

Abstract
The recent clinical availability of the PARP inhibitor olaparib (Lynparza) opens the door for potential therapeutic repurposing for non‐oncological indications. Considering (a) the preclinical efficacy data with PARP inhibitors in non‐oncological diseases and (b) the risk–benefit ratio of treating patients with a compound that inhibits an enzyme that has physiological roles in the regulation of DNA repair, we have selected indications, where (a) the severity of the disease is high, (b) the available therapeutic options are limited, and (c) the duration of PARP inhibitor administration could be short, to provide first‐line options for therapeutic repurposing. These indications are as follows: acute ischaemic stroke; traumatic brain injury; septic shock; acute pancreatitis; and severe asthma and severe acute lung injury. In addition, chronic, devastating diseases, where alternative therapeutic options cannot halt disease development (e.g. Parkinson's disease, progressive multiple sclerosis or severe fibrotic diseases), should also be considered. We present a preclinical and clinical action plan for the repurposing of PARP inhibitors.
Linked Articles
This article is part of a themed section on Inventing New Therapies Without Reinventing the Wheel: The Power of Drug Repurposing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.2/issuetoc
Abbreviations
- HRR
homologous recombination DNA repair
- PBMC
peripheral blood mononuclear cells
- TBI
traumatic brain injury
Tables of Links
| TARGETS |
|---|
| Enzymes |
| PARP, poly(ADP‐ribose) polymerase |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
PARP inhibitors, a novel class of anticancer therapeutics
Activation of PARP (also termed ARTD) catalyses the cleavage of NAD+ into nicotinamide and ADP‐ribose. Initially, this response was linked to DNA damage (DNA single strand breakage) in response to genotoxic agents (Chambon et al., 1963; Durkacz et al., 1980; Shall, 1983; Hottiger et al., 2010). PARylation – the modification of a growing number of cellular proteins by PAR units (Pic et al., 2011; Gibson and Kraus, 2012; Krietsch et al., 2013; Ji and Tulin, 2013; Li and Chen, 2014; Gibson et al., 2016) – is now recognized as an important posttranslational modification that extends beyond DNA damage responses and transcends many aspects of cell signalling. PAR, in fact, is now viewed as an anionic matrix for trapping, recruiting and scaffolding proteins, and thereby affecting many key cellular functions. Some of these functions depend on the catalytic activity of the enzyme, while others depend on its physical presence and consequent protein–protein interactions (Thomas and Tulin, 2013) (Figure 1).
Figure 1.

Overview of key biological functions of PARP1. The top section shows the various domains of PARP, including its DNA‐binding domain, with its zinc fingers (ZnI, ZnII, ZnIII) that are essential for recognition of DNA strand breaks. This domain also contains the nuclear localization signal (NLS). The auto‐modification domain contains the conserved BRCT fold that serves an important protein : protein interaction module in DNA repair and cell signalling. This domain accepts PARP in the context of auto‐PARylation of PARP1. The catalytic domain contains the active site of the enzyme, where binding and cleavage of NAD+ takes place. It also contains the WGR domain, which is one of the domains involved in the RNA‐dependent activation of PARP1. Below the domains, on the right side, the structure of NAD+ is presented, with the nicotinamide part highlighted. The middle part of the figure shows the sequences of the PARylation process catalysed by PARP, starting with recognition of the DNA strand breaks by the DNA‐binding domain (grey ovals depicting the zinc fingers binding to the DNA breaks), followed by the catalytic activation of the enzyme and the cleavage of NAD+ the production of nicotinamide and the generation of PARP, which, in turn, PARylates various acceptor proteins as well as PARP itself. The consumption of NAD+ has metabolic and bioenergetic effects. PARP inhibitors prevent the binding of NAD+ to the active site of PARP and inhibit the catalytic activity of the enzyme. On the left side, the effect of PAR glycohydrolases and ARH3 is shown; these enzymes break down the PARP, leading to the liberation of free PAR.
Identification of the role of PARP in DNA repair provided the rationale for inhibitor development, based on the hypothesis that by inhibiting PARP, the repair of chemotherapy and radiotherapy‐inflicted DNA damage would be suppressed and cancer cell death promoted (see Shall, 1983, Berger et al., 1987; Griffin et al., 1995, Jagtap and Szabo, 2005; Lupo and Trusolino, 2014). A decade ago, it was recognized that cells deficient in the homologous recombination DNA repair (HRR) system ‐ due, for example, to BRCA1 or BRCA2 mutations ‐ are exquisitely sensitive to the cytotoxic effects of PARP inhibition (Bryant et al., 2005; Farmer et al., 2005). As reviewed elsewhere (Curtin and Szabo, 2013; Drew, 2015), in BRCA‐proficient cancer cells, their ‘baseline’ single strand breakage (that occurs as part of endogenous DNA damaging agents, e.g. oxidants) is being repaired by the base excision repair (BER) system, which is recruited by PARP activation. In the absence of efficient BER, however, these single strand breaks persist, collapsing replication forks that are then repaired by the HRR pathway. In BRCA‐deficient cells, the HRR pathway is genetically impaired, and therefore, the collapsed replication forks remain unrepaired, which, ultimately, results in the death of the PARP‐inhibitor‐treated cancer cells. This unique constellation of events, which offers the prospect of selective tumour targeting via induction of synthetic lethality, has vitalized the interest of numerous pharmaceutical companies for PARP inhibitors, to be used in monotherapy, and stimulated extensive drug development that culminated, after dozens of clinical trials, in the approval of the first PARP inhibitor, olaparib (Lynparza) for the therapy of ovarian cancer (Deeks, 2015). In addition, rucaparib has recently been also approved by the FDA for ovarian cancer (https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm533891.htm). Several other PARP inhibitors are currently in various late‐stage clinical trials and clinicaltrials.gov lists over 200 clinical trials with PARP inhibitors, examples of which are shown in Table 1. Some of these trials focus on PARP inhibitor monotherapy and some of them extend beyond BRCA deficiency, because – in addition to the BRCA‐related cytotoxicity mechanism outlined above – there are additional mechanisms through which PARP inhibition can induce cancer cell toxicity (Figure 2 , lower section). The mechanistic aspects of the anticancer effects of PARP inhibitors and the clinical activities and therapeutic perspectives related to the oncological uses of PARP inhibitors are covered in separate review articles (Penning, 2010; Ratner et al., 2012; Lee et al., 2014; OʼSullivan Coyne et al., 2015; Buege and Mahajan, 2015; Sistigu et al., 2015; Sonnenblick et al., 2015; Bao et al., 2016; Crafton et al., 2016; Konecny and Kristeleit, 2016; Parkes and Kennedy, 2016). PARP inhibition in patients has now been achieved with two approved drugs, olaparib (Lynparza) and rucaparib (Rubraca). In addition, it is expected that several other PARP inhibitors will emerge, in coming years, from the pharmaceutical pipelines.
Table 1.
Examples of clinical‐stage PARP inhibitors
| Inhibitor | Structure | Clinical development examples |
|---|---|---|
|
Olaparib (KU59436/AZD2281) AstraZeneca |
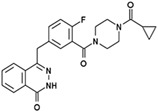
|
Oncology Approved in the US and Europe for the therapy of ovarian cancer as a single agent |
|
Rucaparib (AG‐014699/PF0367338) Clovis |

|
Oncology FDA priority review commenced in 2016 for ovarian cancer as a single agent; decision expected Q1 2017 |
|
Veliparib (ABT‐888) Abbvie |
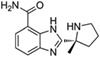
|
Oncology In 2016, the FDA granted Fast Track designation for veliparib, in combination with chemotherapies, for example, carboplatin and paclitaxel, or radiation in advanced squamous non‐small cell lung cancer |
|
E7016 Eisai |

|
Oncology Phases I–II trials in combination with temozolomide melanoma and advanced solid tumours on‐going |
|
Talazoparib (BMN‐673) Biomarin |

|
Oncology Phase III trial in breast cancer as a single agent on‐going |
|
Niraparib (MK4827) Tesaro |

|
Oncology In 2016, the FDA granted Fast Track designation for the treatment of patients with recurrent platinum‐sensitive ovarian, fallopian tube or primary peritoneal cancer as a single agent. Rolling submission under way |
|
CEP‐9722 Checkpoint Therapeutics |

|
Oncology Phase I trials in combination with temozolomide and with gemcitabine + cisplatin in solid tumours |
|
INO‐1001 Inotek/Genentech |
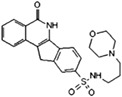
|
Cardiovascular (myocardial infarction) and oncology No longer in development |
Figure 2.
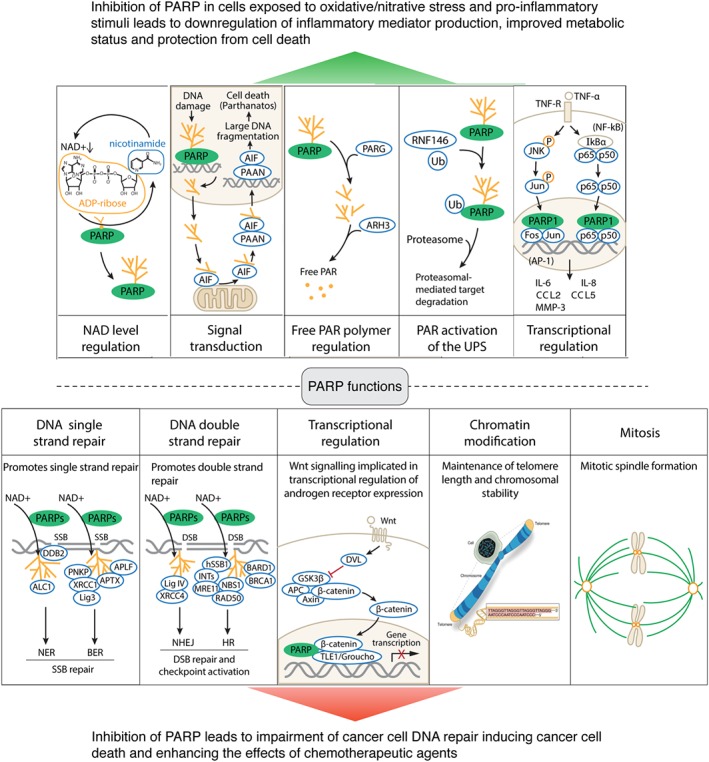
Top section: Mechanisms responsible for the cytoprotective and anti‐inflammatory effects of PARP inhibitors on non‐oncological diseases. From left to right: First subpanel shows PARP activation and consequent NAD+ depletion. These processes can lead to cellular energetic deficit and cell dysfunction; inhibition of PARP prevents these processes and exerts cytoprotective effects (inhibition of cell necrosis). Second subpanel shows the role of PARP activation and free PAR polymers in inducing mitochondrial release of apoptosis‐inducing factor (AIF), which in turn induces cell death (parthanatos). Inhibition of PARP suppresses these processes and inhibits parthanatos. Third subpanel shows the role of PARP in liberating free PAR polymers, which on their own exert cytotoxic effects; inhibition of PARP prevents free PAR polymer formation and suppresses cell death. Fourth subpanel shows that PARylation contributes to activation of the proteasome through an interaction with RNF146; PARP inhibitors suppress these processes. Fifth subpanel shows the role of PARP in contributing to pro‐inflammatory signal transduction via enhancing JNK‐mediated (left sequence) and NF‐κB‐mediated (right sequence) activation of multiple genes and gene products. By inhibiting PARP, these processes are attenuated and inflammatory signalling can be attenuated. The five scenarios shown here can either be cell‐type and stimulus‐ and contex‐ dependent or can also occur simultaneously, depending on the pathophysiological condition. Taken together, PARP inhibitors, by blocking these processes, protect against cell death and suppress inflammatory responses. Bottom section: Mechanisms responsible for the cytotoxic effects of PARP inhibitors on oncological diseases. From left to right: The left side of the first subpanel shows that PARP contributes to single strand break repair, either through facilitating nucleotide excision repair (NER) via interactions with the WD40‐repeat protein DDB2 and the chromatin remodelling enzyme ALC1. The right side of the first subpanel shows that PARP contributes to BER through interaction with a variety of proteins including polynucleotide kinase 3′‐phosphatase (PNKP), X‐ray repair cross‐complementing 1 (XRCC1), aprataxin (APTX), Lig3 (DNA ligase 3) and APLF (a human protein putatively involved in DNA damage response). The second subpanel shows the role of PARP in DNA strand repair; the left side of this subpanel depicts the interactions of PARP with Lig IV (DNA ligase IV) and XRCC4 in the context of NHEJ (nonhomologous end joining); the right side of this subpanel depicts the interactions of PARP with components of the homologous repair (HR). In this context, PAR is recognized by several repair machineries, such as the BRCA1–BARD1 complex, the MRN complex and the hSSB1–INTS complex. The third subpanel depicts the role of PARP in the context of transcriptional regulation of WNT signalling, a pathway implicated in the process of androgen receptor expression. The fourth subpanel depicts the role of PARP in the maintenance of telomere length and chromatin stability, and the fifth subpanel shows the role of PARP in mitotic spindle formation. By inhibiting these processes, PARP inhibitors exert antiproliferative effects and cytotoxic effects, which can be exploited, with beneficial effects, in the therapy of various forms of cancers.
Although the currently on‐going clinical development focuses on oncology, the goal of the current article is to review the potential opportunities of using PARP inhibitors for non‐oncological indications (a repurposing approach), especially for those diseases where DNA damage constitutes part of the pathophysiological response. We will outline the pathomechanisms that make PARP inhibitors attractive for a variety of non‐oncological diseases, review the pros and cons of a repurposing approach and we will recommend a set of indications [including acute ischaemic stroke, severe acute traumatic brain injury (TBI), septic shock, acute pancreatitis, severe acute lung injury (ALI), Parkinson's disease or severe fibrotic diseases] where the expected benefits outweigh the potential risks and where, therefore, clinical trials aimed at repurposing PARP inhibitors are scientifically and medically indicated.
Five decades of advances in the field of PARP
Five decades of intensive work on the biology of PARP have produced thousands of articles, which cannot be reviewed here in detail. PARP (or, as it is now known, PARP1) is now recognized as the first (and most abundant) example of an extensive superfamily of enzymes (reviewed in Amé et al., 2004; Schreiber et al., 2006; Riffell et al., 2012). The physiological role of PARP in the regulation of DNA repair and in the maintenance of genomic integrity has expanded into a field of its own (see Tong et al., 2001; De Vos et al., 2012; Pears et al., 2012; Tallis et al., 2014; Kraus, 2015; Wei and Yu, 2016; Martin‐Hernandez et al., 2017), with novel discoveries showing that nuclear PAR may serve as a substrate for nuclear ATP synthesis required for chromatin remodelling (Wright et al., 2016). PARP is now viewed as a broad regulator of a wide variety of nuclear events, including the regulation of protein‐nucleic acid interactions by means of protein shuttling (Thomas and Tulin, 2013). In addition to nuclear PARP1, the mitochondrial isoform of PARP is receiving more attention (Du et al., 2003; Lai et al., 2008; Bai et al., 2015; Brunyanszki et al., 2016). PAR homeostasis is now viewed as a balance between PAR formation (by PARP superfamily members) and PAR degradation – the latter process being catalysed by PAR glycohydrolases as well as other enzymes such as ARH3 (Gagné et al., 2006; Mashimo et al., 2014; Barkauskaite et al., 2015; Pascal and Ellenberger, 2015).
Another area of intensive research relates to the epigenetic role of PARP as a regulator of gene expression via (1) regulation of chromatin remodelling, (2) functioning as a transcriptional co‐regulator, (3) modulating DNA methylation, (4) poly(ADPribosyl)ation of target proteins involved in gene transcription and/or (5) regulation of RNA metabolism and function (Kraus and Hottiger, 2013; Schiewer and Knudsen, 2014; Bock et al., 2015; Ryu et al., 2015; Jubin et al., 2017; Posavec Marjanović et al., 2017). PARP inhibitors have been found to affect the activation of many transcription factors and the expression of many gene products (Figure 2 , top section). Several levels of crosstalk have been demonstrated between PARP and the sirtuin system (Cantó et al., 2013; Imai and Guarente, 2014; Gueguen et al., 2014; Faraone‐Mennella, 2015; Jęśko and Strosznajder, 2016). In addition to the role of PARP in cell death and in various disease conditions (discussed in the next section), the roles of PARP have been recognized in carcinogenesis (Masutani and Fujimori, 2013) and ageing (Bürkle et al., 2005; Beneke and Bürkle, 2007; Mangerich and Bürkle, 2012; Shilovsky et al., 2013; Imai and Guarente, 2014).
In parallel with the progress made in the field of basic science, the field of pharmacological inhibitors has also advanced significantly. The first generation of inhibitors (3‐aminobenzamide, nicotinamide – active at millimolar concentrations) were followed by second‐generation compounds (e.g. 1,5‐dihydroisoquinoline, 2‐nitro‐6[5H]phenanthridinone, 4‐amino‐1,8‐naphthalimide – active at mid‐micromolar concentrations) and, finally, the third generation, ultrapotent class of inhibitors, (active at low micromolar to high nanomolar concentrations) many of which have progressed into clinical development (see Curtin and Szabo, 2013). Structures of clinical‐stage PARP inhibitors, together with representative examples of their clinical trials, are shown in Table 1.
PARP, an executor of cell necrosis
In parallel with the recognition that PARP activation consumes its substrate, NAD+, Berger put forward the hypothesis that cellular depletion of NAD+, and, secondarily, ATP, in cells exposed to DNA‐damaging agents, may be deleterious to cell viability. Thus, it was proposed that inhibitors of PARP, by preventing the activation of a deleterious bioenergetic cycle, have the potential to sustain vital cellular functions, through the maintenance of cellular bioenergetics and protein synthesis (Sims et al., 1983). The original hypothesis developed in cells exposed to genotoxic agents has subsequently extended to include a diverse set of triggers of DNA damage, such as reactive oxygen species (Schraufstatter et al., 1986), nitric oxide (Heller et al., 1994; Zhang et al., 1994), peroxynitrite (Szabo et al., 1996) and various pathophysiologically relevant triggers, where the oxidative/nitrative stress is produced by NMDA receptor activation in neurons (Zhang et al., 1994), hypoxia/reoxygenation in cardiac myocytes (Gilad et al., 1997), endotoxin stimulation in macrophages (Zingarelli et al., 1996) or elevated extracellular glucose in endothelial cells (Soriano et al., 2001). The mode of cell death triggered by PARP overactivation was found to include mitochondrial dysfunction and is now typically viewed as a regulated (active) form of cell necrosis (Virág et al., 1998; Ha and Snyder, 1999). While the PARP‐mediated cell death processes are diverse, and – depending on the cell type, stimulus and experimental context – range from processes that are more dependent on the energetic deficit, and therefore can be rescued not only by PARP inhibition but also by NAD+ supplementation (Alano et al., 2010; Weidele et al., 2010) to processes that are dependent on the intracellular toxic action of free PAR polymers, a separate mode of cell death termed parthanatos (Andrabi et al., 2006; Fatokun et al., 2014). PARP activation in neurons, in addition to being a downstream effector of NMDA‐receptor activation‐induced neurotoxicity, also increases the expression of calcium‐permeable calcium channels that are responsible for a delayed type of neuronal death (Gerace et al., 2015). By inhibiting PARP, the viability of cells subjected to oxidative/nitrative stress can be improved. In the context of neuroinjury, thus, a therapeutic opportunity exists not only to reduce the delayed loss of neurons associated with brain ischaemia (stroke, cardiac arrest or trauma) but also to decrease the late dementia frequently occurring within a few months of brain ischaemic events. The cytoprotective effects of pharmacological inhibition of PARP are schematically depicted in Figure 2, top section. One of the key aspects that determines whether PARP inhibition has positive or negative effects on cell viability is time. Generally, PARP inhibitors are protective in postmitotic (i.e. non‐replicating) or slowly replicating cells that have sufficient time to repair DNA before it has deleterious biological consequences. On the other hand, in rapidly replicating cells, the impact of PARP inhibitors on cell survival is generally negative. The PARP‐mediated active form of cell death has been reviewed, in detail, in recent articles (Virág and Szabo, 2002; Virág et al., 2013; Baxter et al., 2014; Fatokun et al., 2014).
PARP activation, a facilitator of inflammation and a pathogenetic factor in various non‐oncological diseases
The activators of PARP described in the previous section (e.g. oxidants, free radicals) are produced in a variety of pathophysiological conditions. PARP is not only involved in an active, regulated forms of cell necrosis (see above) but is also involved in signal transduction, including the promotion of various pro‐inflammatory signalling pathways. The processes of cell necrosis and the processes of inflammation are intricately interlinked and form a self‐amplifying positive feedforward cycle (Jagtap and Szabo, 2005) that promote more chronic pathophysiological processes, for example, neurodegeneration and fibrosis, which, in turn, may induce further PARP activation (Strosznajder et al., 2010). Starting with studies in stroke (Zhang et al., 1994; Eliasson et al., 1997) and myocardial infarction (Zingarelli et al., 1997), in the mid 90's, the concept was formulated that PARP inhibitors can be used as cytoprotective and/or anti‐inflammatory agents. As reviewed elsewhere for various specific disease conditions (Pieper et al., 1999; Chiarugi, 2002; Liaudet et al., 2003; Evgenov and Liaudet, 2005; Jagtap and Szabo, 2005; Koh et al., 2005; Komjáti et al., 2005; Kauppinen and Swanson, 2007; Moroni, 2008; Pacher and Szabo, 2008; Besson, 2009; Giansanti et al., 2010; Strosznajder et al., 2010; Szabó and Módis, 2010; Ba and Garg, 2011; Laudisi et al., 2011; Cavone and Chiarugi, 2012; Curtin and Szabo, 2013; Sriram et al., 2014), the list of diseases where preclinical studies demonstrate significant beneficial effects of PARP inhibition includes neurological diseases (e.g. stroke, neurotrauma, neurodegeneration), various forms of critical illness (e.g. septic shock, ALI, acute liver failure), reperfusion injury (e.g. myocardial infarction), inflammatory diseases (arthritis, colitis, asthma) and vascular diseases (diabetic complications, atherosclerosis). In all of these diseases, the likely trigger of PARP activation is DNA strand breakage, which develops, at least in part, due to the formation of reactive species (oxidants and free radicals) produced, as part of disease pathophysiology (reviewed in Pacher and Szabo, 2008). However, other non‐DNA‐damage‐dependent mechanisms of PARP activation are also known (Cohen‐Armon et al., 2007); and it may also be conceivable that basal PARP activity may also have pathophysiological roles as well.
Several PARP inhibitors – including olaparib and veliparib – demonstrated cytoprotective efficacy in an in vitro model of human cortical neuronal death induced by either NMDA receptor activation or oxygen–glucose deprivation (Xu et al., 2016). The body of preclinical data with olaparib in various in vitro and in vivo models of non‐oncological diseases is summarized in Table 2 and includes models of stroke, acute lung, liver and kidney injury, lung inflammation and chronic liver disease (Kapoor et al., 2015; Ghonim et al., 2015a,b; Rom et al., 2016; Teng et al., 2016; Gariani et al., 2017; Mukhopadhyay et al., 2017). With rucaparib – the second PARP inhibitor compound that has been recently approved – other than in vitro data showing its protective effect in human neuronal cultures exposed to NMDA or oxygen/glucose deprivation (Xu et al., 2016), there are no publications available in non‐oncological indications. Although the dose of the PARP inhibitor in the context of cancer therapy can be variable (depending whether it is used as a single agent or in combination with chemo‐ or radiotherapy), generally, in non‐oncological models, the effective doses of the PARP inhibitors (1–5 mg·kg−1 i.p.) are lower than the doses of olaparib used in cancer models (50–200 mg·kg−1·day−1 i.p.) (e.g. To et al., 2014; Henneman et al., 2015; Ter Brugge et al., 2016). This difference in the dose has mechanistic explanations and has clear advantages for therapeutic repurposing (see below).
Table 2.
Examples of the protective actions of olaparib in various in vitro and in vivo models of non‐oncological diseases
| Experimental model | Human disease(s) modelled | Dose or concentration of olaparib | Effects of olaparib | Reference |
|---|---|---|---|---|
| Differentiated human neurons in culture exposed to NMDA receptor ligands or oxygen–glucose deprivation (OGD) | Stroke, neurodegeneration, neuroinflammation | 2 μM (in vitro) | Olaparib protected against NMDA and OGD‐induced neuronal cell death. | Xu et al., 2016 |
| CD3/CD28‐stimulated human CD4 + T‐cells | Asthma, inflammation | 1–5 μM (in vitro) | Olaparib reduced Th2 cytokine production while moderately affecting T‐cell proliferation. It also increased IL‐17 production and increased the T‐reg cell population. | Ghonim et al., 2015b |
| Models of monocyte adhesion and migration across blood brain barrier (BBB) | Blood brain barrier function (relevant for stroke, TBI, neuroinflammation) | Leukocytes were treated ex vivo with olaparib 10 μM for various durations | Olaparib in human primary monocytes diminished their adhesion to and migration across BBB (human primary microvascular endothelium) in in vitro models and prevented barrier injury. In vivo olaparib diminished leukocyte‐endothelial interactions and protected against the deterioration of BBB integrity. | Rom et al. 2016 |
| C2C12 myotubes exposed to H2O2 | Skeletal muscle disorders | 100 nM in vitro | Olaparib rescued the oxidant‐induced decline in cellular NAD+ levels and improved mitochondrial function in myotubes. | Pirinen et al. 2014 |
| Hepatocyte cell line AML12 | Ageing | 100 nM in vitro | Olaparib rescued the oxidant‐induced decline in cellular NAD+ levels and increased mitochondrial biogenesis. | Mouchiroud et al., 2013 |
| Caenorhabditis elegans | Ageing | 1 μM in vitro | Olaparib prevented age‐associated metabolic decline and promoted longevity. It increased mitochondrial biogenesis. | Mouchiroud et al., 2013 |
| Mouse model of transient middle cerebral artery occlusion (2 h ischaemia, 24 h reperfusion) | Acute ischaemic stroke | 1, 3, 5 10 and 25 mg·kg−1, single intraperitoneal dosing | At 3 and 5 mg·kg−1 (but not doses higher or lower) olaparib reduced infarct size, IgG extravasation, and improved neurological scores in some (but not all) tests used. | Teng et al., 2016 |
| Intratracheal administration of endotoxin to mice | ALI, sepsis, acute renal failure | 5 mg·kg−1, single intraperitoneal dosing | Olaparib reduced inflammatory cell (in particular neutrophil) infiltration into the lungs and attenuated pulmonary oedema. It also protected against the endotoxin‐induced secondary kidney injury, shown by improvements in serum urea and creatinine levels. These effects were associated with a reduction in tissue oxidative stress markers and reduced production of various pro‐inflammatory factors (e.g. TNF‐α, IL‐1β and VCAM‐1). | Kapoor et al., 2015 |
| Intraperitoneal administration of endotoxin to mice | Acute hepatitis, sepsis | 50 mg·kg−1, oral dosing | Olaparib reduced plasma levels of hepatic injury markers and improved hepatic NAD+ levels. It also reduced the LPS‐induced hepatic proinflammatory genes Il‐1β and Il‐6. These effects were associated with a reduction of the infiltration of the liver with inflammatory cells. | Gariani et al., 2017 |
| Ovalbumin exposure in ovalbumin‐sensitized mice: pulmonary inflammation and airway hyperreactivity in mice | Asthma bronchiale | 1, 5 and 10 mg·kg−1, single intraperitoneal dosing | At all doses tested, olaparib afforded a dose‐dependent reduction in ovalbumin‐specific IgE production in bronchoalveolar lavage fluid. It also reduced inflammatory cell numbers in bronchoalveolar lavage, and reduced mucus production in the lungs. It improved airway hyperreactivity response to metacholine. Finally, olaparib suppressed several inflammatory mediators including eotaxin, IL‐2, IL‐4, IL‐5, IL‐6, IL‐13 and M‐CSF. | Ghonim et al., 2015a |
| Mice chronically exposed to house dust mite: pulmonary inflammation and airway hyperreactivity | Asthma bronchiale | 5 mg·kg−1, single intraperitoneal dosing | Olaparib prevented the dust mite‐induced increase in overall cellularity, weight and CD4+ T‐cell population in spleens. It also inhibited eotaxin, IL‐4, IL‐5 and IL‐13 production. It reduced IL‐2 and IP‐10 levels and increased the T‐regulatory cell population. In addition, olaparib improved the airway hyperreactivity response to methacholine. | Ghonim et al., 2015b |
| Mice subjected to a high fat high‐sucrose diet for 18 weeks | Non‐alcoholic fatty liver disease (NASH) | 50 mg·kg−1·day−1 orally, either in a preventive mode (starting at 7 weeks of age), or 9 weeks after the start of the diet, in a therapeutic mode (starting at 16 weeks of age) | Olaparib reduced obesity without changes in food intake. It also improved triglyceride and cholesterol status. In post‐treatment mode it reversed hepatic fat deposition, improved hepatic histopathological changes and reduced liver fibrosis. These effects were associated with increased mitochondrial biogenesis and β‐oxidation in the liver; reduction in reactive oxygen species levels and suppression of endoplasmic reticulum stress. Olaparib also improved insulin sensitivity in a glucose tolerance test. | Gariani et al., 2017 |
| Mice subjected to methionine and choline deficient diet (MCD) for 5 weeks | Non‐alcoholic fatty liver disease (NASH) | 50 mg·kg−1·day−1, oral dosing | Olaparib improved hepatic fat deposition, improvement of hepatic histopathological changes and reduction in liver fibrosis. It also reduced plasma levels of hepatic injury markers. Olaparib improved hepatic NAD+ levels and suppressed the expression of multiple pro‐inflammatory genes. | Gariani et al., 2017 |
| Mice subjected to MCD diet for 5 weeks | Non‐alcoholic fatty liver disease (NASH) | 50 mg·kg−1·day−1, oral dosing | Olaparib improved hepatocellular injury, attenuated steatosis and metabolic dysregulation, decreased hepatic inflammation (Ly6G, F4/80, Cxcl2, TNFalpha mRNA expression) and fibrosis. | Mukhopadhyay et al. 2017 |
| Mice subjected to high fat diet for 12 weeks | Non‐alcoholic fatty liver disease (NASH) | 40 mg·kg−1·day−1, oral dosing for 8 days | Olaparib reduced body weight, suppressed liver steatosis and dysregulated hepatic fatty acid metabolism without changing food intake and ambulatory activity. Olaparib also increased O2 consumption, CO2 production and total energy expenditure associated with significantly increased fat oxidation. | Mukhopadhyay et al. 2017 |
| Mice subjected to high fat diet for 12 weeks and a single binge of alcohol | Acute alcoholic hepatitis | 40 mg·kg−1·day−1, oral dosing for 8 days | Olaparib attenuated hepatocellular injury (ALT) and inflammation (decreased hepatic neutrophil infiltration, mRNA expression of Ly6G, ICAM1, Cxcl2, CCR2, IL1β, TNFα). | Mukhopadhyay et al. 2017 |
Expression and activity of PARP is dynamically regulated
Considering pharmacological modulation of PARP for non‐oncological indications, another factor to discuss is the dynamic regulation of PARP activity (as well as PARP expression) by environmental factors or by physiological conditions. If PARP is subject to positive or negative regulation by physiological factors, one can predict that the cells are more likely to tolerate its pharmacological modulation as well. In this respect, several endogenous or natural‐product‐based inhibitors of PARP activity have been described, including nicotinamide, caffeine, hypoxanthine, taurine, xanthurenic acid and kynurenic acid, various naturally occurring diadenosine polyphosphates, the active form of Vitamin D (1α,25‐dihydroxyvitamin D3), Vitamin A and its precursors, 3,5,3′‐triiodothyronine, as well as natural products, like ginsenosides (Banasik et al., 2012; Nabavi et al., 2015). It must be emphasized that most of the above studies are based on in vitro observations only and may have used supra‐physiological concentrations and that the potency of most of these natural/non‐conventional PARP inhibitors is lower than the potency of the state‐of‐the‐art third‐generation PARP inhibitors.
The issue of antibiotic‐mediated PARP inhibition deserves further discussion. In vitro, several antibiotics, including actinomycin D, coumermycin A1, formycin B, novobiocin and showdomycin have been shown to exhibit PARP inhibitory effects (Banasik et al., 2012). In addition, tetracyclines, with the rank order of minocycline > doxycycline > demeclocycline > chlortetracycline, have also been identified as PARP inhibitors. Minocycline also prevented neuronal NAD+ depletion in vitro and neurotoxicity in vivo and exerted the type of anti‐inflammatory action that is expected from a PARP inhibitor (Alano et al., 2006). Minocycline also exerted protective effects in re‐oxygenated cardiac myocytes in vitro, on a cardiopulmonary bypass model in vivo and in a diabetic retinopathy model in vivo. These effects were associated with a suppression of PAR polymer formation (Tao et al., 2010; Dhein et al., 2015; Wu et al., 2015). However, in other models (e.g. in a model of asthma), the PARP inhibitory effects of minocycline were found to involve indirect (i.e. antioxidant) actions, rather than direct effects on the PARP enzyme (Naura et al., 2013).
In addition to regulation at the level of PARP catalytic activity, the expression of PARP1 may also be dynamically regulated. For instance, the expression of PARP1 is regulated during skeletal muscle cell maturation: as myoblasts differentiate into myotubes, PARP1 expression is reduced; this change confers resistance to oxidative stress (Oláh et al., 2015). Also, PARP expression is highly up‐regulated during dendritic cell maturation and contributes to immunocompetence and T cell proliferation (Aldinucci et al., 2007). In other conditions – rodent models of chronic heart failure, renal transplantation or unilateral ureteral obstruction and in the brain of patients with ALS – up‐regulation of PARP1 protein has been demonstrated (Kim et al., 2004; Pillai et al., 2005; O'Valle et al., 2007; Vagnerova et al., 2010; Kim and Padanilam, 2011).
It should be also noted that the expression and activity of PARP shows marked cell‐type differences. For instance, PARP levels are low in resting lymphocytes, high in mitogenically activated lymphocytes and virtually absent in mature granulocytes (Berger et al., 1987). There is also a wide (up to 200‐fold) range in PARP activity between various human subjects (Zaremba et al., 2011). These differences may indicate relative differences in the importance of PARP‐dependent regulation in different cell types or perhaps even in different individual human subjects.
Taken together, PARP activity and expression appears to be regulated by physiological and pathophysiological factors, in response to many commonly used drugs, as well as by gender, as highlighted below. These data, indicating that negative modulation of PARP activity occurs under different physiological settings, indirectly support the notion that pharmacological targeting of PARP may be a therapeutically acceptable strategy in carefully selected non‐oncological indications.
Many of the protective effects of PARP inhibitors are gender‐dependent
The issue of gender differences in the therapeutic effects of PARP inhibitors requires special emphasis. Several studies showed that the efficacy of various PARP inhibitors in rodent models of stroke is preferentially observed in male mice and is also dependent on the age of the animals (Eliasson et al., 1997; Hagberg et al., 2004; McCullough et al., 2005; Yuan et al., 2009); similarly, the protective effect of minocycline is primarily observed in male animals subjected to stroke (Li and McCullough, 2009). However, the gender difference was not observed in all studies. In some studies, PARP inhibitors maintain partial efficacy in female animals (Moroni et al., 2012), and in a primate study of stroke, the PARP inhibitor MP‐124 showed comparable protective effects in both sexes (Matsuura et al., 2011). PARP inhibition was equally protective in sex‐segregated primary cortical neurons exposed to peroxynitrite, but neurons from males were more vulnerable than their female counterparts to nitrosative stress (Du et al., 2004). In addition, in a recent study, olaparib exerted neuroprotective effects on human cortical neurons derived from the human ESC H9 cell line, which is female, as well as in human ESC H1 cell line‐derived cortical cultures, which is male (Xu et al., 2016). Although the mechanism behind the gender effect of PARP inhibitors and the reason why some studies/some PARP inhibitors exhibit sex‐specificity, while others do not, remains unclear, it is interesting that PARP1 appears to become activated in response to stroke in both sexes, but this is associated with a more pronounced decrease of brain NAD+ levels in males than in females (Hagberg et al., 2004).
The sex differences with respect to PARP may also apply to rodent models of shock and inflammation (Mabley et al., 2005). For example, the PARP inhibitor PJ34 decreases the endotoxin‐induced production of TNF‐α and protects against endotoxin‐induced mortality in male, but not female mice. However, the protective effect of PARP inhibitors is restored in female mice by ovariectomy. Moreover, in a porcine model of thoracoabdominal aortic ischaemia–reperfusion injury, PARP inhibitors protect against cardiovascular collapse in male, but not in female animals (Hauser et al., 2006). In addition, PARP inhibition protects in a murine model of autoimmune nephritis in male, but female mice (Jog et al., 2009). Although oestrogen does not appear to inhibit PARP activation in a direct manner, an interaction has been characterized between PARP1 and oestrogen receptor α, whereby a stable complex may sequester PARP1 to specific regions on the DNA making it difficult for its zinc fingers to access and recognize DNA breakpoints (which is essential for the enzymic activation of PARP) (Mabley et al., 2005). In cells generated from male (but not from female) mice, oestrogen was found to inhibit oxidant‐induced PARP activation in vitro (Jog and Caricchio, 2013). One possible explanation for the greater effects of PARP inhibitors in males than females, is the finding that PARP activity in peripheral blood mononuclear cells (PBMCs) and liver tissue was significantly higher in male than female mice (Zaremba et al., 2010). These authors also showed that this effect may be driven by male sex hormones as PARP activity in PBMCs isolated from castrated male mice was lower than in intact males and similar to that in females, and unaffected by oestrogen supplementation. Studies in healthy volunteer humans and cancer patients also revealed that PARP activity in PBMCs isolated from women had significantly lower PARP activity than PMBCs isolated from men. Moreover, women <45 years old (pre‐menopausal) tended to have a lower PARP activity than older women and men (Zaremba et al., 2010). Taken together, PARP activation is influenced by sex and sex hormones, but whether this is androgen driven (in males) or oestrogen suppressed (in females) or both – and the underlying molecular mechanisms – remains to be better characterized. In addition, even though PARP activation is lower in females than in males, the clinical data from olaparib in ovarian cancer clearly indicate that PARP remains functionally relevant in women and can be pharmacologically targeted with clinically relevant therapeutic effects.
On the adverse effect profile of PARP inhibitors
One of the most common objections against the use of PARP inhibitors for non‐oncological indications relates to the question as to whether by ‘artificially’ preventing cell necrosis, do we prolong the life of cells that are ‘doomed’ due to their extensive degree of DNA damage? And if so, what is the potential that these cells later become subject to malignant transformation?
PARP1 is not a DNA repair enzyme per se; it is an enzyme that helps with the recruitment of DNA repair enzymes that execute the DNA repair. PARP1 deficient mice are viable and fertile, although cells from PARP1−/− mice exhibit an increased degree of genomic instability in response to genotoxic agents and PARP1−/− mice exhibit increased mortality in response to irradiation (Shall and de Murcia, 2000). In other studies, BER efficiency was found comparable in wild‐type and PARP1 deficient cells (Vodenicharov et al., 2000). One must keep in mind that in PARP1‐deficient systems, not only the catalytic activity of PARP1 is completely removed but also its structural (scaffolding) function. Furthermore, in PARP1‐deficient systems, the enzyme is absent for the life of the cell, which is not the same as acute treatment of the cells with intermittent or short‐term dosing with PARP inhibitors (which doses perhaps do not induce a 100% inhibition of the catalytic activity of the enzyme).
Many in vitro studies have examined the effect of PARP inhibitors on cellular DNA integrity. Studies with early generation PARP inhibitors (e.g. 3‐aminobenzamide) produced equivocal results, as in some studies, sister chromatid exchange rates and malignant transformations were reduced (Borek et al., 1984), while in other studies, they remained unaffected (D'Souza et al., 1992), and in yet other studies, increases were reported (Shiraishi et al., 1983; Schwartz et al., 1984; D'Souza et al., 1992). In line with the concept that PARP1 deficiency does not equal PARP catalytic inhibition (as already discussed above), Smulson and colleagues compared the tetraploidy inducing potential of PARP1 deficiency with GPI 6150 and found that while PARP1 deficiency increased the percentage of a genetically unstable tetraploid cell population in fibroblasts, the PARP inhibitor (20 μM, 3 weeks of continuous exposure) did not (Simbulan‐Rosenthal et al., 2001). In a number of different cell lines, 24 h exposure to rucaparib (10 μM) resulted in a similar number of H2AX foci as were induced by 2 Gy irradiation (Drew et al., 2011). Ito and colleagues have recently examined the effect of olaparib and veliparib in several human cells, including primary lymphoid cells and non‐tumorigenic and tumorigenic epithelial cell lines. Both olaparib and veliparib, at respective concentrations of 0.1–1 and 1.5–80 μM, induced concentration‐dependent increases in sister chromatid exchange rates and chromatid aberrations (Ito et al., 2016). Similarly, in an in vitro assay where whole chromosomal instability was assessed by using a nonessential human artificial chromosome in the HT1080 human fibrosarcoma cell line, olaparib and veliparib – at concentrations of 5–10 μM (where they inhibited cell proliferation by 50%) – induced significant increases in the rate of human artificial chromosome loss (Lee et al., 2016). In summary, although the above studies did not examine oxidatively stressed cells, and only evaluated a limited number of PARP inhibitors, the data demonstrate that PARP inhibitors can, indeed, exert genotoxic effects in vitro. The predictive power (or lack thereof) of the various in vitro genotoxicity and genetic stability studies on the in vivo tumorigenic potential remains unclear (e.g. Snyder and Green, 2001; Brambilla and Martelli, 2009). As far as direct studies on PARP inhibitors or PARP deficiency on tumourigenesis are concerned, the data are mixed (see Masutani et al., 2003; Masutani and Fujimori, 2013). For instance, in an azoxymethane‐induced colon and liver carcinogenesis model in mice, PARP1 deficiency increased tumour incidence (Nozaki et al., 2003). In a study of age‐related spontaneous carcinogenesis in mice, the incidence of spontaneous tumours was similar in both wild‐type and PARP1‐deficient mice, but the incidence of some malignant tumours (uterine tumours, lung adenocarcinomas and hepatocellular carcinomas) was higher in PARP1‐deficient mice (Piskunova et al., 2008). However, in other models (e.g. nitrosamine‐induced oral and hepatic carcinogenesis) in mice, PARP1 deficiency failed to affect tumour incidence (Gunji et al., 2006; Ogawa et al., 2006). Surprisingly, in some studies, PARP inhibition was even found to reduce carcinogenesis. For instance, when Ela‐myc‐driven pancreatic tumour development in mice was analysed on a PARP1 knockout background, increased tumour necrosis and decreased proliferation, apoptosis and angiogenesis were noted (Martínez‐Bosch et al., 2014). Moreover, inhibition of PARP with PJ34 was found to prevent Helicobacter‐induced gastritis and precancerous lesions in IL‐10−/− mice (Toller et al., 2010). In addition, PARP1‐deficient male mice and mice treated with the PARP inhibitor 3,4‐dihydro‐5‐[4‐(1‐piperidinyl)butoxyl]‐1(2H)‐isoquinolinone showed a marked delay in tumour formation, as well as a dramatic reduction in tumour size and multiplicity in a model of chemically induced skin carcinogenesis, most likely due to its suppressive effect on the expression of several pro‐inflammatory genes (Martin‐Oliva et al., 2004; Martin‐Oliva et al., 2006). Most relevant to the current article is a carcinogenesis study with olaparib and veliparib in a BRCA‐deficient mouse model of breast cancer development. In this model, both veliparib diet (100 mg·kg−1·day−1), or olaparib diet (25, 50, 100 or 200 mg·kg−1·day−1), when administered continuously for up to 43 weeks, exerted antitumor effects. The highest dose of the PARP inhibitor delayed the average age of the first detectable tumour by 2.4 and 6.5 weeks, respectively, compared with controls. Olaparib also increased the average lifespan of these mice by 7 weeks (To et al., 2014). Interestingly, several of the above studies (e.g. the studies by To and the studies by Piskunova and colleagues) utilized female mice only; given the gender‐differences in PARP inhibitors' effects discussed above, it remains to be further investigated whether the effects of PARP inhibitors on carcinogenicity may also be gender‐dependent.
There are additional published studies where animals were treated with PARP inhibitors for extended time periods, in some cases up to 6–9 months (e.g. Bartha et al., 2009; Gariani et al., 2017); these studies have not reported any tumorigenic effects of the PARP inhibitors, although it should be emphasized that these studies did not specifically investigate this issue as part of their original trial design.
Many in vitro studies have been conducted to investigate the effect of PARP inhibitors on different cell populations (normal, necrotic, apoptotic), exposed to various concentrations of genotoxic oxidants (e.g. hydrogen peroxide or peroxynitrite). These oxidants trigger DNA damage, PARP activation, and shift cells from the normal to the necrotic and apoptotic populations (Virág et al., 1998; Ha and Snyder, 1999; Ye, 2008). When cells are treated with PARP inhibitors, the necrotic cell population decreases, while the apoptotic and the normal populations increase. In other words, by inhibiting PARP, necrosis can be prevented and/or shifted to apoptosis, a distinct form of cell death, where the cellular content is not spilled into the extracellular space and, therefore, does not induce further cytotoxicity or inflammation to bystander cells. Through pharmacological suppression of cell necrosis, one can, therefore, not only maintain functional parenchymal cells but also down‐regulate the inflammatory process and prevent self‐amplifying cycles of injury. By pharmacological control of inflammation (either by the direct transcriptional effect of PARP inhibitors discussed above) or by reducing the spillage of necrotic cells into the environment, as well as by interrupting various positive feedback cycles of injury, the degree of genotoxic burden to the cells would be expected to be attenuated (as discussed in Jagtap and Szabo, 2005). Indeed, several studies demonstrate that genetic or pharmacological inactivation of PARP1 reduces tissue myeloperoxidase levels (indicating reduced infiltration of inflammatory cells) and tissue malondialdehyde levels (a marker of tissue oxidative burden) and tissue nitrotyrosine levels (a marker of tissue nitrosative burden) (Soriano et al., 2002; Chatterjee et al., 2003; Esposito et al., 2011; Kapoor et al., 2015). It is, therefore, conceivable that a diseased organism – which is exposed to endogenous genotoxic free radicals and oxidants and exhibits signs of a ‘baseline’ damage to the genetic material (e.g. Bao et al., 2015) – when treated with a PARP inhibitor, may ultimately encounter a reduced ‘burden’ of genotoxic oxidative/nitrative damage than an untreated one. Whether this type of ‘indirect’ genoprotective effect may be able to counteract or compensate for any ‘direct’ genotoxic effect of a PARP inhibitor remains to be determined. In any case, one must emphasize that the effect of a PARP inhibitor on a normal (unperturbed) organism may be drastically different from the effect of the same inhibitor under pathophysiological conditions. For instance, PARP inhibition in normally cultured fibroblasts accelerates the rate of telomere shortening (Cohausz et al., 2008; Boesten et al., 2013). However, under conditions of chronic oxidative stress, PARP inhibition did not accelerate telomere shortening (Boesten et al., 2013). Unfortunately, the vast majority of published studies focusing on the therapeutic effect of PARP inhibitors on various experimental models of disease in vivo do not measure parameters of DNA integrity. However, it must be noted that in a porcine study of thoracic aortic cross‐clamping induced ischaemia–reperfusion model, DNA integrity was monitored in isolated leukocytes ex vivo, and the beneficial cardiovascular effects of the PARP inhibitor INO‐1001 were not associated with increases in DNA damage (Hauser et al., 2006).
Risk/benefit analysis: selection of prime non‐oncological PARP indications
The evidence reviewed above and the list of indications where preclinical data show that PARP inhibitors can exert beneficial therapeutic effects are substantial, both in acute (Table 3) and chronic (Table 4) diseases (Figure 3). Data overviewed in this table (and further discussed in review articles (Virág and Szabo, 2002; Szabó, 2005; Pacher and Szabo, 2008; Curtin and Szabo, 2013; García and Conde, 2015) and other papers (Mota et al., 2005, 2007; Yu et al., 2012; Lee et al., 2013; Li et al., 2013; Mukhopadhyay et al., 2014, 2017; Stoica et al., 2014; Ghonim et al., 2015a,b; Rom et al., 2015; Tao et al., 2015; Ibba et al., 2016; Lehmann et al., 2016; Lucarini et al., 2016; Salluzzo et al., 2016; Wang et al., 2016; Yu et al., 2016; Zhang et al., 2016; Gariani et al., 2017) support the view that PARP inhibitors may be therapeutically advantageous for a variety of non‐oncological indications, if the selection of these indications is supported by appropriate risk/benefit analysis. The ancient Hippocratic oath (Primum nil nocere) calls, first and foremost, for doing no harm. While the risk of increased chromosomal instability – as predicted by the various in vitro assays discussed earlier (e.g. Ito et al., 2016; Lee et al., 2016) – must be kept in mind, below, we outline our method aimed at striking a balance between the expected benefits and potential risks associated with PARP inhibitor therapy for non‐oncological indications.
Table 3.
The therapeutic potential of PARP inhibitors in acute non‐oncological diseases. Overview of acute non‐oncological indications where PARP inhibitors or PARP1 deficiency demonstrated benefit in in vivo preclinical studies
| Disease | Effect of PARP inhibitors in preclinical models | Human evidence for PARP activation | Short duration of drug therapy? | Indication severe? | Therapeutic options limited? | Trial design feasible? |
|---|---|---|---|---|---|---|
| Myocardial infarction | Many classes of PARP inhibitors, as well as PARP1 deficiency exert protective effects, mainly in transient ischaemia models. The time window of intervention is substantial; the beneficial effects are multiple (reduction of infarct size, suppression of inflammation, prevention of later remodelling) is long‐lasting. Efficacy data in large animals also support this indication. | PARP activation was demonstrated in circulating leukocytes in patients with myocardial infarction and therapeutic revascularization. PARP inhibition with INO‐1001 in an exploratory Phase I trial showed a tendency to reduce biomarkers (CRP, IL‐6). |

PARP inhibitors may be administered in single bolus or short infusion at the time of revascularization. |

Revascularization therapy improved acute outcomes (as long as the patient is rapidly transferred into a hospital). However, development of chronic heart failure later on represents a substantial challenge. |

Revascularization is effective. Although there were many preclinical and clinical efforts to combine revascularization with specific therapies to counteract reperfusion injury, no approved or widely used anti‐reperfusion therapy is available. |
Small, exploratory trials, focusing on infarct size and indicators of reperfusion injury trials are feasible. However, large trials (registration endpoints, e.g. survival) require large number of patients and multicenter efforts. |
| Circulatory shock | Multiple classes of PARP inhibitors, as well as PARP1 deficiency exert protective effects, in models of shock induced by bacterial LPS, live bacteria, haemorrhage, burn injury and others. The time window of intervention is substantial; the beneficial effects include improvements in organ function and extension of survival. Efficacy data in large animal models also support this indication. Olaparib protects against renal and pulmonary dysfunction in a rodent model of circulatory shock. | In myocardial sections and circulating leukocytes of patients with circulatory shock, significant PARP activation was observed. The degree of PARP activation showed a close correlation with the degree of sepsis‐induced myocardial dysfunction. There is also evidence of PARP activation in endothelial cells and leukocytes of burn patients. |

PARP inhibitors can be administered orally or parenterally at the time of diagnosis and to continue for several weeks. |
Mortality rates of septic shock remain very high (30–50%). |
The current state‐of‐the‐art is purely supportive; the only specific therapy (activated protein C) has been withdrawn for lack of efficacy. |
Small, exploratory trials, focusing on biomarkers are feasible. However, large trials (registration endpoints, e.g. survival) are expensive, complex and require large number of patients and multicenter efforts. |
| Stroke | Multiple classes of PARP inhibitors, as well as PARP1 deficiency exert protective effects, both in transient and permanent ischaemia models. The time window of intervention is substantial; the beneficial effect is long‐lasting. Efficacy data in non‐human primates also support this indication. Olaparib and veliparib protect against NMDA‐receptor activation induced neurotoxicity in an in vitro model of human neuron injury. | Activation of PARP was shown in brain sections from patients dying from stroke, brain ischaemia due to cardiac arrest. |
PARP inhibitors may be administered in single bolus or short infusion at the time of revascularization. |
Revascularization therapy improved acute outcomes, but significant morbidity and mortality remains even with state‐of‐the‐art therapy and rehabilitation. |
No approved or widely used anti‐reperfusion or neuroprotective therapy is available. |

Small, exploratory trials are feasible. However, large trials (registration endpoint) are complex and expensive. |
| Brain trauma | Multiple classes of PARP inhibitors, as well as PARP1 deficiency exert protective effects in various brain trauma models. The therapeutic window is substantial. The beneficial effect is long‐lasting as it also protects against the breakdown of the blood–brain barrier and prevents the later‐onset neuro‐inflammatory processes. Efficacy data in non‐human primates also support this indication. Olaparib and veliparib protect against NMDA‐receptor activation induced neurotoxicity in an in vitro model of human neuron injury. | Activation of PARP was shown in brain sections from patients dying from brain trauma. Increased PARylated proteins were also found in cerebrospinal fluid samples from adults and children with brain trauma. PARP1 polymorphisms were also identified: a PARP‐1 polymorphism within the automodification‐catalytic domain was associated with neurological outcome, while a polymorphism within the promoter region was associated with cerebrospinal fluid PAR‐modified protein levels. |
PARP inhibitors may be administered in single bolus or short infusion at the time of diagnosis or hospitalization. |
Significant morbidity and mortality remains even with state‐of‐the‐art therapy and rehabilitation. |
No widely used or highly effective neuroprotective therapy is available. |
Small, exploratory trials are feasible. However, large trials (registration endpoint) are complex and expensive. |
| Pancreatitis | The PARP inhibitor 3‐aminobenzamide improves outcomes in several rodent models of pancreatitis. PJ34 was tested in one study (cerulein model) and showed protective effects. PARP1 deficient mice are not protected in one cerulein‐model of pancreatitis, but not in another one. The PARP inhibitor KU0058684 (which is structurally closely related to olaparib) has also demonstrated protective effects in a cerulein model. The therapeutic window of intervention has not yet been carefully explored. | No human data so far |
PARP inhibitors can be administered orally or parenterally at the time of diagnosis and to continue for several weeks. |
Mortality rates of remain very high (30–50%). |
The current state‐of‐the‐art is largely supportive; antibiotics may offer some therapeutic benefit. |
Small, exploratory trials, focusing on biomarkers are readily feasible. Larger trials (registration endpoints) are more involved but nevertheless feasible. |
| Transplant rejection | Treatment of the donor organs and/or the recipient with PARP inhibitors of various structural classes improves organ function, extends transplant life and in some studies also reduced immune‐mediated rejection. | Increased expression of PARP‐1 in transplanted kidneys was detected: it was found to correlate with worsened outcome after transplantation. |
PARP inhibitors would be expected to be administered as part of the storage fluid or to the recipient (acutely or for longer duration to limit rejection). |
Outcomes have substantially improved in recent years; the main problem is the lack of availability of donor organs. If PARP inhibitors could extend the donor pool (e.g. render marginal transplants viable), this would be considered a substantial advance. |
Many effective therapeutic options are available for transplant rejection; less so for organ preservation. |
Small, and larger trials are both feasible as single‐centre or multi‐centre efforts. |
| ALI | Multiple classes of PARP inhibitors, as well as PARP1 deficiency exert protective effects in a variety of ALI models (endotoxin, smoke inhalation, ischaemia–reperfusion, bacterial pneumonia, paraquat). The beneficial effect are multiple (suppression of inflammation, improvement of gas exchange). Efficacy data in large animals also support this indication. Olaparib protects against pulmonary dysfunction in a rodent model of endotoxin‐induced lung injury. | No human evidence so far. |
PARP inhibitors can be administered orally or parenterally at the time of diagnosis and to continue for several weeks. |
Outcomes have improved in recent years with changes in the way supportive care is applied. Nevertheless, morbidity and mortality rates remain high. |
No specific, approved pharmaceutical therapies are available. |
Small, exploratory trials, focusing on physiological parameters and/or biomarkers are feasible. Larger trials (registration endpoints) are more involved but nevertheless feasible. |
In the right 4 columns, attractiveness/feasibility is shown in a graded fashion: indicates least attractive/feasible and indicates most attractive/feasible.
Table 4.
The therapeutic potential of PARP inhibitors in chronic non‐oncological diseases. Overview of chronic non‐oncological indications where PARP inhibitors or PARP1 deficiency demonstrated benefit in in vivo preclinical studies
| Disease | Effect of PARP inhibitors in preclinical models | Human evidence for PARP activation | Short duration of drug therapy? | Indication severe? | Therapeutic options limited? | Trial design feasible? |
|---|---|---|---|---|---|---|
| Chronic neuro‐inflammatory diseases | Several studies, using several different models of neuroinflammation (including EAE) show the protective effect of PARP inhibitors, although some of the data are conflicting; in one study PARP1 deficiency was found to increase, rather than decrease disease severity. Olaparib and veliparib protect against NMDA‐receptor activation induced neurotoxicity in an in vitro model of human neuron injury. | In isolated PBMC relapsing–remitting untreated MS patients, increased PARP1 expression and increased PARylation was found. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
In many neuroinflammatory indications (e.g. MS) significant morbidity and mortality remains. |
In many neuroinflammatory indications (e.g. MS) therapeutic options are limited. |
The complexity and the duration of the trials is substantial. |
| Chronic neuro‐degenerative diseases | Several studies, using several different models of neurodegeneration (including the MPTP model of Parkinson's disease) show the protective effect of PARP inhibitors. PARP1 deficiency is also protective. PARP inhibition protects against dopamine neuron death in AIMP2 transgenic mice. In a Drosophila model of PARKIN induced neurodegeneration, mutation of PARP1 protects against dopaminergic neurodegeneration. Olaparib and veliparib protect against NMDA‐receptor activation induced neurotoxicity in an in vitro model of human neuron injury. | Poly(ADP‐ribosyl)ation in brain sections from patients with Alzheimer's disease, Parkinson's disease and ALS. A PARP1‐410C/T \polymorphism was detected in Sicilian patients with Parkinson's disease and several other PARP1 polymorphisms have also been detected in other patient populations with Parkinson's disease. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
Significant morbidity and mortality remains. |
In many neurodegenerative indications (e.g. Parkinson's) therapeutic options are limit |
The complexity and the overall design of the trials does not represent a substantial hurdle, but and the duration of the trials is substantial. |
| Chronic inflammatory and autoimmune diseases | PARP inhibitors exert protective effects in various models of arthritis, colitis, uveitis, and other models of local inflammation. In some cases, PARP1 deficient systems have also been evaluated and they also afford protective effects. Olaparib protects against pulmonary inflammation in an allergen‐induced pulmonary inflammation model. | Auto‐antibodies against PARP in arthritis, SLE and other autoimmune diseases. Increased poly(ADP‐ribosyl)ation was found in biopsies from patients with colitis. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
In many local inflammatory diseases (e.g. arthritis) outcomes have substantially improved in recent years. |
In many local inflammatory diseases, state‐of‐the‐art and several therapeutic options are available (e.g. arthritis). |
In many local inflammatory diseases, trial designs are well established and feasible. |
| Vascular diseases | PARP inhibitors and/or PARP1 deficiency has been shown to improve vascular contractile and/or relaxant function in vascular dysfunction induced by circulatory shock, diabetes mellitus, atherosclerosis, and physiological ageing. | Activation of PARP has been shown in human atherosclerotic samples and in microvessels present in skin biopsies from type 2 diabetic patients. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
Outcomes have substantially improved in recent years. |
Many effective therapeutic options are available. |
Small, exploratory trials, focusing on markers or functional endpoints are readily feasible. Larger trials are more involved but nevertheless feasible. |
| Chronic heart failure | Multiple classes of PARP inhibitors, as well as PARP1 deficiency exert protective effects in multiple models of heart failure (mechanically induced, chemically induced, diabetes‐associated). PARP inhibitors suppress pathological myocardial remodelling. | Increased poly(ADP‐ribosyl)ation, as well as increased PARP1 expression was demonstrated in human heart samples in end‐stage heart failure and in heart failure followed by left ventricular heart assist device implantation. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
5‐year mortality rates remain very high. |
None of the therapies used currently are efficacious to extend survival. |
Large trials (registration endpoints, e.g. survival) require large number of patients and multicenter efforts. |
| Type I diabetes mellitus | Various PARP inhibitors suppress the onset and severity of streptozotocin‐induced Type I diabetes by preventing pancreatic beta cell necrosis. PARP inhibition also prevents spontaneous and recurrent autoimmune diabetes in NOD mice. | No human evidence so far. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
Outcomes have not improved in recent years. |
No therapies are available to prevent or delay the autoimmune beta‐cell destruction. |
Large trials are necessary to seek demonstration of efficacy; such trials are complex, lengthy and expensive. |
| Chronic liver diseases | PARP inhibitors protect against various forms of chronic liver injury (alcohol‐induced, steatosis‐induced). PARP inhibition suppresses fatty liver disease. PARP inhibition protects against liver fibrosis development. | Increased PARP activation and/or PARP1 expression in human livers from hep B and alcohol‐induced cirrhosis, NASH, and ASH. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
This is a heterogeneous category, but there are many diseases in this category (e.g. liver fibrosis) where morbidity and mortality remains high. |
This is a heterogeneous category, but in some cases (e.g. liver fibrosis) all of the existing therapeutic options remain only marginally effective. |
Both smaller and larger trials are feasible. Trials with meaningful endpoints are long in duration. |
| Chronic pulmonary diseases | In multiple models of asthma, PARP inhibitors of various classes (including olaparib) exert protective effects, including functional parameters and reduced inflammatory mediator production. Hyperoxia‐induced lung injury and bleomycin‐induced fibrosis have also been shown to be attenuated by PARP inhibitors. | Increased poly(ADPribosyl)ation was found in peripheral blood lymphocytes from patients with chronic obstructive pulmonary disease (COPD). Increased poly(ADPribosyl)ation was also demonstrated in PBMCs and lung tissues from asthmatic patients. |
PARP inhibitors would be expected to be administered chronically (years – life‐long). |
This is a heterogeneous category, but there are many diseases in this category (e.g. lung fibrosis) where morbidity and mortality remains high. |
This is a heterogeneous category, but in some cases (e.g. lung fibrosis) all of the existing therapeutic options remain only marginally effective. |
Both smaller and larger trials are feasible. Trials with meaningful endpoints are long in duration. |
In the right 4 columns, attractiveness/feasibility is shown in a graded fashion: indicates least attractive/feasible and indicates most attractive/feasible.
Figure 3.
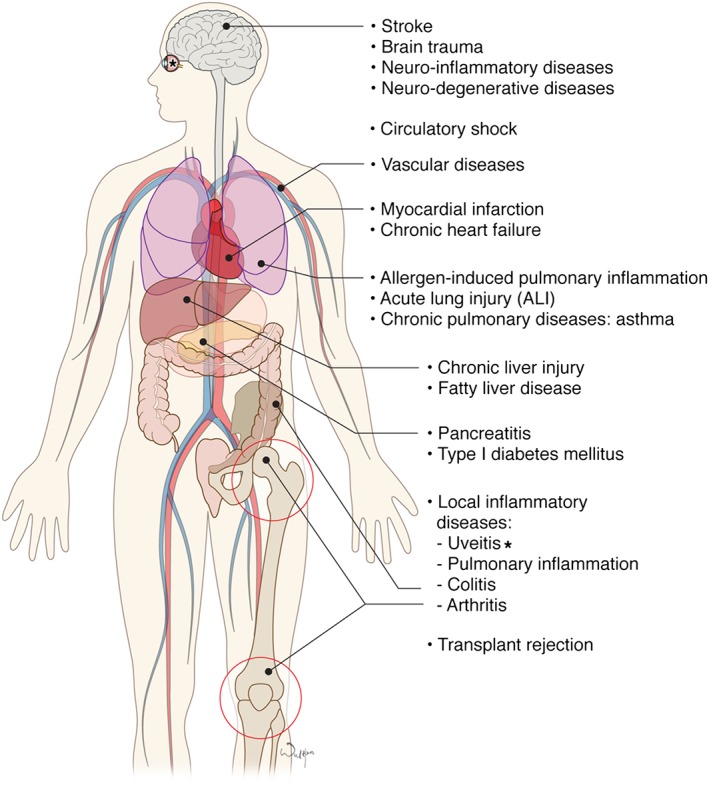
Pathogenetic role of PARP1 in various non‐oncological diseases. See Tables 3 and 4 for additional details.
Olaparib (Lynparza)– similar to a host of other PARP inhibitors that are currently in late‐stage clinical trials – has gone through the expected battery of preclinical safety studies, as well as the early stage human safety studies, even though the required preclinical ‘safety package’ for oncological indications is smaller than for many other disease indications. The product label for Lynparza states that the drug ‘was clastogenic in an in vitro chromosomal aberration assay in mammalian CHO cells and in an in vivo rat bone marrow micronucleus assay’ and that it ‘was teratogenic and caused embryo‐fetal toxicity in rats at exposures below those in patients receiving the recommended human dose of 400 mg twice daily’. Nevertheless, even with the above preclinical toxicity profile, olaparib appears to be well tolerated in cancer patients, nausea and occasional neutropenia being some of the most common side effects (e.g. Kaye et al., 2012; Bixel and Hays, 2015; Bao et al., 2016; Domchek et al., 2016; Ledermann et al., 2016; Leichman et al., 2016; Yonemori et al., 2016). Nevertheless, it must be noted that – to our knowledge – no studies have examined the effect of olaparib on DNA integrity or chromosomal stability in humans. In addition, to our knowledge, the PARP1‐dependent versus PARP1‐independent cellular actions of olaparib (e.g. on DNA stability or cellular viability) have not yet been thoroughly investigated, e.g. by comparing the effect of the drug in wild‐type vs. PARP1‐deficient cell systems.
One way to put the potential side effects of PARP inhibitors into perspective is by comparing their effects with other drugs currently used for non‐oncological indications. For instance, Lee et al. (2016) compared the effect of olaparib and veliparib on chromosomal stability with several classes of drugs, including methotrexate (currently used, in a chronic regimen, in the therapy of arthritis and psoriasis). Methotrexate (tested at 20 μM) increased chromosomal instability to a comparable degree as olaparib and veliparib (tested at 5–10 μM) (Lee et al., 2016). It should also be mentioned that many commonly used drugs have genotoxic effects on various standard toxicological tests (e.g. prazoles, salazines, olmesartan, theobromine) (Brambilla and Martelli, 2006; Brambilla et al., 2010; Brambilla et al., 2013). Examples where many of the currently used drugs (including methotrexate, but also some of the immunotherapies) are known to increase the risk of oncological diseases are chronic rheumatoid arthritis and psoriasis (Solomon et al., 2014; Van Lümig et al., 2015).
Based on the above considerations, one can conclude that genotoxicity (or a risk of genotoxicity) alone should not be a reason to exclude a study drug for testing in a non‐oncological indication. Another way of looking at the question is to consider the alternative therapeutic options. Would one be willing to test a novel, potentially life‐saving therapeutic intervention – even when there is some risk of potential adverse events down the line – if no other drugs were available for the therapy of that particular disease? Consider the parallel with cancer therapy, where therapeutic successes come at a price of increased incidence of malignant diseases in later life (Bhatia and Sklar, 2002; Armstrong et al., 2014).
Based on the above considerations, we conclude that the risk of genotoxicity, alone, should not be sufficient reason to exclude a study drug such as a PARP inhibitor for testing in a non‐oncological indication. On the other hand, it is imperative to minimize the risks and maximize the benefits by identifying the most suitable indications. What common‐sense criteria, then, should be established for our selection process? First of all, there should be preclinical data demonstrating the efficacy of PARP inhibition in clinically relevant preclinical models of disease (see also below). Second, it would be preferred if the selected indication had human data to confirm the activation of PARP in the target organ. Third, the duration of treatment with the PARP inhibitor should be as short as possible, in order to limit potential side effects. Fourth, the selected indication should be severe enough to justify an attempt for novel therapies, especially in light of the potential genotoxic ‘baggage’ that comes with PARP inhibition. Fifth, the existing therapeutic alternatives should be insufficient. Finally, the proposed clinical trial(s) should be logistically feasible.
Based on all of the above criteria (Tables 3 and 4 ), indications where the PARP inhibition would be given chronically and where various therapies (albeit often of insufficient efficacy) already exist (e.g. chronic local inflammatory diseases, vascular diseases, many forms of liver and pulmonary diseases) should be deprioritized. Myocardial infarction – even though it is supported by a vast body of preclinical data and even a small‐scale clinical study with INO‐1001 (Morrow et al., 2009) – should also be de‐prioritized, given the fact that most patients who are transported in time into qualified clinical centres are doing reasonably well with the current standard of therapy. Transplant rejection should also be initially excluded, because of the availability of drugs, which are effective in controlling transplant rejection. Type I diabetes should be excluded at the present stage: the logistics of diabetes prevention trials is very challenging; also, there was a lack of efficacy (Skyler and Type 1 Diabetes TrialNet Study Group, 2008; Simmons and Michels, 2014) of the nicotinamide‐based prevention trials conducted over the last two decades.
Thus, the acute indications, where we recommend repurposing and where the duration of the PARP inhibitor therapy would be expected to be short, are the following: acute ischaemic stroke and acute severe TBI (two acute neurological indications), septic shock, haemorrhagic shock, acute pancreatitis and severe ALI (also termed ARDS). In all of these conditions, many lines of preclinical data support the efficacy of PARP inhibition; the duration of treatment would be expected to be relatively short (days to weeks), the severity of the disease is high, the available specific therapeutic options are limited (stroke, ALI) or, in essence, non‐existent (circulatory shock, pancreatitis). The indication of acute brain injury may be expanded to also include acute spinal cord injury (e.g. in the context of trauma or thoracoabdominal aortic cross‐clamping); the indication of circulatory shock may be expanded to other forms of shock, where PARP inhibition has shown significant benefit in preclinical studies (e.g. haemorrhagic shock, a critical condition with high mortality, and so far, no treatment except from transfusions, and damage control). Haemorrhagic shock – in addition to stroke and TBI – may be prototypical indications for first‐responder/in‐ambulance trials, because these events occur outside the hospital, and the condition of the patients rapidly deteriorates during their transit time to hospital care. One final group of indications relates to the potential use of these agents to attenuate the organ damage induced by chemotherapeutic agents (Ali et al., 2011; Kim and Padanilam, 2011; Mukhopadhyay et al., 2011).
In addition to the acute indications discussed above, there are also a number of severe, debilitating, chronic indications that represent a high unmet need with no alternative therapeutic options (e.g. Parkinson's disease, multiple sclerosis or severe, untreatable forms of metabolic diseases including fibrotic liver, lung and kidney disease), which remain candidates for urgent repurposing, even though the administration of the PARP inhibitor would have to be chronic, and this may be associated with a risk of genotoxicity. In these indications, the currently available therapeutic options are extremely limited and of marginal efficacy. For indications of this type, treatment with PARP inhibitors may be considered in an intermittent fashion (i.e. with ‘drug holidays’). Alternatively, a lower dose of the PARP inhibitor may be attempted. In chronic studies, evaluation of the effects of the PARP inhibitor on DNA integrity may be incorporated into the trials and/or may be incorporated into the routine (e.g. annual) check‐up of these patients. Perhaps chronic PARP inhibitor trials may be delayed until more clinical data are available with PARP inhibitors in oncological indications and in acute non‐oncological indications. Such data (including, for instance, the presence or absence of secondary cancer development in cancer survivors treated with PARP inhibitors) may guide the decision with respect to chronic PARP inhibitor trials. Once sufficient safety data are available in the most severely affected patient populations, additional indications may be considered as well. For instance, one may consider testing low‐dose PARP inhibitor therapy in patients with mild‐to‐moderate or repeat TBI, in order to prevent the onset of subsequent neuroinflammatory or neurodegenerative processes.
We are well aware of the fact that many of disease indications proposed above are viewed as ‘pharmaceutical graveyard’ indications where some very spectacular therapeutic failures (e.g. Opal and Cross, 1999; Moretti et al., 2015; Gruenbaum et al., 2016; Standiford and Ward, 2016) have been encountered over recent decades, but there are also important lessons learned from these failed trials that could be used in the design of future trials. We are also aware of the fact that many of these diseases represent significant unmet medical needs, as well as considerable markets that should make them attractive for development by large pharmaceutical companies. As all PARP inhibitors currently in clinical development are novel chemical entities, the intellectual property status covers both oncological and non‐oncological diseases. In addition, with positive data in non‐oncological indications, label extension as well as additional intellectual property may be created.
The next steps
What, then, are the logical next steps in the field? Are there any activities necessary prior to embarking on clinical trials (with olaparib or rucaparib, or with other PARP inhibitors that are expected to be approved for oncological indications in the future)? First of all, it would be advantageous to generate preclinical efficacy data with the PARP inhibitor that is selected for the clinical trial (rather than relying on data with earlier classes of PARP inhibitors), because one must appreciate the fact that these compounds were primarily designed (and optimized) for cancer therapy, where cytotoxicity (mechanism‐based or perhaps mechanism‐independent, or a combination of the two) may be acceptable. In the context of repurposing them for non‐oncological indications, however, we expect to use the same drugs to elicit cytoprotective and/or anti‐inflammatory therapeutic actions. The preclinical studies should utilize clinically relevant models (typically, large animal models are considered more clinically predictable than rodent models), should include several dose levels (that are reflective of the human doses) and should include animals from both sexes and – if it reflects better the targeted patient population in the planned clinical studies – even aged animals or animals on the background of pre‐existing cardiovascular disease. Such animals are known to be more sensitive to injury than young and healthy animals, and it is likely that the pathophysiological pathways involved in their organ injury processes are not only quantitatively but also qualitatively different (Coletta et al., 2014; Starr and Saito, 2014; Ungvari and Sonntag, 2014).
Currently, the clinically approved PARP inhibitors are olaparib (Lynparza) and rucaparib (Rubraca), but once additional inhibitors are available, the choice of the compound will become important. Although the primary mode of all PARP inhibitors is identical (binding into the active site of PARP1 and thus inhibiting the binding of NAD+, preventing its cleavage to an ADP‐ribosyl unit and nicotinamide plus a proton, thereby blocking the formation of polyADP‐ribose), not all PARP inhibitors are ‘created equal’. They can have variable inhibitory effects on the PARP isoforms other than PARP1, some of them may be more prone to trapping PARP1 at the DNA replication forks than others, some of them may have mechanism‐independent actions (e.g. cytotoxicity), some of them have more off‐target effects on kinases than others, some of them have more inhibitory activity on members of the PARP enzyme family in addition to PARP1 (Wahlberg et al., 2012; Antolín and Mestres, 2014; Pommier et al., 2016). For non‐oncological indications, the safety profile of PARP inhibitor would be expected to be better if it had less PARP trapping activity and less mechanism‐independent cytotoxicity.
The issue of PARP1 inhibition versus effects on other PARP isoforms requires some additional discussion. Olaparib inhibits PARP1 with the highest potency, but it also inhibits PARP2 and other PARP isoforms, while veliparib is a selective inhibitor of PARP1 and PARP2 (and not of other PARP isoforms) (Knezevic et al., 2016; Thorsell et al., 2017). Although PARP1 plays a major role in the pathogenesis of the various non‐oncological diseases discussed in the current review (shown by the protection seen in PARP1‐deficient cells or animals), other PARP isoforms (most notably PARP2) may also contribute to the pathogenesis of some non‐oncological diseases (Popoff et al., 2002; Kofler et al., 2006; Li et al., 2010; Kamboj et al., 2013). Thus, inhibition of PARP2 (and perhaps other PARP isoforms) is likely to contribute to the efficacy of PARP inhibitors. At the same time, effects on PARP2 (or other PARP isoforms) may also affect the safety profile of the PARP inhibitor. As there are still many unknowns with respect to the roles of the minor PARP isoforms in the pathogenesis of both oncological and non‐oncological diseases, from the (practical) standpoint of repurposing, we recommend that we view this issue empirically and should focus on the relevant endpoints (i.e. safety of efficacy of olaparib and veliparib) in various non‐oncological diseases.
Not only the presence of the therapeutic effect of a PARP inhibitor should be established in non‐oncological indications, but the magnitude of this effect and the time window of administration are also very important, in comparison with, or on the background of state‐of‐the‐art therapies (when available). The efficacy of the PARP inhibitor should be comparable or better than any other therapies (or potential development candidates), targeting different pathways or mechanisms of the same disease. All of these issues should be carefully considered in relevant preclinical models of disease, which should be performed to the highest scientific rigour including state‐of‐the‐art blinding and randomization.
In this respect, it is encouraging that the existing body of data (with PARP1‐deficient mice and with various classes of PARP inhibitors) shows marked efficacy in the indications we recommend for repurposing (Tables 3 and 4); the preclinical body of efficacy data include large animal data in state‐of‐the‐art, translationally relevant models of stroke (Matsuura et al., 2011), acute neurological injury (Maier et al., 2007), circulatory shock (Goldfarb et al., 2002) and ALI (Iványi et al., 2003; Shimoda et al., 2003; Murakami et al., 2004). However, there are currently no reports with PARP inhibitors in large animal models of acute brain injury, pancreatitis or Parkinson's disease. Large animal studies – or whatever animal model is considered the most clinically predictable for the disease indication in question – remain to be confirmed, specifically with the choice compound selected for human therapeutic repurposing.
In addition to efficacy endpoints, the preclinical studies with the PARP inhibitor(s) selected for therapeutic repurposing should also incorporate evaluation of multiple parameters of DNA injury and chromosomal integrity, for instance, in circulating leukocytes and in relevant parenchymal tissues.
Another, more long‐term line of thought – in analogy with the BRCA/cancer indications for PARP inhibitors – relates to the identification of potential subsets of patients, where PARP inhibitors would be expected to be most efficacious. For instance, Daemen and colleagues have recently developed an algorithm to predict responsiveness of cancer cells to olaparib therapy, based on the transcriptional levels of seven genes involved in DNA repair pathways (Daemen et al., 2012) and development of similar predictive algorithms may also be feasible for non‐oncological diseases.
Once the clinical indication selection has been finalized, optimal clinical trial design should be implemented, considering issues such as dose selection, timing of administration and concomitant therapies. In addition to the carefully selected endpoints, pharmacokinetic and pharmacodynamic endpoints (to confirm target engagement) should be incorporated. Earlier trials have used measurement of PARP activity (Wang et al., 2015) and/or measurement of PARP‐dependent phosphorylation of histone H2AX in PBMCs ex vivo (Gojo et al., 2017) or the ability of patient plasma to inhibit PARP activity ex vivo (Morrow et al., 2009) to demonstrate that an effective dose of the inhibitor had been achieved. Instead of PBMCs, PARP activity in plucked eyebrow‐hair follicles has also been measured (Fong et al., 2009). When patient tissues are accessible, lowering of cellular NAD+ levels may also be potentially suitable to assess the degree of PARP activation and the effect of PARP inhibitors to restore NAD+. As with to the proposed preclinical studies, the clinical studies should also evaluate the effects of the PARP inhibitor on DNA and chromosome integrity.
In view of the importance of the BRCA/PARP interrelationship in DNA repair, should we consider excluding patients who harbour BRCA mutations (meaning that all potential study subjects would have to undergo rapid BRCA testing prior to the first dosing with the PARP inhibitor)? Considering that BRCA mutant subjects in their somatic cells are heterozygote BRCA mutants (which means that their DNA repair is not compromised) and the BRCA homozygote genotype, which can only be found in cancer cells, produces a loss‐of function mutation, heterozygous BRCA mutation does not confer HRR deficiency and sensitivity to PARP inhibitors – as demonstrated in several studies including experiments in BRCA2+/− mice treated with olaparib (Drew et al., 2011; Kobayashi et al., 2013; Lord and Ashworth, 2016), exclusion of BRCA mutant subjects for PARP inhibitor trials for non‐oncological diseases does not appear to be scientifically justified. At the same time, one should consider excluding patients with other (BRCA‐unrelated) known forms of hyperresponsiveness to DNA damage. What should also be examined, in future clinical trials, is whether the DNA integrity in the BRCA mutant subjects was affected differently from that in the rest of the patient population. However, meaningful data on this question can only be expected from fairly large trials, because the incidence of this mutation in the general population is only about 0.3%.
As far as additional inclusion and exclusion criteria are concerned: females of childbearing potential enrolled into the trial would have to undertake contraception. Male subjects in the trial should also use rigorous contraception. In the acute indications we propose for the first wave of repurposing, the severity of the indications is such that sexual activity of the subjects is unlikely. This issue would be more relevant in some of the first‐line chronic indications such as Parkinson's disease or fibrotic diseases. For safety reasons, initially, the trials should be conducted in adult populations, rather than paediatric or mixed populations.
The issue of sex difference of the protective effect of PARP inhibitors (as discussed earlier) presents an additional challenge of trial design. Because sex difference has not been consistently seen in all preclinical studies, and because the protective effect of PARP inhibitors can be reconstituted in ovariectomized animals (Mabley et al., 2005) that model postmenopausal female patients, the clinical trial should incorporate patients from both sexes, but should be powered sufficiently such that statistical power remains high enough after stratification into three groups: males, premenopausal females and postmenopausal females. The potential effect of hormonal contraception in females on the outcome should also be considered. Post hoc analysis may subsequently evaluate the efficacy of PARP inhibitor therapy, based, for instance, on disease subtype, disease severity, co‐morbidities and the presence or absence of concomitant therapies.
This analysis has, for the most part, focused on repurposing for acute indications. However, we should keep in mind that for the therapy of any patients with serious diseases, there is the consideration of the quality of life and the residual life expectancy and what balance the patient wishes. As discussed above, these choices are made on a daily basis in oncology, and considering many diseases have no disease modifying or preventative treatments (e.g. devastating neurodegenerative or neuroinflammatory diseases, or chronic metabolic diseases where the prognosis is poor and no therapeutic alternatives are known), we believe that these options should not be discarded for fear of a bad outcome, but investigated, monitored and addressed. Industry‐sponsored trials versus investigator‐initiated trials may be more feasible with different endpoints and may also present with different sets of challenges (Table 5). Although the complexities of testing PARP inhibitors for non‐oncological indications are quite substantial, it is hoped that this review – which incorporates the collective knowledge and scientific and medical judgement of many investigators active in the field of PARP – will stimulate renewed interest and further preclinical and clinical research and development activity in this area.
Table 5.
Potential clinical development pathways for repurposing PARP inhibitors for non‐oncological diseases
| Industry path | Academic path | |
|---|---|---|
| Mechanism | Company‐sponsored trials | Investigator‐initiated trials |
| Scale, Approach | Small, mid‐size, large trials. Large trials, targeting the largest commercial indication are feasible. Complex trial designs can be implemented. | Small, mid‐size trials; less complex design, more straightforward endpoints. (But NIH, VA is well suited to run even large‐scale trials.) |
| Results | Registration endpoints completed, Data published, Regulatory approval obtained; on‐label use of the PARP inhibitor. | Meaningful clinical endpoints completed, Data published; the results change of clinical practice, off‐label use of the PARP inhibitor. |
| Pharmaceutical marketing after successful trials | Permitted for on‐label use | Not permitted for off‐label use |
| Challenges | Attract industry interest to consider non‐oncological indications; consider trials in historically challenging indications | Obtain support (e.g. grants) for the trial; clinical trial infrastructure can be a challenge in an academic environment |
| Commercial considerations | The indication must be commercially attractive for the company | Minimal |
| Potential downsides | Trial may not show efficacy; many SAEs are expected in the trials because of the underlying disease; relatedness can be difficult to ascertain; SAEs noted in the trial endanger the already existing (approved) oncology indications | Trial may not show efficacy; unexpected adverse events in patients |
| Most suitable indications | Initial smaller‐scale trials, followed by large, multi‐centre trials are feasible. Indications which require large patient numbers and complex trial designs and survival endpoints (e.g. septic shock and, stroke studies). Chronic studies (e.g. Parkinson's disease). All other indications (ALI, TBI, pancreatitis) are also feasible. | Smaller, single‐centre trials are more feasible. Indications where signs of efficacy may be obtained in smaller trials (e.g. pancreatitis, ALI); focused trials of TBI, exploratory trials in septic shock focusing on biomarker endpoints. |
Author contributions
All authors contributed to writing and editing the paper and are listed in alphabetical order. C.S. prepared the first draft of the manuscript, coordinated the completion of the work with all co‐authors and finalized the paper.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by by P50 NS38377, P50 DA00266 and R37 NS67525 to T.M.D. and V.L.D. and P50 GM060338 to C.S. The work of I‐Hsun Wu for generating the illustrations is appreciated. The editorial assistance of Dr Anita Marton is also appreciated.
Berger, N. A. , Besson, V. C. , Boulares, A. H. , Bürkle, A. , Chiarugi, A. , Clark, R. S. , Curtin, N. J. , Cuzzocrea, S. , Dawson, T. M. , Dawson, V. L. , Haskó, G. , Liaudet, L. , Moroni, F. , Pacher, P. , Radermacher, P. , Salzman, A. L. , Snyder, S. H. , Soriano, F. G. , Strosznajder, R. P. , Sümegi, B. , Swanson, R. A. , and Szabo, C. (2018) Opportunities for the repurposing of PARP inhibitors for the therapy of non‐oncological diseases. British Journal of Pharmacology, 175: 192–222. doi: 10.1111/bph.13748.
References
- Alano CC, Kauppinen TM, Valls AV, Swanson RA (2006). Minocycline inhibits poly(ADP‐ribose) polymerase‐1 at nanomolar concentrations. Proc Natl Acad Sci U S A 103: 9685–9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010). NAD+ depletion is necessary and sufficient for poly(ADP‐ribose) polymerase‐1‐mediated neuronal death. J Neurosci 30: 2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinucci A, Gerlini G, Fossati S, Cipriani G, Ballerini C, Biagioli T et al. (2007). A key role for poly(ADP‐ribose) polymerase‐1 activity during human dendritic cell maturation. J Immunol 179: 305–312. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M, Kamjoo M, Thomas HD, Kyle S, Pavlovska I, Babur M et al. (2011). The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol Cancer Ther 10: 2320–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amé JC, Spenlehauer C, de Murcia G (2004). The PARP superfamily. Bioessays 26: 882–893. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M et al. (2006). Poly(ADP‐ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103: 18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolín AA, Mestres J (2014). Linking off‐target kinase pharmacology to the differential cellular effects observed among PARP inhibitors. Oncotarget 5: 3023–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GT, Kawashima T, Leisenring W (2014). Aging and risk of severe, disabling, life‐threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol 32: 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba X, Garg NJ (2011). Signaling mechanism of poly(ADP‐ribose) polymerase‐1 (PARP‐1) in inflammatory diseases. Am J Pathol 178: 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Nagy L, Fodor T, Liaudet L, Pacher P (2015). Poly(ADP‐ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol Metab 26: 75–83. [DOI] [PubMed] [Google Scholar]
- Banasik M, Stedeford T, Strosznajder RP (2012). Natural inhibitors of poly(ADP‐ribose) polymerase‐1. Mol Neurobiol 46: 55–63. [DOI] [PubMed] [Google Scholar]
- Bao Z, Xiong J, Li W, Chen Z, Shen H, Ying S (2015). Genomic instability in chronic airway inflammatory diseases. Biomed J 38: 117–124. [DOI] [PubMed] [Google Scholar]
- Bao Z, Cao C, Geng X, Tian B, Wu Y, Zhang C et al. (2016). Effectiveness and safety of poly (ADP‐ribose) polymerase inhibitors in cancer therapy: a systematic review and meta‐analysis. Oncotarget 7: 7629–7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskaite E, Jankevicius G, Ahel I (2015). Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP‐dependent protein ADP‐ribosylation. Mol Cell 58: 935–946. [DOI] [PubMed] [Google Scholar]
- Bartha E, Solti I, Kereskai L, Lantos J, Plozer E, Magyar K et al. (2009). PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc Res 83: 501–510. [DOI] [PubMed] [Google Scholar]
- Baxter P, Chen Y, Xu Y, Swanson RA (2014). Mitochondrial dysfunction induced by nuclear poly(ADP‐ribose) polymerase‐1: a treatable cause of cell death in stroke. Transl Stroke Res 5: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneke S, Bürkle A (2007). Poly(ADP‐ribosyl)ation in mammalian ageing. Nucleic Acids Res 35: 7456–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger NA, Berger SJ, Gerson SL (1987). DNA repair, ADP‐ribosylation and pyridine nucleotide metabolism as targets for cancer chemotherapy. Anticancer Drug Des 2: 203–209. [PubMed] [Google Scholar]
- Besson VC (2009). Drug targets for traumatic brain injury from poly(ADP‐ribose)polymerase pathway modulation. Br J Pharmacol 157: 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S, Sklar C (2002). Second cancers in survivors of childhood cancer. Nat Rev Cancer 2: 124–132. [DOI] [PubMed] [Google Scholar]
- Bixel K, Hays JL (2015). Olaparib in the management of ovarian cancer. Pharmgenomics Pers Med 8: 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, Todorova TT, Chang P (2015). RNA regulation by poly(ADP‐ribose) polymerases. Mol Cell 58: 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesten DM, de Vos‐Houben JM, Timmermans L, den Hartog GJ, Bast A, Hageman GJ (2013). Accelerated aging during chronic oxidative stress: a role for PARP‐1. Oxid Med Cell Longev 2013: 680414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek C, Morgan WF, Ong A, Cleaver JE (1984). Inhibition of malignant transformation in vitro by inhibitors of poly(ADP‐ribose) synthesis. Proc Natl Acad Sci U S A 81: 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla G, Martelli A (2006). Genotoxicity and carcinogenicity studies of antihypertensive agents. Mutat Res 612: 115–149. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Martelli A (2009). Update on genotoxicity and carcinogenicity testing of 472 marketed pharmaceuticals. Mutat Res 681: 209–229. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Mattioli F, Martelli A (2010). Genotoxic and carcinogenic effects of gastrointestinal drugs. Mutagenesis 25: 315–326. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Mattioli F, Robbiano L, Martelli A (2013). Genotoxicity and carcinogenicity studies of bronchodilators and antiasthma drugs. Basic Clin Pharmacol Toxicol 112: 302–313. [DOI] [PubMed] [Google Scholar]
- Brunyanszki A, Szczesny B, Virág L, Szabo C (2016). Mitochondrial poly(ADP‐ribose) polymerase: the wizard of Oz at work. Free Radic Biol Med 100: 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E et al. (2005). Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 434: 913–917. [DOI] [PubMed] [Google Scholar]
- Buege M, Mahajan PB (2015). Clinical trials of poly(ADP‐ribose) polymerase inhibitors for cancer therapy: a review. Rev Recent Clin Trials 10: 326–339. [DOI] [PubMed] [Google Scholar]
- Bürkle A, Brabeck C, Diefenbach J, Beneke S (2005). The emerging role of poly(ADP‐ribose) polymerase‐1 in longevity. Int J Biochem Cell Biol 37: 1043–1053. [DOI] [PubMed] [Google Scholar]
- Cantó C, Sauve AA, Bai P (2013). Crosstalk between poly(ADP‐ribose) polymerase and sirtuin enzymes. Mol Aspects Med 34: 1168–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavone L, Chiarugi A (2012). Targeting poly(ADP‐ribose) polymerase‐1 as a promising approach for immunomodulation in multiple sclerosis? Trends Mol Med 18: 92–100. [DOI] [PubMed] [Google Scholar]
- Chambon P, Weill JD, Mandel P (1963). Nicotinamide mononucleotide activation of new DNA‐dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun 11: 39–43. [DOI] [PubMed] [Google Scholar]
- Chatterjee PK, Patel NS, Sivarajah A, Kvale EO, Dugo L, Cuzzocrea S et al. (2003). GW274150, a potent and highly selective inhibitor of iNOS, reduces experimental renal ischemia/reperfusion injury. Kidney Int 63: 853–865. [DOI] [PubMed] [Google Scholar]
- Chiarugi A (2002). Poly(ADP‐ribose) polymerase: killer or conspirator? The 'suicide hypothesis' revisited. Trends Pharmacol Sci 23: 122–129. [DOI] [PubMed] [Google Scholar]
- Cohausz O, Malanga M, Boukamp P, Althaus FR, Bürkle A (2008). Telomere length regulation by poly(ADP‐ribose)polymerase‐1. Nucleic Acids Res 36: 6309–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen‐Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R et al. (2007). DNA‐independent PARP‐1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell 25: 297–308. [DOI] [PubMed] [Google Scholar]
- Coletta C, Módis K, Oláh G, Brunyánszki A, Herzig DS, Sherwood ER et al. (2014). Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care 18: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crafton SM, Bixel K, Hays JL (2016). PARP inhibition and gynecologic malignancies: a review of current literature and on‐going trials. Gynecol Oncol 142: 588–596. [DOI] [PubMed] [Google Scholar]
- Curtin NJ, Szabo C (2013). Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med 34: 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, Wolf DM, Korkola JE, Griffith OL, Frankum JR, Brough R et al. (2012). Cross‐platform pathway‐based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res Treat 135: 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos M, Schreiber V, Dantzer F (2012). The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol 84: 137–146. [DOI] [PubMed] [Google Scholar]
- Deeks ED (2015). Olaparib: first global approval. Drugs 75: 231–240. [DOI] [PubMed] [Google Scholar]
- Dhein S, Grassl M, Gerdom M, Vollroth M, Bakhtiary F, von Salisch S et al. (2015). Organ‐protective effects on the liver and kidney by minocycline in small piglets undergoing cardiopulonary bypass. Naunyn Schmiedebergs Arch Pharmacol 388: 663–676. [DOI] [PubMed] [Google Scholar]
- Domchek SM, Aghajanian C, Shapira‐Frommer R, Schmutzler RK, Audeh MW, Friedlander M et al. (2016). Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol 140: 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew Y (2015). The development of PARP inhibitors in ovarian cancer: from bench to bedside. Br J Cancer 113: S3–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S et al. (2011). Therapeutic potential of poly(ADP‐ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst 103: 334–346. [DOI] [PubMed] [Google Scholar]
- D'Souza D, Thomas IM, Das BC (1992). Effect of inhibitor of poly (ADP‐ribose) polymerase in blood lymphocyte cultures of untreated leprosy patients. Mutat Res 284: 251–255. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC et al. (2003). Intra‐mitochondrial poly‐ADP‐ribosylation contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem 278: 18426–18433. [DOI] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC et al. (2004). Innate gender‐based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem 279: 38563–38570. [DOI] [PubMed] [Google Scholar]
- Durkacz BW, Omidiji O, Gray DA, Shall S (1980). (ADP‐ribose)n participates in DNA excision repair. Nature 283: 593–596. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J et al. (1997). Poly(ADP‐ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095. [DOI] [PubMed] [Google Scholar]
- Esposito E, Mondello S, Di Paola R, Mazzon E, Italiano D, Paterniti I et al. (2011). Glutamine contributes to ameliorate inflammation after renal ischemia/reperfusion injury in rats. Naunyn Schmiedebergs Arch Pharmacol 383: 493–508. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Liaudet L (2005). Role of nitrosative stress and activation of poly(ADP‐ribose) polymerase‐1 in cardiovascular failure associated with septic and hemorrhagic shock. Curr Vasc Pharmacol 3: 293–299. [DOI] [PubMed] [Google Scholar]
- Faraone‐Mennella MR (2015). A new facet of ADP‐ribosylation reactions: SIRTs and PARPs interplay. Front Biosci (Landmark Ed) 20: 458–473. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, Dawson TM (2014). Parthanatos: mitochondrial‐linked mechanisms and therapeutic opportunities. Br J Pharmacol 171: 2000–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui‐Roelvink M et al. (2009). Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134. [DOI] [PubMed] [Google Scholar]
- Gagné JP, Hendzel MJ, Droit A, Poirier GG (2006). The expanding role of poly(ADP‐ribose) metabolism: current challenges and new perspectives. Curr Opin Cell Biol 18: 145–151. [DOI] [PubMed] [Google Scholar]
- García S, Conde C (2015). The role of poly(ADP‐ribose) polymerase‐1 in rheumatoid arthritis. Mediators Inflamm 2015: 837250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariani K, Ryu D, Menzies KJ, Yi HS, Stein S, Zhang H et al. (2017). Inhibiting poly ADP‐ribosylation increases fatty acid oxidation and protects against fatty liver disease. J Hepatol 66: 132–141. [DOI] [PubMed] [Google Scholar]
- Gerace E, Pellegrini‐Giampietro DE, Moroni F, Mannaioni G (2015). Poly(ADP‐ribose) polymerase 1 (PARP‐1) activation and Ca2+ permeable α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) channels in post‐ischemic brain damage: new therapeutic opportunities? CNS Neurol Disord Drug Targets 14: 636–646. [DOI] [PubMed] [Google Scholar]
- Ghonim MA, Pyakurel K, Ibba SV, Al‐Khami AA, Wang J, Rodriguez P et al. (2015a). PARP inhibition by olaparib or gene knockout blocks asthma‐like manifestation in mice by modulating CD4(+) T cell function. J Transl Med 13: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonim MA, Pyakurel K, Ibba SV, Wang J, Rodriguez P, Al‐Khami AA et al. (2015b). PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite‐induced disease in mice. Clin Sci (Lond) 129: 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giansanti V, Donà F, Tillhon M, Scovassi AI (2010). PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol 80: 1869–1877. [DOI] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL (2012). New insights into the molecular and cellular functions of poly(ADP‐ribose) and PARPs. Nat Rev Mol Cell Biol 13: 411–424. [DOI] [PubMed] [Google Scholar]
- Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H et al. (2016). Chemical genetic discovery of PARP targets reveals a role for PARP‐1 in transcription elongation. Science 353: 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad E, Zingarelli B, Salzman AL, Szabó C (1997). Protection by inhibition of poly (ADP‐ribose) synthetase against oxidant injury in cardiac myoblasts In vitro. J Mol Cell Cardiol 29: 2585–2597. [DOI] [PubMed] [Google Scholar]
- Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL et al. (2017). A phase 1 study of the PARP inhibitor veliparib in combination with temozolomide in acute myeloid leukemia. Clin Cancer Res 23: 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb RD, Marton A, Szabó E, Virág L, Salzman AL, Glock D (2002). Protective effect of a novel, potent inhibitor of poly(adenosine 5'‐diphosphate‐ribose) synthetase in a porcine model of severe bacterial sepsis. Crit Care Med 30: 974–980. [DOI] [PubMed] [Google Scholar]
- Griffin RJ, Curtin NJ, Newell DR, Golding BT, Durkacz BW, Calvert AH (1995). The role of inhibitors of poly(ADP‐ribose) polymerase as resistance‐modifying agents in cancer therapy. Biochimie 77: 408–422. [DOI] [PubMed] [Google Scholar]
- Gruenbaum SE, Zlotnik A, Gruenbaum BF, Hersey D, Bilotta F (2016). Pharmacologic neuroprotection for functional outcomes after traumatic brain injury: a systematic review of the clinical literature. CNS Drugs 30: 791–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen C, Palmier B, Plotkine M, Marchand‐Leroux C, Besson VC (2014). Neurological and histological consequences induced by in vivo cerebral oxidative stress: evidence for beneficial effects of SRT1720, a sirtuin 1 activator, and sirtuin 1‐mediated neuroprotective effects of poly(ADP‐ribose) polymerase inhibition. PLoS One 9: e87367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunji A, Uemura A, Tsutsumi M, Nozaki T, Kusuoka O, Omura K et al. (2006). Parp‐1 deficiency does not increase the frequency of tumors in the oral cavity and esophagus of ICR/129Sv mice by 4‐nitroquinoline 1‐oxide, a carcinogen producing bulky adducts. Cancer Lett 241: 87–92. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH (1999). Poly(ADP‐ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 96: 13978–13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M et al. (2004). PARP1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem 90: 1068–1075. [DOI] [PubMed] [Google Scholar]
- Hauser B, Gröger M, Ehrmann U, Albicini M, Brückner UB, Schelzig H et al. (2006). The parp‐1 inhibitor ino‐1001 facilitates hemodynamic stabilization without affecting DNA repair in porcine thoracic aortic cross‐clamping‐induced ischemia/reperfusion. Shock 25: 633–640. [DOI] [PubMed] [Google Scholar]
- Heller B, Bürkle A, Radons J, Fengler E, Jalowy A, Müller M et al. (1994). Analysis of oxygen radical toxicity in pancreatic islets at the single cell level. Biol Chem Hoppe Seyler 375: 597–602. [DOI] [PubMed] [Google Scholar]
- Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP et al. (2015). Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1‐deficient metaplastic breast cancer. Proc Natl Acad Sci U S A 112: 8409–8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch‐Nolte F (2010). Toward a unified nomenclature for mammalian ADP‐ribosyltransferases. Trends Biochem Sci 35: 208–219. [DOI] [PubMed] [Google Scholar]
- Ibba SV, Ghonim MA, Pyakurel K, Lammi MR, Mishra A, Boulares AH (2016). Potential of inducible nitric oxide synthase as a therapeutic target for allergen‐induced airway hyperresponsiveness: a critical connection to nitric oxide levels and PARP activity. Mediators Inflamm 2016: 1984703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Murphy CG, Doubrovina E, Jasin M, Moynahan ME (2016). PARP inhibitors in clinical use induce genomic instability in normal human cells. PLoS One 11: e0159341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iványi Z, Hauser B, Pittner A, Asfar P, Vassilev D, Nalos M et al. (2003). Systemic and hepatosplanchnic hemodynamic and metabolic effects of the PARP inhibitor PJ34 during hyperdynamic porcine endotoxemia. Shock 19: 415–421. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C (2005). Poly(ADP‐ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4: 421–440. [DOI] [PubMed] [Google Scholar]
- Jęśko H, Strosznajder RP (2016). Sirtuins and their interactions with transcription factors and poly(ADP‐ribose) polymerases. Folia Neuropathol 54: 212–233. [DOI] [PubMed] [Google Scholar]
- Ji Y, Tulin AV (2013). Post‐transcriptional regulation by poly(ADP‐ribosyl)ation of the RNA‐binding proteins. Int J Mol Sci 14: 16168–16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jog NR, Caricchio R (2013). Differential regulation of cell death programs in males and females by poly (ADP‐ribose) polymerase‐1 and 17β estradiol. Cell Death Dis 4: e758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jog NR, Dinnall JA, Gallucci S, Madaio MP, Caricchio R (2009). Poly(ADP‐ribose) polymerase‐1 regulates the progression of autoimmune nephritis in males by inducing necrotic cell death and modulating inflammation. J Immunol 182: 7297–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubin T, Kadam A, Gani AR, Singh M, Dwivedi M, Begum R (2017). Poly ADP‐ribose polymerase‐1: beyond transcription and towards differentiation. Semin Cell Dev Biol 63: 167–179. [DOI] [PubMed] [Google Scholar]
- Kamboj A, Lu P, Cossoy MB, Stobart JL, Dolhun BA, Kauppinen TM et al. (2013). Poly(ADP‐ribose) polymerase 2 contributes to neuroinflammation and neurological dysfunction in mouse experimental autoimmune encephalomyelitis. J Neuroinflammation 10: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor K, Singla E, Sahu B, Naura AS (2015). PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol Cell Biochem 400: 153–162. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA (2007). The role of poly(ADP‐ribose) polymerase‐1 in CNS disease. Neuroscience 145: 1267–1272. [DOI] [PubMed] [Google Scholar]
- Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY et al. (2012). Phase II, open‐label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP‐ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol 30: 372–379. [DOI] [PubMed] [Google Scholar]
- Kim J, Padanilam BJ (2011). Loss of poly(ADP‐ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am J Physiol Renal Physiol 301: F450–F459. [DOI] [PubMed] [Google Scholar]
- Kim SH, Engelhardt JI, Henkel JS, Siklós L, Soós J, Goodman C et al. (2004). Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology 62: 319–322. [DOI] [PubMed] [Google Scholar]
- Knezevic CE, Wright G, Remsing Rix LL, Kim W, Kuenzi BM, Luo Y et al. (2016). Proteome‐wide profiling of clinical PARP inhibitors reveals compound‐specific secondary targets. Cell Chem Biol 23: 1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Ohno S, Sasaki Y, Matsuura M (2013). Hereditary breast and ovarian cancer susceptibility genes (review). Oncol Rep 30: 1019–1029. [DOI] [PubMed] [Google Scholar]
- Kofler J, Otsuka T, Zhang Z, Noppens R, Grafe MR, Koh DW et al. (2006). Differential effect of PARP‐2 deletion on brain injury after focal and global cerebral ischemia. J Cereb Blood Flow Metab 26: 135–141. [DOI] [PubMed] [Google Scholar]
- Koh DW, Dawson TM, Dawson VL (2005). Poly(ADP‐ribosyl)ation regulation of life and death in the nervous system. Cell Mol Life Sci 62: 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komjáti K, Besson VC, Szabó C (2005). Poly (adp‐ribose) polymerase inhibitors as potential therapeutic agents in stroke and neurotrauma. Curr Drug Targets CNS Neurol Disord 4: 179–194. [DOI] [PubMed] [Google Scholar]
- Konecny GE, Kristeleit RS (2016). PARP inhibitors for BRCA1/2‐mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer 115: 1157–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL (2015). PARPs and ADP‐Ribosylation: 50 Years … and Counting. Mol Cell 58: 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, Hottiger MO (2013). PARP‐1 and gene regulation: progress and puzzles. Mol Aspects Med 34: 1109–1123. [DOI] [PubMed] [Google Scholar]
- Krietsch J, Rouleau M, Pic É, Ethier C, Dawson TM, Dawson VL et al. (2013). Reprogramming cellular events by poly(ADP‐ribose)‐binding proteins. Mol Aspects Med 34: 1066–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM et al. (2008). Identification of poly‐ADP‐ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem 104: 1700–1711. [DOI] [PubMed] [Google Scholar]
- Laudisi F, Sambucci M, Pioli C (2011). Poly (ADP‐ribose) polymerase‐1 (PARP‐1) as immune regulator. Endocr Metab Immune Disord Drug Targets 11: 326–333. [DOI] [PubMed] [Google Scholar]
- Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G et al. (2016). Overall survival in patients with platinum‐sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Oncol 17: 1579–1589. [DOI] [PubMed] [Google Scholar]
- Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D et al. (2013). Parthanatos mediates AIMP2‐activated age‐dependent dopaminergic neuronal loss. Nat Neurosci 16: 1392–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Ledermann JA, Kohn EC (2014). PARP inhibitors for BRCA1/2 mutation‐associated and BRCA‐like malignancies. Ann Oncol 25: 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Lee NC, Kouprina N, Kim JH, Kagansky A, Bates S et al. (2016). Effects of anticancer drugs on chromosome instability and new clinical implications for tumor‐suppressing therapies. Cancer Res 76: 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann S, Costa AC, Celardo I, Loh SH, Martins LM (2016). Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson's disease. Cell Death Dis 7: e2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichman L, Groshen S, O'Neil BH, Messersmith W, Berlin J, Chan E et al. (2016). Phase II study of olaparib (AZD‐2281) after standard systemic therapies for disseminated colorectal cancer. Oncologist 21: 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Chen J (2014). ADP‐ribosylation: activation, recognition, and removal. Mol Cells 37: 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McCullough LD (2009). Sex differences in minocycline‐induced neuroprotection after experimental stroke. J Cereb Blood Flow Metab 29: 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Klaus JA, Zhang J, Xu Z, Kibler KK, Andrabi SA et al. (2010). Contributions of poly(ADP‐ribose) polymerase‐1 and ‐2 to nuclear translocation of apoptosis‐inducing factor and injury from focal cerebral ischemia. J Neurochem 113: 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Luo C, Chowdhury S, Gao ZH, Liu JL (2013). Parp1 deficient mice are protected from streptozotocin‐induced diabetes but not caerulein‐induced pancreatitis, independent of the induction of Reg family genes. Regul Pept 186: 83–91. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Szabó G, Szabó C (2003). Oxidative stress and regional ischemia‐reperfusion injury: the peroxynitrite‐poly(ADP‐ribose) polymerase connection. Coron Artery Dis 14: 115–122. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2016). BRCAness revisited. Nat Rev Cancer 16: 110–120. [DOI] [PubMed] [Google Scholar]
- Lucarini L, Durante M, Lanzi C, Pini A, Boccalini G, Calosi L et al. (2016). HYDAMTIQ, a selective PARP‐1 inhibitor, improves bleomycin‐induced lung fibrosis by dampening the TGF‐β/SMAD signalling pathway. J Cell Mol Med 20: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo B, Trusolino L (2014). Inhibition of poly(ADP‐ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta 1846: 201–215. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Horváth EM, Murthy KG, Zsengellér Z, Vaslin A, Benko R et al. (2005). Gender differences in the endotoxin‐induced inflammatory and vascular responses: potential role of poly(ADP‐ribose) polymerase activation. J Pharmacol Exp Ther 315: 812–820. [DOI] [PubMed] [Google Scholar]
- Maier C, Scheuerle A, Hauser B, Schelzig H, Szabó C, Radermacher P et al. (2007). The selective poly(ADP)ribose‐polymerase 1 inhibitor INO1001 reduces spinal cord injury during porcine aortic cross‐clamping‐induced ischemia/reperfusion injury. Intensive Care Med 33: 845–850. [DOI] [PubMed] [Google Scholar]
- Mangerich A, Bürkle A (2012). Pleiotropic cellular functions of PARP1 in longevity and aging: genome maintenance meets inflammation. Oxid Med Cell Longev 2012: 321653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Bosch N, Iglesias M, Munné‐Collado J, Martínez‐Cáceres C, Moreno M, Guerra C et al. (2014). Parp‐1 genetic ablation in Ela‐myc mice unveils novel roles for Parp‐1 in pancreatic cancer. J Pathol 234: 214–227. [DOI] [PubMed] [Google Scholar]
- Martin‐Hernandez K, Rodriguez‐Vargas JM, Schreiber V, Dantzer F (2017). Expanding functions of ADP‐ribosylation in the maintenance of genome integrity. Semin Cell Dev Biol 63: 92–101. [DOI] [PubMed] [Google Scholar]
- Martin‐Oliva D, O'Valle F, Munoz‐Gamez JA, Valenzuela MT, Nunez MI, Aguilar M et al. (2004). Crosstalk between PARP‐1 and NF‐nB modulates the promotion of mskin neoplasia. Oncogene 23: 5275–5283. [DOI] [PubMed] [Google Scholar]
- Martin‐Oliva D, Aguilar‐Quesada R, O'Valle F, Munoz‐Gamez JA, Martinez‐Romero R, del Moral RG (2006). Inhibition of poly(ADP‐ribose) polymerase modulates tumor‐related gene expression, including hypoxia‐inducible factor‐1 activation, during skin carcinogenesis. Cancer Res 66: 5744–5756. [DOI] [PubMed] [Google Scholar]
- Mashimo M, Kato J, Moss J (2014). Structure and function of the ARH family of ADP‐ribosyl‐acceptor hydrolases. DNA Repair (Amst) 23: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani M, Fujimori H (2013). Poly(ADP‐ribosyl)ation in carcinogenesis. Mol Aspects Med 34: 1202–1216. [DOI] [PubMed] [Google Scholar]
- Masutani M, Nakagama H, Sugimura T (2003). Poly (ADP‐ribose) and carcinogenesis. Genes Chromosomes Cancer 38: 339–348. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Egi Y, Yuki S, Horikawa T, Satoh H, Akira T (2011). MP‐124, a novel poly(ADP‐ribose) polymerase‐1 (PARP‐1) inhibitor, ameliorates ischemic brain damage in a non‐human primate model. Brain Res 1410: 122–131. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD (2005). Ischemic nitric oxide and poly (ADP‐ribose) polymerase‐1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab 25: 502–512. [DOI] [PubMed] [Google Scholar]
- Moretti A, Ferrari F, Villa RF (2015). Neuroprotection for ischaemic stroke: current status and challenges. Pharmacol Ther 146: 23–34. [DOI] [PubMed] [Google Scholar]
- Moroni F (2008). Poly(ADP‐ribose)polymerase 1 (PARP‐1) and postischemic brain damage. Curr Opin Pharmacol 8: 96–103. [DOI] [PubMed] [Google Scholar]
- Moroni F, Cozzi A, Chiarugi A, Formentini L, Camaioni E, Pellegrini‐Giampietro DE et al. (2012). Long‐lasting neuroprotection and neurological improvement in stroke models with new, potent and brain permeable inhibitors of poly(ADP‐ribose) polymerase. Br J Pharmacol 165: 1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DA, Brickman CM, Murphy SA, Baran K, Krakover R, Dauerman H et al. (2009). A randomized, placebo‐controlled trial to evaluate the tolerability, safety, pharmacokinetics, and pharmacodynamics of a potent inhibitor of poly(ADP‐ribose) polymerase (INO‐1001) in patients with ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of the TIMI 37 trial. J Thromb Thrombolysis 27: 359–364. [DOI] [PubMed] [Google Scholar]
- Mota RA, Sánchez‐Bueno F, Saenz L, Hernández‐Espinosa D, Jimeno J, Tornel PL et al. (2005). Inhibition of poly(ADP‐ribose) polymerase attenuates the severity of acute pancreatitis and associated lung injury. Lab Invest 85: 1250–1262. [DOI] [PubMed] [Google Scholar]
- Mota R, Sánchez‐Bueno F, Berenguer‐Pina JJ, Hernández‐Espinosa D, Parrilla P, Yélamos J (2007). Therapeutic treatment with poly(ADP‐ribose) polymerase inhibitors attenuates the severity of acute pancreatitis and associated liver and lung injury. Br J Pharmacol 151: 998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C et al. (2013). The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154: 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horváth B, Kechrid M, Tanchian G, Rajesh M, Naura AS et al. (2011). Poly(ADP‐ribose) polymerase‐1 is a key mediator of cisplatin‐induced kidney inflammation and injury. Free Radic Biol Med 51: 1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Cao Z, Horváth B, Park O, Wang H et al. (2014). Poly (ADP‐ribose) polymerase‐1 is a key mediator of liver inflammation and fibrosis. Hepatology 59: 1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horváth B, Rajesh M, Varga VZ, Gariani K, Ryu D et al. (2017). PARP inhibition protects against alcoholic and non‐alcoholic steatohepatitis. J Hepatol 66: 589–600. [DOI] [PubMed] [Google Scholar]
- Murakami K, Enkhbaatar P, Shimoda K, Cox RA, Burke AS, Hawkins HK et al. (2004). Inhibition of poly (ADP‐ribose) polymerase attenuates acute lung injury in an ovine model of sepsis. Shock 21: 126–133. [DOI] [PubMed] [Google Scholar]
- Nabavi SF, Sureda A, Habtemariam S, Nabavi SM (2015). Ginsenoside Rd and ischemic stroke; a short review of literatures. J Ginseng Res 39: 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naura AS, Kim H, Ju J, Rodriguez PC, Jordan J, Catling AD et al. (2013). Minocycline blocks asthma‐associated inflammation in part by interfering with the T cell receptor‐nuclear factor κB‐GATA‐3‐IL‐4 axis without a prominent effect on poly(ADP‐ribose) polymerase. J Biol Chem 288: 1458–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki T, Fujihara H, Watanabe M, Tsutsumi M, Nakamoto K, Kusuoka O et al. (2003). Parp‐1 deficiency implicated in colon and liver tumorigenesis induced by azoxymethane. Cancer Sci 94: 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Masutani M, Kato K, Tang M, Kamada N, Suzuki H et al. (2006). Parp‐1 deficiency does not enhance liver carcinogenesis induced by 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in mice. Cancer Lett 236: 32–38. [DOI] [PubMed] [Google Scholar]
- Oláh G, Szczesny B, Brunyánszki A, López‐García IA, Gerö D, Radák Z et al. (2015). Differentiation‐associated downregulation of poly(ADP‐ribose) polymerase‐1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS One 10: e0134227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opal SM, Cross AS (1999). Clinical trials for severe sepsis. Past failures, and future hopes. Infect Dis Clin North Am 13: 285–297. [DOI] [PubMed] [Google Scholar]
- O'Sullivan Coyne G, Chen A, Kummar S (2015). Delivering on the promise: poly ADP ribose polymerase inhibition as targeted anticancer therapy. Curr Opin Oncol 27: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Valle F, Gomez‐Morales M, Del Moral RM, Seron D, Moreso F, Osuna A et al. (2007). Poly(ADP‐ribose) polymerase expression in kidney transplantation: from alfa (alpha) to omega (omega). Transplant Proc 39: 2099–2101. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C (2008). Role of the peroxynitrite‐poly(ADP‐ribose) polymerase pathway in human disease. Am J Pathol 173: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes EE, Kennedy RD (2016). Clinical application of poly(ADP‐ribose) polymerase inhibitors in high‐grade serous ovarian cancer. Oncologist 21: 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal JM, Ellenberger T (2015). The rise and fall of poly(ADP‐ribose): an enzymatic perspective. DNA Repair (Amst) 32: 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pears CJ, Couto CA, Wang HY, Borer C, Kiely R, Lakin ND (2012). The role of ADP‐ribosylation in regulating DNA double‐strand break repair. Cell Cycle 11: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TD (2010). Small‐molecule PARP modulators – current status and future therapeutic potential. Curr Opin Drug Discov Devel 13: 577–586. [PubMed] [Google Scholar]
- Pic E, Gagné JP, Poirier GG (2011). Mass spectrometry‐based functional proteomics of poly(ADP‐ribose) polymerase‐1. Expert Rev Proteomics 8: 759–774. [DOI] [PubMed] [Google Scholar]
- Pieper AA, Verma A, Zhang J, Snyder SH (1999). Poly (ADP‐ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 20: 171–181. [DOI] [PubMed] [Google Scholar]
- Pillai JB, Russell HM, Raman J, Jeevanandam V, Gupta MP (2005). Increased expression of poly(ADP‐ribose) polymerase‐1 contributes to caspase‐independent myocyte cell death during heart failure. Am J Physiol Heart Circ Physiol 288: H486–H496. [DOI] [PubMed] [Google Scholar]
- Pirinen E, Cantó C, Jo YS, Morato L, Zhang H, Menzies KJ et al. (2014). Pharmacological Inhibition of poly(ADP‐ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab 19: 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskunova TS, Yurova MN, Ovsyannikov AI, Semenchenko AV, Zabezhinski MA, Popovich IG et al. (2008). Deficiency in poly(ADP‐ribose) polymerase‐1 (PARP‐1) accelerates aging and spontaneous carcinogenesis in mice. Curr Gerontol Geriatr Res 2008: 754190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O'Connor MJ, de Bono J (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 8: 362ps17. [DOI] [PubMed] [Google Scholar]
- Popoff I, Jijon H, Monia B, Tavernini M, Ma M, McKay R et al. (2002). Antisense oligonucleotides to poly(ADP‐ribose) polymerase‐2 ameliorate colitis in interleukin‐10‐deficient mice. J Pharmacol Exp Ther 303: 1145–1154. [DOI] [PubMed] [Google Scholar]
- Posavec Marjanović M, Crawford K, Ahel I (2017). PARP, transcription and chromatin modeling. Semin Cell Dev Biol 63: 102–113. [DOI] [PubMed] [Google Scholar]
- Ratner ES, Sartorelli AC, Lin ZP (2012). Poly (ADP‐ribose) polymerase inhibitors: on the horizon of tailored and personalized therapies for epithelial ovarian cancer. Curr Opin Oncol 24: 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffell JL, Lord CJ, Ashworth A (2012). Tankyrase‐targeted therapeutics: expanding opportunities in the PARP family. Nat Rev Drug Discov 11: 923–936. [DOI] [PubMed] [Google Scholar]
- Rom S, Zuluaga‐Ramirez V, Dykstra H, Reichenbach NL, Ramirez SH, Persidsky Y (2015). Poly(ADP‐ribose) polymerase‐1 inhibition in brain endothelium protects the blood‐brain barrier under physiologic and neuroinflammatory conditions. J Cereb Blood Flow Metab 35: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom S, Zuluaga‐Ramirez V, Reichenbach NL, Dykstra H, Gajghate S, Pacher P et al. (2016). PARP inhibition in leukocytes diminishes inflammation via effects on integrins/cytoskeleton and protects the blood‐brain barrier. J Neuroinflammation 13: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Kim DS, Kraus WL (2015). New facets in the regulation of gene expression by ADP‐ribosylation and poly(ADP‐ribose) polymerases. Chem Rev 115: 2453–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salluzzo MG, Cosentino FI, Romano C, Scillato F, Morale MC, Rando RG et al. (2016). Poly (ADP‐ribose) polymerase‐1 (PARP‐1) ‐410C/T polymorphism in Sicilian patients with Parkinson's disease. J Neurol Sci 363: 95–96. [DOI] [PubMed] [Google Scholar]
- Schiewer MJ, Knudsen KE (2014). Transcriptional roles of PARP1 in cancer. Mol Cancer Res 12: 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraufstatter IU, Hyslop PA, Hinshaw DB, Spragg RG, Sklar LA, Cochrane CG (1986). Hydrogen peroxide‐induced injury of cells and its prevention by inhibitors of poly(ADP‐ribose) polymerase. Proc Natl Acad Sci U S A 83: 4908–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G (2006). Poly(ADP‐ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7: 517–528. [DOI] [PubMed] [Google Scholar]
- Schwartz JL, Morgan WF, Wolff S (1984). Reduction of sister chromatid exchange frequency with time after mutagen exposure in Chinese hamster ovary cells in the presence of 3‐aminobenzamide. Environ Mutagen 6: 203–210. [DOI] [PubMed] [Google Scholar]
- Shall S (1983). ADP‐ribosylation, DNA repair, cell differentiation and cancer. Princess Takamatsu Symp 13: 3–25. [PubMed] [Google Scholar]
- Shall S, de Murcia G (2000). Poly(ADP‐ribose) polymerase‐1: what have we learned from the deficient mouse model? Mutat Res 460: 1–15. [DOI] [PubMed] [Google Scholar]
- Shilovsky GA, Khokhlov AN, Shram SI (2013). The protein poly(ADP‐ribosyl)ation system: its role in genome stability and lifespan determination. Biochemistry (Mosc) 78: 433–444. [DOI] [PubMed] [Google Scholar]
- Shimoda K, Murakami K, Enkhbaatar P, Traber LD, Cox RA, Hawkins HK et al. (2003). Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am J Physiol Lung Cell Mol Physiol 285: L240–L249. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y, Tanaka Y, Kato M, Miwa M, Sugimura T (1983). Effect of poly(ADP‐ribose) polymerase inhibitors on the frequency of sister‐chromatid exchanges in Bloom syndrome cells. Mutat Res 122: 223–228. [DOI] [PubMed] [Google Scholar]
- Simbulan‐Rosenthal CM, Rosenthal DS, Luo D, Li JH, Zhang J, Smulsom ME (2001). Inhibition of poly(ADP‐ribose) polymerase activity is insufficient to induce tetrapolidity. Nucl Acid Res 29: 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons K, Michels AW (2014). Lessons from type 1 diabetes for understanding natural history and prevention of autoimmune disease. Rheum Dis Clin North Am 40: 797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JL, Berger SJ, Berger NA (1983). Poly(ADP‐ribose) polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5'‐triphosphate pools in DNA‐damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochem 22: 5188–5194. [DOI] [PubMed] [Google Scholar]
- Sistigu A, Manic G, Obrist F, Vitale I (2015). Trial watch – inhibiting PARP enzymes for anticancer therapy. Mol Cell Oncol 3: e1053594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skyler JS, Type 1 Diabetes TrialNet Study Group (2008). Update on worldwide efforts to prevent type 1 diabetes. Ann N Y Acad Sci 1150: 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder RD, Green JW (2001). A review of the genotoxicity of marketed pharmaceuticals. Mutat Res 488: 151–169. [DOI] [PubMed] [Google Scholar]
- Solomon DH, Kremer JM, Fisher M, Curtis JR, Furer V, Harrold LR et al. (2014). Comparative cancer risk associated with methotrexate, other non‐biologic and biologic disease‐modifying anti‐rheumatic drugs. Semin Arthritis Rheum 43: 489–497. [DOI] [PubMed] [Google Scholar]
- Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M (2015). An update on PARP inhibitors‐‐moving to the adjuvant setting. Nat Rev Clin Oncol 12: 27–41. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Virág L, Jagtap P, Szabó E, Mabley JG, Liaudet L et al. (2001). Diabetic endothelial dysfunction: the role of poly(ADP‐ribose) polymerase activation. Nat Med 7: 108–113. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Liaudet L, Szabó E, Virág L, Mabley JG, Pacher P et al. (2002). Resistance to acute septic peritonitis in poly(ADP‐ribose) polymerase‐1‐deficient mice. Shock 17: 286–292. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram CS, Jangra A, Kasala ER, Bodduluru LN, Bezbaruah BK (2014). Targeting poly(ADP‐ribose)polymerase1 in neurological diseases: a promising trove for new pharmacological interventions to enter clinical translation. Neurochem Int 76: 70–81. [DOI] [PubMed] [Google Scholar]
- Standiford TJ, Ward PA (2016). Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl Res 167: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr ME, Saito H (2014). Sepsis in old age: review of human and animal studies. Aging Dis 5: 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, Kabadi SV, Hanscom M, Byrnes KR et al. (2014). PARP‐1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma 31: 758–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB (2010). Poly(ADP‐ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol 41: 187–196. [DOI] [PubMed] [Google Scholar]
- Szabó C (2005). Roles of poly(ADP‐ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res 52: 60–71. [DOI] [PubMed] [Google Scholar]
- Szabó C, Módis K (2010). Pathophysiological roles of peroxynitrite in circulatory shock. Shock 34: S4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Zingarelli B, O'Connor M, Salzman AL (1996). DNA strand breakage, activation of poly (ADP‐ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A 93: 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallis M, Morra R, Barkauskaite E, Ahel I (2014). Poly(ADP‐ribosyl)ation in regulation of chromatin structure and the DNA damage response. Chromosoma 123: 79–90. [DOI] [PubMed] [Google Scholar]
- Tao R, Kim SH, Honbo N, Karliner JS, Alano CC (2010). Minocycline protects cardiac myocytes against simulated ischemia–reperfusion injury by inhibiting poly(ADP‐ribose) polymerase‐1. J Cardiovasc Pharmacol 56: 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Chen X, Hao S, Hou Z, Lu T, Sun M et al. (2015). Protective actions of PJ34, a poly(ADP‐ribose)polymerase inhibitor, on the blood‐brain barrier after traumatic brain injury in mice. Neuroscience 291: 26–36. [DOI] [PubMed] [Google Scholar]
- Teng F, Zhu L, Su J, Zhang X, Li N, Nie Z et al. (2016). Neuroprotective effects of poly(ADP‐ribose)polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem Res 41: 1516–1526. [DOI] [PubMed] [Google Scholar]
- Ter Brugge P, Kristel PP, van der Burg E, Boon U, de Maaker M, Lips E et al. (2016). Mechanisms of therapy resistance in patient‐derived xenograft models of BRCA1‐deficient breast cancer. J Natl Cancer Inst 108: djw148. [DOI] [PubMed] [Google Scholar]
- Thomas C, Tulin AV (2013). Poly‐ADP‐ribose polymerase: machinery for nuclear processes. Mol Aspects Med 34: 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell AG, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L et al. (2017). Structural basis for potency and promiscuity in poly(ADP‐ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem 60: 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C, Kim E‐H, Royce DB, Williams CR, Collins RM, Risingsong R et al. (2014). The PARP inhibitors, veliparib and olaparib, are effective chemopreventive agents for delaying mammary tumor development in BRCA1‐deficient mice. Cancer Prev Res 7: 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toller IM, Altmeyer M, Kohler E, Hottiger MO, Müller A (2010). Inhibition of ADP ribosylation prevents and cures helicobacter‐induced gastric preneoplasia. Cancer Res 70: 5912–5922. [DOI] [PubMed] [Google Scholar]
- Tong WM, Cortes U, Wang ZQ (2001). Poly(ADP‐ribose) polymerase: a guardian angel protecting the genome and suppressing tumorigenesis. Biochim Biophys Acta 1552: 27–33. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Sonntag WE (2014). Brain and cerebrovascular aging – new mechanisms and insights. J Gerontol A Biol Sci Med Sci 69: 1307–1310. [DOI] [PubMed] [Google Scholar]
- Vagnerova K, Liu K, Ardeshiri A, Cheng J, Murphy SJ, Hurn PD et al. (2010). Poly (ADP‐ribose) polymerase‐1 initiated neuronal cell death pathway – do androgens matter? Neuroscience 166: 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lümig PP, Menting SP, van den Reek JM, Spuls PI, van Riel PL, van de Kerkhof PC et al. (2015). An increased risk of non‐melanoma skin cancer during TNF‐inhibitor treatment in psoriasis patients compared to rheumatoid arthritis patients probably relates to disease‐related factors. J Eur Acad Dermatol Venereol 29: 752–760. [DOI] [PubMed] [Google Scholar]
- Virág L, Szabo C (2002). The therapeutic potential of poly(ADP‐ribose) polymerase inhibitors. Pharmacol Rev 54: 375–429. [DOI] [PubMed] [Google Scholar]
- Virág L, Salzman AL, Szabó C (1998). Poly(ADP‐ribose) synthetase activation mediates mitochondrial injury during oxidant‐induced cell death. J Immunol 161: 3753–3759. [PubMed] [Google Scholar]
- Virág L, Robaszkiewicz A, Rodriguez‐Vargas JM, Oliver FJ (2013). Poly(ADP‐ribose) signaling in cell death. Mol Aspects Med 34: 1153–1167. [DOI] [PubMed] [Google Scholar]
- Vodenicharov MD, Sallmann FR, Satoh MS, Poirier GG (2000). Base excision repair is efficient in cells lacking poly(ADP‐ribose) polymerase 1. Nucleic Acids Res 28: 3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG et al. (2012). Family wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol 30: 283–288. [DOI] [PubMed] [Google Scholar]
- Wang DD, Li C, Sun W, Zhang S, Shalinsky DR, Kern KA et al. (2015). PARP activity in peripheral blood lymphocytes as a predictive biomarker for PARP inhibition in tumor tissues – a population pharmacokinetic/pharmacodynamic analysis of rucaparib. Clin Pharmacol Drug Dev 4: 89–98. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Y, Parapanov R, Abdelnour E, Gronchi F, Perentes JY et al. (2016). Pharmacological reconditioning of marginal donor rat lungs using inhibitors of peroxynitrite and poly (ADP‐ribose) polymerase during ex vivo lung perfusion. Transplant 100: 1465–1473. [DOI] [PubMed] [Google Scholar]
- Wei H, Yu X (2016). Functions of PARylation in DNA damage repair pathways. Genom Proteom Bioinform 14: 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidele K, Kunzmann A, Schmitz M, Beneke S, Bürkle A (2010). Ex vivo supplementation with nicotinic acid enhances cellular poly(ADP‐ribosyl)ation and improves cell viability in human peripheral blood mononuclear cells. Biochem Pharmacol 80: 1103–1112. [DOI] [PubMed] [Google Scholar]
- Wright RH, Lioutas A, Le Dily F, Soronellas D, Pohl A, Bonet J et al. (2016). ADP‐ribose‐derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 352: 1221–1225. [DOI] [PubMed] [Google Scholar]
- Wu Y, Chen Y, Wu Q, Jia L, Du X (2015). Minocycline inhibits PARP‐1 expression and decreases apoptosis in diabetic retinopathy. Mol Med Rep 12: 4887–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JC, Fan J, Wang X, Eacker SM, Kam TI, Chen L et al. (2016). Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med 8: 333ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K (2008). PARP inhibitor tilts cell death from necrosis to apoptosis in cancer cells. Cancer Biol Ther 7: 942–944. [DOI] [PubMed] [Google Scholar]
- Yonemori K, Tamura K, Kodaira M, Fujikawa K, Sagawa T, Esaki T et al. (2016). Safety and tolerability of the olaparib tablet formulation in Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol 78: 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Deng W, Wang W, Ding Y, Jin H, Chen C et al. (2012). Inhibition of poly(ADP‐ribose) polymerase attenuates acute kidney injury in sodium taurocholate‐induced acute pancreatitis in rats. Pancreas 41: 1299–1305. [DOI] [PubMed] [Google Scholar]
- Yu J, Zuo T, Deng W, Shi Q, Ma P, Chen C et al. (2016). Poly(ADP‐ribose) polymerase inhibition suppresses inflammation and promotes recovery from adrenal injury in a rat model of acute necrotizing pancreatitis. BMC Gastroenterol 16: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD (2009). Sex differences in the response to activation of the poly (ADP‐ribose) polymerase pathway after experimental stroke. Exp Neurol 217: 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba T, Thomas H, Cole M, Plummer ER, Curtin NJ (2010). Doxorubicin‐induced suppression of poly(ADP‐ribose) polymerase‐1 (PARP1) activity and expression and its implication for PARP inhibitors in clinical trials. Cancer Chemother Pharmacol 66: 807–812. [DOI] [PubMed] [Google Scholar]
- Zaremba T, Thomas HD, Cole M, Coulthard SA, Plummer ER, Curtin NJ (2011). Poly(ADP‐ribose) polymerase‐1 (PARP‐1) pharmacogenetics, activity and expression analysis in cancer patients and healthy volunteers. Biochem J 436: 671–679. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH (1994). Nitric oxide activation of poly(ADP‐ribose) synthetase in neurotoxicity. Science 263: 687–689. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang C, Tian Y, Zhang F, Xu W, Li X et al. (2016). Inhibition of poly(ADP‐ribose) polymerase‐1 protects chronic alcoholic liver injury. Am J Pathol 186: 3117–3130. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, O'Connor M, Wong H, Salzman AL, Szabó C (1996). Peroxynitrite‐mediated DNA strand breakage activates poly‐adenosine diphosphate ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J Immunol 156: 350–358. [PubMed] [Google Scholar]
- Zingarelli B, Cuzzocrea S, Zsengellér Z, Salzman AL, Szabo C (1997). Protection against myocardial ischemia and reperfusion injury by 3‐aminobenzamide, an inhibitor of poly (ADP‐ribose) synthetase. Cardiovasc Res 36: 205–215. [DOI] [PubMed] [Google Scholar]