

Abstract
Background and Purpose
Parkinson's disease is characterized by progressive decline in motor function due to degeneration of nigrostriatal dopaminergic neurons, as well as other deficits including cognitive impairment and behavioural abnormalities. Mitochondrial dysfunction, leading to loss of ATP‐dependent cellular functions, calcium overload, excitotoxicity and oxidative stress, is implicated in the pathophysiology of Parkinson's disease. Using the 5‐HT1F receptor agonist LY344864, a known inducer of mitochondrial biogenesis (MB), we investigated the therapeutic efficacy of stimulating MB on dopaminergic neuron loss in a mouse model of Parkinson's disease.
Experimental Approach
Male C57BL/6 mice underwent bilateral intrastriatal 6‐hydroxydopamine or saline injections and daily treatment with 2 mg·kg−1 LY344864 or vehicle for 14 days beginning 7 days post‐lesion. Tyrosine hydroxylase immunoreactivity (TH‐ir) and MB were assessed in the brains of all groups following treatment, and locomotor activity was evaluated prior to lesioning, 7 days post‐lesion and after treatment.
Key Results
Increased mitochondrial DNA content and nuclear‐ and mitochondrial‐encoded mRNA and protein expression was observed in specific brain regions of LY344864‐treated naïve and lesioned mice, indicating augmented MB. LY344864 attenuated TH‐ir loss in the striatum and substantia nigra compared to vehicle‐treated lesioned animals. LY344864 treatment also increased locomotor activity in 6‐hydroxydopamine lesioned mice, while vehicle treatment had no effect.
Conclusions and Implications
These data revealed that LY344864‐induced MB attenuates dopaminergic neuron loss and improves behavioural endpoints in this model. We suggest that stimulating MB may be beneficial for the treatment of Parkinson's disease and that the 5‐HT1F receptor may be an effective therapeutic target.
Abbreviations
- 6‐OHDA
6‐hydroxydopamine
- COX1
cytochrome c oxidase 1
- MB
mitochondrial biogenesis
- mtDNA
mitochondrial DNA
- ND1
NADH dehydrogenase 1
- NDUFS1
NADH:ubiquinone oxidoreductase core subunit S1
- Nrf2
nuclear respiratory factor 2
- PGC‐1α
peroxisomal proliferator γ coactivator‐1α
- TFAM
mitochondrial transcription factor A
- TH‐ir
TH immunoreactivity
Introduction
Parkinson's disease is the most common neurodegenerative movement disorder, affecting over 1% of individuals over 60 and 5% of those over 85 (de Lau and Breteler, 2006; Lezi and Swerdlow, 2012; Reeve et al., 2014). Parkinson's disease is progressive, resulting in loss of motor control and cognitive impairment (Cooper et al., 1991). Symptoms arise from degeneration of dopaminergic neurons in the substantia nigra pars compacta and the formation of Lewy bodies in surviving neurons (Lezi and Swerdlow, 2012). The current care for Parkinson's disease is symptomatic, as no therapy exists capable of slowing progression. While this treatment can help maintain quality of life during early stages of the disease, over time, the side effects of treatment begin to outweigh the benefits (Bezard et al., 2013). Therefore, therapeutics that can reduce or halt the progression of Parkinson's disease through prevention of neuronal cell death and/or restoration of neuronal function are of dire need.
Mitochondrial dysfunction has long been implicated in the pathophysiology of Parkinson's disease, stemming, in part, from reduced complex I activity in the substantia nigra of Parkinson's disease patients (Schapira et al., 1990). Disruption of mitochondrial function leads to increased ROS, decreased ATP synthesis, Ca2+ overload and excitotoxicity, all of which are characteristic of many neurodegenerative diseases, including Parkinson's disease (Lezi and Swerdlow, 2012). Neurons are particularly reliant on mitochondrial homeostasis due to their strict regulation of ATP‐dependent processes, high energy demand and lack of antioxidant defences (Adibhatla and Hatcher, 2010). Several genetic mutations converging on mitochondrial pathways are associated with both familial and age‐related Parkinson's disease (Lezi and Swerdlow, 2012; Chaturvedi and Flint Beal, 2013). Furthermore, excitotoxicity, ROS‐induced damage and mitochondrial DNA (mtDNA) mutations have been observed in the brains of Parkinson's disease patients (Schapira, 1999; 2008; Bender et al., 2006). Additionally, variants in somatic mtDNA or nuclear‐encoded mitochondrial genes, namely, complex I genes, resulting in mitochondrial defects have been suggested to influence predisposition to Parkinson's disease (Coskun et al., 2012). These data implicate mitochondrial dysfunction as an integral contributor to the development and progression of Parkinson's disease.
Mitochondria exist in a balance of degradation and mitochondrial biogenesis (MB) to maintain high‐quality mitochondria and minimize oxidative stress. MB is a transcriptional programme defined as the repair, growth and/or division of pre‐existing mitochondria (Ventura‐Clapier et al., 2008) and involves an intricate network of several transcriptional pathways for both nuclear‐ and mtDNA‐encoded genes governed by peroxisomal proliferator γ coactivator‐1α (PGC‐1α) (Kelly and Scarpulla, 2004; Ventura‐Clapier et al., 2008).
Our laboratory developed a drug discovery programme to ascertain inducers of MB and identified 5‐HT receptors, including 5‐HT1F, as novel mediators of MB (Beeson et al., 2010; Rasbach et al., 2010). While not extensively characterized, the 5‐HT1F receptor is found in multiple tissues, including the kidney and various brain regions (Bruinvels et al., 1994; Fugelli et al., 1997; Lucaites et al., 2005; Garrett et al., 2014). We previously showed that treatment with the selective 5‐HT1F receptor agonist LY344864 increased MB, restoring kidney function following the onset of maximal ischaemia/reperfusion injury (Garrett et al., 2014). Furthermore, siRNA knockdown of 5‐HT1F receptors in renal proximal tubule cells not only decreased mitochondrial protein expression, indicating that 5‐HT1F receptors may regulate overall mitochondrial content, but also prevented LY344864‐induced MB (Garrett et al., 2014). Additional studies have revealed that LY344864‐induced MB occurs via a Gβγ‐dependent pathway in these cells (Gibbs et al., 2017). Given the pivotal role of 5‐HT receptors in the CNS (Barnes and Sharp, 1999), and the involvement of mitochondrial dysfunction in Parkinson's disease (Schapira, 1999; 2008; Lin and Beal, 2006; Lezi and Swerdlow, 2012; Chaturvedi and Flint Beal, 2013), this study sought to determine the therapeutic potential of 5‐HT1F receptor‐mediated MB via LY344864 treatment on disease progression in a 6‐hydroxydopamine (6‐OHDA) mouse model of Parkinson's disease.
Methods
Animals
Three‐month‐old male wild‐type C57Bl/6 mice were obtained from The Jackson Laboratory (Stock No: 000664, Bar Harbor, ME, USA) and housed singly at 20–22°C with a 12 h light/dark cycle and food and water available ad libitum. Animals were allowed to habituate for 7 days prior to use. All studies were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina in accordance with the guidelines set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Experimental design
Naïve mice were treated i.p. with either 2 mg·kg−1 LY344864 (n = 8) or saline (n = 8) for 14 days. This dose of LY344864 was based on previous dose–response studies (Garrett et al., 2014). Mice underwent spontaneous locomotor assessment prior to (‘initial’) and after completion of (‘final’) treatment to determine the effect of LY344864 on locomotor activity.
For studies using 6‐OHDA lesioning, additional mice were randomized into three treatment groups: saline controls plus vehicle (‘Saline’, n = 11), 6‐OHDA lesioning plus vehicle (‘6‐OHDA + Vehicle’, n = 14) and 6‐OHDA plus daily i.p. administration of 2 mg·kg−1 LY344864 (‘6‐OHDA + LY344864’, n = 14). Prior to any procedural intervention, all mice underwent locomotor activity assessment (see below) to determine gross baseline pre‐lesion behaviour. A second behavioural assessment was performed 7 days following 6‐OHDA lesioning. Mice were then treated with LY344864 or saline (vehicle) for 14 days, after which a final motor assessment was performed and tissue collected for analysis. See the schematic representation of the experimental design below.
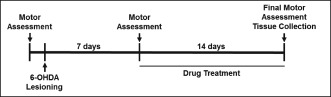
6‐OHDA lesioning
Mice were anaesthetized with 10 mg·kg−1 ketamine and 6 mg·kg−1 xylazine via i.p. injection and continuously monitored for spontaneous breathing and depth of anesthesia using the toe‐pinch method. Body temperature was maintained using a heat mat throughout the procedure. Mice were placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA), and two burr holes were drilled into the skull using a Dremel electric drill at the following coordinates: AP: +0.9, ML: ±1.5 DV: −3.0 relative to Bregma. Mice then underwent bilateral intrastriatal injections of 6‐OHDA (inducing subclinical lesions of nigrostriatal dopamine neurons, which result in slow, progressive neuronal degeneration) via Hamilton syringe at a concentration of 5 μg·μL−1 per injection site, containing 0.9% NaCl and 0.02% ascorbate. The syringe was left in place for 5 min after infusion and then retracted slowly.
Spontaneous locomotion
Spontaneous locomotor activity was assessed in mice of all groups in a Digiscan Animal Activity Monitor system (Omnitech Electronics Model RXYZCM(8) TAO, Columbus, OH, USA), as described previously (Boger et al., 2006). Mice were placed in plexiglass chambers with photocell emitters and receptors spaced equally along the perimeter, creating a grid of infrared beams measuring total distance and vertical activity. Vertical activity was determined by number of beam breaks, and total distance was determined by centimetres travelled. Mice were tested in a darkened environment, and data were collected at 5 min intervals for 60 min at the same time of day for each group (9:00 to 12:00 h).
Tissue collection
Following 14 days of LY344864/vehicle treatment, mice were killed by an overdose of isofluorane, or anaesthetized using isopropanol and killed by decapitation. The brains were quickly removed, and the striatum, frontal cortex, hippocampus and substantia nigra from each hemisphere were dissected and individually flash frozen in liquid nitrogen. A subset of animals was used for immunohistochemical detection of tyrosine hydoxylase for which the left hemisphere was post‐fixed in 4% paraformaldehyde for 48 h and then transferred to 30% sucrose in 0.1 M PBS for 24 h prior to sectioning.
mtDNA and RNA expression
DNA was isolated from brain samples using the Qiagen DNeasy Blood and Tissue Kit (Valencia, CA, USA), and 5 ng was used for qPCR quantification of relative mitochondrial DNA (mtDNA) content. NADH dehydrogenase 1 (ND1), a mitochondrial gene, was measured and normalized to the nuclear encoded gene β‐actin. Total RNA was extracted from unpooled brain regions using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) based on the manufacturer's protocol. cDNA was synthesized using the iScript cDNA Synthesis Kit and qPCR performed using 500 ng of cDNA template and SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad, Hercules, CA, USA). Technical replicates were used to ensure reliability of single values. Fold changes were calculated using the ∆∆Ct method.
The primers used were as follows: PGC‐1α sense: 5′‐AGGAAATCCGAGCGGAGCTGA‐3′, antisense: 5′‐GCAAGAAGGCGACACATCGAA‐3′; ND1: sense: 5′‐TGAATCCGAGCATCCTACC‐3′, antisense: 5′‐ATTCCTGCTAGGAAAATTGG‐3′; COX1 sense: 5′‐CCTGAGCAGGAATAGTAGGG‐3′, antisense: 5′‐AGTGGTACAAGTCAGTTCCC‐3′; NDUFS1 sense: 5′‐AGATGATTTGGGAACAACGG‐3′, antisense: 5′‐TAAGGCTTAGAGGTTAGGGC‐3′; β‐actin sense: 5′‐TAAGGAACAACCCAGCATCC‐3′, antisense: 5′‐CAGTGAGGCCAGGATAGAGC‐3′.
Immunohistochemistry
Forty‐five micrometre sections of the left hemisphere of the brain were obtained using a cryostat (Microm, Zeiss, Thornwood, NY, USA). As previously described (Boger et al., 2006; 2007; Farrand et al., 2016), to determine the effects of LY344864 on nigrostriatal dopaminergic neurons, serial sections of the striatum (every sixth) and substantia nigra (every third) were processed for free‐floating immunohistochemistry using a rabbit polyclonal antibody against tyrosine hydroxylase (1:5000; Pel‐Freeze Inc., Roger, AZ, USA). Free‐floating serial sections were exposed to 1:2:7 H2O2 :methanol : 0.01 M TBS (pH 7.6) for 15 min to quench endogenous peroxidase activity. Sections were permeabilized with Tris‐buffered saline solution with Tween (TBST), followed by 20 min treatment with 0.1 M sodium m‐peroxidase in TBS. Blocking was performed with 10% normal goat serum obtained from Sigma‐Aldrich (St. Louis, MO, USA) in TBST for 30 min at room temperature. Sections were incubated in primary antibody for 24 h, washed in TBST, incubated with biotin‐conjugated goat anti‐rabbit IgG (1:200; Vector Labs, Burlingame, CA, USA) and subsequently incubated in an avidin‐biotin complex using the Vector Labs ABC kit. Vector Labs VIP was used to develop the reaction, yielding a purple product. The sections were then mounted on slides and coverslips applied using DPX.
Semi‐quantitation was performed by determining the intensity of TH‐immunoreactivity (TH‐ir) in the dorsolateral striatum using NIH Image® program to measure a grey scale value within the range of 0–256, where 0 represents white and 256 represents black, as described previously (Boger et al., 2007). Images were captured with a Nikon Eclipse E‐600 microscope, an Olympus‐750 video camera system, and a computer. Background staining was subtracted, and analysis was performed blinded from 5–6 sections per brain and averaged to obtain a single value per animal.
Stereological cell counts
Quantitative estimation of the total number of TH‐ir neurons in the substantia nigra was achieved using unbiased stereological cell counting, as described previously (Boger et al., 2006). Stereo Investigatory stereological software (MicroBrightfield, Colchester, VT, USA) in combination with a Prior H128 computer‐controlled x–y–z motorized stage was the optical fractionator system used for this study. Briefly, the substantia nigra was outlined under 10× magnification, and the outlined region was measured with a systematic random design of dissector‐counting frames with an area of 2500 μm2 and a sampling grid of 100 × 100 μm. The outline excluded the substantia nigra pars reticulata. Neurons with a clear nucleus, at least one axonal/dendritic process and stained positive for TH‐ir were counted using a 40× objective lens with 1.4 numerical aperture. Cell number was counted blinded on every third section throughout the substantia nigra in all groups. Settings were adjusted to result in a minimum of 200 neurons counted per brain.
Immunoblot analysis
Proteins were extracted from the various brain regions using radioimmunoprecipitation assay buffer (50 mM Tris–HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X‐100, pH 7.4) with protease inhibitor cocktail (1:100), 1 mM sodium fluoride and 1 mM sodium orthovanadate (Sigma‐Aldrich). Equal amounts of protein (3–10 μg) were loaded onto 4–15% SDS‐PAGE gels, resolved by gel electrophoresis and transferred onto nitrocellulose or PVDF membranes (Bio‐Rad). Membranes were blocked in 5% BSA in TBST and incubated overnight with primary antibody at 4°C with gentle agitation. Primary antibodies used in these studies included nuclear respiratory factor 2 (Nrf2; 1:500; Cell Signaling, Danvers, MA, USA), PGC1α (1:1000; Abcam, Cambridge, MA, USA), mitochondrial transcription factor A (TFAM; 1:1000; Abcam) and α‐tubulin (1:1000; Abcam). Membranes were incubated with the appropriate secondary antibody before visualization using enhanced chemiluminescence (Thermo Scientific, Waltham, MA, USA) and the GE ImageQuant LAS4000 (GE Life Sciences, Pittsburgh, PA, USA). Optical density was determined using the ImageJ software from the National Institutes of Health.
Statistical analysis
Tissue isolated from a single animal or a single animal's behaviour represented n = 1. Behavioural assays were performed using multiple experimental cohorts of n = 2–6 per group each to obtain n = 11–14 per group. As the same tissue cannot be used for both TH‐ir and the mitochondrial assays, each group was divided into subsets for additional analyses. Differences in expression between treatment groups was analysed using two‐way ANOVA followed by Tukey's post hoc test, with eight mice in the Saline + Vehicle group (n = 8) and nine mice in all other groups (n = 9). Total final behavioural activity was compared to post‐lesion activity using Student's t‐test. Striatal TH‐ir and substantia nigra TH‐positive cells were analysed using one‐way ANOVA followed by Fisher's post hoc test, with eight mice in the 6‐OHDA + LY344864 group (n = 8) and six mice in all other groups (n = 6). In all cases, GraphPad Prism software (La Jolla, CA, USA) was used and a P < 0.05 was considered indicative of a statistically significant difference between mean values. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
The 6‐OHDA was obtained from Sigma‐Aldrich (Cat. No: H4381). LY344864 hydrochloride was obtained from Tocris (Cat. No: 2451, Bristol, UK). Ketamine (Mfg #05098916106) and xylazine (Mfg #50989‐149‐11) were obtained from Patterson Veterinary (Devens, MA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Effect of LY344864 on naïve mice
Naïve mice underwent a 14 day daily treatment with 2 mg·kg−1 LY344864 or vehicle. Following treatment, the frontal cortex, striatum, hippocampus and substantia nigra were extracted and analysed for mitochondrial endpoints, including mtDNA copy number (Figure 1A), PGC‐1α mRNA (Figure 1B) and mRNA expression of the mitochondrially encoded complex IV subunit cytochrome c oxidase 1 (COX1, Figure 1C). While no differences were observed between LY344864‐ and vehicle‐treated mice in the frontal cortex or hippocampus, increased mtDNA copy number and COX1 expression were seen in the striatum and substantia nigra of mice exposed to LY344864. Daily treatment with LY344864 also resulted in increased PGC‐1α expression in the striatum, compared to vehicle treatment. Locomotor assessment was performed in these animals both prior to LY344864 exposure (‘initial’) and after 14 days of daily treatment (‘final’), with no difference being observed between the two groups (Figure 1D).
Figure 1.
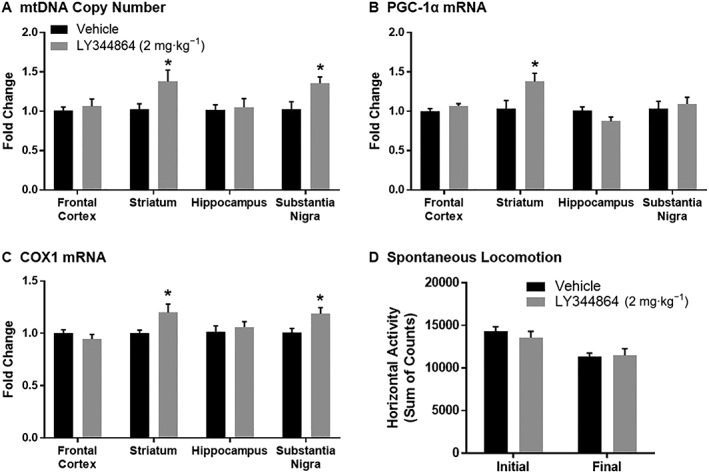
Effect of LY344864 on mitochondrial biogenesis and spontaneous locomotion in naïve animals. Mice were exposed to vehicle or LY344864 (2 mg·kg−1) daily for 14 days. The frontal cortex, striatum, hippocampus and substantia nigra were extracted from mice from each group and analysed for mtDNA content (A) and mRNA expression of PGC‐1α (B) and COX1 (C). Total horizontal activity was assessed prior to treatment (initial) and following 14 days of treatment with vehicle or LY344864 (final). Data represent eight mice per group and are expressed as mean ± SEM (*P < 0.05 compared to Vehicle).
TH immunohistochemistry and stereological cell counts
Mice underwent bilateral intrastriatal 6‐OHDA lesioning followed by 14 day treatment with 2 mg·kg−1 LY344864 or vehicle beginning 7 day post‐lesion. Dorsal striatal TH‐ir was examined in a subset of animals from each group (n = 6–8 per group) following treatment (21 day post‐lesion). Saline‐injected control mice had robust TH‐ir throughout the striatum, while 6‐OHDA‐lesioned mice had reduced TH‐ir in the dorsal striatum (Figure 2). Daily administration of LY344864 following 6‐OHDA lesioning resulted in greater TH‐ir in the dorsal striatum compared to vehicle‐treated 6‐OHDA mice, with levels being comparable to control mice (Figure 2D).
Figure 2.
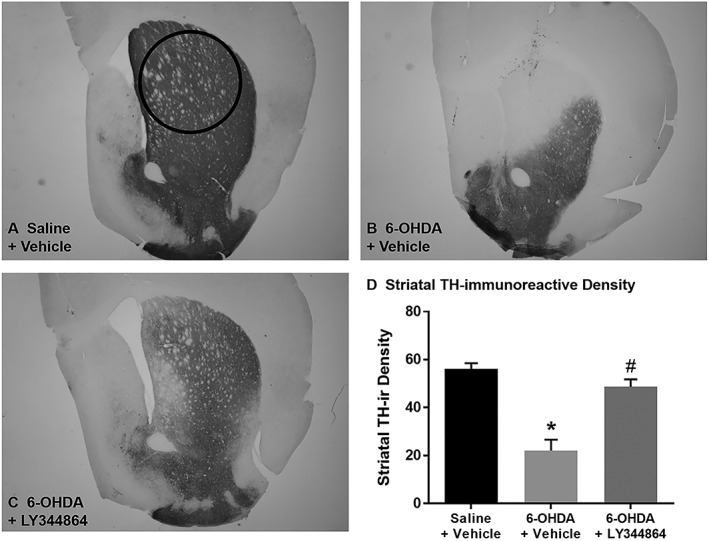
Effect of LY344864 on striatal TH‐ir in 6‐OHDA‐lesioned mice. Mice were subjected to bilateral intrastriatal 6‐OHDA lesions followed by 14 days of daily exposure to vehicle or LY344864 (2 mg·kg−1) beginning 7 days after lesioning. The left hemisphere of a subset of control (A) and lesioned mice following daily vehicle (B) or LY344864 (C) treatment was assessed for striatal TH‐ir density (D, *P < 0.05 compared to Saline + Vehicle, #P < 0.05 compared to 6‐OHDA + Vehicle). All images are shown at a magnification of 4×, and data represent six to eight mice per group and are expressed as mean ± SEM.
The number of TH‐positive neurons was measured in the substantia nigra pars compacta of the same subset of animals (Figure 3). Intrastriatal 6‐OHDA injections resulted in a 50% loss in substantia nigra TH‐positive cells when compared to saline‐injected control animals (Figure 3D). While 6‐OHDA‐lesioned mice treated with LY344864 still depicted fewer substantia nigra TH‐positive cells than control animals, this group had a greater number of substantia nigra TH‐positive cells than 6‐OHDA mice exposed to vehicle.
Figure 3.
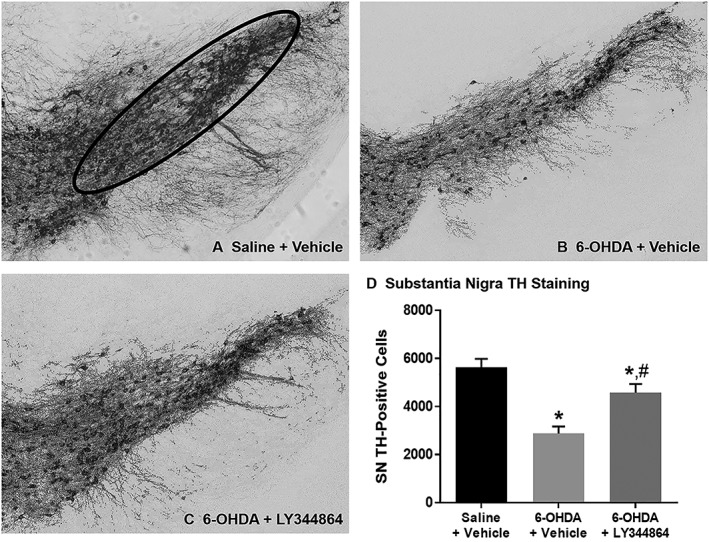
Effect of LY344864 on substantia nigra TH‐ir in 6‐OHDA‐lesioned mice. Mice were subjected to bilateral intrastriatal 6‐OHDA lesions followed by 14 days of daily exposure to vehicle or LY344864 (2 mg·kg−1) beginning 7 days after lesioning. The left hemisphere of a subset of control (A) and lesioned mice following daily vehicle (B) or LY344864 (C) treatment was assessed for substantia nigra TH staining (D, *P < 0.05 compared to Saline + Vehicle, #P < 0.05 compared to 6‐OHDA + Vehicle). All images are shown at a magnification of 10×, and data represent six to eight mice per group and are expressed as mean ± SEM.
Spontaneous locomotor activity
Spontaneous locomotor activity was assessed in all mice prior to 6‐OHDA lesioning (pre‐lesion), 7 days after lesioning (post‐lesion) and after 14 days of treatment with either LY344864 or vehicle (final). Bilateral intrastriatal 6‐OHDA lesioning consistently resulted in a decrease in movement in all groups, with no differences in pre‐lesion activity observed (Figure 4). 6‐OHDA mice treated for 14 days with vehicle alone depicted no change in final total distance travelled compared to that observed post‐lesion (Figure 4A). In contrast, LY344864‐treated mice depicted a near 35% increase in total final distance travelled compared to that observed post‐lesion in this group (Figure 4A).
Figure 4.
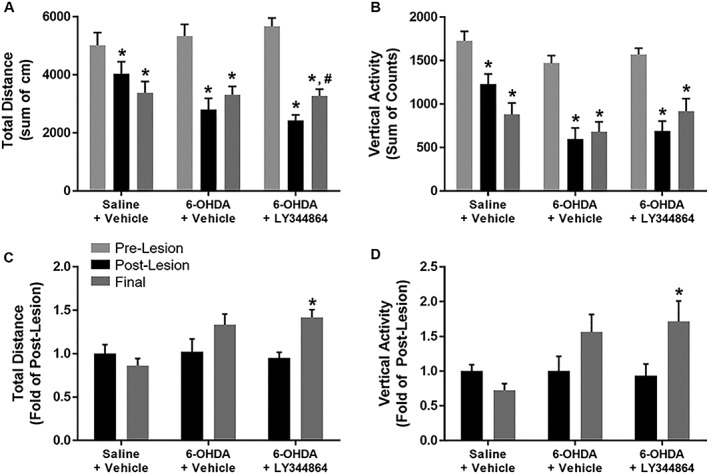
Effect of LY344864 on spontaneous locomotor activity in 6‐OHDA‐lesioned mice. Mice were subjected to bilateral intrastriatal 6‐OHDA lesions followed by 14 days of daily exposure to vehicle or LY344864 (2 mg·kg−1) beginning 7 days after lesioning. Total horizontal (A, C) and vertical activity (B, D) were assessed prior to lesioning (pre‐lesion), 7 days later (post‐lesion) and following 14 days of daily treatment with vehicle or LY344864 (final). The sum of locomotor activity across the full 60 min period was assessed for each group (A and B, *P < 0.05 compared to pre‐lesion, #P < 0.05 compared to post‐lesion), and the change in activity between post‐lesion and final movement was determined for each animal per group (C and D, *P < 0.05 compared to post‐lesion). In all cases, data represent 11–14 mice per group and are expressed as mean ± SEM.
Similar to that observed with horizontal activity, 14 days of vehicle treatment did not alter vertical activity compared to that measured post‐lesion (Figure 4B). The final total vertical activity of 6‐OHDA mice treated with LY344864, while greater, was not statistically different from that post‐lesion. Using paired comparisons between final and post‐lesion activity of each animal, however, total distance (Figure 4C) and vertical activity (Figure 4D) were found to increase following LY344864 treatment.
mtDNA copy number
The frontal cortex, striatum, hippocampus and substantia nigra were extracted after 14 days of treatment from mice of all groups (n = 8–9 per group) and analysed for mitochondrial endpoints, including mtDNA copy number (Figure 5A). mtDNA copy number decreased approximately 25% in the substantia nigra of 6‐OHDA mice compared to control mice. Daily LY344864 treatment, however, increased substantia nigra mtDNA content compared to vehicle, returning copy number to levels comparable to control mice. Lesioned animals also depicted a trend towards decreased mtDNA in the frontal cortex and striatum compared to controls, with content again returning to control levels upon daily exposure to LY344864.
Figure 5.
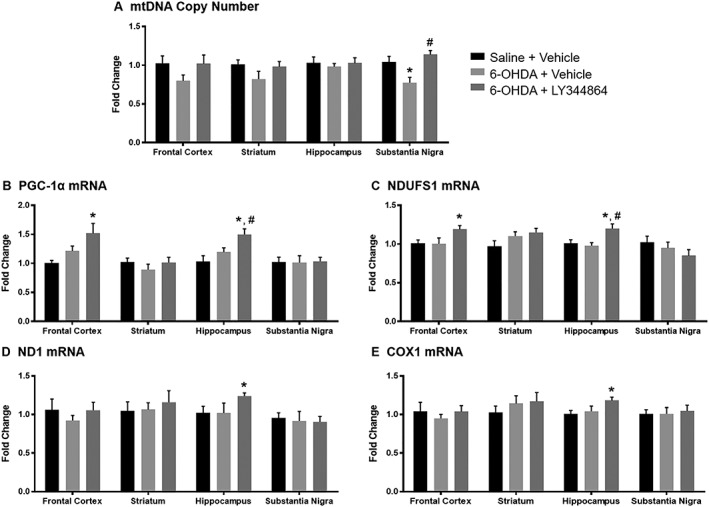
Effect of LY344864 on mtDNA content and mitochondrial mRNA in 6‐OHDA‐lesioned mice. Mice were subjected to bilateral intrastriatal 6‐OHDA lesions followed by 14 days of daily exposure to vehicle or LY344864 (2 mg·kg−1) beginning 7 days after lesioning. The frontal cortex, striatum, hippocampus and substantia nigra were extracted from a subset of mice from each group and analysed for mtDNA content (A) and mRNA expression of PGC1α (B) and NDUFS1 (C), two nuclear‐DNA encoded mitochondrial genes, and of ND1 (D) and COX1 (E), two mitochondrial‐DNA encoded mitochondrial genes. Data represent eight to nine mice per group and are expressed as mean ± SEM (*P < 0.05 compared to Saline + Vehicle, #P < 0.05 compared to 6‐OHDA + Vehicle).
Expression of mitochondrial genes
Daily LY344864 treatment increased PGC‐1α mRNA expression compared to controls in the frontal cortex and increased expression compared to controls and vehicle in the hippocampus (Figure 5B). LY344864‐treated animals displayed increased expression of the nuclear‐encoded mitochondrial gene NADH:ubiquinone oxidoreductase core subunit S1 (NDUFS1), a subunit of complex I of the electron transport chain, compared to controls in the frontal cortex, and compared to controls and vehicle‐treated mice in the hippocampus (Figure 5C).
Alterations in two mitochondrial‐encoded mitochondrial genes following 6‐OHDA lesioning and LY344864 or vehicle treatment were also investigated: the electron transport chain complex I subunit ND1 and COX1. Mice treated daily with LY344864 portrayed increased expression of both mitochondrial‐encoded genes in the hippocampus compared to control mice (Figure 5D, E). There was no difference in mitochondrial gene expression observed between saline control animals and vehicle‐treated 6‐OHDA‐lesioned mice 21 day post‐lesion in any brain regions analysed (Figure 5B–E).
Contrary to mRNA expression, PGC‐1α protein was decreased in the frontal cortex and hippocampus of both vehicle‐ and LY344864‐treated lesioned mice compared to sham controls (Figure 6A, C). Conversely, striatum PGC‐1α protein was decreased approximately 40% in vehicle‐treated lesioned mice yet returned to sham levels with LY344864 treatment (Figure 6B). A similar effect was also observed in the substantia nigra (Figure 6D), where PGC‐1α decreased in the vehicle‐treated mice yet increased in LY344864‐treated animals. TFAM protein was also increased in the striatum of lesioned mice treated with LY344864 compared to vehicle‐treated mice. Protein of the transcription factor Nrf2, which is a coactivator of PGC‐1α and has been linked to transcriptional control of multiple mitochondrial genes including TFAM (Wu et al., 1999), was decreased in the frontal cortex and striatum of lesioned animals treated with either vehicle or LY344864.
Figure 6.
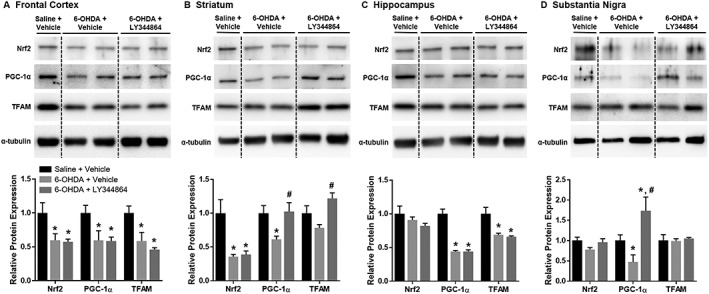
Effect of LY344864 on mitochondrial proteins in 6‐OHDA‐lesioned mice. Mice were subjected to bilateral intrastriatal 6‐OHDA lesions followed by 14 days of daily exposure to Vehicle or LY344864 (2 mg·kg−1) beginning 7 days after lesioning. The frontal cortex (A), striatum (B), hippocampus (C) and substantia nigra (D) were extracted from a subset of mice from each group and analysed for protein expression of Nrf2, PGC‐1α and TFAM. Data represent four to five mice per group and are expressed as mean ± SEM (*P < 0.05 compared to Saline + Vehicle, #P < 0.05 compared to 6‐OHDA + Vehicle).
Discussion
Neurons are highly dependent upon mitochondrial function due to the necessity for strict regulation of ATP‐dependent processes and Ca2+ concentration (Dubinsky, 2005). Although the full aetiology of Parkinson's disease remains unknown, a large amount of research has indicated that mitochondrial dysfunction is integral to the pathophysiology of the disease. Substantia nigra dopaminergic neurons are particularly susceptible to mitochondrial dysfunction and oxidative damage due to high amounts of pro‐oxidant iron and low amounts of the antioxidant glutathione (Bender et al., 2006). Unfortunately, pharmacological agents aimed at minimizing oxidative stress in Parkinson's disease patients have proven largely ineffective (Kamat et al., 2008). It has been suggested that increasing neuronal mitochondrial content could compensate for neurodegeneration‐induced bioenergetic deficits observed with Parkinson's disease (Mattson et al., 2008; Burbulla et al., 2010; Yan et al., 2013).
In recent years, up‐regulation of MB has become a popular therapeutic target for the treatment of many diseases, including neurodegenerative disorders. Several studies have implicated PGC‐1α in Parkinson's disease, with PGC‐1α knockout mice having increased vulnerability to dopaminergic neuronal degeneration while increasing PGC‐1α protects neuronal cells from oxidative stress and cell death (St‐Pierre et al., 2006). Additionally, PPARγ agonists, including pioglitazone and resveratrol, which activate PGC‐1α and subsequently stimulate MB, have proven protective in rodent models of Parkinson's disease (Khan et al., 2010; Pinto et al., 2016). The majority of these studies, however, began treatment either prior to injury or prior to the onset of parkinsonian symptoms. This study examined the effect of pharmacological enhancement of MB 7 days after bilateral intrastriatal 6‐OHDA lesioning in mice using the mitochondrial biogenic compound LY344864 and a heretofore unexplored pathway via 5‐HT1F agonism.
A prominent increase in both striatal and substantia nigra pars compacta TH‐ir was observed in mice that received 2 mg·kg−1 LY344864 i.p. daily for 14 days following 6‐OHDA lesioning compared to vehicle‐treated 6‐OHDA mice, with the degree of striatal TH‐ir being comparable to that of control mice. The increase in nigrostriatal TH‐ir was an exciting finding, given that the loss of nigrostriatal dopaminergic neurons are fundamental to the development and progression of Parkinson's disease pathology. Interestingly, LY344864‐treated mice also exhibited increased final locomotion compared to pre‐lesion activity, unlike vehicle‐treated 6‐OHDA mice. Decreased activity was observed during each behavioural assessment in the saline control animals, which is likely due to repeated exposure to the behavioural apparatus leading to familiarization with the chamber and intersession habituation (Bolivar, 2009).
There is evidence indicating the importance of mitochondria for the pre and postsynaptic function of neurons, as well as neuroplasticity. Mitochondria within neurons are mobile in response to physiological changes, collecting in regions with high metabolic demands, including synaptic terminals and axonal branches (Sheng and Cai, 2012). Additionally, mitochondrial mobility is integral during the formation of new axons; loss of mitochondria prior to axogenesis inhibits axon development (Mattson and Partin, 1999; Ruthel and Hollenbeck, 2003). Based on these and other published data indicating the significance of mitochondria in neuronal function, differentiation and outgrowth (Cheng et al., 2010), the increased TH‐ir observed following daily administration of LY344864 could be attributed to MB‐induced improvement in mitochondrial homeostasis within the central nervous system, encouraging neuronal growth and repair. Additionally, 5‐HT has been shown to boost neuronal mitochondrial mobility within axons via the 5‐HT1A receptor (Chen et al., 2007). Therefore, direct agonism of the 5‐HT1F receptor may have a similar effect, also contributing to neural outgrowth. Additional experimentation is necessary to address this possibility.
LY344864 is a selective 5‐HT1F receptor agonist with a binding affinity of 6 nM (K i) and EC50 of 3 nM. During the early characterization of LY344864, it was observed that both plasma and brain levels were well in excess of these concentrations for at least 8 h in rats after a single 1 mg·kg−1 i.v. injection (Phebus et al., 1997). Plasma levels, however, dropped nearly 85% after 8 h (260 to 40 nM), while brain levels remained fairly constant around 60 nM (Phebus et al., 1997). Moderate increases in mtDNA and nuclear‐ and mitochondrial‐encoded mitochondrial mRNA were observed in certain brain regions following 14 days of LY344864 treatment. Given the high efficacy and persistent brain presence of LY344864, in addition to the 2 week duration of the dosing protocol, we hypothesize that these markers of MB increased to a larger extent at an earlier time point, enhancing protein expression and facilitating the repair/injury prevention of nigrostriatal dopaminergic neurons, then declined over time, potentially due to receptor desensitization. Further analysis of the time course of LY344864‐induced MB in the Parkinson's disease brain is needed to assess this hypothesis.
While LY344864 treatment consistently increased nuclear‐ and mitochondrial‐encoded mRNA expression in the hippocampus and frontal cortex, protein expression was not altered in either of these regions. Conversely, no effect on mRNA was observed in the striatum, while PGC‐1α and TFAM protein expression was increased in this region with LY344864 treatment. The existence of a discrepancy between mRNA and protein expression is not uncommon, including with regard to mitochondrial genes (Tian et al., 2004; Margineantu et al., 2007). Multiple cellular processes could contribute to this occurrence. For example, PGC‐1α protein has a relatively short half‐life but can be subjected to various posttranslational modifications to increase stability, including acetylation, phosphorylation and ubiquitination, affecting expression (Fernandez‐Marcos and Auwerx, 2011). Despite the lack of change in mRNA expression, the increased PGC‐1α and TFAM protein observed in the striatum of LY344864‐treated mice indicates enhanced MB in this region, which likely contributed to the increased TH‐ir and locomotor activity observed in this group.
As previously mentioned, Parkinson's disease is characterized by cognitive impairment. While cognition was not assessed here, the finding that nuclear‐ and mitochondrial‐encoded mRNA was increased in the hippocampus and frontal cortex, brain regions associated with cognitive function, warrants further investigation into the effect of LY344864 on cognitive changes in this model. While there is limited information regarding 5‐HT1F receptor localization in the brain, multiple studies have reported high densities in the cortex and hippocampus of both rodents and primates (Bruinvels et al., 1994; Lucaites et al., 2005; Shukla et al., 2014); therefore, these data could be attributed to drug sensitivity in areas with enhanced receptor density (Kenakin, 1997; 2002). Given the consistent increase in mRNA expression in these regions, it is plausible that increased protein expression could be observed with longer exposure (i.e. 21 days).
While there was no difference in gene expression in the substantia nigra, there was a decrease in mtDNA content in this region following 6‐OHDA lesioning, which returned to control levels following LY344864 treatment. Studies have indicated the presence of 5‐HT1F receptors in the substantia nigra of both rodents and humans, but at much lower levels than that of other areas, including the hippocampus and cortical regions (Waeber and Moskowitz, 1995; Castro et al., 1997). Therefore, these findings may be due to the susceptibility of substantia nigra neurons to mitochondrial damage and mtDNA deletions with Parkinson's disease (Bender et al., 2006), increasing their sensitivity to LY344864‐induced MB. Consistent with mtDNA content, PGC‐1α protein expression was decreased in vehicle‐treated lesioned mice yet increased in LY344864‐treated mice, surpassing that of sham control mice. A similar trend, though not significant, was observed with Nrf2 protein expression in this region.
A potential limitation of this therapeutic strategy is that enhancing MB may not exclusively increase healthy mitochondrial content. In diseases with an abundance of defective or mutated mitochondria, such as Parkinson's disease, it is possible that increasing MB will exacerbate the negative consequences of mitochondrial dysfunction. However, defective or mutated mitochondria may not be able to undergo MB because of their dysfunction. This, in combination with the variability of the pathophysiology of the disease between patients, is a potential explanation for why the limited number of clinical trials that have investigated mitochondrial biogenic compounds for the treatment of Parkinson's disease have not provided conclusive positive results (Aviles‐Olmos et al., 2013; Schapira et al., 2014; Simuni et al., 2015). It is important to note that these clinical trials reported no evidence of the presence or absence of drug‐induced MB.
No perfect model of Parkinson's disease exists, with the various toxin and transgenic models having disparate benefits. For example, the intrastriatal 6‐OHDA model of Parkinson's disease used here, in contrast to the more aggressive medial forebrain bundle and intranigral 6‐OHDA models, presents with gradual dopaminergic neuron degeneration; however, this model is not characterized by the formation of Lewy bodies (Beal, 2001). While the results presented here are promising, future studies should be performed using alternative models of Parkinson's disease and assessing other therapeutic endpoints, including cognitive function. Nevertheless, these data not only provide evidence for LY344864‐induced MB in the brain of 6‐OHDA‐lesioned mice but also suggest a previously unknown therapeutic role for 5‐HT1F receptor agonism for the treatment of Parkinson's disease.
Author contributions
N.E.S. contributed to the conception and design of this study, acquisition and analysis of data and drafting the manuscript and figures. M.K.L contributed to the acquisition and analysis of data. D.C. contributed to the acquisition and analysis of data. H.A.B. contributed to the conception and design of this study and acquisition and analysis of data. R.G.S. contributed to the conception and design of this study, acquisition and analysis of data and drafting the manuscript and figures.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by the MUSC Barmore Foundation (R.G.S. and H.A.B.), the National Institute of General Medical Sciences: GM084147 (R.G.S) and the Biomedical Laboratory Research and Development, VA Office of Research and Development: BX: 000851 (R.G.S.).
Scholpa, N. E. , Lynn, M. K. , Corum, D. , Boger, H. A. , and Schnellmann, R. G. (2018) 5‐HT1F receptor‐mediated mitochondrial biogenesis for the treatment of Parkinson's disease. British Journal of Pharmacology, 175: 348–358. doi: 10.1111/bph.14076.
Contributor Information
Heather A Boger, Email: boger@musc.edu.
Rick G Schnellmann, Email: schnell@pharmacy.arizona.edu.
References
- Adibhatla RM, Hatcher JF (2010). Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 12: 125–169. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviles‐Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T et al (2013). Exenatide and the treatment of patients with Parkinson's disease. J Clin Invest 123: 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes NM, Sharp T (1999). A review of central 5‐HT receptors and their function. Neuropharmacology 38: 1083–1152. [DOI] [PubMed] [Google Scholar]
- Beal MF (2001). Experimental models of Parkinson's disease. Nat Rev Neurosci 2: 325–334. [DOI] [PubMed] [Google Scholar]
- Beeson CC, Beeson GC, Schnellmann RG (2010). A high‐throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem 404: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH et al (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38: 515–517. [DOI] [PubMed] [Google Scholar]
- Bezard E, Yue Z, Kirik D, Spillantini MG (2013). Animal models of Parkinson's disease: limits and relevance to neuroprotection studies. Mov Disord 28: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Huang P, Zaman V, Smith AC, Hoffer BJ et al (2006). A partial GDNF depletion leads to earlier age‐related deterioration of motor function and tyrosine hydroxylase expression in the substantia nigra. Exp Neurol 202: 336–347. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Patrick KS, Ramamoorthy S, Denehy ED, Zhu H et al (2007). Long‐term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line‐derived neurotrophic factor heterozygous mice. J Neurosci 27: 8816–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolivar VJ (2009). Intrasession and intersession habituation in mice: from inbred strain variability to linkage analysis. Neurobiol Learn Mem 92: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinvels AT, Landwehrmeyer B, Gustafson EL, Durkin MM, Mengod G, Branchek TA et al (1994). Localization of 5‐HT1B, 5‐HT1D alpha, 5‐HT1E and 5‐HT1F receptor messenger RNA in rodent and primate brain. Neuropharmacology 33: 367–386. [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Krebiehl G, Kruger R (2010). Balance is the challenge – the impact of mitochondrial dynamics in Parkinson's disease. Eur J Clin Invest 40: 1048–1060. [DOI] [PubMed] [Google Scholar]
- Castro ME, Pascual J, Romon T, del Arco C, del Olmo E, Pazos A (1997). Differential distribution of [3H]sumatriptan binding sites (5‐HT1B, 5‐HT1D and 5‐HT1F receptors) in human brain: focus on brainstem and spinal cord. Neuropharmacology 36: 535–542. [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK, Flint Beal M (2013). Mitochondrial diseases of the brain. Free Radic Biol Med 63: 1–29. [DOI] [PubMed] [Google Scholar]
- Chen S, Owens GC, Crossin KL, Edelman DB (2007). Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol Cell Neurosci 36: 472–483. [DOI] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP (2010). Mitochondria and neuroplasticity. ASN Neuro 2: e00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Sagar HJ, Jordan N, Harvey NS, Sullivan EV (1991). Cognitive impairment in early, untreated Parkinson's disease and its relationship to motor disability. Brain 114 (Pt 5): 2095–2122. [DOI] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner S, Chen H‐W, Marciniack C, LaFerla F et al (2012). A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta 1820: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM (2005). CNS mitochondria in neurodegenerative disorders. Antioxid Redox Signal 7: 1089–1091. [DOI] [PubMed] [Google Scholar]
- Farrand AQ, Gregory RA, Backman CM, Helke KL, Boger HA (2016). Altered glutamate release in the dorsal striatum of the MitoPark mouse model of Parkinson's disease. Brain Res 1651: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Marcos PJ, Auwerx J (2011). Regulation of PGC‐1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93: 884s–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugelli A, Moret C, Fillion G (1997). Autoradiographic localization of 5‐HT1E and 5‐HT1F binding sites in rat brain: effect of serotonergic lesioning. J Recept Signal Transduct Res 17: 631–645. [DOI] [PubMed] [Google Scholar]
- Garrett SM, Whitaker RM, Beeson CC, Schnellmann RG (2014). Agonism of the 5‐hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther 350: 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs WS, Garrett SM, Beeson CC, Schnellmann RG (2017). Identification of Dual Mechanisms Mediating 5‐hydroxytryptamine Receptor 1F Induced Mitochondrial Biogenesis. American journal of physiology Renal physiology: ajprenal.00324.02017. [DOI] [PMC free article] [PubMed]
- Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN, Hensley K (2008). Antioxidants in central nervous system diseases: preclinical promise and translational challenges. J Alzheimers Dis 15: 473–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC (2004). Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18: 357–368. [DOI] [PubMed] [Google Scholar]
- Kenakin T (1997). Pharmacologic analysis of drug±receptor interaction, 3rd edn. Raven Press: New York. [Google Scholar]
- Kenakin T (2002). Drug efficacy at G protein‐coupled receptors. Annu Rev Pharmacol Toxicol 42: 349–379. [DOI] [PubMed] [Google Scholar]
- Khan MM, Ahmad A, Ishrat T, Khan MB, Hoda MN, Khuwaja G et al (2010). Resveratrol attenuates 6‐hydroxydopamine‐induced oxidative damage and dopamine depletion in rat model of Parkinson's disease. Brain Res 1328: 139–151. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LML, Breteler MMB (2006). Epidemiology of Parkinson's disease. Lancet Neurol 5: 525–535. [DOI] [PubMed] [Google Scholar]
- Lezi E, Swerdlow RH (2012). Mitochondria in neurodegeneration. Adv Exp Med Biol 942: 269–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443: 787–795. [DOI] [PubMed] [Google Scholar]
- Lucaites VL, Krushinski JH, Schaus JM, Audia JE, Nelson DL (2005). [3H]LY334370, a novel radioligand for the 5‐HT1F receptor. II. Autoradiographic localization in rat, guinea pig, monkey and human brain. Naunyn Schmiedebergs Arch Pharmacol 371: 178–184. [DOI] [PubMed] [Google Scholar]
- Margineantu DH, Emerson CB, Diaz D, Hockenbery DM (2007). Hsp90 inhibition decreases mitochondrial protein turnover. PLoS One 2: e1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Partin J (1999). Evidence for mitochondrial control of neuronal polarity. J Neurosci Res 56: 8–20. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60: 748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phebus LA, Johnson KW, Zgombick JM, Gilbert PJ, Van Belle K, Mancuso V et al (1997). Characterization of LY344864 as a pharmacological tool to study 5‐HT1F receptors: binding affinities, brain penetration and activity in the neurogenic dural inflammation model of migraine. Life Sci 61: 2117–2126. [DOI] [PubMed] [Google Scholar]
- Pinto M, Nissanka N, Peralta S, Brambilla R, Diaz F, Moraes CT (2016). Pioglitazone ameliorates the phenotype of a novel Parkinson's disease mouse model by reducing neuroinflammation. Mol Neurodegener 11: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasbach KA, Funk JA, Jayavelu T, Green PT, Schnellmann RG (2010). 5‐hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J Pharmacol Exp Ther 332: 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve A, Simcox E, Turnbull D (2014). Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res Rev 14: 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthel G, Hollenbeck PJ (2003). Response of mitochondrial traffic to axon determination and differential branch growth. J Neurosci 23: 8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH (1999). Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia. Biochim Biophys Acta 1410: 159–170. [DOI] [PubMed] [Google Scholar]
- Schapira AH (2008). Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol 7: 97–109. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990). Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54: 823–827. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW, Greenamyre JT, Bezard E (2014). Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet (London, England) 384: 545–555. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13: 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R, Watakabe A, Yamamori T (2014). mRNA expression profile of serotonin receptor subtypes and distribution of serotonergic terminations in marmoset brain. Front Neural Circuits 8: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simuni T, Kieburtz K, Tilley B, Elm JJ, Ravina B, Babcock D et al (2015). Pioglitazone in early Parkinson's disease: a phase 2, multicentre, double‐blind, randomised trial. The Lancet Neurology 14: 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St‐Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S et al (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC‐1 transcriptional coactivators. Cell 127: 397–408. [DOI] [PubMed] [Google Scholar]
- Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ et al (2004). Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol Cell Proteomics 3: 960–969. [DOI] [PubMed] [Google Scholar]
- Ventura‐Clapier R, Garnier A, Veksler V (2008). Transcriptional control of mitochondrial biogenesis: the central role of PGC‐1alpha. Cardiovasc Res 79: 208–217. [DOI] [PubMed] [Google Scholar]
- Waeber C, Moskowitz MA (1995). [3H]sumatriptan labels both 5‐HT1D and 5‐HT1F receptor binding sites in the guinea pig brain: an autoradiographic study. Naunyn Schmiedebergs Arch Pharmacol 352: 263–275. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V et al (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell 98: 115–124. [DOI] [PubMed] [Google Scholar]
- Yan MH, Wang X, Zhu X (2013). Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62: 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]