

Abstract
Background and Purpose
Olaparib, rucaparib and niraparib, potent inhibitors of poly(ADP‐ribose) polymerase (PARP) are approved as anti‐cancer drugs in humans. Considering the previously demonstrated role of PARP in various forms of acute and chronic myocardial injury, we tested the effects of olaparib in in‐vitro models of oxidative stress in cardiomyocytes, and in an in vivo model of cardiac transplantation.
Experimental Approach
H9c2‐embryonic rat heart‐derived myoblasts pretreated with vehicle or olaparib (10μM) were challenged with either hydrogen peroxide (H2O2) or with glucose oxidase (GOx, which generates H2O2 in the tissue culture medium). Cell viability assays (MTT, lactate dehydrogenase) and Western blotting for PARP and its product, PAR was performed. Heterotopic heart transplantation was performed in Lewis rats; recipients were treated either with vehicle or olaparib (10 mg kg‐1). Left ventricular function of transplanted hearts was monitored via a Millar catheter. Multiple gene expression in the graft was measured by qPCR.
Key Results
Olaparib blocked autoPARylation of PARP1 and attenuated the rapid onset of death in H9c2 cells, induced by H2O2, but did not affect cell death following chronic, prolonged oxidative stress induced by GOx. In rats, after transplantation, left ventricular systolic and diastolic function were improved by olaparib. In the transplanted hearts, olaparib also reduced gene expression for c‐jun, caspase‐12, catalase, and NADPH oxidase‐2.
Conclusions and Implications
Olaparib protected cardiomyocytes against oxidative stress and improved graft contractility in a rat model of heart transplantation. These findings raise the possibility of repurposing this clinically approved oncology drug, to be used in heart transplantation.
Linked Articles
This article is part of a themed section on Inventing New Therapies Without Reinventing the Wheel: The Power of Drug Repurposing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.2/issuetoc
Abbreviations
- dP/dtmax
maximal slope of the systolic pressure increment
- dP/dtmin
maximal slope of the diastolic pressure decrement
- GOx
glucose oxidase
- NAD
nicotinamide adenine dinucleotide
- Niraparib
MK‐4827; (S)‐2‐(4‐(piperidin‐3‐yl)phenyl)‐2H‐indazole‐7‐carboxamide
- NOX2
NADPH oxidase 2
- NOX4
NADPH oxidase 4
- Olaparib
4‐[[3‐[4‐(cyclopropanecarbonyl)piperazine‐1‐carbonyl]‐4‐fluoro‐phenyl]methyl]‐2H‐phthalazin‐1‐one
- Rucaparib
(8‐fluoro‐1,3,4,5‐tetrahydro‐2‐[4‐[(methylamino)methyl]phenyl]6H‐azepino[5,4,3‐cd]indol‐6‐one)
- Tau
left‐ventricular pressure decay
Introduction
The concept that activation of the nuclear enzyme poly (ADP‐ribose) polymerase (PARP) plays a pathophysiological role in myocardial ischaemia/reperfusion injury was put forward 20 years ago (Zingarelli et al., 1997). Multiple lines of follow‐up studies have confirmed and extended these findings and demonstrated the protective effects of PARP1 genetic deficiency, as well as of various classes of PARP inhibitors (including 3‐aminobenzamide, nicotinamide, PJ34 and INO‐1001) in various models of acute and chronic myocardial insults, including ischaemia/reperfusion injury, transplantation and chronic heart failure (see: Szabó et al., 2004; Booz, 2007; Pacher and Szabó, 2008; Berger et al., 2018). The concept of cardioprotection mediated by PARP inhibition has also been tested in a small clinical trial, where trends for anti‐inflammatory and cardioprotective effects were noted in patients with myocardial infarction (Morrow et al., 2009).
Although, in the late 90s and early 2000s much work has been conducted in the field of PARP and reperfusion injury and cell death, the clinical translation of PARP inhibitors has taken on a different direction. Exploiting a growing body of preclinical data, showing that PARP inhibitors can suppress DNA repair, and thereby enhancing the anticancer efficacy of various DNA‐damaging anticancer therapeutic agents, especially on the background of certain breast cancer mutations, intensive research and development work commenced in the mid 2000s to translate these findings into clinical benefit (see Curtin and Szabó, 2013, Drew, 2015). These efforts resulted in the clinical approval of three PARP inhibitors, olaparib, marketed as Lynparza (Deeks, 2015), rucaparib, marketed as Rubraca and niraparib (MK‐4827), marketed as Zejula.
Although the current pharmaceutical and clinical focus of PARP inhibitors is in oncology , the approval and clinical availability of ultrapotent PARP inhibitors may also open the door for repurposing of these compounds for non‐oncological indications (see Berger et al., 2018). ROS play an important part in ischaemia/induced tissue damage and reperfusion injury. The deleterious effects of the oxidative stress occur as the result of both an impairment of the antioxidant defence system and increased production of ROS. The underlying mechanisms of cold preservation injury include secretion of various oxidant and free radical species (including ROS and peroxynitrite). Furthermore, when a donor heart is reperfused following a period of ischaemic cold crystalloid storage, the acute ischaemic myocardium is also subjected to several abrupt biochemical and metabolic changes including intracellular calcium overload, energy depletion, acidosis, activation of granulocytes and generation of ROS inducing organ dysfunction (Hearse and Bolli, 1992). In view of the pathogenetic role of PARP in various forms of cardiac injury, in the current study, we have tested the effect of the first clinically approved PARP inhibitor, olaparib, in models of oxidative stress in cardiomyocytes in vitro and in a model of cardiac transplantation in vivo.
Methods
In vitro models of oxidative stress
Cell culture studies
H9c2 embryonic rat heart‐derived myoblasts obtained from ATCC (CRL‐1446) were cultured in DMEM, supplemented with 10% fetal bovine serum and 100 IU·mL−1 penicillin and 100 μg·mL−1 streptomycin at 37°C in 5% CO2. The effect of olaparib was investigated in H9c2 cells treated with H2O2 or glucose oxidase (GOx) as described (Szczesny et al., 2014). Briefly, in majority of experiments, H9c2 cells were pretreated with 10 μM olaparib for 30 min at 37°C in 5% CO2 followed by treatment with either (a) 300 μM or 1 mM H2O2 for 15 min–24 h at 37°C in 5% CO2 (b) 0.05 U·mL−1 GOx for 1–24 h or (c) 0.05 U·mL−1 GOx for 1 h, followed by incubation for another 24 h.
LDH assay
LDH release was used as a cytotoxicity assay, for the determination of necrotic cell death, as described (Szczesny et al., 2014). Changes in absorbance were read kinetically at 492 nm for 15 min on a monochromator‐based reader (Powerwave HT, Biotek) at 37°C. LDH activity values are shown as Vmax for kinetic assays in mOD per min.
MTT assay
Metabolically active cells were determined by their conversion of tetrazolium dye, MTT, (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) to its insoluble formazan, and was used for the determination of cell viability, as described by Szczesny et al., (2014).
Western blot analysis
H9c2 cells were lysed using NP‐40 lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP‐40), and protein concentration was determined with DC Protein Assay (Bio‐Rad). Western blot analysis was performed as described (Olah et al., 2015) with the membranes sequentially probed using antibodies against PARP1, HRP‐conjugated actin (Cell Signaling Technology, Inc, Danvers, MA, USA) and PAR, the product of PARP (BD Biosciences Pharmingen, San Diego, USA).
Animals
All animal care and experimental procedures were in compliance with the ‘Principles of Laboratory Animal Care’, formulated by the National Society for Medical Research, and the ‘Guide for the Care and Use of Laboratory Animals’, prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH Publication No. 86‐23, revised 1996). This investigation was reviewed and approved by the appropriate institutional review committees. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Donor rats
Male adult Lewis rats, to be used as donors, were purchased (Janvier Lab, Le Genest‐Saint‐Isle, France) at 2 months of age. Animals were housed in a room at 22 ± 2°C under 12 h light/dark cycles and were fed standard laboratory diet ad libitum.
Experimental groups
Rats were randomly divided into two groups: (1) control group: the donor rats were treated with DMSO/10% 2‐hydroxy‐propyl‐ß‐cyclodextrin‐PBS vehicle and (2) olaparib‐treated group: the donor rats received olaparib. Haemodynamic measurements were assessed 1.5 h after a short intravenous infusion of either olaparib (10 mg·kg−1) or the corresponding vehicle. Six donor rats were used in each group. The experimental protocol is shown in Figure 1.
Figure 1.
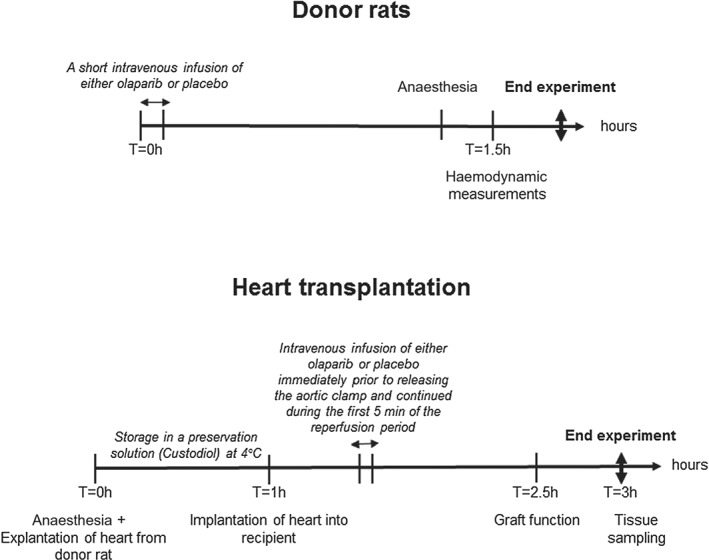
Experimental protocol. Donor rats: haemodynamic measurements were assessed 1.5 h after a short intravenous infusion of either olaparib (10 mg·kg−1) or the corresponding vehicle. Heart transplantation: a short intravenous infusion of either olaparib (10 mg·kg−1) or the corresponding vehicle through the inferior vena cava was started immediately prior to releasing the aortic clamp and continued during the first 5 min of the reperfusion period. One and half hours after transplantation, left‐ventricular graft function was assessed, and tissue and blood samples were collected.
Left ventricular cardiac function
The rats were tracheotomised, intubated and artificially ventilated with 1.5% isoflurane mixed with 100% oxygen. A polyethylene catheter was inserted into the left external jugular vein for fluid administration. A 2F microtip pressure‐volume catheter was inserted into the right carotid artery and advanced into the ascending aorta. After a 5 min stabilization period, the arterial blood pressure was recorded, the catheter was advanced into the left‐ventricle under pressure control. With the use of a special pressure‐volume analysis programme (PVAN, Millar Instruments, Houston, TX, USA), heart rate, systolic and diastolic blood pressures, mean arterial pressure, LV end‐systolic pressure, LV end‐diastolic pressure, effective arterial elastance (Ea), maximal slope of systolic pressure increment (dP/dtmax) and diastolic pressure decrement (dP/dtmin), and time constant of the left ventricular pressure decay (Tau‐g; according to the Glantz method (Pacher et al., 2008)) were calculated. LV pressure‐volume relations were assessed by transiently compressing the inferior vena cava. The slope Emax and Ees of the LV end‐systolic pressure–volume relationship, preload recruitable stroke work (PRSW), maximal elastance, and the dP/dtmax/end‐diastolic volume relation were calculated as a load‐independent index of LV contractility indexes. The slope of the LV end‐diastolic pressure‐volume relationship (EDPVR) was calculated as a reliable index of LV stiffness.
In vivo model of heart transplantation
Animals
Male adult donor Lewis rats (Janvier Lab, Le Genest‐Saint‐Isle, France) were purchased at 8 months of age and studied at the age of 15 months. Sex‐matched young rats, age 2 months, were used as recipients. Animals were housed in a room at 22 ± 2°C under 12 h light/dark cycles and were fed standard laboratory diet ad libitum.
Experimental rat model of heterotopic heart transplantation
Experimental groups
Rats were randomly divided into two groups: (1) control group: hearts explanted from old donor rats were subjected to 1 h of cold ischaemic preservation and transplanted into young recipient rats receiving a vehicle during reperfusion and (2) olaparib‐treated group: hearts explanted from old donors were submitted to 1 h of cold ischaemic preservation and transplanted into young recipient rats receiving olaparib during reperfusion. Olaparib was dissolved in DMSO and then added to 10% 2‐hydroxy‐propyl‐ß‐cyclodextrin‐PBS solution. A short intravenous infusion of either olaparib (10 mg·kg−1) or the corresponding DMSO/10% 2‐hydroxy‐propyl‐ß‐cyclodextrin‐PBS vehicle through the inferior vena cava was started immediately prior to releasing the aortic clamp and continued during the first 5 min of the reperfusion period. Six donor (15 months) and six recipient (2 months) rats were used in each group. The experimental protocol is shown in Figure 1.
Surgical technique of heterotopic heart transplantation
The transplantations were performed in an isogenic Lewis to Lewis rat strain using the experimental model previously described (Loganathan et al., 2015). Briefly, the donor rats were anaesthetized with a single i.p. injection of xylazine (3 mg·kg−1) and ketamine (100 mg·kg−1). Cardiac arrest was induced by Custodiol (histidine‐tryptophane‐ketoglutarate) solution (Dr Franz Köhler, Chemie GmbH, Germany), and the superior and inferior caval veins, and the pulmonary veins, were tied en masse with a 4‐0 single silk suture. Then, the heart was explanted and immediately placed into cold Custodiol solution (4°C).
The recipient rats were anaesthetized (as described above) and then heparinized (400 IU·kg−1). The aorta and the pulmonary artery of the donor heart were anastomosed end to side to the abdominal aorta and the vena cava of the recipient rat respectively. To minimize variability between experiments, the duration between explanation and reperfusion has been standardized to 1 h. After completing the anastomoses, the heart was reperfused with blood in situ for 1.5 h.
Functional measurement in the graft
One and half hours after transplantation, a small incision was made into the apex using a 0.9 mm cannula. A 3F latex balloon catheter (Edwards Lifesciences Corporation, Irvine, CA, USA) was then introduced into the left ventricle via this incision. Before starting the measurement and inflating the balloon, we waited until no arrhythmias, that could disturb the measurement, occurred. This period usually lasted 1–5 min. The latex balloon was only inflated during the measurement and was deflated between each measurement. Heart rate, left ventricular (LV) systolic pressure, LV end‐diastolic pressure, maximal slope of the systolic pressure increment dP/dtmax and maximal slope of the diastolic pressure decrement dP/dtmin were measured by a Millar micromanometer (Millar Instruments, Houston, TX, USA), and the rate pressure product was calculated. The data were analysed offline with PVAN 3.6 software (Millar Instruments, Houston, TX, USA).
Gene expression by qPCR analysis in the graft
Myocardial gene expression of Bax, caspase‐12, caspase‐3, catalase, c‐Jun, glutathione peroxidase (GPX)‐4, NADPH oxidase (NOX)‐2, NOX‐4 and SOD‐1 were performed by quantitative PCR (qPCR), as described (Loganathan et al., 2015). Sample quantifications were normalized to GAPDH expression. Primers were obtained from TIB Molbiol (Berlin, Germany), and their sequences and UPL probes used are represented in Table 1. Evaluation was performed with LightCycler 480 SW 1.5 software (Roche, Mannheim, Germany).
Table 1.
The sequence for forward (F) and reverse (R) primers (from 5′ to 3′) and Universal Probe Library (UPL) probes
| Assay | Sequence | UPL probes |
|---|---|---|
| Bax | F: 5′‐TAGCAAACTGGTGCTCAAGG‐3′ | 130 |
| R: 5′‐GCCACCCTGGTCTTGGAT‐3′ | ||
| Caspase‐12 | F: 5′‐TGGATACTCAGTGGTGATAAAGGA‐3′ | 94 |
| R: 5′‐ACGGCCAGCAAACTTCATTA‐3′ | ||
| Caspase‐3 | F: 5′‐CCGACTTCCTGTATGCTTACTCTA‐3′ | 80 |
| R: 5′‐CATGACCCGTCCCTTGAA‐3′ | ||
| Catalase | F: 5′‐ATCAGGGATGCCATGTTGTT‐3′ | 129 |
| R: 5′‐GGGTCCTTCAGGTGAGTTTG‐3′ | ||
| c‐Jun | F: 5′‐TACCGGCCAGCAACTTTC‐3′ | 115 |
| R: 5′‐TAGGCGCAGAAGAGGTTTTG‐3′ | ||
| GAPDH | F: 5′‐CTACCCACGGCAAGTTCAAT‐3′ | 111 |
| R: 5′‐ATTTGATGTTAGCGGGATCG‐3′ | ||
| GPX4 | F: 5′‐TGGGAAATGCCATCAAATG‐3′ | 25 |
| R: 5′‐CGGCAGGTCCTTCTCTATCA‐3′ | ||
| NOX2 | F: 5′‐TGAAGAGTATCTCAATTTTGCTAGAGA‐3′ | 5 |
| R: 5′‐TGATGACAATTCCTGTGATGC‐3′ | ||
| NOX4 | F: 5′‐GCATCGATACTAAACACTCTACTGGA‐3′ | 115 |
| R: 5′‐CTGGAATGATTGGATGTCTCTG‐3′ | ||
| SOD1 | F: 5′‐GGTCCAGCGGATGAAGAG‐3′ | 5 |
| R: 5′‐GGACACATTGGCCACACC‐3′ |
Immunohistochemistry
Immunohistochemistry for myeloperoxidase (1:200, Abcam Cambridge, UK) was performed to determine neutrophil accumulation, and the evaluation was done in four randomized non‐overlapping fields of the myocardium, and an average value was calculated.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The operators who measured myocyte death, cardiac function and expression of various factors in the myocardial samples were blinded with respect to the identity of the samples. Data are expressed as the mean ± SEM. The normal distribution of the datasets was tested by D'Agostino–Pearson or Sharipo–Wilk test. In the case of normal distribution, Student's t‐test was used to analyse the differences between the groups. Datasets that failed to show normal distribution were analysed by the non‐parametric Mann–Whitney or Kolmogorov–Smirnov test. *P < 0.05 represents statistically significant differences between group means.
Materials
Olaparib was obtained from LC Laboratories (Woburn, MA, USA). Unless indicated otherwise, all other all chemicals were obtained from Sigma‐Aldrich (St. Louis, MO, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Effects of olaparib in cardiac myocytes exposed to oxidative stress in vitro
The effect of various concentration of olaparib on cell viability was assessed in rat heart‐derived myoblasts (H9c2 cells) using MTT and LDH assays; no significant cell injury was detected with 1–100 μM of olaparib after 15 and 30 min incubation (Figure 2A, B). Next, the effect of various concentrations of olaparib was tested in H9c2 cells treated with 300 μM or 1 mM of H2O2. Cytoprotective effects of 1–10 μM of olaparib – assessed by MTT and LDH assays – were observed after both 15 and 30 min incubation (Figure 2A, B).
Figure 2.
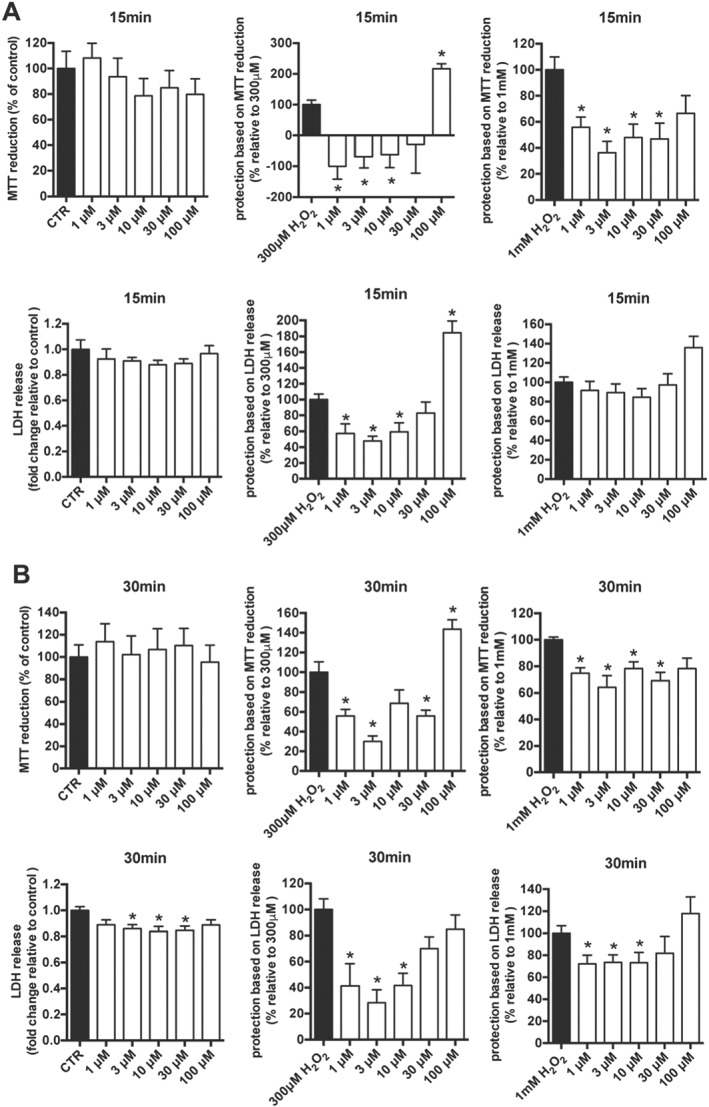
Olaparib protects heart myoblasts against oxidant‐induced damage. The viability of H9c2 cells treated with various concentration of olaparib (1–100 μM) alone or in combination with 300 μM or 1 mM H2O2 for 15 min (A) or 30 min (B) was assessed using MTT and LDH assays. Data shown are means ± SEM; *P < 0.05, significant protective effect of olaparib; n = 5 per group.
In subsequent experiments, we decided to use 10 μM olaparib against cell injury induced by various forms of oxidative stress. We have employed two different types of oxidative injury: short, high‐dose of bulk oxidants (extracellular H2O2; experimental design shown in Figure 3A) and prolonged but constant low level of oxidative stress (extracellular H2O2 generated by GOx); experimental design shown in Figure 4A and 5A in H9c2 cells. As shown in Figure 3, we observed protection against the H2O2‐induced loss of cellular viability. Treatment with olaparib restored MTT conversion (Figure 3B) and reduced the release of LDH into the culture medium (Figure 3C), up to 60 min after the addition of H2O2. These were particularly marked with 1 mM H2O2 treatment. In contrast, no beneficial effect of olaparib was detected at 24 h after H2O2 (Figure 3B, C). As shown in Figure 3D, olaparib markedly inhibited the autoPARylation of PARP1, 15 min after H2O2 challenge. However, neither 300 μM nor 1 mM H2O2 induced any detectable cleavage or degradation of PARP1 (Figure 3E).
Figure 3.
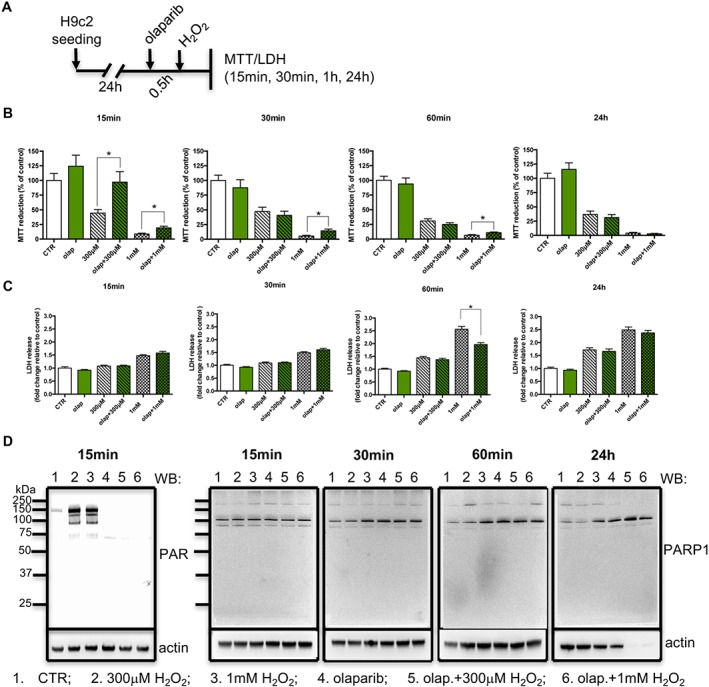
Olaparib protects cardiac myoblasts against acute, severe oxidative stress. (A) Experimental design. Reduction of MTT (B) and release of LDH (C) in H9c2 cells treated with 300 μM or 1 mM H2O2 in the absence or presence of 10 μM olaparib (olap). (D) Western blot analysis of PARylation , at 15 min after H2O2. ( E ) Western blot analysis of PARP1 expression, at 15min ‐24h, after H2O2. Data shown are means ± SEM. *P < 0.05, significant protective effect of olaparib; n = 5 per group.
Figure 4.
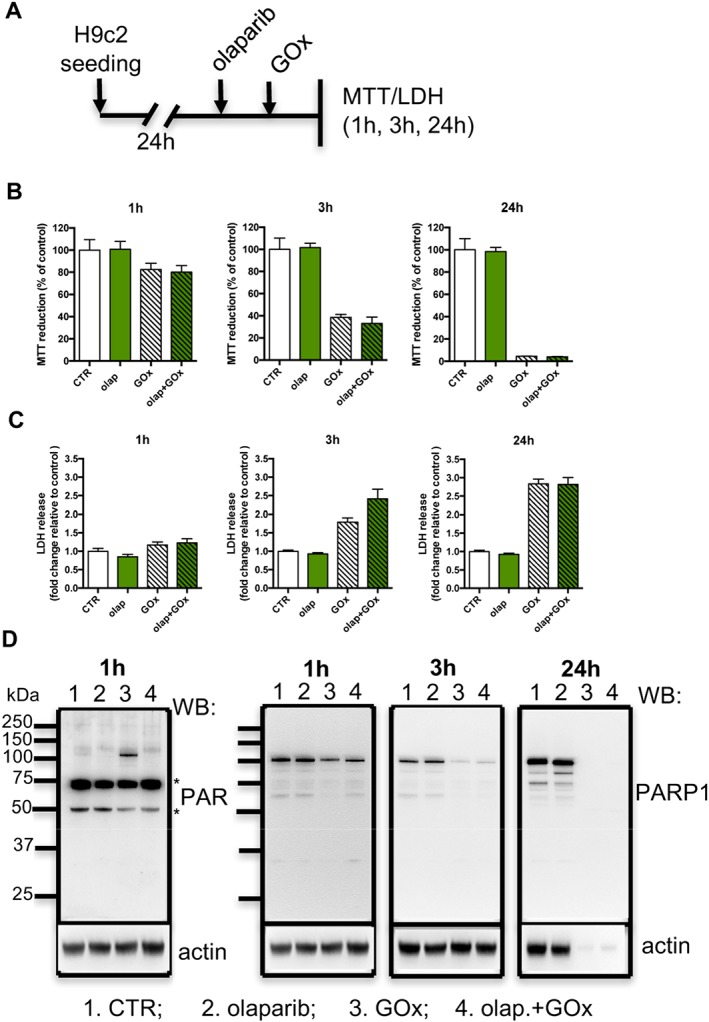
Olaparib does not protect cardiac myoblasts against prolonged, low‐level oxidative stress. (A) Experimental design. Reduction of MTT (B) and release of LDH (C) in H9c2 cells treated with 0.05 U·mL−1 GOx in the absence or presence of 10 μM olaparib (olap). (D) Western blot analysis of PARylation and PARP1 (* indicates a non‐specific band). Data shown are means ± SEM. n = 5 per group.
Figure 5.
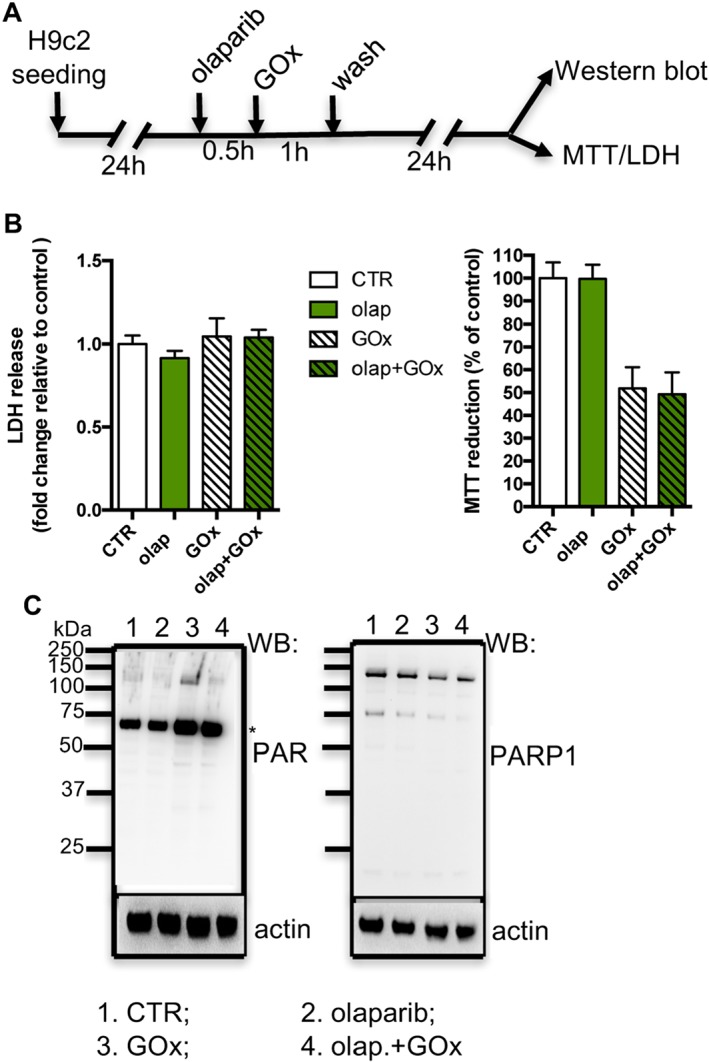
Olaparib does not protect cardiac myoblasts against prolonged, low‐level oxidative stress at later time points. (A) Experimental design. (B) Reduction of MTT and release of LDH in H9c2 cells treated with 0.05 U·mL−1 GOx for 1 h followed by washout of GOx and incubated for additional 24 h in the absence or presence of 10 μM olaparib (olap). (C) Western blot analysis of PARylation and PARP1 (* indicates non‐specific band). Data shown are means ± SEM. n = 5 per group.
Next, we investigated effect of prolonged oxidative stress (a lower‐level, steady flux of H2O2 was generated enzymically by GOx, which utilizes the glucose – present in the culture medium – as its substrate). As shown in Figure 4 , we did not detect any protective effect of olaparib against the GOx‐induced loss of cell viability – at 1, 3 or 24 h (Figure 4B, C respectively). Interestingly, we observed an inhibition of autoPARylation of PARP1 induced by GOx in H9c2 cells pretreated with olaparib at 1 h. Moreover, the level of PARP1 protein was also significantly reduced at 3 and 24 h of GOx treatment (Figure 4D). Quantification of PARP1/actin densitometry at 3 h after GOx treatment showed that PARP1 levels were decreased to 19 ± 6% of control (n = 5) and after 24 h of GOx incubation, no bands corresponding to PARP1 could be detected.
Next, in an ‘exposure/washout’ protocol, we tested the effect of olaparib in H9c2 cells treated with GOx for 1 h at subsequent time points (experimental design in Figure 5A). Although we detected a reduction of PARP1 autoPARylation at 24 h post‐injury in H9c2 cells treated with olaparib (Figure 5C), the viability of H9c2 cells was not affected by olaparib (Figure 5B). Once again, PARP1 levels decreased, to a lesser extent than in the study where GOx was present for the entire duration of the experiments. Quantification of PARP1/actin densitometry at 3 h after GOx treatment showed that PARP1 levels were decreased to 63 ± 23% of control (n = 5).
Taken together, the in vitro studies suggest that olaparib protects cardiac myocytes against injury induced by acute severe oxidative stress, but not from injury elicited by prolonged, lower‐level oxidative stress. In addition, the long‐term oxidant treatment also down‐regulated PARP1 protein.
Effects of olaparib on cardiac function in donors in vivo
Cardiac indexes derived from pressure‐volume analysis are shown in Table 2. Treatment of donor rats with olaparib had no effect on heart rate, systolic and diastolic pressures, LV end‐systolic and end‐diastolic pressures, stroke volume, load‐dependent (dP/dtmax) and load‐independent (slope of dP/dtmax‐end‐diastolic volume, PRSW, Ees, Emax) contractility parameters, ejection phase indices (ejection fraction and cardiac output), indices of the active phase of relaxation (dP/dtmin, Tau‐Glant and Tau‐Weiss) and end‐diastolic stiffness (EDPVR).
Table 2.
Cardiac catheterization parameters in vehicle‐ and olaparib‐treated donor rats
| Control | Olaparib | |
|---|---|---|
| Heart rate (beats per min) | 383 ± 4 | 375 ± 13 |
| Systolic blood pressure (mmHg) | 132 ± 8 | 138 ± 11 |
| Diastolic blood pressure (mmHg) | 110 ± 7 | 110 ± 8 |
| Mean arterial pressure (mmHg) | 117 ± 7 | 120 ± 9 |
| LVSP (mmHg) | 126 ± 6 | 143 ± 12 |
| LVEDP (mmHg) | 7.8 ± 1.0 | 7.0 ± 0.5 |
| Ejection fraction (%) | 56 ± 5 | 56 ± 4 |
| Cardiac output (mL·min−1) | 69 ± 7 | 74 ± 5 |
| Stroke volume (μL) | 181 ± 16 | 197 ± 14 |
| Arterial elastance, Ea (mmHg*μL) | 0.74 ± 0.10 | 0.75 ± 0.09 |
| Systemic vascular resistance (mmHg·mL−1·min−1) | 1.8 ± 0.2 | 1.7 ± 0.2 |
| dP/dtmax (mmHg·s−1) | 7944 ± 563 | 8850 ± 1104 |
| dP/dtmin (mmHg·s−1) | 10 365 ± 924 | 10 972 ± 1050 |
| Tau (Glantz) (s) | 12.0 ± 0.8 | 12.5 ± 0.6 |
| Tau (Weiss) (s) | 12.0 ± 0.8 | 12.5 ± 0.6 |
| EDPVR slope (mmHg·μL−1) | 10.3 ± 0.4 | 10.1 ± 0.4 |
| Ees (mmHg·μL−1) | 0.54 ± 0.07 | 0.46 ± 0.08 |
| Emax (mmHg·μL−1) | 2.0 ± 0.2 | 1.6 ± 0.2 |
| PRSW (mmHg) | 128 ± 15 | 159 ± 14 |
| dP/dtmax/EDV slope [(mmHg·s−1)·μL−1] | 48 ± 11 | 42 ± 9 |
Values shown are means ± SEM. n = 6 rats in each group. dP/dtmax, maximal slope of the systolic pressure increment; dP/dtmin, maximal slope of the diastolic pressure decrement; dP/dtmax/EDV, dP/dtmax/end diastolic volume; Ees and Emax, slope of the end‐systolic pressure–volume relationship; LVEDP, left‐ventricular end‐diastolic pressure; LVSP indicates left‐ventricular systolic pressure; PRSW, preload recruitable stroke; Tau, time constant of left ventricular pressure decay calculated by Glantz method (Tau Glantz) or by Weiss method (Tau Weiss).
Effects of olaparib in transplanted hearts
After transplantation, LV systolic function (shown by increased dP/dtmax) and diastolic function (shown by increased dP/dtmin and prolonged Tau) were significantly improved in the olaparib group compared with the control group (Figure 6), indicating that olaparib improved post‐transplant cardiac performance. Heart rate, left ventricular systolic pressure, left ventricular end‐diastolic pressure (used for estimating LV preload) and the rate pressure product (used to determine the myocardial work) did not show any major differences among the groups (Table 3).
Figure 6.
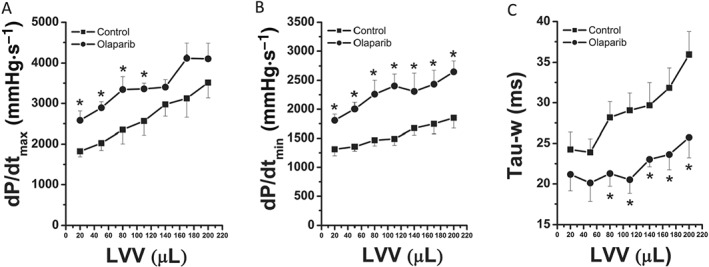
Olaparib improves left‐ventricular systolic and diastolic function of transplanted rat hearts. (A) Maximal slope of the systolic pressure increment (dP/dtmax)‐left‐ventricular volume, (B) maximal slope of the diastolic pressure decrement (dP/dtmin)‐left‐ventricular volume and (C) time constant of left‐ventricular pressure decay (Tau‐w). Data shown are means ± SEM. n = 6 rats per group. *P < 0.05, significantly different from control.
Table 3.
Haemodynamic parameters at an intraventricular volume of 80 μL after 1.5 h of reperfusion
| Control | Olaparib | |
|---|---|---|
| Heart rate (beats per min) | 150 ± 10 | 131 ± 11 |
| LVSP (mmHg) | 78 ± 3 | 93 ± 8 |
| LVEDP (mmHg) | 0.68 ± 0.31 | 0.73 ± 0.14 |
| RPP (mmHg per beats per min) | 11 543 ± 696 | 11 903 ± 953 |
LVSP, left‐ventricular systolic pressure, LVEDP, left‐ventricular end‐diastolic pressure; RPP, rate pressure product. Data shown are means ± SEM. n = 6 rats per group.
After transplantation, quantitative real‐time PCR from myocardium RNA extracts revealed that mRNA levels for c‐Jun, caspase‐12, catalase and NOX2 were significantly lower in the olaparib group than the values in the vehicle‐treated transplantation group (Figure 7). No statistically significant effects were noted on caspase‐3, NOX4, GPX4, Bax and SOD1 (Figure 7).
Figure 7.
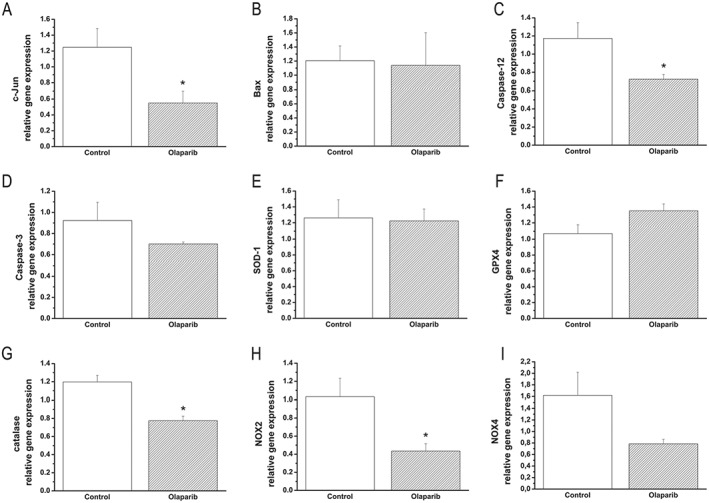
Olaparib affects gene expression in transplanted rat hearts. (A) c‐Jun, (B) bax, (C) caspase‐12, (D) caspase‐3, (E) SOD‐1, (F) GPX4, (G) catalase, (H) NOX2 and (I) NOX4. Data shown are means ± SEM. n = 6 rats per group. *P < 0.05, significantly different from control.
Additionally, after transplantation, the number of myeloperoxidase‐expressing cells was significantly lower in the olaparib‐treated hearts than in the vehicle‐treated transplantation group (Figure 8).
Figure 8.
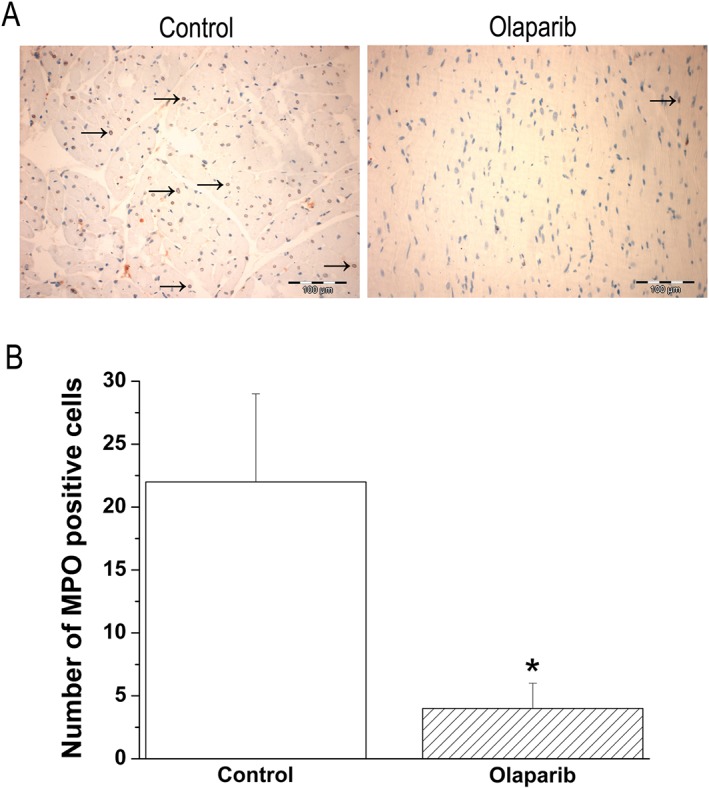
Olaparib treatment reduces neutrophil infiltration in the graft. (A) Representative photomicrographs of myeloperoxidase (MPO, magnification ×200; scale bar: 100 μm) and histological scores of (B) total number of myeloperoxidase‐expressing cells. Black arrows indicate myeloperoxidase positive cells (not all are marked). Data shown are as means ± SEM. n = 6 rats per group. *P < 0.05, significantly different from control.
Discussion
The key findings of the study are the following: (1) olaparib‐treated cells are protected from cell dysfunction and cell death induced by an acute, severe burst of oxidant species, but not from cell dysfunction and cell death elicited by a longer‐term, lower‐level exposure to oxidants and (2) olaparib‐treated rat heart transplant recipients are protected against the transplantation‐associated suppression of myocardial contractility, and this is associated with changes in the expression of several genes, including inflammatory genes (c‐Jun) and cell death effectors (caspase‐12), and with a decreased neutrophil accumulation. These data demonstrate that the PARP inhibitor olaparib – although it was originally developed and optimized for oncological indications and has not previously been tested in models of cardiac dysfunction – exerted significant cardioprotective effects. Therefore, the current study raises the possibility of therapeutic repurposing of olaparib in heart transplantation, and perhaps also in various other indications associated with myocardial damage and cardiac dysfunction.
PARP is an abundant enzyme present throughout the phylogenetic spectrum. Although it is primarily studied in the nucleus (Virág and Szabó, 2002; Jagtap and Szabó, 2005; Gibson and Kraus, 2012; Gupte et al., 2017), emerging evidence also demonstrates its localization and multiple roles in mitochondria (see Brunyanszki et al., 2016). PARP plays a variety of important physiological roles in DNA repair, chromatin remodelling, cell differentiation, transcriptional regulation, cell cycle control and programmed cell death (reviewed in Virág and Szabó, 2002; Jagtap and Szabó, 2005; Gibson and Kraus, 2012; Gupte et al., 2017). The main poly(ADP‐ribosylating) enzyme is PARP1; it is the first discovered, original member of a growing family of enzymes. Multiple lines of evidence, beginning with Nathan Berger's studies in the 80s, identified the activation or over‐activation of PARP as a key pathway of oxidant‐ and free‐radical mediated cell injury (see Berger, 1985; Berger et al., 1995). Studies by Zingarelli and colleagues, 20 years ago, implicated PARP overactivation in the pathogenesis of myocardial reperfusion injury (Zingarelli et al., 1997). In vitro studies in cardiac myocytes showed that, when exposed to cytotoxic concentrations of various free radicals and oxidants (nitric oxide, H2O2, hydroxyl radical and peroxynitrite), cells develop DNA single strand breaks, and, in turn, PARP activation triggers an energy‐consuming, inefficient cellular metabolic cycle by transferring ADP ribose units to nuclear protein acceptors. This process leads to the depletion of intracellular nicotinamide adenine dinucleotide (NAD+) and ATP pools leading to cellular mitochondrial dysfunction and cell death, via the necrotic route (Gilad et al., 1997; Zingarelli et al., 1997; Zingarelli et al., 1998). Follow‐on studies extended these findings, both using pharmacological PARP inhibitors of various classes (Thiemermann et al., 1997; Bowes et al., 1998; Docherty et al., 1999; Liaudet et al., 2001; Zingarelli et al., 2003; Zingarelli et al., 2004; Kaplan et al., 2005; Eltze et al., 2008; Oh et al., 2009; Roesner et al., 2010; Gerö et al., 2014), as well as genetically modified mice deficient in PARP1 (Zingarelli et al., 1998; Grupp et al., 1999; Pieper et al., 2000; Yang et al., 2000). The cardiac protective effect of PARP inhibitors was subsequently extended into various models of heart transplantation. Treatment of the recipients with PARP inhibitors resulted in improved myocardial contractility and longer survival time of the transplanted hearts (Fiorillo et al., 2002, 2003; Szabó et al., 2002, 2005, 2006; Liu et al., 2004; Gao et al., 2007).
The mode of action of PARP inhibitors, as cardioprotective agents, appears to involve a combination of mechanisms. The first one relates to cellular bioenergetics. Myocytes depend on an efficient conversion of chemical into mechanical energy for their contractile function. Disruption of cellular energetics has adverse effects on myocardial contraction and excitability, and more severe disturbances trigger myocyte death. Direct measurements demonstrate that NAD+ and ATP levels are depleted in myocytes exposed to various forms of oxidative stress, as well as in ischaemic/reperfused hearts; and these alterations are attenuated by PARP1 deficiency or PARP inhibition (Docherty et al., 1999; Pieper et al., 2000; Fiorillo et al., 2003). A second mechanism relates to the modulation by PARP of various inflammatory mediator pathways. PARP exerts pluripotent actions in regulating the expression, activation and nuclear translocation of several key pro‐inflammatory genes and proteins. For instance, inhibition or deletion of PARP inhibits the activation of MAP kinases, AP‐1 and NF‐κB, which, in turn, suppresses the expression of various pro‐inflammatory genes, including TNF‐α, the inducible NO synthase and intercellular adhesion molecule‐1 (Zingarelli et al., 1999; Zingarelli et al., 2003; Song et al., 2013). A third mechanism of cardiac protection by PARP inhibitors may be related to maintenance of endothelial integrity and endothelial function, either by direct protective effects – because PARP activation, in response to various forms of oxidative insults, induces endothelial dysfunction (Garcia Soriano et al., 2001; Radovits et al., 2007; Horváth et al., 2009; Módis et al., 2012) – or by secondary effects involving the inhibition of adhesion, activation and tissue infiltration of various mononuclear and polymorphonuclear cell types into the myocardium (Zingarelli et al., 1997). All of the above processes may also influence each other in a variety of ways, forming a positive feed‐forward network of myocardial injury (see Jagtap and Szabó, 2005).
The current results with olaparib appear to confirm and extend the above findings, generated with various generations of PARP inhibitors over the years. What makes olaparib special is that – in contrast to the previously studied PARP inhibitors, with the exception of nicotinamide, (a vitamin with low PARP inhibitory potency and many additional pharmacological actions) – it is clinically approved and is available to be used in patients. Olaparib monotherapy is generally well tolerated. The most common side effects (>10%) include fatigue, nausea and vomiting, diarrhoea, dyspepsia, headache, decreased appetite and dizziness (Goulooze et al., 2016). Although the clear focus with olaparib – both preclinically and clinically – has been the therapy of various cancers, several groups have started conducting preclinical studies with olaparib in the context of various non‐oncological applications, with an eye on future therapeutic repurposing. These efforts, so far, have demonstrated (1) protective effects of olaparib in vitro against NMDA receptor stimulation induced or oxygen–glucose deprivation induced death in differentiated human neurons (Xu et al., 2016) and in retinal pigment epithelial cell lines exposed to H2O2 (Jang et al., 2017) and in cisplatin‐induced injury in chronic myeloid leukaemia cells (Xiao and Kan, 2017), (2) protective effects of olaparib in a mouse model of transient middle cerebral artery occlusion and reperfusion (Teng et al., 2016), (3) protective effects of olaparib in various models of model of lung injury/lung inflammation, either induced by endotoxin (Kapoor et al., 2015) or in asthma models elicited by senzitization to ovalbumin (Ghonim et al., 2015a) or to house dust mites (Ghonim et al., 2015b), (4) protective effects of olaparib in various models of multiple organ dysfunction, either elicited by bacterial lipopolysaccharide (Kapoor et al., 2015) or by third‐degree thermal injury (Ahmad et al., 2018), and (5) protective effects of olaparib in rodent models of acute and chronic liver failure (Gariani et al., 2017; Mukhopadhyay et al., 2017). The current data confirm and extend these findings and demonstrate the protective effect of olaparib in oxidant‐challenged cardiac myocytes and in transplanted rat hearts. Additionally, olaparib administration in healthy donor rats had no effects on LV cardiac function compared to vehicle‐treated controls.
The findings that only the acute cell death, but not the prolonged cell death induced by lower‐level oxidative stress, was attenuated by PARP inhibition is consistent with the mode of action of PARP inhibitors, as these compounds, typically, are known to protect against severe cell injury (necrotic type), but not against the chronic, apoptotic forms of cell death (Virág et al., 1998, 2013; Ha and Snyder, 1999; Sosna et al., 2014). An additional factor may be the fact that long‐term oxidant exposure (or even a short‐term exposure to oxidants, followed by a longer follow‐up period) results in the down‐regulation of PARP1 protein. Clearly, if the pharmacological target of olaparib is no longer present in the cell, then no pharmacological (cytoprotective or otherwise) effects can be expected. The actual mechanism of oxidant‐mediated PARP1 down‐regulation requires further investigation. It is important to mention that in earlier studies, PARP1 is down‐regulated in vitro – for example, in myoblasts during differentiation (Olah et al., 2015).
As heart transplantation is associated with an acute burst of oxidant species, the protective effects in vivo may be, at least in part, also related to protection against oxidant‐mediated energetic alterations and associated cellular bioenergetic dysfunction. The fact that the improved cardiac function in the transplant model was also associated with changes in the expression of some genes (inflammatory, cell death effector and oxidant/antioxidant) but not others, requires further analysis. Nevertheless, the down‐regulation of c‐Jun is consistent with the results from many studies using PARP inhibitors or PARP silencing (Song et al., 2008; Huang et al., 2009; Mester et al., 2009; Kim et al., 2012; Radnai et al., 2012). Moreover, inhibition of neutrophil infiltration is consistent with previous findings showing similar effect of PARP inhibitors of other structural classes, as well as PARP1 deficiency in various models of ischaemia–reperfusion and inflammation (Szabó et al., 1997; Szabó, 1998; Zingarelli et al., 1998; Liaudet et al., 2001, 2002; Mabley et al., 2001).
We hypothesize that the down‐regulation of catalase may be due to the fact that the transplanted hearts may be exposed to a lower overall level of oxidative stress (also shown by the lower expression of NOX2 and a tendency for lower expression of NOX4), which, in turn, only triggers a lower degree of compensatory up‐regulation of various antioxidant mechanisms. Caspase‐12 is an enzyme which has been involved in endoplasmic reticulum‐associated apoptosis (García de la Cadena and Massieu, 2016). It is also an enzyme that is known to degrade PARP1 (Bajaj and Sharma, 2006) but the significance of its down‐regulation in olaparib‐treated hearts remains to be determined in future studies. Additionally, after transplantation, inflammatory cell infiltrates, as shown by immunohistochemical analysis of myeloperoxidase positive‐cells, were significantly lower in the olaparib‐treated hearts than in the vehicle‐administered hearts.
Due to possible adverse effects of olaparib on DNA repair (see Berger et al., 2018), further work is needed to test whether olaparib affects DNA integrity and chromosomal stability in cardiac myocytes during transplantation or reperfusion injury. However – as discussed, in detail in Berger et al., 2018 – it must be emphasized that the therapeutically effective dose of olaparib in non‐oncological indications is likely to be lower than in oncological indications, because the effects of PARP inhibitors on NAD+ levels and cellular bioenergetics are gradual and concentration‐dependent, while the inhibition of DNA repair requires a complete or near‐complete inhibition of PARP activity, which can only be achieved at the top end of the concentration–response curve. This is also shown in the current results, where the effective dose of olaparib (10 mg·kg−1) was lower than the effective dose of olaparib in rodent models of cancer (100 mg·kg−1·day−1 or higher) (see also Berger et al., 2018).
We are aware of the fact that the current study has a number of limitations. Firstly, maintenance of cellular ATP levels is an important strategy to reduce ischaemia/reperfusion injury, because it counteracts osmotic swelling, sarcolemmal rupture and necrosis (Ferdinandy et al., 2007). Although, in previous studies, PARP inhibitors or PARP1 deficiency has been shown to maintain myocardial NAD+ and ATP levels in ischaemic/reperfused hearts (e.g. Docherty et al., 1999; Grupp et al., 1999), in the current study, we did not determine NAD+ or high‐energy phosphate levels. Secondly, the dose–response relationship of olaparib has not been examined. Indeed, the heterotopic heart transplantation model is not a suitable model for the determination of an optimal dose regimen for olaparib. Finally, in our experimental model, ischaemia/reperfusion is imposed on the background of one comorbidity including ageing, a major risk factor for cardiovascular disease. However, in the clinical setting, multiple comorbidities often coexist and cardiovascular risk factors may interfere with ischaemia/reperfusion injury and cardioprotection (Ferdinandy et al., 2014). Although animal data can only be extrapolated to humans with caution, our results, nevertheless, show that pharmacological inhibition of PARP (with a potentially repurposable drug, olaparib) improves contractile function during early reperfusion after heart transplantation from older donors.
We have recommended (Berger et al., 2018) that – at least initially – the therapeutic repurposing of PARP inhibitors currently clinically used for oncological indications should focus on those non‐oncological indications where the disease is associated with a high risk of mortality, the alternative therapeutic options are limited and the expected duration of drug administration is relatively short. Based on the results of the current study, severe forms of cardiac injury may also be considered in the future for the therapeutic repurposing of olaparib or other clinically used PARP inhibitors.
Author contributions
S.K.‐I., B.S., M.M., S.L., M.R., F.L. and S.L. were responsible for the conduct of the experiments, analysis of data and preparation of the manuscript; C.S. and G.S. did the experimental design, data analysis and preparation of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by a grant from the National Institutes of Health; 2P50GM060338 to C.S. and R21ES024648 to B.S.
Korkmaz‐Icöz, S. , Szczesny, B. , Marcatti, M. , Li, S. , Ruppert, M. , Lasitschka, F. , Loganathan, S. , Szabó, C. , and Szabó, G. (2018) Olaparib protects cardiomyocytes against oxidative stress and improves graft contractility during the early phase after heart transplantation in rats. British Journal of Pharmacology, 175: 246–261. doi: 10.1111/bph.13983.
References
- Ahmad A, Olah G, Herndon DN, Szabo C (2018). The clinically used PARP inhibitor olaparib improves organ function, suppresses inflammatory responses and accelerates wound healing in a murine model of third‐degree burn injury. Br J Pharmacol 175: 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj G, Sharma RK (2006). TNF‐alpha‐mediated cardiomyocyte apoptosis involves caspase‐12 and calpain. Biochem Biophys Res Commun 345: 1558–1564. [DOI] [PubMed] [Google Scholar]
- Berger NA (1985). Poly(ADP‐ribose) in the cellular response to DNA damage. Radiat Res 101: 4–15. [PubMed] [Google Scholar]
- Berger NA, Whitacre CM, Hashimoto H, Berger SJ, Chatterjee S (1995). NAD and poly(ADP‐ribose) regulation of proteins involved in response to cellular stress and DNA damage. Biochimie 77: 364–367. [DOI] [PubMed] [Google Scholar]
- Berger NA, Besson VC, Boulares AH, Bürkle A, Chiarugi A, Clark RS et al (2018). Opportunities for the repurposing of PARP inhibitors for the therapy of non‐oncological diseases. Br J Pharmacol 175: 192–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booz GW (2007). PARP inhibitors and heart failure – translational medicine caught in the act. Congest Heart Fail 13: 105–112. [DOI] [PubMed] [Google Scholar]
- Bowes J, Ruetten H, Martorana PA, Stockhausen H, Thiemermann C (1998). Reduction of myocardial reperfusion injury by an inhibitor of poly (ADP‐ribose) synthetase in the pig. Eur J Pharmacol 359: 143–150. [DOI] [PubMed] [Google Scholar]
- Brunyanszki A, Szczesny B, Virág L, Szabó C (2016). Mitochondrial poly(ADP‐ribose) polymerase: the Wizard of Oz at work. Free Radic Biol Med 100: 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, Szabó C (2013). Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med 34: 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks ED (2015). Olaparib: first global approval. Drugs 75: 231–240. [DOI] [PubMed] [Google Scholar]
- Docherty JC, Kuzio B, Silvester JA, Bowes J, Thiemermann C (1999). An inhibitor of poly (ADP‐ribose) synthetase activity reduces contractile dysfunction and preserves high energy phosphate levels during reperfusion of the ischaemic rat heart. Br J Pharmacol 127: 1518–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew Y (2015). The development of PARP inhibitors in ovarian cancer: from bench to bedside. Br J Cancer 113: S3–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltze T, Boer R, Wagner T, Weinbrenner S, McDonald MC, Thiemermann C (2008). Imidazoquinolinone, imidazopyridine, and isoquinolindione derivatives as novel and potent inhibitors of the poly(ADP‐ribose) polymerase (PARP): a comparison with standard PARP inhibitors. Mol Pharmacol 74: 1587–1598. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF (2007). Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59: 418–458. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R (2014). Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev 66: 1142–1174. [DOI] [PubMed] [Google Scholar]
- Fiorillo C, Pace S, Ponziani V, Nediani C, Perna AM, Liguori P et al (2002). Poly(ADP‐ribose) polymerase activation and cell injury in the course of rat heart heterotopic transplantation. Free Radic Res 36: 79–87. [DOI] [PubMed] [Google Scholar]
- Fiorillo C, Ponziani V, Giannini L, Cecchi C, Celli A, Nediani C et al (2003). Beneficial effects of poly (ADP‐ribose) polymerase inhibition against the reperfusion injury in heart transplantation. Free Radic Res 37: 331–339. [DOI] [PubMed] [Google Scholar]
- Gao L, Kwan JC, Macdonald PS, Yang L, Preiss T, Hicks M (2007). Improved poststorage cardiac function by poly (ADP‐ribose) polymerase inhibition: role of phosphatidylinositol 3‐kinase Akt pathway. Transplantation 84: 380–386. [DOI] [PubMed] [Google Scholar]
- García de la Cadena S, Massieu L (2016). Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase‐12. Apoptosis 21: 763–777. [DOI] [PubMed] [Google Scholar]
- Garcia Soriano F, Virág L, Jagtap P, Szabó E, Mabley JG, Liaudet L et al (2001). Diabetic endothelial dysfunction: the role of poly(ADP‐ribose) polymerase activation. Nat Med 7: 108–113. [DOI] [PubMed] [Google Scholar]
- Gariani K, Ryu D, Menzies KJ, Yi HS, Stein S, Zhang H et al (2017). Inhibiting poly ADP‐ribosylation increases fatty acid oxidation and protects against fatty liver disease. J Hepatol 66: 132–141. [DOI] [PubMed] [Google Scholar]
- Gerö D, Szoleczky P, Chatzianastasiou A, Papapetropoulos A, Szabó C (2014). Modulation of poly(ADP‐ribose) polymerase‐1 (PARP‐1)‐mediated oxidative cell injury by ring finger protein 146 (RNF146) in cardiac myocytes. Mol Med 20: 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonim MA, Pyakurel K, Ibba SV, Al‐Khami AA, Wang J, Rodriguez P et al (2015a). PARP inhibition by olaparib or gene knockout blocks asthma‐like manifestation in mice by modulating CD4(+) T cell function. J Transl Med 13: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonim MA, Pyakurel K, Ibba SV, Wang J, Rodriguez P, Al‐Khami AA et al (2015b). PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite‐induced disease in mice. Clin Sci (Lond) 129: 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL (2012). New insights into the molecular and cellular functions of poly(ADP‐ribose) and PARPs. Nat Rev Mol Cell Biol 13: 411–424. [DOI] [PubMed] [Google Scholar]
- Gilad E, Zingarelli B, Salzman AL, Szabó C (1997). Protection by inhibition of poly (ADP‐ribose) synthetase against oxidant injury in cardiac myoblasts in vitro. J Mol Cell Cardiol 29: 2585–2597. [DOI] [PubMed] [Google Scholar]
- Goulooze SC, Cohen AF, Rissmann R (2016). Olaparib. Br J Clin Pharmacol 81: 171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp IL, Jackson TM, Hake P, Grupp G, Szabó C (1999). Protection against hypoxia‐reoxygenation in the absence of poly (ADP‐ribose) synthetase in isolated working hearts. J Mol Cell Cardiol 31: 297–303. [DOI] [PubMed] [Google Scholar]
- Gupte R, Liu Z, Kraus WL (2017). PARPs and ADP‐ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31: 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HC, Snyder SH (1999). Poly(ADP‐ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 96: 13978–13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearse DJ, Bolli R (1992). Reperfusion induced injury: manifestations, mechanisms, and clinical relevance. Cardiovasc Res 26: 101–108. [DOI] [PubMed] [Google Scholar]
- Horváth EM, Benko R, Kiss L, Murányi M, Pék T, Fekete K et al (2009). Rapid 'glycaemic swings' induce nitrosative stress, activate poly(ADP‐ribose) polymerase and impair endothelial function in a rat model of diabetes mellitus. Diabetologia 52: 952–961. [DOI] [PubMed] [Google Scholar]
- Huang D, Wang Y, Yang C, Liao Y, Huang K (2009). Angiotensin II promotes poly(ADP‐ribosyl)ation of c‐Jun/c‐Fos in cardiac fibroblasts. J Mol Cell Cardiol 46: 25–32. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabó C (2005). Poly(ADP‐ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4: 421–440. [DOI] [PubMed] [Google Scholar]
- Jang KH, Do YJ, Son D, Son E, Choi JS, Kim E (2017). AIF‐independent parthanatos in the pathogenesis of dry age‐related macular degeneration. Cell Death Dis 8: e2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, O'Connor M, Hake PW, Zingarelli B (2005). Inhibitors of poly (ADP‐ribose) polymerase ameliorate myocardial reperfusion injury by modulation of activator protein‐1 and neutrophil infiltration. Shock 23: 233–238. [PubMed] [Google Scholar]
- Kapoor K, Singla E, Sahu B, Naura AS (2015). PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol Cell Biochem 400: 153–162. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Long KE, Tang K, Padanilam BJ (2012). Poly(ADP‐ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int 82: 193–203. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Szabó E, Timashpolsky L, Virág L, Cziráki A, Szabó C (2001). Suppression of poly (ADP‐ribose) polymerase activation by 3‐aminobenzamide in a rat model of myocardial infarction: long‐term morphological and functional consequences. Br J Pharmacol 133: 1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet L, Pacher P, Virág L, Soriano FG, Hasko G, Szabó C (2002). Activation of poly(ADP‐ribose) polymerase is a central mechanism of lipopolysaccharide‐induced acute pulmonary inflammation. Am J Respir Crit Care Med 165: 372–377. [DOI] [PubMed] [Google Scholar]
- Liu Y, Son NH, Szabólcs MJ, Ma N, Sciacca RR, Albala A (2004). Effects of inhibition of poly(adenosine diphosphate‐ribose) synthase on acute cardiac allograft rejection. Transplantation 78: 668–674. [DOI] [PubMed] [Google Scholar]
- Loganathan S, Korkmaz‐Icoz S, Radovits T, Li S, Mikles B, Barnucz E et al (2015). Effects of soluble guanylate cyclase activation on heart transplantation in a rat model. J Heart Lung Transplant 34: 1346–1353. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Jagtap P, Perretti M, Getting SJ, Salzman AL, Virág L et al (2001). Anti‐inflammatory effects of a novel, potent inhibitor of poly (ADP‐ribose) polymerase. Inflamm Res 50: 561–569. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester L, Szabó A, Atlasz T, Szabadfi K, Reglodi D, Kiss P et al (2009). Protection against chronic hypoperfusion‐induced retinal neurodegeneration by PARP inhibition via activation of PI‐3‐kinase Akt pathway and suppression of JNK and p38 MAP kinases. Neurotox Res 16: 68–76. [DOI] [PubMed] [Google Scholar]
- Módis K, Gero D, Erdélyi K, Szoleczky P, DeWitt D, Szabó C (2012). Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem Pharmacol 83: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DA, Brickman CM, Murphy SA, Baran K, Krakover R, Dauerman H et al (2009). A randomized, placebo‐controlled trial to evaluate the tolerability, safety, pharmacokinetics, and pharmacodynamics of a potent inhibitor of poly(ADP‐ribose) polymerase (INO‐1001) in patients with ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of the TIMI 37 trial. J Thromb Thrombolysis 27: 359–364. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horváth B, Rajesh M, Varga ZV, Gariani K, Ryu D et al (2017). PARP inhibition protects against alcoholic and non‐alcoholic steatohepatitis. J Hepatol 66: 589–600. [DOI] [PubMed] [Google Scholar]
- Oh KS, Lee S, Yi KY, Seo HW, Koo HN, Lee BH (2009). A novel and orally active poly(ADP‐ribose) polymerase inhibitor, KR‐33889 [2‐[methoxycarbonyl(4‐methoxyphenyl) methylsulfanyl]‐1H‐benzimidazole‐4‐carboxylic acid amide], attenuates injury in in vitro model of cell death and in vivo model of cardiac ischemia. J Pharmacol Exp Ther 328: 10–18. [DOI] [PubMed] [Google Scholar]
- Olah G, Szczesny B, Brunyánszki A, López‐García IA, Gerö D, Radák Z et al (2015). Differentiation‐associated downregulation of poly(ADP‐ribose) polymerase‐1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS One 10: e0134227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Szabó C (2008). Role of the peroxynitrite‐poly(ADP‐ribose) polymerase pathway in human disease. Am J Pathol 173: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA (2008). Measurement of cardiac function using pressure‐volume conductance catheter technique in mice and rats. Nat Protoc 3: 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Walles T, Wei G, Clements EE, Verma A, Snyder SH et al (2000). Myocardial postischemic injury is reduced by polyADPripose polymerase‐1 gene disruption. Mol Med 6: 271–282. [PMC free article] [PubMed] [Google Scholar]
- Radnai B, Antus C, Racz B, Engelmann P, Priber JK, Tucsek Z et al (2012). Protective effect of the poly(ADP‐ribose) polymerase inhibitor PJ34 on mitochondrial depolarization‐mediated cell death in hepatocellular carcinoma cells involves attenuation of c‐Jun N‐terminal kinase‐2 and protein kinase B/Akt activation. Mol Cancer 11: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovits T, Zotkina J, Lin LN, Bömicke T, Arif R, Gerö D et al (2007). Poly(ADP‐Ribose) polymerase inhibition improves endothelial dysfunction induced by hypochlorite. Exp Biol Med (Maywood) 232: 1204–1212. [DOI] [PubMed] [Google Scholar]
- Roesner JP, Mersmann J, Bergt S, Bohnenberg K, Barthuber C, Szabó C et al (2010). Therapeutic injection of PARP inhibitor INO‐1001 preserves cardiac function in porcine myocardial ischemia and reperfusion without reducing infarct size. Shock 33: 507–512. [DOI] [PubMed] [Google Scholar]
- Song ZF, Ji XP, Li XX, Wang SJ, Wang SH, Zhang Y (2008). Inhibition of the activity of poly (ADP‐ribose) polymerase reduces heart ischaemia/reperfusion injury via suppressing JNK‐mediated AIF translocation. J Cell Mol Med 12: 1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song ZF, Chen DY, Du B, Ji XP (2013). Poly (ADP‐ribose) polymerase inhibitor reduces heart ischaemia/reperfusion injury via inflammation and Akt signalling in rats. Chin Med J (Engl) 126: 1913–1917. [PubMed] [Google Scholar]
- Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P et al (2014). TNF‐induced necroptosis and PARP‐1‐mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci 71: 331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C (1998). Role of poly (ADP‐ribose) synthetase in inflammation. Eur J Pharmacol 350: 1–19. [DOI] [PubMed] [Google Scholar]
- Szabó C, Lim LH, Cuzzocrea S, Getting SJ, Zingarelli B, Flower RJ et al (1997). Inhibition of poly (ADP‐ribose) synthetase attenuates neutrophil recruitment and exerts anti‐inflammatory effects. J Exp Med 186: 1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó G, Bährle S, Stumpf N, Sonnenberg K, Szabó EE, Pacher P et al (2002). Poly(ADP‐Ribose) polymerase inhibition reduces reperfusion injury after heart transplantation. Circ Res 90: 100–106. [DOI] [PubMed] [Google Scholar]
- Szabó G, Liaudet L, Hagl S, Szabó C (2004). Poly(ADP‐ribose) polymerase activation in the reperfused myocardium. Cardiovasc Res 61: 471–480. [DOI] [PubMed] [Google Scholar]
- Szabó G, Soós P, Heger U, Flechtenmacher C, Bährle S, Zsengellér Z et al (2005). Poly(ADP‐ribose) polymerase inhibition attenuates biventricular reperfusion injury after orthotopic heart transplantation. Eur J Cardiothorac Surg 27: 226–234. [DOI] [PubMed] [Google Scholar]
- Szabó G, Bährle S, Sivanandam V, Stumpf N, Gerö D, Berger I et al (2006). Immunomodulatory effects of poly(ADP‐ribose) polymerase inhibition contribute to improved cardiac function and survival during acute cardiac rejection. J Heart Lung Transplant 25: 794–804. [DOI] [PubMed] [Google Scholar]
- Szczesny B, Módis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A et al (2014). AP39, a novel mitochondria‐targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 41: 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng F, Zhu L, Su J, Zhang X, Li N, Nie Z et al (2016). Neuroprotective effects of poly(ADP‐ribose)polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem Res 41: 1516–1526. [DOI] [PubMed] [Google Scholar]
- Thiemermann C, Bowes J, Myint FP, Vane JR (1997). Inhibition of the activity of poly(ADP ribose) synthetase reduces ischemia‐reperfusion injury in the heart and skeletal muscle. Proc Natl Acad Sci U S A 94: 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virág L, Szabó C (2002). The therapeutic potential of poly(ADP‐ribose) polymerase inhibitors. Pharmacol Rev 54: 375–429. [DOI] [PubMed] [Google Scholar]
- Virág L, Scott GS, Cuzzocrea S, Marmer D, Salzman AL, Szabó C (1998). Peroxynitrite‐induced thymocyte apoptosis: the role of caspases and poly (ADP‐ribose) synthetase (PARS) activation. Immunology 94: 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virág L, Robaszkiewicz A, Rodriguez‐Vargas JM, Oliver FJ (2013). Poly(ADP‐ribose) signaling in cell death. Mol Aspects Med 34: 1153–1167. [DOI] [PubMed] [Google Scholar]
- Xiao LY, Kan WM (2017). Poly ADP‐ribose polymerase inhibition suppresses cisplatin toxicity in chronic myeloid leukemia cells. Anticancer Drugs 28: 316–321. [DOI] [PubMed] [Google Scholar]
- Xu JC, Fan J, Wang X, Eacker SM, Kam TI, Chen L et al (2016). Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med 8: 333ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zingarelli B, Szabó C (2000). Effect of genetic disruption of poly (ADP‐ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia‐reperfusion injury. Shock 13: 60–66. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Cuzzocrea S, Zsengellér Z, Salzman AL, Szabó C (1997). Protection against myocardial ischemia and reperfusion injury by 3‐aminobenzamide, an inhibitor of poly (ADP‐ribose) synthetase. Cardiovasc Res 36: 205–215. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Salzman AL, Szabó C (1998). Genetic disruption of poly (ADP‐ribose) synthetase inhibits the expression of P‐selectin and intercellular adhesion molecule‐1 in myocardial ischemia/reperfusion injury. Circ Res 83: 85–94. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Szabó C, Salzman AL (1999). Blockade of Poly(ADP‐ribose) synthetase inhibits neutrophil recruitment, oxidant generation, and mucosal injury in murine colitis. Gastroenterology 116: 335–345. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Hake PW, O'Connor M, Denenberg A, Kong S, Aronow BJ (2003). Absence of poly(ADP‐ribose)polymerase‐1 alters nuclear factor‐kappa B activation and gene expression of apoptosis regulators after reperfusion injury. Mol Med 9: 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, Hake PW, O'Connor M, Denenberg A, Wong HR, Kong S et al (2004). Differential regulation of activator protein‐1 and heat shock factor‐1 in myocardial ischemia and reperfusion injury: role of poly(ADP‐ribose) polymerase‐1. Am J Physiol Heart Circ Physiol 286: H1408–H1415. [DOI] [PubMed] [Google Scholar]