

Abstract
Background and Purpose
Protease‐activated receptor 1 (PAR1) has been demonstrated to be involved in the pathogenesis of viral diseases. However, its role remains controversial. The goal of our study was to investigate the contribution of PAR1 to respiratory syncytial virus (RSV) and human metapneumovirus (hMPV) infections.
Experimental Approach
Pharmacological approaches were used to investigate the role of PAR1 during RSV and hMPV infection, in vitro using epithelial A549 cells and in vivo using a mouse model of virus infection.
Key Results
In vitro, the PAR1 antagonist RWJ‐56110 reduced the replication of RSV and hMPV in A549 cells. In agreement with these results, RWJ‐56110‐treated mice were protected against RSV and hMPV infections, as indicated by less weight loss and mortality. This protective effect in mice correlated with decreased lung viral replication and inflammation. In contrast, hMPV‐infected mice treated with the PAR1 agonist TFLLR‐NH2 showed increased mortality, as compared to infected mice, which were left untreated. Thrombin generation was shown to occur downstream of PAR1 activation in infected mice via tissue factor exposure as part of the inflammatory response, and thrombin inhibition by argatroban reduced the pathogenicity of the infection with no additive effect to that induced by PAR1 inhibition.
Conclusion and Implications
These data show that PAR1 plays a detrimental role during RSV and hMPV infections in mice via, at least, a thrombin‐dependent mechanism. Thus, the use of PAR1 antagonists and thrombin inhibitors may have potential as a novel approach for the treatment of RSV and hMPV infections.
Abbreviations
- hMPV
human metapneumovirus
- PAR1
protease‐activated receptor 1
- RSV
respiratory syncytial virus
- TAT
thrombin‐antithrombin
- TF
tissue factor
Introduction
Human metapneumovirus (hMPV) and respiratory syncytial virus (RSV) are members of the new Pneumoviridae family of non‐segmented, negative‐stranded, enveloped RNA viruses. hMPV is a member of Metapneumovirus genus whereas RSV belongs to the Orthopneumovirus genus (Afonso et al., 2016). Both viruses are major causes of acute respiratory tract infections. RSV is the second most important cause of death worldwide among children below the age of 5 years, responsible for up to 250 000 deaths a year (Lozano et al., 2012). Unfortunately, there is currently no licensed vaccine to prevent RSV and hMPV infections and their treatment is still limited to the use of Ribavirin, a weakly effective antiviral agent, and immunoglobulins (Drysdale et al., 2016; Esposito and Mastrolia, 2016). The most severe forms of RSV and hMPV infections are associated with both significant inflammatory pathology as well as excessive virus replication (DeVincenzo et al., 2010; Rosenberg and Domachowske, 2012; Roussy et al., 2014). In such a context, anti‐inflammatory and/or antiviral therapies may lead to more effective control of RSV and hMPV infections.
Protease‐activated receptors (PARs) belong to the superfamily of GPCRs and has four members: PAR1–4. PAR1 was the first family member discovered, consisting of 425 amino acids with a thrombin cleavage site between residues 41 and 42, and is known as the highest affinity thrombin receptor (Vu et al., 1991; Sidhu et al., 2014). It was first identified for its role in platelet activation and initiation of the coagulation cascade (Vu et al., 1991). Subsequently, PAR1 has been demonstrated to be involved in multiple physiological and pathological processes, such as thrombosis, wound healing, inflammation, embryonic development and cancer progression (Vergnolle, 2009; Gieseler et al., 2013).
The involvement of PAR1 in viral infections has been reported in the case of herpesvirus, influenza virus, coxsackievirus, dengue virus and hMPV (Scholz et al., 2004; Sutherland et al., 2007; Cabello‐Gutiérrez et al., 2009; Aerts et al., 2013; Antoniak et al., 2013; Khoufache et al., 2013; Antoniak and Mackman, 2014; Mackman and Antoniak, 2014). However, this receptor seems to exert a dual role, notably in herpesvirus and influenza virus infections. Indeed, Scholz et al. (2004) suggested that thrombin/PAR1‐mediated Sp1 phosphorylation inhibits human cytomegalovirus replication in retinal pigment epithelial cells, whereas Pryzdial's team found a significant increase of herpesvirus replication in endothelial cells and fibroblasts after PAR1 activation (Sutherland et al., 2007). With regard to influenza, Riteau's team reported that PAR1 contributed to the deleterious inflammatory response in the lungs and increased the pathogenesis of the virus (Khoufache et al., 2013). In contrast, Antoniak et al. (2013) showed that PAR1 increased the immune response to influenza virus infection and thereby inhibited virus replication.
In most studies, TFLLR‐NH2 and SCH79797 have been used as PAR1 agonist and antagonist, respectively, for assessing the involvement of PAR1 in viral infections (Sutherland et al., 2012; Aerts et al., 2013; Khoufache et al., 2013). However, while SCH79797 has been demonstrated as a specific human PAR1 antagonist, its specificity in mice is controversial (Di Serio et al., 2007; Pawlinski et al., 2007). The use of non‐specific tools might explain the divergent results on the role of PAR1 in infectious diseases. PAR1 is of great interest in drug development (French et al., 2015) and particularly for the design of novel antivirals targeting the host instead of the virus. Thus, it is of crucial importance to better understand its role during viral infections. Here, we aimed to investigate the role of PAR1 during RSV and hMPV infections using more specific tools. In addition to the PAR1‐specific agonist TFLLR‐NH2, we used for the first time RWJ‐56110, an indole‐based peptide mimetic and highly specific PAR1 antagonist (Andrade‐Gordon et al., 1999; Derian et al., 2003; Maryanoff et al., 2003), which has also been shown to specifically inhibit the murine PAR1 (Andrade‐Gordon et al., 1999; Kaufmann et al., 2005).
We showed that PAR1 has a deleterious effect during RSV and hMPV infections, acting both on viral replication and inflammation. Thus, these results suggest that PAR1 is a promising target for the development of novel antivirals against RSV and hMPV infections.
Methods
Cells and viruses
Hep‐2 and LLC‐MK2 cells (ATCC, Manassas, VA, USA) were maintained in minimal essential medium (Thermo Fisher Scientific, Burlington, ON, Canada) supplemented with 10% FBS (Wisent, Saint‐Jean‐Baptiste, QC, Canada) and HEPES buffer (2.5 g·L−1). A549 and NIH/3T3 cells (ATCC) were maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS and 1% penicillin–streptomycin (Thermo Fisher Scientific).
The RSV strains A2 (ATCC) were grown in Hep‐2 cells and concentrated by ultracentrifugation at 141 000× g for 2 h at 4°C using Optima™ L‐90 ultracentrifuge with SW 28 Ti rotor (Beckman Counter, Montreal, QC, Canada). The hMPV strain C85473, a clinical isolate, was grown in LLC‐MK2 cells and concentrated in a similar manner and schedule at room temperature. The RSV and hMPV virus titres were determined by immunostaining as previously described (Deffrasnes et al., 2008; Persson et al., 2014). Virus titres are expressed as plaque‐forming units (PFU) mL−1.
Cytotoxicity assessment
The 50% cytotoxic (CC50) of the PAR1 agonist TFLLR‐NH2 (GenScript, Piscataway, NJ, USA) and antagonist RWJ‐56110 (Tocris Bioscience, Bristol, UK), PAR4 agonist AY‐NH2 (Bachem AG, Bubendorf, Switzerland) and thrombin inhibitor argatroban (Toronto Research Chemicals, Toronto, ON, Canada) were determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA) according to the manufacturer's instructions.
Selectivity of PAR1 antagonist RWJ‐56110
NIH/3T3 cells were pretreated with 10 μM of the PAR1 antagonist RWJ‐56110 and incubated at 37°C for 1 h. The cells were then treated with or without 50 μM of PAR1 agonist TFLLR‐NH2 or 200 μM of PAR4 agonist AY‐NH2 and incubated at 37°C for 5 min (Khoufache et al., 2013). DMEM medium served as mock PAR1 agonist and antagonist treatments. Thereafter, the cells were lysed in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific) according to the manufacturer's instructions. Cell lysates were collected, and the concentrations of protein were determined using the Quick Start Bradford Protein Assay (Bio‐Rad Laboratories, Mississauga, ON, Canada) for Western blot analysis of ERK phosphorylation.
For siRNA inhibition study, A549 cells were transfected with Thrombin R siRNA specifically targeting PAR1 (Santa Cruz Biotechnology, Dallas, TX, USA) or MISSION siRNA Universal Negative Control (Sigma Aldrich, Oakville, ON, Canada) in the presence of Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. After 24 h, the cells were treated with or without 10 μM RWJ‐56110 and incubated at 37°C for 1 h. DMEM medium served as control. The cells were then inoculated with 30 PFU of RSV or 300 PFU of hMPV per well and incubated at 37°C. Three days later, virus titres were determined by immunostaining and expressed as PFU mL−1.
In vitro viral infections
A549 cells were treated with TFLLR‐NH2 and incubated at 37°C for 5 min or with RWJ‐56110 and incubated at 37°C for 1 h at different doses of 0, 5, 10, 20, 40, 80 and 160 μM. The cells were then inoculated with 30 PFU of RSV or 300 PFU of hMPV per well and incubated at 37°C. In another experiment, Hep‐2 or LLC‐MK2 cells were treated with argatroban at different doses of 0, 31.25, 62.5, 125, 250 and 500 μM and incubated at 37°C for 1 h. The cells were then inoculated with 30 PFU of RSV or hMPV per well and incubated at 37°C. Three days later, virus titers were determined by immunostaining and expressed as PFU mL−1.
To determine the effect of TFLLR‐NH2, RWJ‐56110 or argatroban on ERK phosphorylation during RSV and hMPV infections in vitro, A549 cells were treated with or without 50 μM of TFLLR‐NH2 and incubated at 37°C for 5 min, 10 μM of RWJ‐56110 or 100 μM of argatroban and incubated at 37°C for 1 h. DMEM medium or DMSO (Sigma Aldrich) served as mock PAR1 agonist and antagonist or argatroban treatments respectively. The cells were then uninfected or infected with 30 PFU of RSV or 300 PFU of hMPV. Cell lysates were collected at 5 and 30 min post‐infection for Western blot analysis of ERK phosphorylation.
To investigate the capacity of RSV and hMPV to activate PAR1, A549 cells were infected with 30 PFU of RSV or 300 PFU of hMPV per well. DMEM medium served as uninfected control. Cells were harvested at 5 and 30 min and lysed for Western blot analysis of PAR1 activation.
Western blot analysis
Equal protein amounts were separated on 10% SDS‐PAGE gels and then transferred to nitrocellulose membranes (GE HealthCare Life Sciences, Mississauga, ON, Canada) and blocked using 5% BSA (Sigma Aldrich). Primary antibodies were used at a dilution of 1:1000 rabbit anti‐phospho‐ERK, rabbit anti‐ERK (Cell Signaling Technology, Boston, MA, USA), mouse anti‐PAR1 (R&D Systems, Minneapolis, MN, USA), rabbit anti‐cleaved‐Ser42 PAR1 (Biorbyt, San Francisco, CA, USA) and mouse anti‐α‐tubulin (Sigma Aldrich). Secondary antibodies were used at a dilution of 1:5000 HRP‐conjugated mouse anti‐rabbit, rabbit anti‐mouse (Cell Signaling Technology). Signal detection was carried out using the West Pico Plus Chemiluminescent Substrate (Thermo Fisher Scientific).
Ethics statement
Six‐week‐old female BALB/c mice with a body weight of 16.5–18 g were purchased from Charles River Laboratories (Senneville, QC, Canada). This animal strain is widely used for the study of RSV and hMPV infections (Domachowske et al., 2004; Schildgen et al., 2007). Mice were housed under pathogen‐free conditions in the animal research facility of the Quebec University Health Centre (Quebec City, QC, Canada) and allowed to acclimatize for 1 week prior to the start of experiments. All in vivo studies were approved by the Animal Protection Committee of the Quebec University Health Centre in accordance with the guidelines of the Canadian Council on Animal Care (Protocol number: CPAC 2013‐082‐3). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The animals were killed by CO2 inhalation if humane end points were reached (weight loss ≥20% of pre‐experimental body weight).
In vivo viral infections, PAR1 treatments and thrombin inhibition
Mice were anaesthetized by inhalation of isoflurane vaporized at concentrations of 3–4% and oxygen flow rate adjusted to 1.5 L·min−1. The assessment of anesthetic depth is based on six parameters recommended by the Animal Protection Committee of the Quebec University Health Centre including the following: heart rate, respiratory rate, capillary refill time, body temperature, mucous membrane color and palpebral reflex. They were then infected intranasally with RSV (3 × 106 PFU per mouse) or hMPV at two different doses: 0.5 × 106 (non‐lethal dose) or 106 (LD50 dose) PFU per mouse. Equal volumes of Opti‐MEM medium (25 μl) served as mock infection. At the same time, mice were treated, by intranasal instillation, with 10 μl of 500 μM of TFLLR‐NH2 or RWJ‐56110. Equivalent dilutions of Opti‐MEM medium served as control. These treatments were repeated once a day for four consecutive days. PAR1 agonist TFLLR‐NH2 and antagonist SCH79797 have been previously tested at two doses: 50 and 500 μM. We found that the higher dose (500 μM) was better than the low dose (50 μM) for investigating the role of PAR1 in hMPV infection in mice (Aerts et al., 2013). Therefore, we decided to use this dose for both PAR1 agonist TFLLR‐NH2 and antagonist RWJ‐56110 in this study.
For thrombin inhibition, mice were injected i.p. with argatroban at a dose of 9 mg·kg−1 (Asanuma et al., 2004). They were then immediately infected with RSV or hMPV. The treatment with argatroban was repeated once a day for four consecutive days during infections. Control mice were given DMSO in a similar manner. Individual body weight and clinical signs were used to monitor animal health and response to infection and were recorded daily for 2 weeks.
Lung viral titrations
To evaluate virus titres on day 5 post‐infection, mice were killed by cervical dislocation under isoflurane anaesthesia and whole lungs were harvested and then homogenized in PBS (1 mL per sample) using a TH Tissue Homogenizer (Omni International, Kennesaw, GA, USA). The supernatants were collected after centrifugation at 350× g for 10 min at 4°C. The RSV and hMPV virus titres were determined by immunostaining and expressed as PFU g−1 of the lung.
Broncho‐alveolar lavage and cell counting
On day 5 post‐infection, mice were killed by cervical dislocation under isoflurane anaesthesia, and broncho‐alveolar lavage (BAL) was performed with PBS. The cells in the lavage fluid were pelleted by centrifugation at 300× g for 5 min at 4°C and then suspended in PBS, whereas BAL supernatants were collected for evaluating inflammatory and coagulation parameters with the exception of tissue factor (TF). The measurement of TF was carried out in the fluids of BAL with and without centrifugation.
Viable cell number was determined using a haemocytometer and expressed as number mL−1 of BAL. For differential cell counts, 100 μL of suspended cells were spun onto a slide by using a Shandon Cytospin™ 3 cytocentrifuge (Thermo Fisher Scientific) at 100× g for 5 min at room temperature. Slides were then air‐dried and stained with May‐Grunewald Giemsa solutions (Sigma Aldrich) according to the manufacturer's instructions. Differences in cell counts were made by using standard morphological criteria and counting at least 300 cells per sample. The results are expressed as different percentages.
BAL cytokine and total protein quantification
The concentrations of 23 cytokines and chemokines [eotaxin (CCL11), G‐CSF, GM‐CSF, IFN‐γ, IL‐1α, IL‐1β, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐9, IL‐10, IL‐12(p40), IL‐12(p70), IL‐13, IL‐17, KC, MCP‐1, MIP‐1α, MIP‐1β, RANTES and TNF‐α] in BAL supernatants were determined using the Bio‐Plex Pro™ Mouse Cytokine 23‐plex panel (Bio‐Rad Laboratories) according to the manufacturer's instructions. Cytokine and chemokine levels are expressed as pg·mL−1 of BAL.
Total protein levels in BAL supernatants were determined using Quick Start™ Bradford Protein Assay according to the manufacturer's instructions. The results are expressed as mg·mL−1 of BAL.
Measurement of thrombin‐antithrombin (TAT) and TF levels
The levels of TAT and TF in BAL supernatants were determined using a TAT mouse elisa kit (Abcam, Toronto, ON, Canada) and Mouse Coagulation Factor III/TF DuoSet elisa (R&D Systems) following the manufacturer's instructions and the results are expressed as ng or pg·mL−1 of BAL supernatants.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology Curtis et al. (2015). All group data subjected to statistical analysis have a minimum of n = 5 independent samples from different individuals per group for reliable P values. All statistical tests were conducted using the GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA, USA). The results are expressed as the mean ± SEM for each group. Survival data were analysed by comparing Kaplan–Meier curves using the log–rank test. Virus titres, cytokines, chemokines, total protein levels, immune cell recruitment, cell differentiation, TAT and TF levels were analysed using two‐way ANOVA followed by Tukey's post hoc test. Differences were considered statistically significant when P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Cytotoxicity of PAR1 agonist TFLLR‐NH2 and antagonist RWJ‐56110, PAR4 agonist AY‐NH2 and thrombin inhibitor argatroban
The CC50 concentrations of these compounds are presented in Figure 1A. In the case of PAR1 and PAR4 agonists, the CC50 concentrations could not be determined because no cell death was observed at the highest tested dose of these two peptides (0.8 mM). In a previous study, we demonstrated that the PAR1 antagonist SCH79797, which is currently used in the majority of models of viral infections, was extremely toxic for cells (Aerts et al., 2013). Herein, we showed that the PAR1 antagonist RWJ‐56110 has almost no toxic effects on at least two cell types: Hep‐2 and LLC‐MK2 (CC50 > 0.7 mM).
Figure 1.
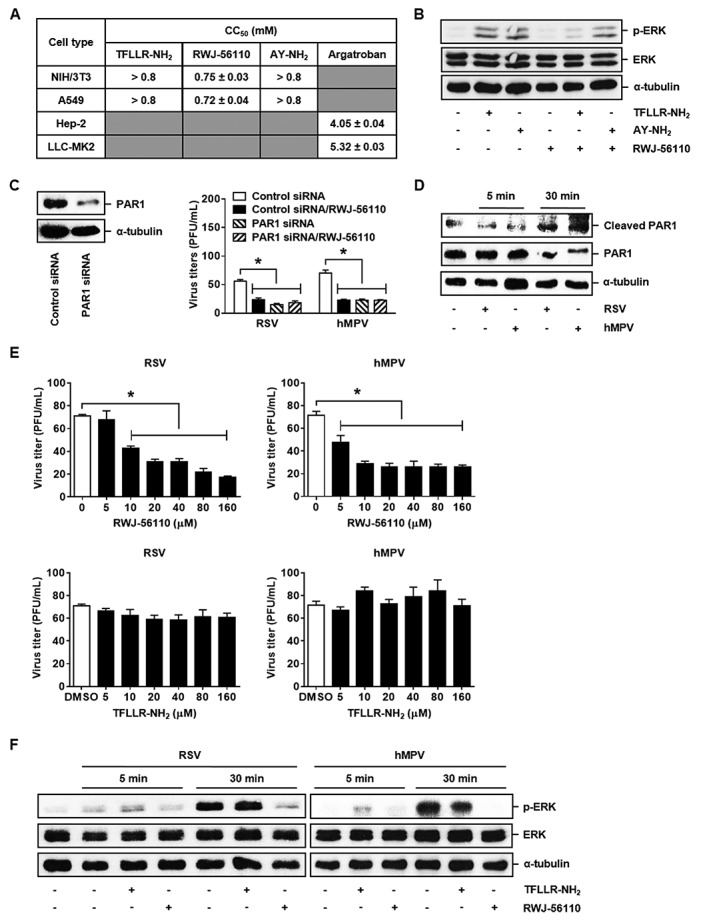
In vitro antiviral effect of RWJ‐56110. (A) Cytotoxicity of TFLLR‐NH2, AY‐NH2, RWJ‐56110 and argatroban was determined by the MTS test. (B, C) Selectivity of RWJ‐56110 to PAR1 was evaluated by studying its capacity to inhibit PAR1 activation‐induced ERK phosphorylation in NIH/3T3 cells and to modulate the viral replication in A549 cells transfected with siRNA targeting PAR1 (n = 6 per group, *P < 0.05 control siRNA/RWJ‐56110; PAR1 siRNA or PAR1 siRNA/RWJ‐56110 vs. control siRNA). (D) The capacity of RSV and hMPV to activate PAR1 was investigated in A549 cells by Western blot analysis. (E, F) Antiviral effect of RWJ‐56110 was investigated in A549 cells. The cells were treated with DMSO, TFLLR‐NH2 or RWJ‐56110 and then infected with RSV or hMPV. Virus‐infected cells were identified by immunostaining and counted [n = 6 per group, *P < 0.05 RWJ‐56110 5, 10, 20, 40, 80 or 160 μM vs. DMSO (0 μM)]. In addition to the evaluation of viral replication, Western blot analysis was used to study ERK phosphorylation.
The selectivity of RWJ‐56110 to PAR1 has been demonstrated previously (Andrade‐Gordon et al., 1999). Herein, the selectivity of this compound was confirmed by evaluating its capacity to inhibit PAR4 activation‐induced ERK phosphorylation in NIH/3T3 cells since PAR1 activation has been demonstrated to phosphorylate ERK (Khoufache et al., 2013). We found that RWJ‐56110 inhibited ERK phosphorylation induced by the PAR1 agonist TFLLR‐NH2, but not by PAR4 agonist AY‐NH2 (Figure 1B).
Western blot analysis revealed a decreased PAR1 gene expression resulting from siRNA transfection in A549 cells, which was consistent with previous findings (Liu et al., 2004). RSV and hMPV replication in control negative siRNA‐transfected cells treated with RWJ‐56110 did not significantly differ from that of cells undergoing siRNA knockdown of PAR1 gene expression. Moreover, RWJ‐56110 treatment did not modulate the viral titres in siRNA‐transfected cells (Figure 1C). Thus, RWJ‐56110 can be used as a novel potential PAR1 antagonist for studying the involvement of PAR1 in various physiological or pathological processes.
RWJ‐56110 exhibited an antiviral effect against RSV and hMPV in A549 cells
To investigate the role of PAR1 in RSV and hMPV replication, A549 cells were infected with 30 PFU of RSV or 300 PFU of hMPV and treated with the indicated concentrations of TFLLR‐NH2 or RWJ‐56110. By counting infected cells identified by immunostaining, we showed that RWJ‐56110 treatment significantly reduced both RSV and hMPV titres in a dose‐dependent manner. In contrast, PAR1 activation had no influence on the replication of these viruses (Figure 1E). These results show that RSV and hMPV replication is increased via PAR1 in vitro.
In parallel, we detected that ERK was phosphorylated in RSV‐ and hMPV‐infected cells after 30 min of infection (Figure 1F). These findings were consistent with those showing that both RSV and hMPV induce PAR1 activation after 30 min of infection, as indicated by the cleavage of PAR1 (Figure 1D). In other words, RSV and hMPV promoted ERK activation by activating PAR1, as previously reported (Wang et al., 2002; Khoufache et al., 2013). In agreement with Figure 1E, TFLLR‐NH2 did not have an additive effect to that of the viruses on ERK phosphorylation whereas RWJ‐56110 inhibited virus‐induced ERK phosphorylation (Figure 1F). The inhibition of virus‐triggered ERK phosphorylation could be related to the decreased viral replication resulting from RWJ‐56110 treatment, and the lack of effect of TFLLR‐NH2 in these conditions is because ERK phosphorylation has been demonstrated to be required for RSV infection (Monick et al., 2001; Pazdrak et al., 2002; Kong et al., 2004).
RWJ‐56110 reduced weight loss and prevented mortality in RSV‐ and hMPV‐infected mice
In vivo, TFLLR‐NH2‐triggered PAR1 activation did not significantly affect RSV infection. However, there was an increased mortality of hMPV‐infected mice receiving this agonist, as compared to untreated animals. Consistent with a deleterious role of PAR1 in these settings, RWJ‐56110 reduced weight loss for both RSV‐ and hMPV‐infected mice at non‐lethal doses (3 × 106 PFU of RSV and 0.5 × 106 PFU of hMPV per mouse) (Figure 2A). TFLLR‐NH2 treatment reduced only and moderately the survival of hMPV‐infected mice. Consistent with other reports, infections with RSV, even at very high dose of infection, did not increase mouse mortality (Meng et al., 2014); no protection by RSV could thus be observed, and TLLFR‐NH2 did not affect the survival of these treated mice. However, when mice were challenged with hMPV at an LD50 dose (106 PFU per mouse), RWJ‐56110 treatment fully protected the mice from a fatal outcome (Figure 2B). Altogether, these results show that PAR1 plays a deleterious role in the pathogenesis of RSV and hMPV.
Figure 2.
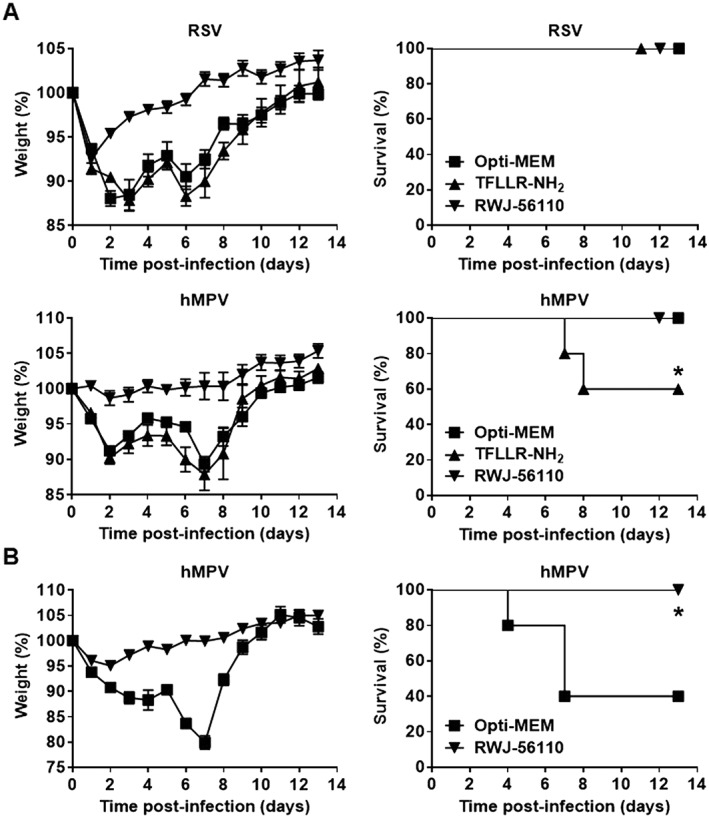
Time course of RSV‐ and hMPV‐induced pathogenesis and survival rate in mice in response to RWJ‐56110 treatment. (A) Mice were infected with RSV or hMPV at sublethal doses and treated with Opti‐MEM medium, TFLLR‐NH2 or RWJ‐56110. (B) Mice were infected with hMPV at an LD50 dose and treated with Opti‐MEM medium or RWJ‐56110. They were monitored for weight loss for 14 days after infection and killed if humane end points were reached. Results are average % weight loss ± SEM or average % survival (n = 10 per group, *P < 0.05 TFLLR‐NH2 vs. control or RWJ‐56110 vs. Opti‐MEM).
Protective effect of RWJ‐56110 treatment on viral replication and inflammation
To determine the effect of RWJ‐56110 treatment on viral replication and inflammation, mice were infected with 3 × 106 PFU of RSV or 106 PFU of hMPV and the lung homogenates as well as BAL fluids were harvested at day 5 post‐infection to assess lung viral titres, leukocyte recruitment, alteration of alveolar‐capillary permeability as well as inflammatory cytokine and chemokine production. The results showed that RWJ‐56110 treatment substantially reduced viral loads in the lungs as well as the total concentration of proteins in BAL of both RSV‐ and hMPV‐infected mice (Figure 3A, B). Although total leukocyte recruitment was influenced by RWJ‐56110 treatment (Figure 3C), no difference in the proportion of leukocytes (macrophages, lymphocytes and neutrophils) between untreated and RWJ‐56110‐treated groups was observed (Figure 3D). Thus, RWJ‐56110 decreases the recruitment of all leukocytes instead of a particular cell type.
Figure 3.
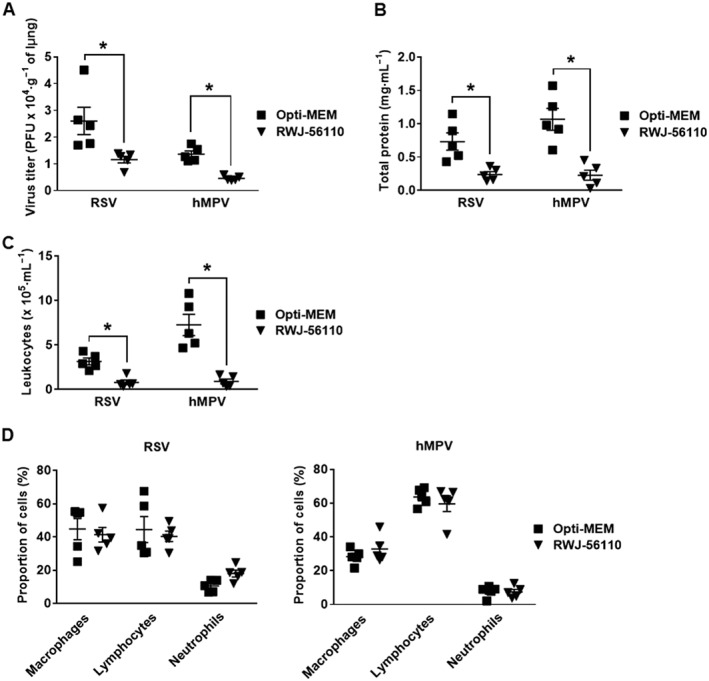
Protective effect of RWJ‐56110 after virus challenge. Mice were infected with RSV or hMPV and treated with Opti‐MEM medium, TFLLR‐NH2 or RWJ‐56110. Five days after infection, mice were killed and the lungs and BAL fluids were collected for evaluating (A) viral replication in the lungs, (B) BAL total protein, (C) BAL leukocyte recruitment and (D) BAL cell differentiation (n = 5 per group, *P < 0.05 RWJ‐56110 vs. Opti‐MEM).
In addition, the levels of the cytokines G‐CSF, IL‐3, IL‐4, IL‐6, IL‐10, IL‐12(p40), IL‐12(p70), IL‐17, KC and MCP‐1 were decreased in the BAL of RSV‐ and hMPV‐infected mice treated with RWJ‐56110 in comparison to infected/untreated mice (Figure 4). In contrast, RWJ‐56110 treatment specifically induced a decrease in IL‐5 levels during RSV infection whereas a decrease in IFN‐γ levels and an increase in IL‐9 levels were seen during hMPV infection (Figure 5). The concentrations of other cytokines and chemokines (eotaxin, GM‐CSF, IL‐1α, IL‐1β, IL‐2, IL‐13, MIP‐1α, MIP‐1β, RANTES and TNF‐α) were not influenced by the RWJ‐56110 treatment.
Figure 4.
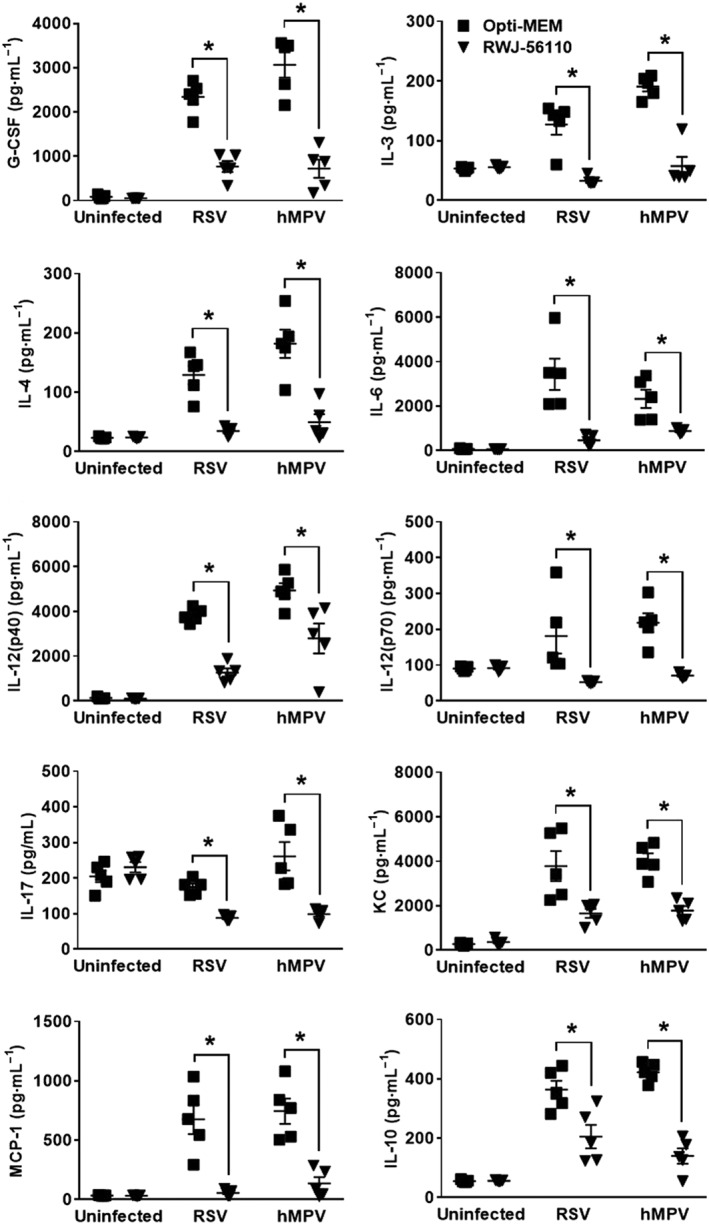
Profile of cytokines and chemokines in response to RWJ‐56110 during RSV and hMPV infections. Mice were infected or not with RSV or hMPV and treated with Opti‐MEM medium, TFLLR‐NH2 or RWJ‐56110. Five days after infection, mice were killed and BAL fluids were collected for evaluating the profile of cytokines and chemokines (n = 5 per group, *P < 0.05 RWJ‐56110 vs. Opti‐MEM).
Figure 5.
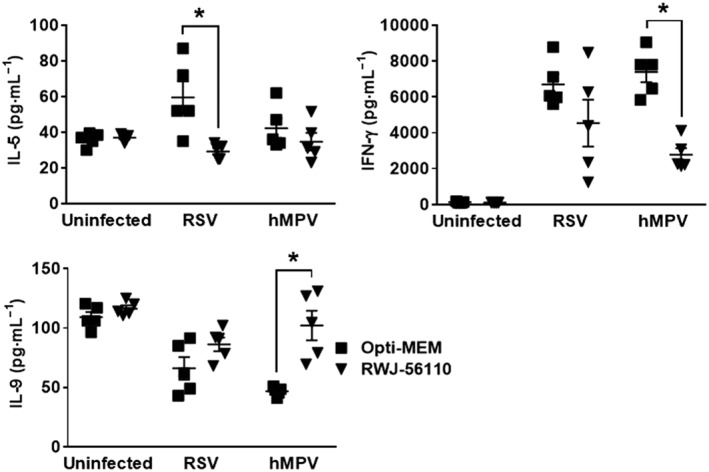
Profile of IFN‐γ, IL‐5 and IL‐9 in response to RWJ‐56110 during RSV and hMPV infections. Mice were infected or not with RSV or hMPV and treated with Opti‐MEM medium, TFLLR‐NH2 or RWJ‐56110. Five days after infection, mice were killed and BAL fluids were collected for evaluating the profile of cytokines and chemokines (n = 5 per group, *P < 0.05 RWJ‐56110 vs. Opti‐MEM).
RWJ‐56110 affects thrombin generation
Since PAR1 is a thrombin receptor (Kahn et al., 1998), we next examined whether coagulation is initiated upon RSV and hMPV infections. To this end, mice were infected with 3 × 106 PFU of RSV or 106 PFU of hMPV, and at day 5 post‐infection, BAL fluids were harvested to analyse TAT levels, a surrogate marker of thrombin generation. The results presented in Figure 6A show that TAT levels are increased upon RSV and hMPV infections in comparison to uninfected mice. Thus, both RSV and hMPV could activate the coagulation cascade, leading to thrombin generation. Interestingly, when infected mice were treated with RWJ‐56110, a significant decrease in TAT levels was observed in comparison with untreated mice. These findings led us to hypothesize that RSV‐ and hMPV‐triggered activation of PAR1 occurs upstream of thrombin generation that, in turn, increases the pathogenesis of these infections.
Figure 6.
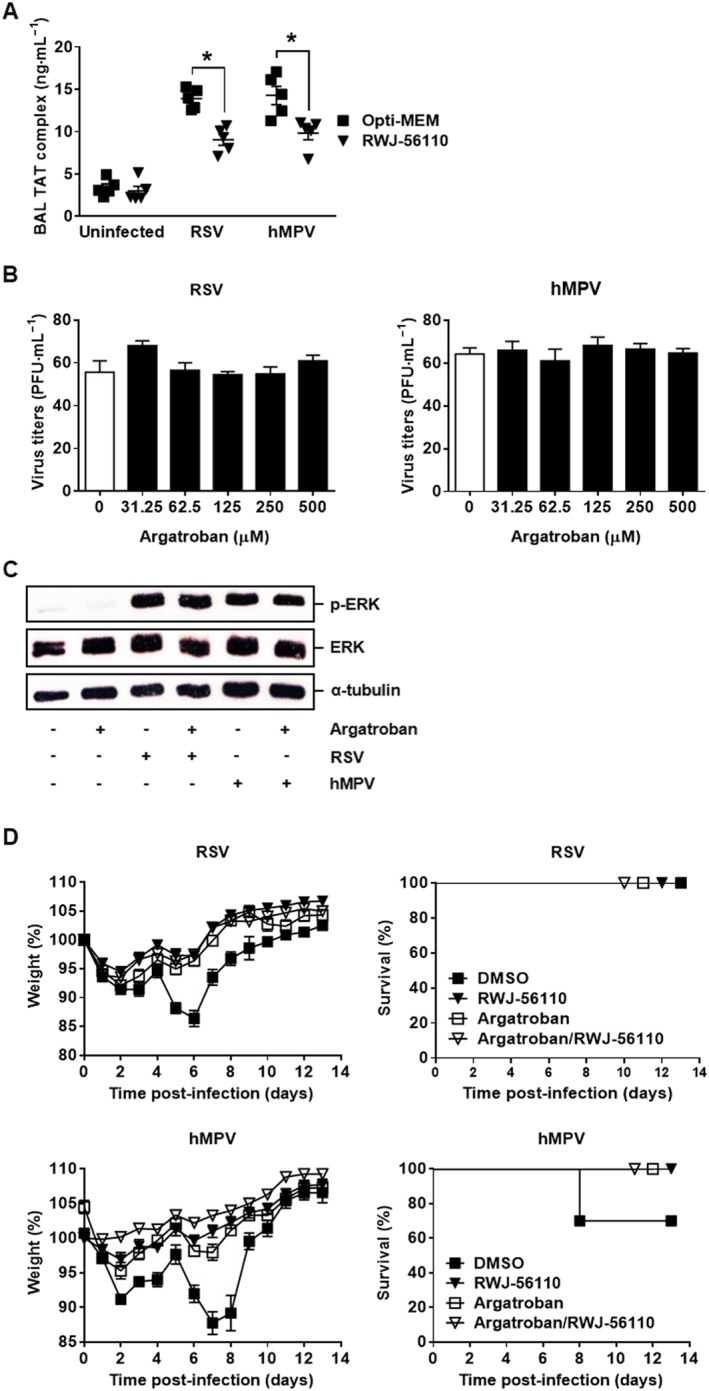
Thrombin is involved in the pathogenesis of RSV and hMPV. (A) Mice were infected with RSV or hMPV and treated with Opti‐MEM medium or RWJ‐56110. Five days after infection, mice were killed and the BAL fluids were collected for evaluating TAT levels (n = 5 per group, *P < 0.05 RWJ‐56110 vs. Opti‐MEM). (B, C) Antiviral effect of argatroban was investigated in vitro. Hep‐2 or LLC‐MK2 cells were treated with argatroban and then infected with RSV or hMPV. Virus‐infected cells were identified by immunostaining and counted (n = 6 per group). In parallel, A549 cells were treated or not with argatroban and then infected with RSV or hMPV. The cells were harvested for Western blot analysis of ERK phosphorylation. (D) Mice were injected with DMSO or argatroban and infected with RSV or hMPV. Animals were treated in parallel with Opti‐MEM medium or RWJ‐56110. They were monitored for weight loss for 14 days after infection and killed if humane end points were reached. Results are average % weight loss ± SEM or average % survival (n = 10 per group).
To test this possibility, thrombin was inhibited by using the direct specific thrombin inhibitor argatroban that blocks thrombin‐catalysed or ‐induced reactions (Yoshinaga et al., 2003; Dhillon, 2009). As expected, argatroban had no effect on the replication of RSV and hMPV, in vitro (Figure 6B), since there is no source of prothrombin, TF or factor X in these conditions. Furthermore, argatroban treatment also did not increase the level of ERK phosphorylation in virus‐infected cells (Figure 6C), which was shown to be required for the viral replication (Figure 1F). In contrast, argatroban treatment provided a full protection from RSV‐ and hMPV‐induced pathogenesis and death. Interestingly, co‐treatment of the mice with argatroban and RWJ‐56110 did not improve the protective effect of argatroban, reinforcing our hypothesis that PAR1 might lead to thrombin generation (Figure 6D).
Argatroban protected mice from viral replication and inflammation
To better define the effect of the treatment with argatroban, viral replication and inflammation were assessed in the lung homogenates and BAL fluids of RSV‐ or hMPV‐infected mice treated or not with argatroban. The results show that argatroban treatment significantly reduced pulmonary virus loads in vivo as well as leukocyte recruitment and concentrations of total protein in the lungs (Figures 7A–C). Interestingly, argatroban treatment also induced a decreased in the pulmonary levels of G‐CSF, IL‐3, IL‐4, IL‐6, IL‐1α, IL‐12(p40), IL‐12(p70), IL‐17, KC and MCP‐1 upon RSV and hMPV infections (Figure 8). In contrast, argatroban treatment specifically induced a decrease in IL‐5 levels during RSV infection whereas a specific decrease in IFN‐γ and IL‐10 levels and an increase in IL‐9 levels were observed during hMPV infection (Figure 9). Notably, we observed that in all these experiments, the effect of argatroban treatment was very similar to that of RWJ‐56110, with the exception to its effects on IL‐1α and IL‐10, and that co‐treatment did not provide an additional inhibitory effect on these parameters. The differences in IL‐1α and IL‐10 secretion in response to RWJ‐56110 treatment in two separate experiments might be due to the slightly different virulence of the virus stocks despite similar quantities of virus administered to the animals.
Figure 7.
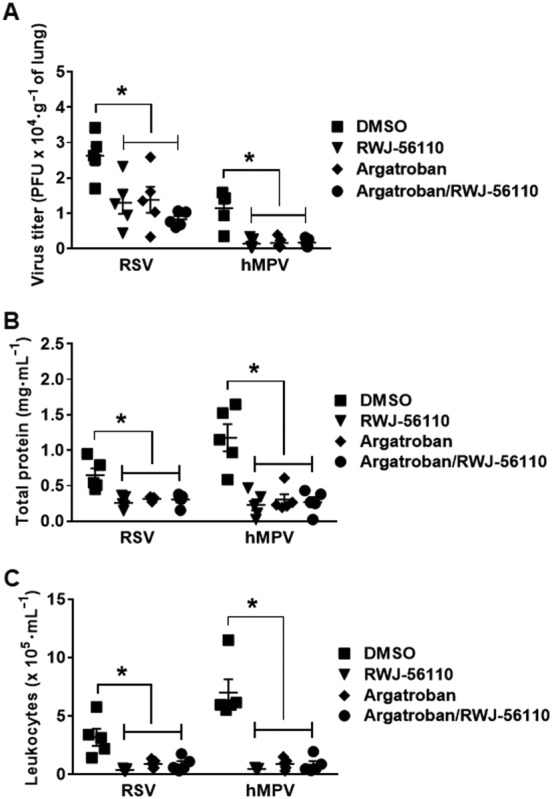
Protective effect of thrombin inhibition after virus challenge. Mice were injected with DMSO or argatroban and infected with RSV or hMPV. Animals were treated in parallel with Opti‐MEM medium or RWJ‐56110. Five days after infection, mice were killed and the lungs and BAL fluids were collected for evaluating (A) viral replication in the lungs, (B) BAL total protein and (C) leukocyte recruitment (n = 5 per group, *P < 0.05 RWJ‐56110; argatroban or argatroban/RWJ‐56110 vs. DMSO).
Figure 8.
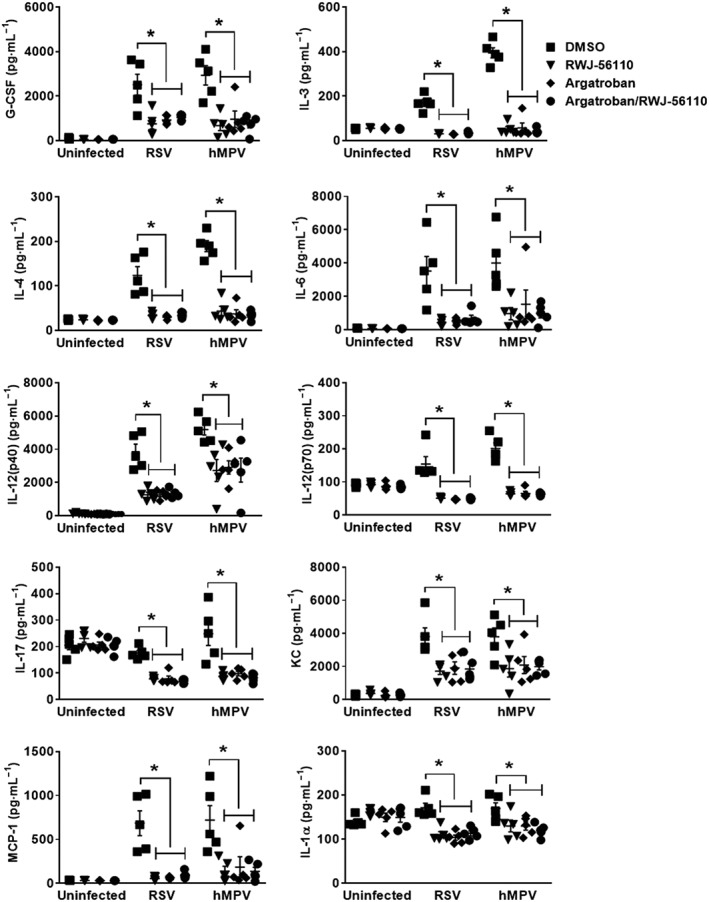
Profile of cytokines and chemokines in response to thrombin inhibition during RSV and hMPV infections. Mice were injected with DMSO or argatroban. Thereafter, they were infected or not with RSV or hMPV. They were treated in parallel with Opti‐MEM medium or RWJ‐56110. Five days after infection, mice were killed and the BAL fluids were collected for evaluating the profile of cytokines and chemokines (n = 5 per group, *P < 0.05 RWJ‐56110; argatroban or argatroban/RWJ‐56110 vs. DMSO).
Figure 9.
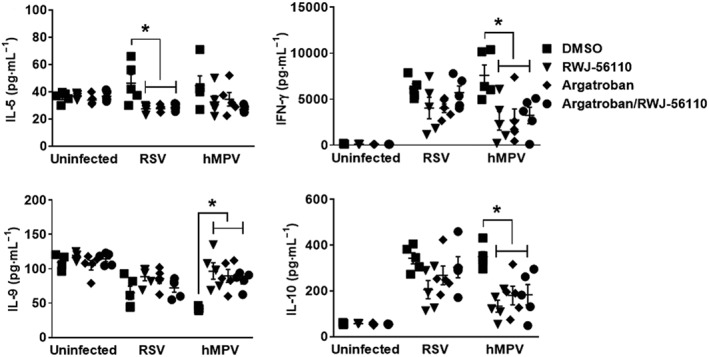
Profile of IFN‐γ, IL‐5, IL‐9 and IL‐10 in response to thrombin inhibition during RSV and hMPV infections. Mice were injected with DMSO or argatroban. Thereafter, they were infected or not with RSV or hMPV. They were treated in parallel with Opti‐MEM medium or RWJ‐56110. Five days after infection, mice were killed and the BAL fluids were collected for evaluating the profile of cytokines and chemokines (n = 5 per group, *P < 0.05 RWJ‐56110; argatroban or argatroban/RWJ‐56110 vs. DMSO).
PAR1 activation was not dependent on thrombin generation during viral infections
To firmly establish the mechanism by which PAR1 plays a deleterious role in viral infections, TAT and TF were evaluated in the fluids of BAL. Figure 10A shows that treatments with RWJ‐56110 alone or combined treatment with RWJ‐56110 and argatroban significantly diminished TAT levels in RSV‐ and hMPV‐infected mice. Moreover, TAT levels were not different between RWJ‐56110‐treated groups and argatroban‐treated groups. These data again confirm that the effect of RWJ‐56110 occurs upstream of thrombin generation. Furthermore, TF levels were elevated in the non‐centrifuged BAL of infected groups (but not after centrifugation), as compared to control uninfected groups, consistent with the expression of TF at the surface of inflammatory cells infiltrating the lungs. Treatment with RWJ‐56110 alone significantly decreased TF levels in both RSV‐ and hMPV‐infected groups whereas treatment with argatroban alone did not affect the quantity of TF. Finally, a combination of argatroban with RWJ‐56110 had no additional effect over that of RWJ‐56110 alone (Figure 10B). These data indicate that PAR1 activation does not depend on thrombin generation during viral infections and also suggest that PAR1 activation is involved in the virus pathogenesis upstream of thrombin generation via inflammation‐triggered TF activity.
Figure 10.
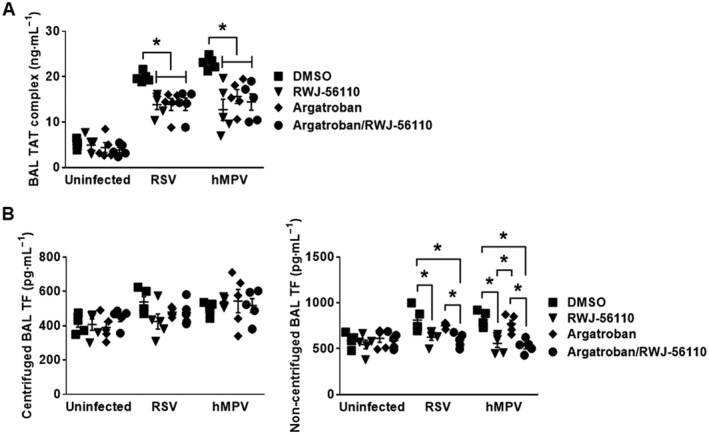
Profile of TAT and TF in response to thrombin inhibition during RSV and hMPV infections. Mice were injected with DMSO or argatroban. Thereafter, they were infected or not with RSV or hMPV. They were treated in parallel with Opti‐MEM medium or RWJ‐56110. Five days after infection, mice were killed and the BAL fluids were collected for evaluating the profile of TAT and TF (n = 5 per group, *P < 0.05 RWJ‐56110 or argatroban/RWJ‐56110 vs. DMSO; argatroban vs. argatroban/RWJ‐56110; RWJ‐56110 vs. argatroban).
Discussion
In the present study it was shown that PAR1 plays a detrimental role during hMPV and RSV infections. In vitro, blocking PAR1 by using the specific antagonist RWJ‐56110 inhibited virus replication. Thus, PAR1 increases the infectivity of both hMPV and RSV. In agreement with these in vitro results, stimulating PAR1 with its specific agonist TFLLR‐NH2 increased hMPV pathogenesis in mice, while treating the mice with the antagonist RWJ‐56110 had the opposite effect. Thus, our findings show that PAR1 plays a detrimental role during RSV and hMPV infections. These results are consistent with our previous findings showing that PAR1 inhibition by SCH79797 exerts an antiviral effect against this virus by preventing furin‐induced cleavage of hMPV fusion protein (Aerts et al., 2013). To our knowledge, this is the first time that PAR1 has been demonstrated to be involved in RSV infections. RSV and hMPV use distinct mechanisms to interfere with the host's innate and adaptive immune responses (Guerrero‐Plata et al., 2005; Guerrero‐Plata et al., 2006). Despite these differences, blocking PAR1 activation had beneficial effects on both viruses.
In the present study, TFLLR‐NH2 did not appear to provide a substantial increase in viral replication, in vitro. One possible explanation for this result is that this PAR1 agonist does not add appropriate signals in nature or intensity to those induced by the viruses. The absence of an effect of TFLLR‐NH2 on the level of ERK phosphorylation observed in these infected cells favours this hypothesis. Indeed, although there are no findings to support a relationship between ERK phosphorylation and hMPV replication, it has been demonstrated that ERK phosphorylation is critical for RSV infection (Monick et al., 2001; Pazdrak et al., 2002; Kong et al., 2004). In contrast, PAR1 inhibition by RWJ‐56110 displayed a potent antiviral effect. This could presumably be explained by the fact that RSV and hMPV can effectively activate PAR1 by themselves, as demonstrated in Figure 1D. We were able to demonstrate the protective effect of PAR1 inhibition on hMPV‐induced mortality, in vivo. Unfortunately, we could not do this for RSV because the BALB/c mice are only semi‐permissive to RSV with it having no effect on the survival rate of these mice (Meng et al., 2014). However, the ability of PAR1 inhibition to protect these mice from weight loss allowed us to conclude that PAR1 inhibition by RWJ‐56110 may provide protection against RSV and hMPV infections.
PAR1 is a thrombin receptor whose activation is triggered by proteolytic cleavage of its extracellular N‐terminal domain, notably by thrombin or other proteases (Vu et al., 1991). Thrombin has been reported to contribute to the pathogenesis of several viruses (Dubovi et al., 1983; Sutherland et al., 2007, 2012; Hsue et al., 2012; Lin et al., 2012; Antoniak et al., 2013; Boilard et al., 2014; Bondu et al., 2015). To our knowledge, little is known about the involvement of thrombin in the pathogenesis of RSV and hMPV infections. Indeed, only one study has shown that thrombin increases RSV‐induced cell fusion in A549 cells (Dubovi et al., 1983), whereas there is no report about the involvement of thrombin in hMPV infection. It was evident that, similar to PAR1 inhibition by RWJ‐56110, thrombin inhibition by argatroban resulted in significantly less weight loss and mortality and decreased viral replication and inflammation in infected mice. Therefore, we can conclude that thrombin plays an essential role in the pathogenesis of RSV and hMPV infections. There is a strong bidirectional link between inflammation and the activation of the coagulation cascade with inflammatory cells providing TF, and in turn thrombin generation itself is responsible for leukocyte recruitment and, therefore, exacerbates inflammation during infections (Chen and Dorling, 2009). Moreover, no difference in the antiviral effect was found between single and combined RWJ‐56110 and argatroban treatments.
The coagulation system is schematically divided into three pathways: the extrinsic, intrinsic and common pathways. In the extrinsic pathway, the TF/FVIIa complex is the key initiator of the coagulation protease cascade and activates both FIX and VX, leading to the formation of thrombin (Mackman, 2009). Here, we measured TF levels in the non‐centrifuged and centrifuged fluids of BAL and found that RSV and hMPV induced an elevation of TF levels in the non‐centrifuged samples. Thus, the major sources of TF in RSV and hMPV infections were probably leukocytes recruited in infected lungs or microparticles, as previously reported (Bastarache et al., 2009; Pawlinski et al., 2010). Interestingly, TF levels were decreased by PAR1 inhibition, but not by thrombin inhibition, indicating that PAR1 activation occurred via an unknown thrombin‐independent pathway. Further studies are required to clarify the mechanisms of RSV‐ and hMPV‐induced PAR1 activation. Moreover, decreased TF levels due to PAR1 inhibition correlated with decreased TAT levels. In other words, PAR1 activation occurs upstream of thrombin and modulates TF exposure. This results in uncontrolled thrombin generation, which in turn plays a detrimental role during RSV and hMPV infections.
Previous reports have shown that PAR1 induces an adverse effect during influenza virus infections through plasminogen (Khoufache et al., 2013), which itself acts through fibrinolysis (Berri et al., 2013). Thus, altogether, these results suggest that PAR1 activation upon viral infection dysregulates haemostasis, leading to a more severe disease. Once activated, PAR1 generally exerts a pro‐inflammatory effect such as mast cell degranulation, increased vascular permeability, cytokine release and leukocyte recruitment, which may exacerbate inflammation (Vergnolle et al., 2002). In contrast to our study, PAR1 has been reported to protect against pathogens in some instances (Scholz et al., 2004; Kaneider et al., 2007; Wee et al., 2010; Antoniak et al., 2013; Chionh et al., 2015). Potentially, PAR1 may have different roles according to the pathogens and host models. Alternatively, it is known that a well‐regulated immune response to infection plays a protective role in contrast to the dysregulated excessive inflammation. Thus, as discussed by Berri et al. (2014), the role of PAR1 in viral infection may be dual, protective during a mild viral infection and deleterious during a severe viral infection.
In summary, we demonstrate here, for the first time, that PAR1 inhibition by RWJ‐56110 exerts an antiviral effect against both RSV and hMPV. We also showed that the antiviral effect of RWJ‐56110 results in the blockade of TF‐mediated thrombin generation during pneumovirus infections. Finally, the present study highlights the importance of thrombin depletion by using its inhibitor argatroban or PAR1 inhibition by using its antagonists RWJ‐56110 for attenuating the consequences of RSV and hMPV infections, including weight loss, mortality, virus loads, lung tissue injuries and inflammation in mice. Thus, the use of thrombin inhibitors or PAR1 antagonists may have potential as a novel and efficacious strategy for the prevention and/or treatment of RSV and hMPV infections that warrant additional studies.
Author contributions
V.L., M.J.P. and M.E.H. conceived and designed this study. V.L., C.C. and C.R. performed the experiments. V.L., B.R., M.C.A., M.J.P., M.E.H. and G.B. were involved in the discussions of the data. V.L. and B.R. wrote the manuscript. G.B. reviewed and edited this manuscript. All the authors read and approved this manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was funded by the Canadian Institutes of Health Research (grant 273261 to G.B.). The authors are thankful to Julie‐Christine Lévesque (Technological platforms of Imaging and Cytometry, Infectious Disease Research Centre, Quebec, QC, Canada) for the assistance in the use of the Luminex system.
Lê, V. B. , Riteau, B. , Alessi, M.‐C. , Couture, C. , Jandrot‐Perrus, M. , Rhéaume, C. , Hamelin, M.‐È. , and Boivin, G. (2018) Protease‐activated receptor 1 inhibition protects mice against thrombin‐dependent respiratory syncytial virus and human metapneumovirus infections. British Journal of Pharmacology, 175: 388–403. doi: 10.1111/bph.14084.
References
- Aerts L, Hamelin ME, Rhéaume C, Lavigne S, Couture C, Kim W et al (2013). Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. PLoS ONE 8: e72529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonso CL, Amarasinghe GK, Bányai K, Bào Y, Basler CF, Bavari S et al (2016). Taxonomy of the order Mononegavirales: update 2016. Arch Virol 161: 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade‐Gordon P, Maryanoff BE, Derian CK, Zhang H‐C, Addo MF, Darrow AL et al (1999). Design, synthesis, and biological characterization of a peptide‐mimetic antagonist for a tethered‐ligand receptor. Proc Natl Acad Sci U S A 96: 12257–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniak S, Mackman N (2014). Multiple roles of the coagulation protease cascade during virus infection. Blood 123: 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniak S, Owens AP, Baunacke M, Williams JC, Lee RD, Weithäuser A et al (2013). PAR‐1 contributes to the innate immune response during viral infection. J Clin Invest 123: 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma K, Wakabayashi H, Hayashi T, Okuyama N, Seto M, Matsumine A et al (2004). Thrombin inhibitor, argatroban, prevents tumor cell migration and bone metastasis. Oncology 67: 166–173. [DOI] [PubMed] [Google Scholar]
- Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB (2009). Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 297: L1035–L1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault‐Grunenwald ML, Lê VB et al (2013). Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog 9: e1003229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berri F, Lê VB, Jandrot‐Perrus M, Lina B, Riteau B (2014). Switch from protective to adverse inflammation during influenza: viral determinants and hemostasis are caught as culprits. Cell Mol Life Sci 71: 885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T et al (2014). Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 123: 2854–2863. [DOI] [PubMed] [Google Scholar]
- Bondu V, Schrader R, Gawinowicz MA, McGuire P, Lawrence DA, Hjelle B et al (2015). Elevated cytokines, thrombin and PAI‐1 in severe HCPS patients due to Sin Nombre virus. Virus 7: 559–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello‐Gutiérrez C, Manjarrez‐Zavala ME, Huerta‐Zepeda A, Cime‐Castillo J, Monroy‐Martínez V, Correa BB et al (2009). Modification of the cytoprotective protein C pathway during Dengue virus infection of human endothelial vascular cells. Thromb Haemost 101: 916–928. [PubMed] [Google Scholar]
- Chen D, Dorling A (2009). Critical roles for thrombin in acute and chronic inflammation. J Thromb Haemost 7: 122–126. [DOI] [PubMed] [Google Scholar]
- Chionh YT, Ng GZ, Ong L, Arulmuruganar A, Stent A, Saeed MA et al (2015). Protease‐activated receptor 1 suppresses Helicobacter pylori gastritis via the inhibition of macrophage cytokine secretion and interferon regulatory factor 5. Mucosal Immunol 8: 68–79. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffrasnes C, Hamelin ME, Prince GA, Boivin G (2008). Identification and evaluation of a highly effective fusion inhibitor for human metapneumovirus. Antimicrob Agents Chemother 52: 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derian CK, Maryanoff BE, Andrade‐Gordon P, Zhang HC (2003). Design and evaluation of potent peptide‐mimetic PAR1 antagonists. Drug Dev Res 59: 355–366. [Google Scholar]
- DeVincenzo JP, Wilkinson T, Vaishnaw A, Cehelsky J, Meyers R, Nochur S et al (2010). Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med 182: 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S (2009). Argatroban: a review of its use in the management of heparin‐induced thrombocytopenia. Am J Cardiovasc Drugs 9: 261–282. [DOI] [PubMed] [Google Scholar]
- Di Serio C, Pellerito S, Duarte M, Massi D, Naldini A, Cirino G et al (2007). Protease‐activated receptor 1‐selective antagonist SCH79797 inhibits cell proliferation and induces apoptosis by a protease‐activated receptor 1‐independent mechanism. Basic Clin Pharmacol Toxicol 101: 63–69. [DOI] [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Rosenberg HF (2004). Animal models for studying respiratory syncytial virus infection and its long term effects on lung function. Pediatr Infect Dis J 23: S228–S234. [DOI] [PubMed] [Google Scholar]
- Drysdale SB, Green CA, Sande CJ (2016). Best practice in the prevention and management of paediatric respiratory syncytial virus infection. Ther Adv Infect Dis 3: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubovi EJ, Geratz JD, Tidwell RR (1983). Enhancement of respiratory syncytial virus‐induced cytopathology by trypsin, thrombin, and plasmin. Infect Immun 40: 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito S, Mastrolia MV (2016). Metapneumovirus infections and respiratory complications. Semin Respir Crit Care Med 37: 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SL, Arthur JF, Tran HA, Hamilton JR (2015). Approval of the first protease‐activated receptor antagonist: rationale, development, significance, and considerations of a novel anti‐platelet agent. Blood Rev 29: 179–189. [DOI] [PubMed] [Google Scholar]
- Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R (2013). Proteinase‐activated receptors (PARs) – focus on receptor‐receptor‐interactions and their physiological and pathophysiological impact. Cell Commun Signal 11: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Plata A, Casola A, Garofalo RP (2005). Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J Virol 79: 14992–14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME et al (2006). Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol 34: 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsue PY, Scherzer R, Grunfeld C, Nordstrom SM, Schnell A, Kohl LP et al (2012). HIV infection is associated with decreased thrombin generation. Clin Infect Dis 54: 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S et al (1998). A dual thrombin receptor system for platelet activation. Nature 394: 690–694. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C et al (2007). ‘Role reversal’ for the receptor PAR1 in sepsis‐induced vascular damage. Nat Immunol 8: 1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann R, Schulze B, Krause G, Mayr LM, Settmacher U, Henklein P (2005). Proteinase‐activated receptors (PARs)—the PAR3 Neo‐N‐terminal peptide TFRGAP interacts with PAR1. Regul Pept 125: 61–66. [DOI] [PubMed] [Google Scholar]
- Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E et al (2013). PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest 123: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, San Juan H, Behera A, Peeples ME, Wu J, Lockey RF et al (2004). ERK‐1/2 activity is required for efficient RSV infection. FEBS Lett 559: 33–38. [DOI] [PubMed] [Google Scholar]
- Lin SW, Chuang YC, Lin YS, Lei HY, Liu HS, Yeh TM (2012). Dengue virus nonstructural protein NS1 binds to prothrombin/thrombin and inhibits prothrombin activation. J Infect 64: 325–334. [DOI] [PubMed] [Google Scholar]
- Liu J, Schuff‐Werner P, Steiner M (2004). Double transfection improves small interfering RNA‐induced thrombin receptor (PAR‐1) gene silencing in DU 145 prostate cancer cells. FEBS Lett 577: 175–180. [DOI] [PubMed] [Google Scholar]
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V et al (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N (2009). The role of tissue factor and factor VIIa in hemostasis. Anesth Analg 108: 1447–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N, Antoniak S (2014). Roles of PAR1 and PAR2 in viral myocarditis. Thromb Res 133: S18–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanoff BE, Zhang HC, Andrade‐Gordon P, Derian CK (2003). Discovery of potent peptide‐mimetic antagonists for the human thrombin receptor, protease‐activated receptor‐1 (PAR‐1). Curr Med Chem Cardiovasc Hematol Agents 1: 13–36. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Stobart CC, Hotard AL, Moore ML (2014). An overview of respiratory syncytial virus. PLoS Pathog 10: e1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monick MM, Staber JM, Thomas KW, Hunninghake GW (2001). Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen‐activated protein kinase. J Immunol 166: 2681–2687. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM et al (2007). Protease‐activated receptor‐1 contributes to cardiac remodeling and hypertrophy. Circulation 116: 2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlinski R, Wang JG, Owens AP, Williams J, Antoniak S, Tencati M et al (2010). Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood 116: 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazdrak K, Olszewska‐Pazdrak B, Liu T, Takizawa R, Brasier AR, Garofalo RP et al (2002). MAPK activation is involved in posttranscriptional regulation of RSV‐induced RANTES gene expression. Am J Physiol Lung Cell Mol Physiol 283: L364–L372. [DOI] [PubMed] [Google Scholar]
- Persson BD, Jaffe AB, Fearns R, Danahay H (2014). Respiratory syncytial virus can infect basal cells and alter human airway epithelial differentiation. PLoS ONE 9: e102368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg HF, Domachowske JB (2012). Inflammatory responses to respiratory syncytial virus (RSV) infection and the development of immunomodulatory pharmacotherapeutics. Curr Med Chem 19: 1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussy JF, Carbonneau J, Ouakki M, Papenburg J, Hamelin MÈ, De Serres G et al (2014). Human metapneumovirus viral load is an important risk factor for disease severity in young children. J Clin Virol 60: 133–140. [DOI] [PubMed] [Google Scholar]
- Schildgen O, Simon A, Williams J (2007). Animal models for human metapneumovirus (HMPV) infections. Vet Res 38: 117–126. [DOI] [PubMed] [Google Scholar]
- Scholz M, Vogel JU, Höver G, Prösch S, Kotchetkov R, Cinatl J et al (2004). Thrombin induces Sp1‐mediated antiviral effects in cytomegalovirus‐infected human retinal pigment epithelial cells. Med Microbiol Immunol 193: 195–203. [DOI] [PubMed] [Google Scholar]
- Sidhu T, French S, Hamilton J (2014). Differential signaling by protease‐activated receptors: implications for therapeutic targeting. Int J Mol Sci 15: 6169–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland MR, Friedman HM, Pryzdial EL (2007). Thrombin enhances herpes simplex virus infection of cells involving protease‐activated receptor 1. J Thromb Haemost 5: 1055–1061. [DOI] [PubMed] [Google Scholar]
- Sutherland MR, Ruf W, Pryzdial EL (2012). Tissue factor and glycoprotein C on herpes simplex virus type 1 are protease‐activated receptor 2 cofactors that enhance infection. Blood 119: 3638–3645. [DOI] [PubMed] [Google Scholar]
- Vergnolle N (2009). Protease‐activated receptors as drug targets in inflammation and pain. Pharmacol Ther 123: 292–309. [DOI] [PubMed] [Google Scholar]
- Vergnolle N, Derian CK, D'Andrea MR, Steinhoff M, Andrade‐Gordon P (2002). Characterization of thrombin‐induced leukocyte rolling and adherence: a potential proinflammatory role for proteinase‐activated receptor‐4. J Immunol 169: 1467–1473. [DOI] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR (1991). Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64: 1057–1068. [DOI] [PubMed] [Google Scholar]
- Wang H, Ubl JJ, Stricker R, Reiser G (2002). Thrombin (PAR‐1)‐induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am J Physiol Cell Physiol 283: C1351–C1364. [DOI] [PubMed] [Google Scholar]
- Wee JL, Chionh YT, Ng GZ, Harbour SN, Allison C, Pagel CN et al (2010). Protease‐activated receptor‐1 down‐regulates the murine inflammatory and humoral response to Helicobacter pylori . Gastroenterology 138: 573–582. [DOI] [PubMed] [Google Scholar]
- Yoshinaga M, Sunagawa M, Shimada S, Nakamura M, Murayama S, Kosugi T (2003). Argatroban, specific thrombin inhibitor, induced phenotype change of cultured rabbit vascular smooth muscle cells. Eur J Pharmacol 461: 9–17. [DOI] [PubMed] [Google Scholar]