 300: L167–L175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Lee NC, Kouprina N, Kim JH, Kagansky A, Bates S et al. (2016). Effects of anticancer drugs on chromosome instability and new clinical implications for tumor‐suppressing therapies. Cancer Res 76: 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet L, Pacher P, Mabley JG, Virág L, Soriano FG, Haskó G et al. (2002). Activation of poly(ADP‐ribose) polymerase‐1 is a central mechanism of lipopolysaccharide‐induced acute lung inflammation. Am J Respir Crit Care Med 165: 372–377. [DOI] [PubMed] [Google Scholar]
- Liu SB, Liu J, Liu DW, Wang XT, Yang RL (2015). Inhibition of poly‐(ADP‐ribose) polymerase protects the kidney in a canine model of endotoxic shock. Nephron 130: 281–292. [DOI] [PubMed] [Google Scholar]
- Luo X, Kraus WL (2012). On PAR with PARP: cellular stress signaling through poly(ADP‐ribose) and PARP‐1. Genes Dev 26: 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabley JG, Horváth EM, Murthy KG, Zsengellér Z, Vaslin A, Benko R et al. (2005). Gender differences in the endotoxin‐induced inflammatory and vascular responses: potential role of poly(ADP‐ribose) polymerase activation. J Pharmacol Exp Ther 315: 812–820. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD (2005). Ischemic nitric oxide and poly (ADP‐ribose) polymerase‐1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab 25: 502–512. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecott GA, Al‐Mousawi AM, Gauglitz GG, Herndon DN, Jeschke MG (2010). The role of hyperglycemia in burned patients: evidence‐based studies. Shock 33: 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunbileje JO, Porter C, Herndon DN, Chao T, Abdelrahman DR, Papadimitriou A et al. (2016). Hypermetabolism and hypercatabolism of skeletal muscle accompany mitochondrial stress following severe burn trauma. Am J Physiol Endocrinol Metab 311: E436–E448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oláh G, Finnerty CC, Sbrana E, Elijah I, Gerö D, Herndon DN et al. (2011). Increased poly(ADP‐ribosyl)ation in skeletal muscle tissue of pediatric patients with severe burn injury: prevention by propranolol treatment. Shock 36: 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oláh G, Szczesny B, Brunyánszki A, López‐García IA, Gerö D, Radák Z et al. (2015). Differentiation‐associated downregulation of poly(ADP‐ribose) polymerase‐1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS One 10: e0134227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oumouna M, Datta R, Oumouna‐Benachour K, Suzuki Y, Hans C, Matthews K et al. (2006). Poly(ADP‐ribose) polymerase‐1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL‐5. J Immunol 177: 6489–6496. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C (2008). Role of the peroxynitrite‐poly(ADP‐ribose) polymerase pathway in human disease. Am J Pathol 173: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J, Cui J, Gong H, Xi C, Zhang TM (2015). Effect of NAD on PARP‐mediated insulin sensitivity in oleic acid treated hepatocytes. J Cell Physiol 230: 1607–1613. [DOI] [PubMed] [Google Scholar]
- Porter C, Tompkins RG, Finnerty CC, Sidossis LS, Suman OE, Herndon DN (2016). The metabolic stress response to burn trauma: current understanding and therapies. Lancet 388: 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyriochou A, Olah G, Deitch EA, Szabó C, Papapetropoulos A (2008). Inhibition of angiogenesis by the poly(ADP‐ribose) polymerase inhibitor PJ‐34. Int J Mol Med 22: 113–118. [PubMed] [Google Scholar]
- Rae L, Fidler P, Gibran N (2016). The physiologic basis of burn shock and the need for aggressive fluid resuscitation. Crit Care Clin 32: 491–505. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Godlewski G, Bátkai S, Haskó G, Liaudet L et al. (2006). Poly(ADP‐ribose)polymerase inhibition decreases angiogenesis. Biochem Biophys Res Commun 350: 1056–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray JJ, Meizoso JP, Allen CJ, Teisch LF, Yang EY, Foong HY et al. (2016). Admission hyperglycemia predicts infectious complications after burns. J Burn Care Res in press. [DOI] [PubMed] [Google Scholar]
- Rosenthal MD, Moore FA (2015). Persistent inflammatory, immunosuppressed, catabolic syndrome (PICS): A new phenotype of multiple organ failure. J Adv Nutr Hum Metab 1: e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarras MP Jr, Mason S, McAllister G, Intine RV (2014). Inhibition of poly‐ADP ribose polymerase enzyme activity prevents hyperglycemia‐induced impairment of angiogenesis during wound healing. Wound Repair Regen 22: 666–670. [DOI] [PubMed] [Google Scholar]
- Schraufstatter IU, Hyslop PA, Hinshaw DB, Spragg RG, Sklar LA, Cochrane CG (1986). Hydrogen peroxide‐induced injury of cells and its prevention by inhibitors of poly(ADP‐ribose) polymerase. Proc Natl Acad Sci U S A 83: 4908–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shall S (1983). ADP‐ribosylation, DNA repair, cell differentiation and cancer. Princess Takamatsu Symp 13: 3–25. [PubMed] [Google Scholar]
- Shimoda K, Murakami K, Enkhbaatar P, Traber LD, Cox RA, Hawkins HK et al. (2003). Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am J Physiol Lung Cell Mol Physiol 285: L240–L249. [DOI] [PubMed] [Google Scholar]
- Sims JL, Berger SJ, Berger NA (1983). Poly(ADP‐ribose) polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5'‐triphosphate pools in DNA‐damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochem 22: 5188–5194. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Liaudet L, Szabó E, Virág L, Mabley JG, Pacher P et al. (2002). Resistance to acute septic peritonitis in poly(ADP‐ribose) polymerase‐1‐deficient mice. Shock 17: 286–292. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Nogueira AC, Caldini EG, Lins MH, Teixeira AC, Cappi SB et al. (2006). Potential role of poly(adenosine 5'‐diphosphate‐ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit Care Med 34: 1073–1079. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Virág L, Jagtap P, Szabó E, Mabley JG, Liaudet L et al. (2001). Diabetic endothelial dysfunction: the role of poly(ADP‐ribose) polymerase activation. Nat Med 7: 108–113. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui H, Shi C, Yan Z, Li H (2015). Combination of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor xenografts due to increased autophagy. Drug Des Devel Ther 9: 3183–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Zingarelli B, O'Connor M, Salzman AL (1996). DNA strand breakage, activation of poly (ADP‐ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A 93: 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Haskó G, Zingarelli B, Németh ZH, Salzman AL, Kvetan V et al. (1997). Isoproterenol regulates tumour necrosis factor, interleukin‐10, interleukin‐6 and nitric oxide production and protects against the development of vascular hyporeactivity in endotoxaemia. Immunology 90: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (2007). Poly (ADP‐ribose) polymerase activation and circulatory shock. Novartis Found Symp 280: 92–103. [DOI] [PubMed] [Google Scholar]
- Szczesny B, Brunyánszki A, Ahmad A, Oláh G, Porter C, Toliver‐Kinsky T et al. (2015). Time‐dependent and organ‐specific changes in mitochondrial function, mitochondrial DNA integrity, oxidative stress and mononuclear cell infiltration in a mouse model of burn injury. PLoS One 10: e0143730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng F, Zhu L, Su J, Zhang X, Li N, Nie Z et al. (2016). Neuroprotective effects of poly(ADP‐ribose) polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem Res 41: 1516–1526. [DOI] [PubMed] [Google Scholar]
- To C, Kim EH, Royce DB, Williams CR, Collins RM, Risingsong R et al. (2014). The PARP inhibitors, veliparib and olaparib, are effective chemopreventive agents for delaying mammary tumor development in BRCA1‐deficient mice. Cancer Prev Res (Phila) 7: 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zingarelli B, Szabo C (2000). Effect of genetic disruption of poly (ADP‐ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia–reperfusion injury. Shock 13: 60–66. [DOI] [PubMed] [Google Scholar]
- Vassallo JD, Janovitz EB, Wescott DM, Chadwick C, Lowe‐Krentz LJ, Lehman‐McKeeman LD (2009). Biomarkers of drug‐induced skeletal muscle injury in the rat: troponin I and myoglobin. Toxicol Sci 111: 402–412. [DOI] [PubMed] [Google Scholar]
- Virág L, Salzman AL, Szabo C (1998). Poly(ADP‐ribose) synthetase activation mediates mitochondrial injury during oxidant‐induced cell death. J Immunol 161: 3753–3759. [PubMed] [Google Scholar]
- Virág L, Szabo C (2002). The therapeutic potential of poly(ADP‐ribose) polymerase inhibitors. Pharmacol Rev 54: 375–429. [DOI] [PubMed] [Google Scholar]
- Walko TD 3rd, Di Caro V, Piganelli J, Billiar TR, Clark RS, Aneja RK (2015). Poly(ADP‐ribose) polymerase 1‐sirtuin 1 functional interplay regulates LPS‐mediated high mobility group box 1 secretion. Mol Med 20: 612–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Huang X, Li Y, Guo K, Ning P, Zhang Y (2013). PARP‐1 inhibitor, DPQ, attenuates LPS‐induced acute lung injury through inhibiting NF‐κB‐mediated inflammatory response. PLoS One 8: e79757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JC, Fan J, Wang X, Eacker SM, Kam TI, Chen L et al. (2016). Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med 8: 333ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Tanaka M, Sato T, Ida C, Ohta N, Hamada T et al. (2016). Effect of mild temperature shift on poly(ADP‐ribose) and γH2AX levels in cultured cells. Biochem Biophys Res Commun 476: 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaalishvili G, Zaldastanishvili E, Karapetian M, Zaalishvili T (2012). Increased PARP‐1 levels in nuclear matrix isolated from heat shock treated rat liver. Biochem (Mosc) 77: 105–110. [DOI] [PubMed] [Google Scholar]
- Zaremba T, Thomas HD, Cole M, Coulthard SA, Plummer ER, Curtin NJ (2011). Poly(ADP‐ribose) polymerase‐1 (PARP‐1) pharmacogenetics, activity and expression analysis in cancer patients and healthy volunteers. Biochem J 436: 671–679. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH (1994). Nitric oxide activation of poly(ADP‐ribose) synthetase in neurotoxicity. Science 263: 687–689. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yao J, Wang X, Li H, Liu T, Zhao W (2015). Poly (ADP‐ribose) synthetase inhibitor has a heart protective effect in a rat model of experimental sepsis. Int J Clin Exp Pathol 8: 9824–9835. [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Patel D, Sen S, Shanmugam V, Sidawy A, Mishra L et al. (2016). Poly‐ADP‐ribose polymerase inhibition enhances ischemic and diabetic wound healing by promoting angiogenesis. J Vasc Surg in press. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, O'Connor M, Wong H, Salzman AL, Szabó C (1996). Peroxynitrite‐mediated DNA strand breakage activates poly‐adenosine diphosphate ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J Immunol 156: 350–358. [PubMed] [Google Scholar]