

Abstract
Background and Purpose
Nonsteroidal anti‐inflammatory drugs (NSAIDs) are administered to manage the pain typically found in patients suffering from pancreatitis. NSAIDs also display anti‐proliferative activity against cancer cells; however, their effects on normal, untransformed cells are poorly understood. Here, we evaluated whether NSAIDs inhibit the proliferation of pancreatic acinar cells during the development of acute pancreatitis.
Experimental Approach
The NSAIDs ibuprofen and diclofenac were administered to C57BL/6 mice after induction of pancreatitis with serial injections of cerulein. In addition, ibuprofen was administered concomitantly with 3,5,3‐L‐tri‐iodothyronine (T3), which induces acinar cell proliferation in the absence of tissue inflammation. The development of pancreatic inflammation, acinar de‐differentiation into metaplastic lesions and acinar proliferation were quantified by histochemical, biochemical and RT‐PCR approaches.
Key Results
Therapeutic ibuprofen treatment selectively reduced pancreatic infiltration of activated macrophages in vivo, and M1 macrophage polarization and pro‐inflammatory cytokine expression both in vivo and in vitro. Reduced macrophage activation was accompanied by reduced acinar de‐differentiation into acinar‐to‐ductal metaplasia. Acinar proliferation was significantly impaired in the presence of ibuprofen and diclofenac, as demonstrated at both the level of proliferation markers and expression of cell cycle regulators. Ibuprofen also reduced acinar cell proliferation induced by mitogenic stimulation with T3, a treatment that does not elicit pancreatic inflammation.
Conclusions and Implications
Our study provides evidence that the NSAIDs ibuprofen and diclofenac inhibit pancreatic acinar cell division. This suggests that prolonged treatment with these NSAIDs may negatively affect the regeneration of the pancreas and further studies are needed to confirm these findings in a clinical setting.
Linked Articles
This article is part of a themed section on Inventing New Therapies Without Reinventing the Wheel: The Power of Drug Repurposing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.2/issuetoc
Abbreviations
- ADM
acinar‐to‐ductal metaplasia
- NSAIDs
nonsteroidal anti‐inflammatory drugs
- T3
3,5,3‐L‐tri‐iodothyronine
Introduction
Nonsteroidal anti‐inflammatory drugs (NSAIDs) are a broad family of compounds primarily used as analgesics to treat pains of different origin and to control inflammation. At the molecular level, NSAIDs exert these effects by inhibiting the activity of COX1 and COX2 (Chavez and Dekorte, 2003), enzymes that catalyse the synthesis of PGs and thromboxanes. In addition to the established and well‐exploited analgesic and anti‐inflammatory effects of these compounds, numerous studies have demonstrated that NSAIDs are also effective in the prevention of many common cancers (Taketo, 1998; Vainio, 2001). This is particularly evident in the case of ibuprofen, the most commonly used over‐the‐counter NSAID (Bushra and Aslam, 2010). Specifically, this drug showed a superior effectiveness compared with other NSAIDs in suppressing proliferation and inducing apoptosis of human prostate cancer cells at clinically relevant concentrations (Andrews et al., 2002). Similar inhibition of proliferation exerted by ibuprofen, alone or in combination therapy, was also observed in the case of gastric (Bonelli et al., 2011), lung (Endo et al., 2014), colon (Reddy et al., 1992; Greenspan et al., 2011) and breast cancer, suggesting that ibuprofen may be useful in the chemoprevention of different malignancies (reviewed in Piazza et al., 2010, Gurpinar et al., 2014).
While the anti‐proliferative effect of ibuprofen is well documented in the case of cancer cells, the impact of this drug on the proliferation of normal cells in the absence of malignant transformation is poorly elucidated. Ibuprofen is amongst the therapeutic interventions administered to relieve the pain typically experienced by patients suffering from mild forms of acute pancreatitis or, in the most severe cases of the disease, when patients are weaned off narcotic therapy. In addition, it is also used to manage chronic pancreatitis, where the permanent inflammation of the organ is associated with chronic pain (reviewed in Banks et al., 2010). NSAID treatment has also been shown to have a prophylactic effect; it prevents post‐operative pancreatitis when given prior to endoscopic retrograde cholangiopancreatography (Murray et al., 2003; Sotoudehmanesh et al., 2007). Surprisingly, extensive studies to evaluate the efficacy of NSAID during acute pancreatitis are lacking. A recent review of the available literature highlighted the fact that, while NSAIDs are able to control pain in acute pancreatitis patients, the use of NSAIDs is also associated with the risk for developing acute pancreatitis (reviewed in Pezzilli et al., 2010). Thus, further clinical trials are needed to identify the optimal NSAID to be used in the management of pancreatitis.
In this study, we evaluated whether the therapeutic administration of the NSAIDs ibuprofen and diclofenac affects the course of acute pancreatitis in terms of progression of inflammation and regeneration of the organ, using the most widespread murine model of the disease based on cerulein treatment. In addition, we investigated whether ibuprofen, administered in the absence of inflammatory insult, is able to directly inhibit mitogen‐induced proliferation of pancreatic acinar cells.
Methods
Animal experiments
All animal experiments were performed in accordance with Swiss Federal animal regulations and approved by the cantonal veterinary office of Zurich. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Mice used in this study were adult 8–12‐week‐old C57BL/6 mice in a weight range of 25–30 g (Envigo Laboratories, Horst, The Netherlands). Groups of four to five mice were kept in standard individually ventilated cages in an specific pathogen free (SPF) facility under standardized conditions (12 h light/dark cycle, 23°C, humidity of 50–60%) with food and water ad libitum. Only male mice were used in this study. Pancreatitis was induced via six i.p. injections of 50 μg·kg−1 cerulein, at hourly intervals. In the ‘staggered’ protocol, cerulein treatment was performed on three alternate days (Monday, Wednesday and Friday), and specimens were harvested from the animals on the following Monday, 7 days after the initial cerulein injection. In the ‘consecutive’ protocol, cerulein treatment was performed on two consecutive days (Monday and Tuesday), and specimens were harvested from the animals on the following Monday, 7 days after the initial cerulein injection. Ibuprofen and diclofenac (Sigma) were injected twice daily, 2 h apart, i.p. at 25 and 10 mg·kg−1 respectively. Control animals for cerulein, ibuprofen and diclofenac treatments received vehicle (saline solution, 0.9% NaCl) injections. In the ‘staggered’ protocol of pancreatitis, ibuprofen was administered for 5 days starting 2 h after the second set of cerulein injections. In the ‘consecutive’ protocol of pancreatitis, ibuprofen and diclofenac were administered for 5 days starting 24 h after the second set of cerulein injections.
3,5,3‐L‐Tri‐iodothyronine (T3) was administered daily i.p. at 400 mg·kg−1. Stock solution of 2 mg·mL−1 T3 were prepared in 0.1 M NaOH, pH 7.4 and freshly diluted in saline to the final concentration required for the in vivo experiments. Control animals received vehicle injections. Ibuprofen was injected twice daily, i.p. at 25 mg·kg−1, 1 h before and 1 h after T3 injections. Schematic representations of the different study groups are depicted in the relevant figures.
Specimens were harvested from the animals under isoflurane anaesthesia. Groups of five animals were tested for each experiment and time point. Animals were assigned randomly to different experimental groups for all in vivo studies. Data collection and evaluation of all in vivo and in vitro experiments were performed blindly of the group identity.
Mammalian cell cultures
The RAW264.7 macrophage cell line was maintained in DMEM + GlutaMAX supplemented with 10% FBS, 50 U·mL−1 penicillin and 50 μg·mL−1 streptomycin at 37°C in a 5% CO2 atmosphere. Cells were pre‐incubated with 800 μM ibuprofen for 30 min and stimulated with 10 ng·mL−1 LPS for 16 h in the presence or absence of ibuprofen.
The AR42J acinar cell line was maintained in F12K medium supplemented with 20% FBS, 50 U·mL−1 penicillin and 50 μg·mL−1 streptomycin at 37°C in a 5% CO2 atmosphere. For proliferation experiments, cells were seeded in 96‐well plates and incubated with different concentrations of ibuprofen for 72 h.
Cell number, cell viability and cell diameter were determined using an automated cell counter (NucleoCounter® NC‐200™, Chemometec, Allerod, Denmark).
Transcript analysis
Total RNA was extracted from pancreatic tissue, as described previously (Graf et al., 2002), or cell lines and reverse‐transcribed with qScript™ cDNA SuperMix (Quanta Biosciences). Gene expression was measured by real‐time PCR on a 7500 Fast Real‐Time PCR System (Applied Biosystems) using Taqman probes (Applied Biosystems). Transcript levels were normalized using 18S RNA as a reference and expressed as ΔΔCt relative to the value of control samples.
Immunohistochemistry
Pancreas specimens were fixed overnight in neutral buffered formalin, dehydrated through a series of graded ethanol baths and embedded in paraffin. For histological analyses, 3 μm tissue sections were deparaffinized in xylol and rehydrated in graded ethanol. Following antigen retrieval in citrate buffer and neutralization of endogenous peroxidase activity with 0.3% hydrogen peroxide in methanol, sections were incubated with protein block solution (Dako), primary and secondary antibodies and stained with 3,3′‐diaminobenzidine tetrahydrochloride (Vectastain® ABC HRP Kit, Vector Laboratories, Peterborough, UK), according to the manufacturer's protocol. Primary antibodies used in this study were as follows: rabbit anti‐Ki67 (#ab16667, Abcam, Cambridge, UK); rabbit anti‐phospho‐histone 3 (#2066052, Millipore, MA, USA); rabbit anti‐amylase (#A8273‐1VL, Sigma‐Aldrich, Buchs, Switzerland); rabbit anti‐PU.1 (#2266, Cell Signalling Technologies, Danvers, MA); rabbit anti‐γH2A.X (Ser139) (#9718, Cell Signalling Technologies, Danvers, MA); rat anti‐F4/80 (#T‐2006 BMA Biomedicals, Switzerland); rabbit anti‐CD3 (#A 0452, Dako); rabbit anti‐cleaved caspase‐3 (Asp175) (#9661, Cell Signalling Technologies, Danvers, MA); mouse anti‐YM1/Chitinase 3‐like 3 (#AF2446, R&D Systems, MN, USA); and rabbit anti‐IRF‐5 (#10547–1‐AP, Proteintech, Manchester, UK).
Secondary antibodies used in this study were biotinylated goat anti‐rabbit IgG (H + L), included in the Vectastain® ABC HRP Kit. Nuclei were visualized with DAPI.
Detection of DNA fragmentation in apoptotic cells was performed with a TUNEL assay using an ApopTag peroxidase Kit (MP Biomedicals, Illkirch, France), according to the manufacturer's instructions.
For quantitative analysis of acinar‐ductal metaplasia (ADM), paraffin‐embedded pancreas specimens were immunostained for amylase, and slides were scanned with a NDP NanoZoomer Digital Pathology Slide Scanner (Hamamatsu) and analysed for ADM lesions in a blinded fashion. ADM present in the entire pancreas slide of five mice from each treatment condition were quantified by manual counting. ADM were identified according to the following: (i) loss of amylase content; (ii) structural re‐organization into tubular complexes; and (iii) stromal reaction characterized by presence of cell infiltrates. The area occupied by ADM was expressed as percentage of total pancreatic area present in each slide.
The amount of amylase and F4/80 expressing macrophages present in the tissue was determined by densitometric quantification in at least 10 random high‐power fields per slide using the Cell^P analysis software (Olympus). Positive area was expressed as percentage of total pancreatic area present in each power field. Pancreatic ducts and vessels were excluded from the analysis.
Microscopy analyses were performed on a wide‐field Nikon Eclipse Ti (Amsterdam, The Netherlands). Quantification of labelled cells was performed in at least 10 randomly selected high‐power fields (×200) per slide using the NIS Elements BR Analysis and Cell^P analysis software.
Data and analysis
The data are expressed as means ± SEM. The statistical significance of differences in the means of experimental groups was determined using Student's unpaired, two‐tailed t‐test (GraphPad Prism 4.0c; GraphPad Software, Inc.), and a probability value <0.05 was considered statistically significant. Data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Ibuprofen treatment reduces pancreatic recruitment of activated macrophages following induction of acute pancreatitis
To assess the effect of ibuprofen during the progression of pancreatitis, we administered the drug in a therapeutic manner starting 2 days after induction of the disease via cerulein injections (regimen scheme depicted in Figure 1A). In this experimental setting, the initial acinar injury is comparable in the control and ibuprofen‐treated groups. Ibuprofen was applied at a dose similar to the range used in human therapy (Janssen and Venema, 1985) and which was demonstrated to inhibit PG production and pain perception in mice (Salama et al., 2016). Animals were harvested 7 days after the first cerulein injection. Following induction of pancreatitis, ibuprofen‐treated mice displayed histological alterations of pancreatic parenchyma similar to vehicle‐treated mice (Figure 1B) and a comparable pattern of weight loss (Figure S1). We then investigated whether ibuprofen reduced the infiltration of inflammatory cells. The total number of leukocytes was only slightly lowered in the presence of the drug (Figure 1C). However, ibuprofen reduced the level of macrophages recruited in the pancreas, tested both via F4/80 immunostaining, detecting macrophages, and gene expression levels (Figure 1D, F). On the contrary, the number of CD3‐positive T cells was comparable in control and treated samples (Figure 1D, F). These data suggest that ibuprofen targets the infiltration of selected sub‐populations of inflammatory cells. Concomitant with reduced macrophage infiltration, ibuprofen treatment also reduced the expression of selected cytokines, chemokines and adhesion molecules normally up‐regulated in the pancreas following induction of pancreatitis (Figure 1G). To test whether ibuprofen directly inhibits macrophage activation and cytokine expression, we treated RAW 264.7 macrophages with the drug in vitro and quantified their activation upon LPS stimulation. RAW cells responded to 16 h of LPS treatment, as evidenced by a change in cell shape to a flattened, pancake‐like morphology (McWhorter et al., 2013) (Figure 2A), increased cell diameter (Figure 2B) and reduced proliferation (Figure 2C). Ibuprofen did not alter these parameters; however, similarly to what was observed in vivo, it reduced the expression of selected cytokines in LPS‐treated macrophages (Figure 2D). This suggests that ibuprofen directly counteracts the functional activation of macrophages.
Figure 1.
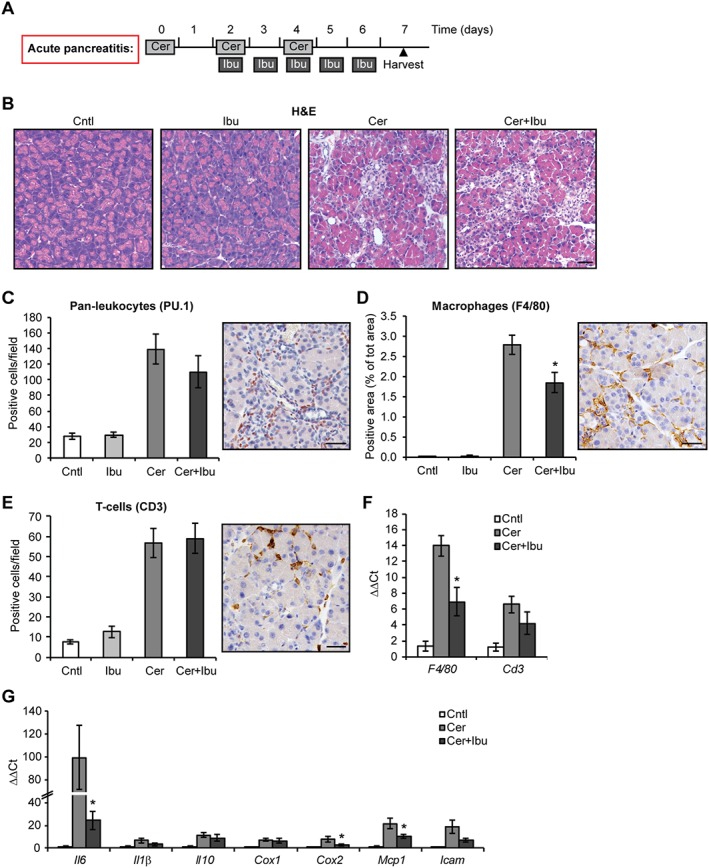
Ibuprofen treatment reduces macrophage infiltration following acute pancreatitis. (A) Schematic representation of ibuprofen treatment using the ‘staggered’ protocol of cerulein‐induced acute pancreatitis. Light grey boxes represent six i.p. injections of 50 μg·kg−1 cerulein (Cer) administered hourly on alternate days. Dark grey boxes represent two i.p. injections of 25 mg·kg−1 ibuprofen (Ibu) administered daily 4 h apart. Black triangle indicates the time of animal harvest, counting from the first cerulein injection. (B) Haematoxylin and eosin (H&E) staining of pancreata after the indicated treatments. (C) Quantification of leukocytes, positive for the pan‐leukocytes PU.1, infiltrating the pancreas. Right panel, representative microphotograph of stained cells. (D) Quantification of F4/80‐positive activated macrophages infiltrating the pancreas. Right panel, representative microphotograph of stained cells. (E) Quantification of CD3‐positive T‐cells infiltrating the pancreas. Right panel, representative microphotograph of stained cells. (F) qPCR of F4/80 and Cd3 expression. (G) qPCR of inflammatory cytokines, chemokine and adhesion molecule expression. Results are average ± SEM (n = 5), *P < 0.05. Scale bars: 50 μm.
Figure 2.
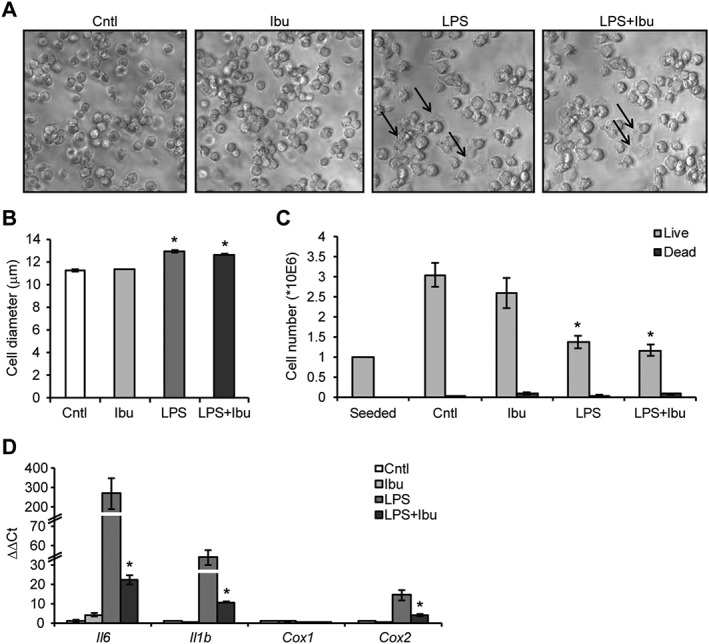
Ibuprofen treatment reduces cytokine expression in LPS‐activated macrophages. (A) Microphotograph showing morphological alterations and flattened shape (arrows) of RAW264.7 macrophages upon activation with 10 ng·mL−1 LPS for 16 h in the presence of 800 μM ibuprofen. (B) Cell diameter of LPS‐treated RAW264.7 macrophages in the presence of ibuprofen. (C) Quantification of live and dead RAW264.7 macrophages treated with LPS in the presence of ibuprofen. (D) qPCR of pro‐inflammatory markers in RAW264.7 macrophages treated with LPS in the presence of ibuprofen. Results are average ± SEM (n = 5), *P < 0.05. Scale bars: 50 μm.
Ibuprofen treatment reduces ADM formation following induction of acute pancreatitis
We then further analysed the inhibitory effect of ibuprofen on macrophages, as these cells play an essential role in the context of pancreatitis. Indeed, macrophages orchestrate both the initiation and the resolution of inflammation (reviewed in Shrivastava and Bhatia, 2010). In addition, M1‐polarized macrophages directly influence the status of acinar cells, as they can initiate and drive the transient trans‐differentiation of acinar cells into ADM, which occurs during the regenerative phase of pancreatitis (Liou et al., 2013; Liou and Storz, 2015). Specifically, F4/80‐positive macrophages are abundantly found associated with ADM lesions, where M1 macrophages are more abundant than M2 macrophages (Figure 3A). Ibuprofen decreased the expression of M1 macrophage markers (Cd40, iNos2), without altering the levels of M2 macrophage markers (Arg1, Clec10) (Figure 3B). Amongst the cytokines secreted by M1 macrophages, TNF and RANTES/CCL5 have been previously identified as the major inducers of acinar cell reprogramming into ADM (Liou et al., 2013). We found that ibuprofen treatment limited the expression of both cytokines in the pancreas upon induction of pancreatitis (Figure 3C).
Figure 3.
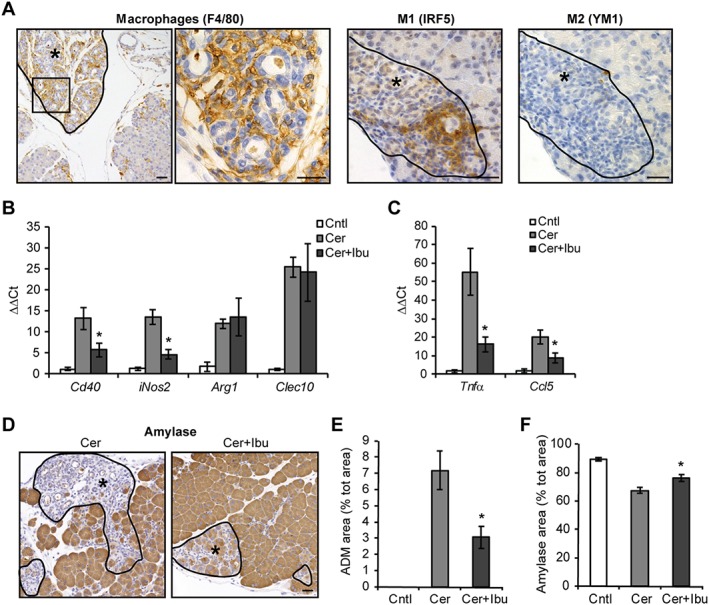
Ibuprofen treatment alters macrophage polarization and reduces ADM formation following acute pancreatitis. (A) Staining of F4/80‐positive macrophages and M1, M2 polarized macrophages in ADM lesions (asterisks). (B) qPCR of M1 and M2 macrophage markers in pancreata. (C) qPCR of ADM‐promoting pro‐inflammatory cytokines. (D) Staining of amylase revealing intact acinar cells (brown areas) and ADM lesions with loss of amylase (asterisks). (E) Quantification of ADM areas following amylase staining, expressed as percentage of total area. (F) Quantification of intact acinar cell areas following amylase staining, expressed as percentage of total area. Results are average ± SEM (n = 5), *P < 0.05. Scale bars: 50 μm.
Finally, we analysed whether the reduced amount of M1 macrophages and ADM‐promoting cytokines observed in the pancreas following ibuprofen treatment was associated with and reduced ADM formation. Ibuprofen‐treated mice showed a decreased trend of mature ADM lesions, morphologically defined by loss of amylase expression, structural re‐organization into tubular complexes and stromal reaction with robust cell infiltration (Figure 3D, E) and decreased trend, albeit not significant, of the ADM marker CK19 expression (Figure S2). In addition, densitometric quantification of amylase levels, which allows the determination of tissue with intact acinar cells, showed larger area of amylase‐expressing acinar cells devoid of ADM upon ibuprofen treatment (Figure 3F). Collectively, these data indicate that inhibition of macrophage recruitment/function mediated by ibuprofen treatment is likely to limit acinar de‐differentiation into metaplastic lesions following induction of pancreatitis.
Ibuprofen treatment reduces acinar cell proliferation following induction of acute pancreatitis
Adult pancreatic acinar cells have the ability to initiate a proliferation programme following pancreatitis to regenerate damaged tissue. As ibuprofen limits the proliferation of different cancer cell types, we tested whether the drug also reduced the proliferation of untransformed acinar cells. Ibuprofen treatment decreased the number of acinar cells expressing the proliferation markers Ki67 and phosphor‐histone 3 (pH 3) (Figure 4A). Expression of both early and late cyclins was also reduced (Figure 4B). Reduced acinar proliferation was not caused by up‐regulation of cyclin‐dependent kinase inhibitors (CDKi). Indeed, ibuprofen‐treated pancreata showed a general decrease in CDKi and p53 expression (Figure 4C). Ibuprofen exerts its anti‐proliferative actions on several immortalized cell lines, including microglia (Elsisi et al., 2005), glioma (Gomes and Colquhoun, 2012) and gastric cancer cells (Bonelli et al., 2011), through induction of apoptosis. Thus, we tested whether apoptotic cell death was induced in acinar cells upon treatment with the drug. Quantification of cleaved caspase 3 (Figure 4D) and DNA fragmentation by TUNEL assay (Figure S3A) showed that ibuprofen did not increase apoptosis of acinar cells in the context of pancreatitis. Similarly, ibuprofen treatment did not increase the levels of DNA damage in acinar cells (Figure S3B) or the expression of heat shock protein 72 and Bcl2 (Figure 4E), which exert a protective effect against cell death in the pancreas (Saluja and Dudeja, 2008). Collectively, these results showed that ibuprofen administration reduced acinar cell proliferation in the context of pancreatitis without engaging a cell death programme previously reported in ibuprofen‐treated cancer cells.
Figure 4.
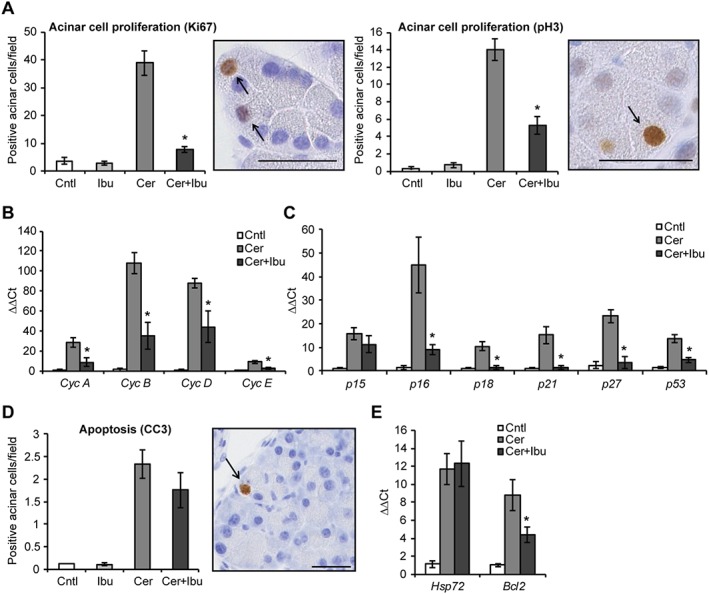
Ibuprofen treatment reduces acinar cell proliferation following acute pancreatitis. (A) Quantification of proliferating acinar cells upon staining with the general proliferation marker Ki67 and with the mitosis‐specific marker pH 3. Right panels, representative microphotographs of stained cells. qPCR of cyclins (B) and cell cycle inhibitors (C) in pancreata after induction of pancreatitis. (D) Quantification of cleaved caspase 3 (CC3)‐positive apoptotic acinar cells after induction of pancreatitis. Right panel, representative microphotograph of stained cells. (E) qPCR of Hsp72 and Bcl2 expression in pancreata. Results are average ± SEM (n = 5), *P < 0.05. Scale bars: 50 μM.
Finally, we tested whether ibuprofen was effective in reducing acinar proliferation when administered after the completion of cerulein treatment. To this aim, cerulein was applied on two consecutive days followed by 5 days of ibuprofen (scheme in Figure 5A). Ibuprofen reduced proliferation of acinar and interstitial cells also in this protocol of acute pancreatitis (Figure 5B, C). In addition, similar inhibition was achieved by the non‐selective COX inhibitor diclofenac (Figure 5A–C), suggesting that different NSAIDs share the ability to interfere with acinar cell proliferation.
Figure 5.
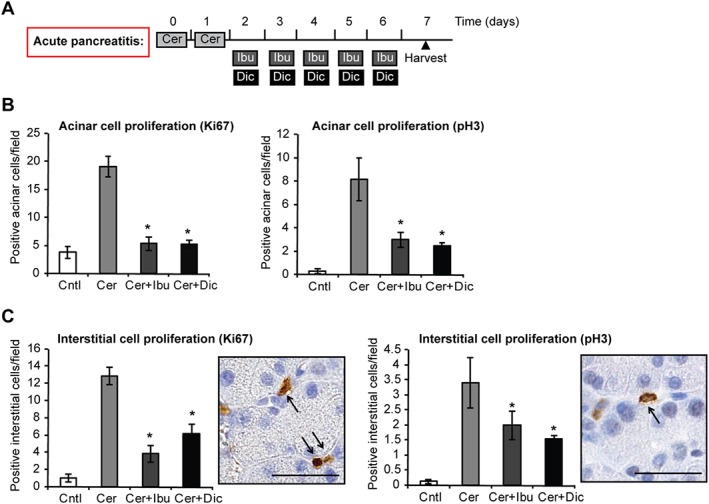
Ibuprofen and diclofenac treatments reduce acinar cell proliferation in a second model of acute pancreatitis. (A) Schematic representation of NSAID treatment using the ‘consecutive’ protocol of cerulein‐induced acute pancreatitis. Light grey boxes represent six i.p. injections of 50 μg·kg−1 cerulein (Cer) administered hourly on two consecutive days. Dark grey boxes represent two i.p. injections of 25 mg·kg−1 ibuprofen (Ibu) administered daily 4 h apart. Black boxes represent two i.p. injections of 10 mg·kg−1 diclofenac (Dic) administered daily 4 h apart. Black triangle indicates the time of animal harvest, counting from the first cerulein injection. (B) Quantification of proliferating acinar cells upon staining with the general proliferation marker Ki67 and with the mitosis‐specific marker pH 3. (C) Quantification of proliferating interstitial cells upon staining with the general proliferation marker Ki67 and with the mitosis‐specific marker pH 3. Right panels, representative microphotographs of stained cells. Results are average ± SEM (n = 5), *P < 0.05.
Ibuprofen treatment reduces acinar cell proliferation upon T3‐induced mitogenic stimulation
While our results point out an anti‐proliferative effect of ibuprofen in the context of pancreatitis, the altered inflammatory milieu observed in the presence of the drug may constitute a confounding factor that prevents the direct assessment of the inhibitory properties of ibuprofen in acinar cells. Thus, we tested the effect of ibuprofen on acinar cell proliferation in an experimental model independent from inflammation. To this aim, mice were treated for 4 days with the thyroid hormone T3, which acts as a mitogen for pancreatic acinar cells (Ledda‐Columbano et al., 2005; Kowalik et al., 2010), concomitantly with ibuprofen administration (scheme depicted in Figure 6A). As expected, T3 did not induce leukocyte recruitment in the pancreas (Figure 6B) or up‐regulation of COX enzymes (Figure 6C). Importantly, ibuprofen treatment did not change basal leukocyte levels (Figure 6B), but selectively decreased proliferation of acinar cells without affecting interstitial cells (Figure 6D). Similar to what was observed in the context of pancreatitis, reduction of acinar cell proliferation was not associated with increased apoptotic cell death of acinar cells (Figure S4). In a further experiment, we tested whether ibuprofen delayed acinar cell division resulting in a long‐lasting proliferation index. Four days after the last T3 administration, acinar proliferation was reduced to near baseline levels in both groups, suggesting that ibuprofen‐treated mice responded to T3 withdrawal without residual proliferation activity at the time point analysed (Figure 6E, F).
Figure 6.
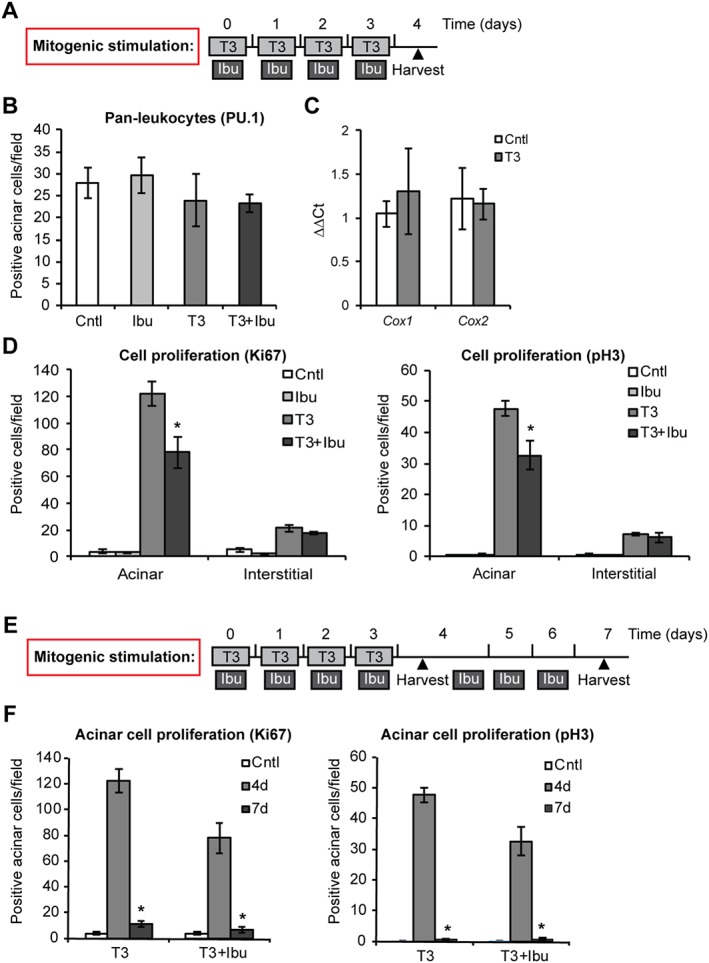
Ibuprofen treatment reduces acinar cell proliferation following mitogenic stimulation. (A) Schematic representation of ibuprofen treatment during stimulation with T3. Light grey boxes represent daily i.p. injections of 400 mg·kg−1 T3. Dark grey boxes represent two i.p. injections of 25 mg·kg−1 ibuprofen (Ibu) administered daily 4 h apart. Black triangle indicates the time of animal harvest, counting from the first T3 injection. (B) Quantification of PU.1‐positive pan‐leukocytes infiltrating the pancreas. (C) qPCR of Cox1, 2 expression in pancreata. (D) Quantification of proliferating acinar and interstitial cells upon staining with the general proliferation marker Ki67 and with the mitosis‐specific marker pH 3. (E) Schematic representation of prolonged ibuprofen treatment after stimulation with T3. Symbols are as in (A). (F) Quantification of proliferating acinar cells upon staining with the general proliferation marker Ki67 and with the mitosis‐specific marker pH 3. Results are average ± SEM (n = 5), *P < 0.05.
Collectively, these results show that ibuprofen is a bona fide inhibitor of acinar cell proliferation induced by T3 stimulation.
Finally, ibuprofen was also tested in vitro on the pancreatic acinar cell line AR42J. Despite their tumour origin, these cells are quite unique in retaining morphological and functional characteristics typical of adult acinar cells, albeit harbouring an unrestrained proliferative ability. Similar to what observed in vivo, in vitro treatment with clinically relevant concentrations of ibuprofen (Andrews et al., 2002) reduced the proliferation of AR42J cells in a dose‐dependent manner (Figure S5).
Discussion
Current treatment of pancreatitis primarily involves supportive therapy, which includes pain relief and prevention of infection (Bang et al., 2008; Paisley and Kinsella, 2014). In this context, NSAIDs are frequently used by virtue of their analgesic as well as anti‐inflammatory effects. However, preclinical studies demonstrated that NSAIDs are also effective in inhibiting the proliferation of a wide range of cancer cells in vitro, either alone or in combination with other cancer therapies. This is further supported by clinical trials demonstrating that long‐term use of NSAIDs, including ibuprofen, significantly reduces the risk of colorectal, breast, lung, prostate and gastric cancer and inhibits proliferation of cancer cells, including glioma, neuroblastoma and bladder cancer cells (Baron and Sandler, 2000; Ulrich et al., 2006; Johnson et al., 2010; Ikegaki et al., 2014; Chai et al., 2015; Fajardo and Piazza, 2015; Leidgens et al., 2015), thus increasing the interest for a novel therapeutic application of these drugs. However, this observation raises the question whether the inhibition of cell proliferation mediated by NSAIDs is also observed in non‐malignant cells, with the consequence that the drug treatment results in delayed regeneration of tissues following injury.
In the present study, we investigated the effect of the NSAIDs ibuprofen and diclofenac on the regeneration of pancreatic tissue in vivo following induction of pancreatitis. Our results showed that administration of these NSAIDs significantly reduced the proliferation of acinar cells. Given the anti‐inflammatory properties of ibuprofen, we then investigated whether reduced acinar proliferation was derived from a reduced inflammatory response. We did not observe a general decrease in pan‐leukocyte infiltration in the pancreas upon ibuprofen treatment. However, macrophage levels and pro‐inflammatory cytokine/chemokine expression were reduced, indicating that the drug affected the recruitment of selected leukocyte populations and probably their activation. This was further confirmed in in vitro experiments where ibuprofen treatment hampered LPS‐induced activation and cytokine expression in macrophages. Reduced inflammation may reduce inflammation‐dependent damage of acinar cells and consequently the need of acinar cell proliferation. However, this direct correlation was not observed, as DNA damage and apoptosis were unchanged in acinar cells in the presence of the drug.
In this context, it is worth mentioning that expression of the anti‐apoptotic marker Bcl2 was reduced during pancreatitis upon ibuprofen treatment. While the exact mechanisms of this lower expression are not completely defined, it is likely that this phenotype is a consequence of the reduced inflammation observed following NSAID administration, as inflammatory mediators have been reported to regulate Bcl2 expression in various types of cells (Minshall et al., 1997; Pugazhenthi et al., 1999). However, another intriguing possibility is that lower Bcl2 levels reflect the reduced proliferation of acinar cells. Indeed, the cellular role of Bcl2 is not restricted to regulating apoptosis, but it also influences cell proliferation by restraining entry into the cell cycle and promoting quiescence (reviewed in Cory et al., 2003). Thus, reduced acinar proliferation in the presence of ibuprofen may result in reduced Bcls2 induction to limit its negative control of cell division.
In addition, ibuprofen treatment reduced acinar cell proliferation also in an inflammation‐independent setting, suggesting that the anti‐inflammatory and anti‐proliferative effects are two independent outcomes of ibuprofen treatment. On the other hand, the anti‐inflammatory effect of ibuprofen is likely to impact on the formation of metaplastic lesions triggered by induction of pancreatitis. In this respect, ibuprofen treatment limited pancreatic infiltration of M1 macrophages. The critical role of this cell type and their secreted cytokines in the trans‐differentiation of acinar cells into ADM was shown recently (Liou et al., 2013). Amongst numerous cytokines produced by macrophages, only TNF and RANTES/CCL5 were able to drive acinar cell trans‐differentiation through activation of NF‐kB activity and NF‐kB‐induced target genes. Importantly, expression of these cytokines was also reduced in our ibuprofen‐treated pancreata, thus probably explaining the reduced trend of ADM formation observed upon induction of pancreatitis.
The striking inhibition of acinar proliferation we observed upon ibuprofen and diclofenac treatment raises the question on the identity of molecular mechanisms underlining the phenotype. As the drugs target COXs and consequently reduce PG synthesis (Rome and Lands, 1975), one possibility is that PGs play a direct role in promoting cell proliferation. In support of this hypothesis, elevated COX2 activity and PG production have been found in various hyper‐proliferating cancer cells. Importantly, overexpression of this enzyme or exogenous administration of PGEs resulted in increased proliferation of different cancer cells (Sheng et al., 2001; Gu et al., 2008; Wang and Dubois, 2010; Gomes and Colquhoun, 2012; Menter and Dubois, 2012), indicating that COX2 activity and PGE synthesis are able to drive the cellular replicative programme. In addition, signalling pathways, which are often mutated and activated during cancer development, including Wnt and Ras pathways, have been shown to up‐regulate COX2 expression (Araki et al., 2003), thus directly implicating COX2 activity as a key factor promoting tumourigenesis and cancer progression.
However, the reduced acinar cell proliferation observed following ibuprofen treatment is likely to be, at least partially, independent of COX2 activity. The key approach to answer this question was provided by our analysis of T3‐induced acinar proliferation, an experimental setting that does not trigger the development of an inflammatory response. Indeed, we showed that COX2 was not expressed during the pronounced proliferation of acinar cells induced by T3 administration. Nevertheless, ibuprofen treatment reduced T3‐induced acinar proliferation. In addition, we showed previously that pancreatitis is characterized by increased pancreatic expression of COX2, but acinar proliferation does not robustly decrease upon either genetic ablation or selective pharmacological inhibition of COX2 (Silva et al., 2011).
Collectively, these data indicate that ibuprofen limits acinar proliferation independently from its anti‐inflammatory effect and that this inhibition of proliferation is likely to be mediated by a COX2‐independent mechanisms targeted by ibuprofen. In this context, COX‐independent effects have been evoked also to explain the effectiveness of NSAIDs to inhibit cancer cell proliferation (reviewed in Matos and Jordan, 2015). Accumulating evidence that supports this hypothesis includes the fact that (i) the anti‐tumour effects of NSAIDs are typically seen at concentrations higher than those required to inhibit PGE synthesis (Tegeder et al., 2001; Grosch et al., 2006) and (ii) NSAIDs still have antineoplastic effects when used against COX1‐ and COX2‐deficient cells (Zhang et al., 1999).
One concept that emerges from the numerous reports describing the anti‐proliferative actions of ibuprofen and other COX inhibitors in cancer cells is that the drug treatment induces cellular death through apoptosis. While this effect is undoubtedly important for the potential of NSAIDs as anti‐cancer therapy, induction of cell death may not be a common outcome of ibuprofen administration to normal untransformed cells. In fact, we did not detect increased apoptosis in the presence of ibuprofen in mice treated with the drug alone, following induction of pancreatitis or upon mitogenic stimulation with T3. Thus, it is likely that ibuprofen reduced acinar proliferation mainly via control of cell cycle. In support of this hypothesis, a very interesting study cross‐examining the effect of ibuprofen in different eukaryotic model organisms showed that the drug delays cell cycle progression through the G1 phase, in the absence of cancer‐related pathologies. Importantly, this led to increased lifespan conserved in multiple species (He et al., 2014), even in yeast cells that are devoid of COX enzymes (Simmons et al., 2004). Further studies also demonstrated that ibuprofen reduced the proliferation of non‐cancer cells, including endothelial and human coronary artery smooth muscle cells (Dannoura et al., 2014; Wiktorowska‐Owczarek et al., 2015).
Conclusion
Modulation of the immune system has been proposed as a valid therapeutic strategy to mitigate the inflammatory response and consequently the severity of pancreatitis. Our work raised a caveat regarding the use of the commonly prescribed anti‐inflammatory agents ibuprofen and diclofenac as it revealed that the drugs exert a significant anti‐proliferative effect in acinar cells, as demonstrated in both an inflammatory situation following induction of pancreatitis and non‐inflammatory situation upon T3‐induced mitogenic stimulation. Given that COX2 inhibitors have been reported to impair regeneration in a murine model of skin wound (Gourevitch et al., 2014), bone fracture healing (Simon and O'connor, 2007) and tendon healing (Connizzo et al., 2014), further studies are warranted to assess whether the anti‐proliferative effect of these NSAIDs detected in acinar cells is detrimental for regeneration of the injured pancreas. In this regard, special interest should be focused not only on long‐term inflammatory damage of the organ in the context of chronic pancreatitis but also on regeneration from tissue loss following pancreatectomy.
Author contributions
The authors of this manuscript contributed to the study design, acquisition, analysis, interpretation of data, drafting and critical revision of the manuscript. M.B., E.M., A.R. and R.C. performed experiments, generated and analysed data, revised the manuscript; R.G. revised the manuscript; S.S. designed the study and wrote the manuscript. All authors approved the submitted version.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Quantification of weight loss, expressed as percentage of initial weight, in mice upon induction of acute pancreatitis. Results are average ± SEM (n = 5), *P < 0.05.
Figure S2 qPCR of ADM marker Ck19. Results are average ± SEM (n = 5), *P < 0.05.
Figure S3 (A) Quantification of TUNEL‐positive apoptotic acinar cells after induction of pancreatitis. Right panel, representative microphotograph of stained cells. (B) Quantification of DNA damage in acinar cells upon γH2A.X staining after induction of pancreatitis. Right panel, representative microphotograph of stained cells. Results are average ± SEM (n = 5), *P < 0.05. Scale bars: 50 μM.
Figure S4 Quantification of cleaved‐caspase 3 (CC3)‐positive apoptotic acinar cells following mitogenic stimulation with T3. Results are average ± SEM (n = 5), *P < 0.05.
Figure S5 Quantification of live cell number after incubating AR42J acinar cells with different concentrations of ibuprofen for 72 h. ‘Seeded’ indicates the initial number of seeded cells. Results are average ± SEM (n = 3), *P < 0.05.
Acknowledgements
This research received grants from the Swiss National Science Foundation (Grant no. 310030–146725) and the Amélie Waring Foundation.
Bombardo, M. , Malagola, E. , Chen, R. , Rudnicka, A. , Graf, R. , and Sonda, S. (2018) Ibuprofen and diclofenac treatments reduce proliferation of pancreatic acinar cells upon inflammatory injury and mitogenic stimulation. British Journal of Pharmacology, 175: 335–347. doi: 10.1111/bph.13867.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews J, Djakiew D, Krygier S, Andrews P (2002). Superior effectiveness of ibuprofen compared with other NSAIDs for reducing the survival of human prostate cancer cells. Cancer Chemother Pharmacol 50: 277–284. [DOI] [PubMed] [Google Scholar]
- Araki Y, Okamura S, Hussain SP, Nagashima M, He P, Shiseki M et al. (2003). Regulation of cyclooxygenase‐2 expression by the Wnt and ras pathways. Cancer Res 63: 728–734. [PubMed] [Google Scholar]
- Bang UC, Semb S, Nojgaard C, Bendtsen F (2008). Pharmacological approach to acute pancreatitis. World J Gastroenterol 14: 2968–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks PA, Conwell DL, Toskes PP (2010). The management of acute and chronic pancreatitis. Gastroenterol Hepatol (N Y) 6: 1–16. [PMC free article] [PubMed] [Google Scholar]
- Baron JA, Sandler RS (2000). Nonsteroidal anti‐inflammatory drugs and cancer prevention. Annu Rev Med 51: 511–523. [DOI] [PubMed] [Google Scholar]
- Bonelli P, Tuccillo FM, Calemma R, Pezzetti F, Borrelli A, Martinelli R et al. (2011). Changes in the gene expression profile of gastric cancer cells in response to ibuprofen: a gene pathway analysis. Pharmacogenomics J 11: 412–428. [DOI] [PubMed] [Google Scholar]
- Bushra R, Aslam N (2010). An overview of clinical pharmacology of ibuprofen. Oman Med J 25: 155–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai AC, Robinson AL, Chai KX, Chen LM (2015). Ibuprofen regulates the expression and function of membrane‐associated serine proteases prostasin and matriptase. BMC Cancer 15: 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez ML, Dekorte CJ (2003). Valdecoxib: a review. Clin Ther 25: 817–851. [DOI] [PubMed] [Google Scholar]
- Connizzo BK, Yannascoli SM, Tucker JJ, Caro AC, Riggin CN, Mauck RL et al. (2014). The detrimental effects of systemic ibuprofen delivery on tendon healing are time‐dependent. Clin Orthop Relat Res 472: 2433–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, Huang DC, Adams JM (2003). The Bcl‐2 family: roles in cell survival and oncogenesis. Oncogene 22: 8590–8607. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannoura A, Giraldo A, Pereira I, Gibbins JM, Dash PR, Bicknell KA et al. (2014). Ibuprofen inhibits migration and proliferation of human coronary artery smooth muscle cells by inducing a differentiated phenotype: role of peroxisome proliferator‐activated receptor gamma. J Pharm Pharmacol 66: 779–792. [DOI] [PubMed] [Google Scholar]
- Elsisi NS, Darling‐Reed S, Lee EY, Oriaku ET, Soliman KF (2005). Ibuprofen and apigenin induce apoptosis and cell cycle arrest in activated microglia. Neurosci Lett 375: 91–96. [DOI] [PubMed] [Google Scholar]
- Endo H, Yano M, Okumura Y, Kido H (2014). Ibuprofen enhances the anticancer activity of cisplatin in lung cancer cells by inhibiting the heat shock protein 70. Cell Death Dis 5: e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo AM, Piazza GA (2015). Chemoprevention in gastrointestinal physiology and disease. Anti‐inflammatory approaches for colorectal cancer chemoprevention. Am J Physiol Gastrointest Liver Physiol 309: G59–G70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes RN, Colquhoun A (2012). E series prostaglandins alter the proliferative, apoptotic and migratory properties of T98G human glioma cells in vitro. Lipids Health Dis 11: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourevitch D, Kossenkov AV, Zhang Y, Clark L, Chang C, Showe LC et al. (2014). Inflammation and its correlates in regenerative wound healing: an alternate perspective. Adv Wound Care (New Rochelle) 3: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf R, Schiesser M, Lussi A, Went P, Scheele GA, Bimmler D (2002). Coordinate regulation of secretory stress proteins (PSP/reg, PAP I, PAP II, and PAP III) in the rat exocrine pancreas during experimental acute pancreatitis. J Surg Res 105: 136–144. [DOI] [PubMed] [Google Scholar]
- Greenspan EJ, Madigan JP, Boardman LA, Rosenberg DW (2011). Ibuprofen inhibits activation of nuclear {beta}‐catenin in human colon adenomas and induces the phosphorylation of GSK‐3{beta}. Cancer Prev Res (Phila) 4: 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosch S, Maier TJ, Schiffmann S, Geisslinger G (2006). Cyclooxygenase‐2 (COX‐2)‐independent anticarcinogenic effects of selective COX‐2 inhibitors. J Natl Cancer Inst 98: 736–747. [DOI] [PubMed] [Google Scholar]
- Gu P, Su Y, Guo S, Teng L, Xu Y, Qi J et al. (2008). Over‐expression of COX‐2 induces human ovarian cancer cells (CAOV‐3) viability, migration and proliferation in association with PI3‐k/Akt activation. Cancer Invest 26: 822–829. [DOI] [PubMed] [Google Scholar]
- Gurpinar E, Grizzle WE, Piazza GA (2014). NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res 20: 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Tsuchiyama SK, Nguyen QT, Plyusnina EN, Terrill SR, Sahibzada S et al. (2014). Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet 10: e1004860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaki N, Hicks SL, Regan PL, Jacobs J, Jumbo AS, Leonhardt P et al. (2014). S(+)‐ibuprofen destabilizes MYC/MYCN and AKT, increases p53 expression, and induces unfolded protein response and favorable phenotype in neuroblastoma cell lines. Int J Oncol 44: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen GM, Venema JF (1985). Ibuprofen: plasma concentrations in man. J Int Med Res 13: 68–73. [DOI] [PubMed] [Google Scholar]
- Johnson CC, Hayes RB, Schoen RE, Gunter MJ, Huang WY, Team PT (2010). Non‐steroidal anti‐inflammatory drug use and colorectal polyps in the prostate, lung, colorectal, and ovarian cancer screening trial. Am J Gastroenterol 105: 2646–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik MA, Perra A, Pibiri M, Cocco MT, Samarut J, Plateroti M et al. (2010). TRbeta is the critical thyroid hormone receptor isoform in T3‐induced proliferation of hepatocytes and pancreatic acinar cells. J Hepatol 53: 686–692. [DOI] [PubMed] [Google Scholar]
- Ledda‐Columbano GM, Perra A, Pibiri M, Molotzu F, Columbano A (2005). Induction of pancreatic acinar cell proliferation by thyroid hormone. J Endocrinol 185: 393–399. [DOI] [PubMed] [Google Scholar]
- Leidgens V, Seliger C, Jachnik B, Welz T, Leukel P, Vollmann‐Zwerenz A et al. (2015). Ibuprofen and diclofenac restrict migration and proliferation of human glioma cells by distinct molecular mechanisms. PLoS One 10: e0140613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M et al. (2013). Macrophage‐secreted cytokines drive pancreatic acinar‐to‐ductal metaplasia through NF‐kappaB and MMPs. J Cell Biol 202: 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Storz P (2015). Inflammatory macrophages in pancreatic acinar cell metaplasia and initiation of pancreatic cancer. Oncoscience 2: 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos P, Jordan P (2015). Beyond COX‐inhibition: ‘side‐effects’ of ibuprofen on neoplastic development and progression. Curr Pharm Des 21: 2978–2982. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF (2013). Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A 110: 17253–17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menter DG, Dubois RN (2012). Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol 2012: 723419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall C, Arkins S, Straza J, Conners J, Dantzer R, Freund GG et al. (1997). IL‐4 and insulin‐like growth factor‐I inhibit the decline in Bcl‐2 and promote the survival of IL‐3‐deprived myeloid progenitors. J Immunol 159: 1225–1232. [PubMed] [Google Scholar]
- Murray B, Carter R, Imrie C, Evans S, O'suilleabhain C (2003). Diclofenac reduces the incidence of acute pancreatitis after endoscopic retrograde cholangiopancreatography. Gastroenterology 124: 1786–1791. [DOI] [PubMed] [Google Scholar]
- Paisley P, Kinsella J (2014). Pharmacological management of pain in chronic pancreatitis. Scott Med J 59: 71–79. [DOI] [PubMed] [Google Scholar]
- Pezzilli R, Morselli‐Labate AM, Corinaldesi R (2010). NSAIDs and acute pancreatitis: a systematic review. Pharmaceuticals 3: 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza GA, Keeton AB, Tinsley HN, Whitt JD, Gary BD, Mathew B et al. (2010). NSAIDs: old drugs reveal new anticancer targets. Pharmaceuticals (Basel) 3: 1652–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Miller E, Sable C, Young P, Heidenreich KA, Boxer LM et al. (1999). Insulin‐like growth factor‐I induces bcl‐2 promoter through the transcription factor cAMP‐response element‐binding protein. J Biol Chem 274: 27529–27535. [DOI] [PubMed] [Google Scholar]
- Reddy BS, Tokumo K, Kulkarni N, Aligia C, Kelloff G (1992). Inhibition of colon carcinogenesis by prostaglandin synthesis inhibitors and related compounds. Carcinogenesis 13: 1019–1023. [DOI] [PubMed] [Google Scholar]
- Rome LH, Lands WE (1975). Structural requirements for time‐dependent inhibition of prostaglandin biosynthesis by anti‐inflammatory drugs. Proc Natl Acad Sci U S A 72: 4863–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama RA, El Gayar NH, Georgy SS, Hamza M (2016). Equivalent intraperitoneal doses of ibuprofen supplemented in drinking water or in diet: a behavioral and biochemical assay using antinociceptive and thromboxane inhibitory dose‐response curves in mice. PeerJ 4: e2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluja A, Dudeja V (2008). Heat shock proteins in pancreatic diseases. J Gastroenterol Hepatol 23 (Suppl 1): S42–S45. [DOI] [PubMed] [Google Scholar]
- Sheng H, Shao J, Washington MK, Dubois RN (2001). Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem 276: 18075–18081. [DOI] [PubMed] [Google Scholar]
- Shrivastava P, Bhatia M (2010). Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J Gastroenterol 16: 3995–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Weber A, Bain M, Reding T, Heikenwalder M, Sonda S et al. (2011). COX‐2 is not required for the development of murine chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 300: G968–G975. [DOI] [PubMed] [Google Scholar]
- Simmons DL, Botting RM, Hla T (2004). Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56: 387–437. [DOI] [PubMed] [Google Scholar]
- Simon AM, O'connor JP (2007). Dose and time‐dependent effects of cyclooxygenase‐2 inhibition on fracture‐healing. J Bone Joint Surg Am 89: 500–511. [DOI] [PubMed] [Google Scholar]
- Sotoudehmanesh R, Khatibian M, Kolahdoozan S, Ainechi S, Malboosbaf R, Nouraie M (2007). Indomethacin may reduce the incidence and severity of acute pancreatitis after ERCP. Am J Gastroenterol 102: 978–983. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketo MM (1998). Cyclooxygenase‐2 inhibitors in tumorigenesis (part I). J Natl Cancer Inst 90: 1529–1536. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Pfeilschifter J, Geisslinger G (2001). Cyclooxygenase‐independent actions of cyclooxygenase inhibitors. FASEB J 15: 2057–2072. [DOI] [PubMed] [Google Scholar]
- Ulrich CM, Bigler J, Potter JD (2006). Non‐steroidal anti‐inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer 6: 130–140. [DOI] [PubMed] [Google Scholar]
- Vainio H (2001). Is COX‐2 inhibition a panacea for cancer prevention? Int J Cancer 94: 613–614. [DOI] [PubMed] [Google Scholar]
- Wang D, Dubois RN (2010). Eicosanoids and cancer. Nat Rev Cancer 10: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiktorowska‐Owczarek A, Namiecinska M, Owczarek J (2015). The effect of ibuprofen on bFGF, VEGF secretion and cell proliferation in the presence of LPS in HMEC‐1 cells. Acta Pol Pharm 72: 889–894. [PubMed] [Google Scholar]
- Zhang X, Morham SG, Langenbach R, Young DA (1999). Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase‐null embryo fibroblasts. J Exp Med 190: 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Quantification of weight loss, expressed as percentage of initial weight, in mice upon induction of acute pancreatitis. Results are average ± SEM (n = 5), *P < 0.05.
Figure S2 qPCR of ADM marker Ck19. Results are average ± SEM (n = 5), *P < 0.05.
Figure S3 (A) Quantification of TUNEL‐positive apoptotic acinar cells after induction of pancreatitis. Right panel, representative microphotograph of stained cells. (B) Quantification of DNA damage in acinar cells upon γH2A.X staining after induction of pancreatitis. Right panel, representative microphotograph of stained cells. Results are average ± SEM (n = 5), *P < 0.05. Scale bars: 50 μM.
Figure S4 Quantification of cleaved‐caspase 3 (CC3)‐positive apoptotic acinar cells following mitogenic stimulation with T3. Results are average ± SEM (n = 5), *P < 0.05.
Figure S5 Quantification of live cell number after incubating AR42J acinar cells with different concentrations of ibuprofen for 72 h. ‘Seeded’ indicates the initial number of seeded cells. Results are average ± SEM (n = 3), *P < 0.05.