

Abstract
Deuterium labeling is of great value in organic synthesis and the pharmaceutical industry. However, the state-of-the-art C–H/C–D exchange using noble metal catalysts or strong bases/acids suffers from poor functional group tolerances, poor selectivity and lack of scope for generating molecular complexity. Herein, we demonstrate the deuteration of halides using heavy water as the deuteration reagent and porous CdSe nanosheets as the catalyst. The deuteration mechanism involves the generation of highly active carbon and deuterium radicals via photoinduced electron transfer from CdSe to the substrates, followed by tandem radicals coupling process, which is mechanistically distinct from the traditional methods involving deuterium cations or anions. Our deuteration strategy shows better selectivity and functional group tolerances than current C–H/C–D exchange methods. Extending the synthetic scope, deuterated boronic acids, halides, alkynes, and aldehydes can be used as synthons in Suzuki coupling, Click reaction, C–H bond insertion reaction etc. for the synthesis of complex deuterated molecules.
Developing convenient deuterium labeling procedures is important in organic synthesis and the pharmaceutical industry. Here, the authors report a mild photocatalytic strategy for controllable deuteration of halides using D2O as the reagent and porous CdSe nanosheets as the catalyst.
Introduction
Owing to the kinetic isotope effect, deuterium- and tritium-labeled compounds find important applications in the investigation of reaction mechanism1, the modification of reaction selectivity in total synthesis2, as well as the preparation of advanced materials with enhanced performance3. In particular, D/T labeling has attracted growing interests in pharmaceutical industry because the incorporation of heavy hydrogen can change the absorption, distribution, and toxicological properties of the drugs, while retaining their original potency and selectivity owing to the longer residence time of the more inert C–D bonds compared with the C–H isotopologues4–6. Since the first deuteration of bioactive molecules to promote improved pharmacokinetic profile in the 1960s7, tremendous efforts have been devoted to the synthesis of deuterium- or tritium-labeled pharmaceuticals. Owing to the prevalence of aryl moiety in drug molecules8, direct deuteration of aryl ring would be promising for generating new drugs with high therapeutic values. Indeed, the first deuterated drug has been approved by the US Food and Drug Administration in April 20179.
The prevalent methods for deuterium incorporation are metal-catalyzed or acid/base promoted C–H/C–D exchanges10, which allow direct incorporation of deuterium without the need to pre-functionalize the starting materials and without substantially altering the structure of the molecules. Nevertheless, conventional C–H/C–D exchanges suffer from several drawbacks. First, C–H bonds are usually difficult to activate. Industrial C–H/C–D exchanges relying on the weak acidity or basicity of C–H bonds require the use of high temperature and strong acid/base, leading to significant safety risks and poor functional group tolerances11,12. Multiple exchange processes are also necessary to achieve high deuteration content13. Second, approaches using homogeneous noble metal catalysts to activate C–H bonds are restricted by the complexity of catalyst synthesis and high cost14,15. Third, the selective deuteration of C–H bonds remains challenging; the position of deuteration is difficult to control and requires directing groups to install deuterium at the ortho positions on aromatic and hetero-aromatic rings16. Therefore, undesired deuteration at multiple reactive sites is difficult to prevent17,18. The development of a universal deuteration strategy with wide functional group tolerance will be highly valuable in synthetic chemistry and pharmaceutical industry.
C–X (where X is a halogen) bonds are ubiquitous in many important organic molecules and natural products19. In fact, well-developed halogen chemistry can be exploited for selective deuteration20. For example, Onomura group developed a Pd-NHC system for effective deuterodechlorination of aryl/heteroaryl chlorides21. However, C–X to C–D transformations generally require the use of noble metal catalysts, complex ligands and special deuterium donors. Recently, photoinduced electron transfer from excited photocatalysts to halides (C–I, C–Br, C–Cl, and C–F) has emerged as a powerful strategy to generate highly reactive carbon radicals, which are useful intermediates for noble metal-free substitution, addition, or coupling reactions to produce value-added compounds22. A comprehensive review on the photoredox reactions used in organic and polymer synthesis is provided by Xu and Boyer et al23. Owing to their mild reaction conditions and high efficiency, photocatalytic methods have been successfully applied in total synthesis or drug discovery process24,25. Despite some early studies on photocatalytic hydrogenation of Ar–X using amines, alcohols, or Hantzsch ester as hydrogen donors26–28, photochemical deuteration has not been widely studied owing to the difficulty in accessing suitable deuterium sources and the lack of a suitable catalytic system. Heavy water (D2O) is an ideal deuteration reagent from the perspective of safety, cost and handling and it is worthwhile exploring photocatalytic hydrogen evolution (PHE) as a potential route for generating active D radicals for deuteration reactions.
Herein, we report a universal C–X to C–D transformation using D2O as deuterium source and porous 2D-CdSe as photocatalyst; the latter can catalyze C–X bonds cleavage as well as split D2O to generate deuterium radicals. The subsequent coupling between carbon and deuterium radicals results in the formation of deuterated products. Compared with conventional C–H/C–D exchange technology, this method is distinguished by its high selectivity, good functional group tolerance, and ability to undergo intramolecular tandem reaction in mild conditions.
Results
Porous CdSe nanosheet photocatalyst
The C–X to C–D transformation proposed here relies on the effective photocatalytic splitting of D2O to generate active D radicals (Fig. 1 and Supplementary Table 1). CdSe is selected for its appropriate bandgap for solar energy absorption and a conduction band edge that is more negative in potential than water reduction potential29,30. Reducing its dimension from bulk to nanoscale further improves its catalytic performance owing to quantum confinement effect. Current synthetic methods for CdSe nanosheets utilize organic ligands to control the shape and size of the nanocrystals. However, such ligands invariably block or deactivate the active catalytic sites. To increase the density of catalytic sites, porosity can be introduced into 2D-CdSe, as the edges of these pores can boost catalytic activity owing to their lower coordination numbers and modified electronic structures31,32.
Fig. 1.

Proposed deuteration strategy. a State-of-the-art C–H to C–D exchange; b photocatalytic C–X to C–D transformation in this work
Ultrathin CdSe nanosheets can be synthesized in bulk quantities by a colloidal method based on previously reported procedure33. To improve the catalytic activity, 2D-CdSe nanosheets were corroded under acidic condition to increase porosity. The porous CdSe nanosheets were characterized by scanning transmission electron microscopy in the annular dark field mode (STEM-ADF). As shown in Fig. 2 and Supplementary Fig. 5b, as-synthesized CdSe nanosheets consist of {002} planes; the typical thickness of these sheets as measured by atomic force microscopy (AFM, Supplementary Fig. 4) is 1.7 nm (~seven layers). The porous CdSe nanosheets contain nanocrystalline domains as well as irregular pores, as shown in Fig. 2c, Supplementary Fig. 1–3 and Supplementary Table 3. STEM-ADF image and selected area electron diffraction pattern in Fig. 2d reveal that the CdSe nanosheets have hexagonal wurtzite structure. Electron energy loss spectroscopy elemental mapping and XPS data of Cd and Se (Fig. 2b, Supplementary Fig. 5 & Notes 1 ~ 3) validate that the CdSe nanosheets consists of nanocrystalline phase with a Cd/Se ratio of ~1:0.9.
Fig. 2.

Porous CdSe nanosheets. a STEM-ADF image of CdSe nanosheets, scale bar: 500 nm; b STEM-ADF image and corresponding EELS mapping of CdSe nanosheets, scale bar: 20 nm; c atomic resolution STEM-ADF image of CdSe nanocrystalline domains, scale bar: 2 nm; and d zooming in on a nanocrystal domain: (ii) enlarged filtered image of the region enclosed by the yellow box in (i) with its corresponding (iii) simulated image, (iv) atomic structure, (v) fast Fourier transform (FFT) pattern, and (vi) simulated FFT pattern, scale bar: 1 nm
The porous CdSe nanosheet consists of nanocrystalline domains, which are likely to have its optical properties modified by zero-dimensional quantum confinement34; the presence of Cd sites with unsaturated coordination further increases the electron density at the Fermi level and enhances its catalytic properties (Supplementary Fig. 10). Figure 3a compares the UV-Vis spectra of porous and non-porous CdSe nanosheets. The absorption peaks of the lowest energy electron-light hole and electron-heavy hole excitation in porous CdSe are blue-shifted by 12 nm, which are manifestations of the domain size reduction after the chemical treatment to generate porosity35. Porous CdSe also shows two red-shifted components in the photoluminescence (PL) spectra, which are absent in the non-porous sample. The red-shifted peaks are assigned to emission from trap or edge sites populated by the non-radiative relaxation of photo-excited electrons, which increases the probability of subsequent electron transfer to chemical species in the solution, thus resulting in approximately three to four times higher H2 evolution rate for our porous CdSe nanosheets (Fig. 3c) compared with commercial CdSe nanoparticles and nanoplates. The highly efficient PHE is the key to achieve successful C–X mediated deuteration, as discussed in the following section36. The band edge alignments in porous, 2D-CdSe and other photocatalysts are provided in Fig. 3b and Supplementary Fig. 6.
Fig. 3.

Band structure and photocatalytic performance of porous CdSe nanosheets. a UV-Vis and PL spectra of porous and non-porous CdSe nanosheets; b band edge energies of various photocatalysts compared with redox energy levels for water splitting and for the reduction of aryl iodides; c a comparison of the PHE activities of porous CdSe, commercial CdSe nanoparticles, and nanoplates; and d comparison of the yields and conversions for the hydrodehalogenation of p-iodoanisole using various photocatalysts
Photocatalytic C–X to C–D transformations
A core feature of our strategy is using the in situ photo-generated D radicals to realize deuterium incorporation in aryl ring through C–X bonds cleavage. The hydrodehalogenation of halides is chosen as the model reaction for studying C–X to C–D transformation. To dissolve the organic substrates, acetonitrile (CH3CN) is used to form a 1:1 mixture with water, in which case nearly quantitative yield is achieved (Supplementary Table 5, entry 1). Isotope labeling by deuterium was performed to determine the origin of H. In the case of D2O/CH3CN, the reduction product 2ao was obtained in 91% isolated yield with a deuteration ratio of 93% as determined from NMR (Supplementary Fig. 9 (b)), whereas no deuterated product was detected when H2O/CD3CN mixture was used as the solvent (Supplementary Fig. 9 (c)). These results prove convincingly that water, rather than CH3CN, is the primary H donor for this C–I hydrogenation reaction. The highest yield was obtained by combining 5 mg of CdSe with 0.25 M of Na2SO3; complete conversion took place within 2 h (Supplementary Table 5, entry 1). Control experiments using catalysts such as CdSe nanoparticles, CdSe nanoplates, commercial CdS, TiO2 nanoparticles or other sacrificing agents give lower catalytic performance for hydrodehalogenation as compared with our porous CdSe nanosheets. The presence of light, photocatalyst and a sacrificial agent are essential for the hydrodehalogenation reaction (Fig. 3d, Supplementary Tables 5 and 6). Although UV light is sufficient to dehalogenate many organic halides, we did not observe any hydrogenated product in the absence of CdSe photocatalyst (Supplementary Table 4). It is important to note that hydrogen gas cannot be directly used for hydrodehalogenation. No formation of 2c’ was detected in the dark in the presence of 1 atm. H2 (all other conditions remaining identical) even after a reaction time of 24 h (Supplementary Table 5, entry 11). Therefore, the hydrogenation of aryl iodide should be owing solely to H radicals generated from photocatalytic water splitting. It should be pointed out that all the substrates tested can be photocatalysed using porous CdSe under visible-light excitation, the reaction time is typically 2 ~ 3 times longer than with UV excitation (see Table 1 and Supplementary Table 5, entry 12 for a comparison of performances using UV and visible light), but comparable yields can be obtained.
Table 1.
Scope of photocatalytic C–X to C–D transformationa
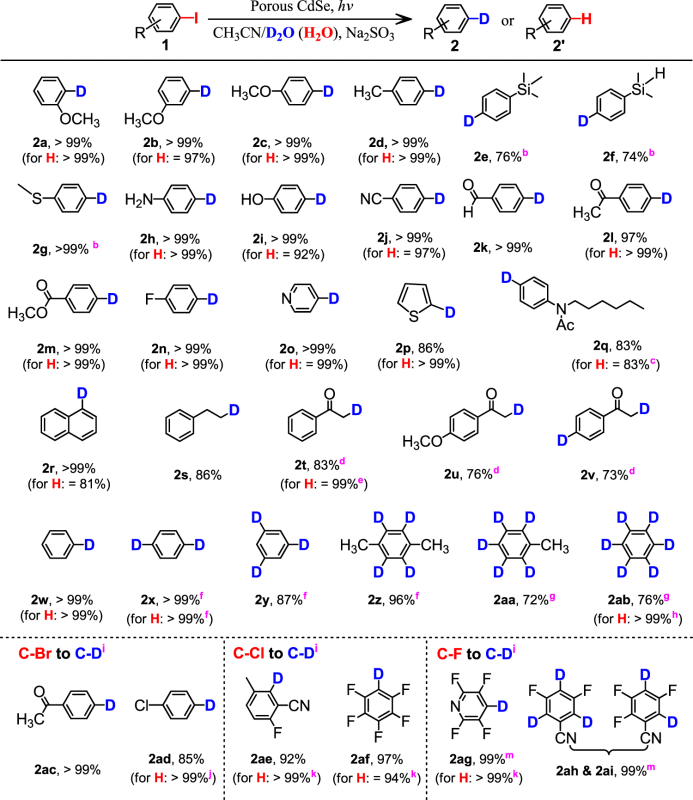
|
a Reaction conditions: halides substrate (0.1 mmol), porous CdSe (5 mg), Na2SO3 (0.25 M), CH3CN/D2O (2.5 mL/1.5 mL), irradiation 8 h (>420 nm), r.t. For hydrodehalogenation, the reaction was conducted under UV light (>280 nm) for 2 h in CH3CN/H2O (2.5 mL/2.5 mL)
b >420 nm, 12 h
c isolated yield
d 6 h, isolated yield
e >420 nm, 6–8 h
f 0.05 mmol of aryl iodides
g 0.0375 mmol of aryl iodides, CdSe (7.5 mg), Na2SO3 (0.375 M)
h 0.025 mmol of 1ab, 10 mg CdSe, 0.375 M Na2SO3, 6 h
i >280 nm, 12 h
j >420 nm, 12 h
k >280 nm, 10 h
m conversion yields were reported. Yields (Error ca. 5%) were calculated from GC measurements using standard curve
To validate the universality of this photocatalytic hydrodehalogenation of halides with water as the hydrogen source, a wide range of halogen-containing substrates were examined. For the sake of investigating the substrate scopes, some experiments were conducted in UV light owing to its shorter reaction time. As listed in Table 1, a wide range of iodinated substrates, including aryl, heteroaryl, alkyl, and alkynyl iodides are amenable to our strategy, producing the corresponding hydrogenated products in good to excellent yields with good functional groups tolerance. Obvious chemoselective hydrogenation of C–I bonds can be seen in the presence of other halogen substituents (F, Cl, and Br) on aryl rings (Table 1, 2n and Table 2, 2aj, 2ak). This chemo-selectivity may be attributed to the different reduction potentials and bond dissociation energies of C–X bonds (Supplementary Table 2)37. For 1,3,5-triiodobenzene substrate, we can obtain benzene, iodobenzene, and 1,3-diiodobenezene as the major hydrogenated product in a stepwise manner through minor modifications of standard conditions (Supplementary Table 7), which attests to the remarkable flexibility of our methodology. The C–Br, C–Cl, and C–F bonds can be also be hydrogenated in Table 1 and Supplementary Table 7 using longer reaction time.
Table 2.
D-labeled tools box from photocatalytic C–I to C–D tansformationa
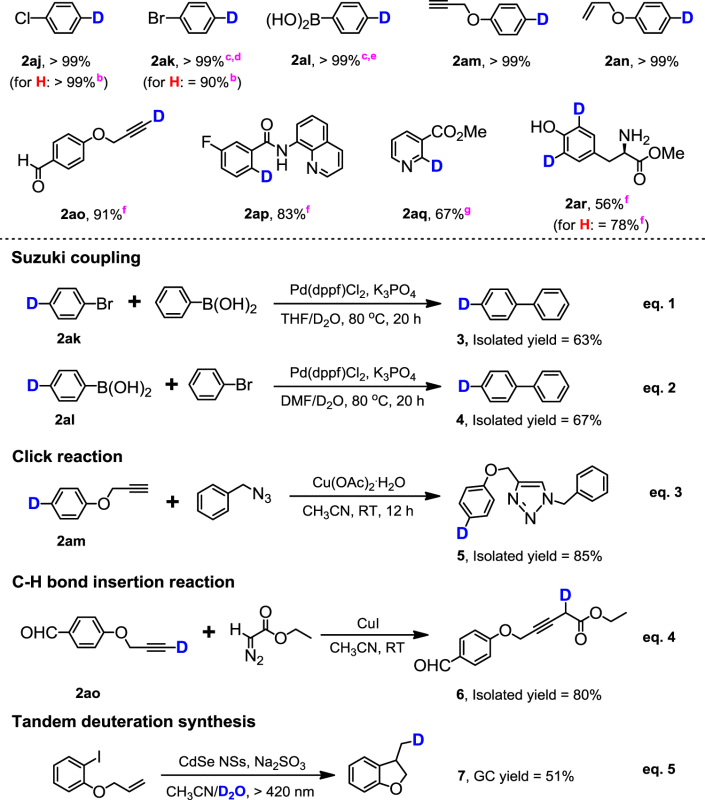
|
a Reaction conditions: aryl iodides (0.1 mmol), porous CdSe (5 mg), Na2SO3 (0.25 M), CH3CN/D2O (2.5 mL/1.5 mL), irradiation 8 h (>420 nm), r.t. For hydrodehalogenation, the reaction was conducted under UV light (>280 nm) for 2 h in CH3CN/H2O (2.5 mL/2.5 mL)
b >280 nm, 2 h
c conversion yield was reported
d THF/D2O (2.5 mL/1.5 mL)
e DMF/D2O (2.5 mL/1.5 mL)
f isolated yield
g aryl bromide, >280 nm, 12 h. Yields (Error ca. 5%) were calculated from GC-MS using standard curves
Based on the optimum conditions of hydrodehalogenation, the substrate scopes for deuteration were investigated with D2O as deuterium source. As expected, deuteration only occurs at the C–X position with good efficiency, as shown in Table 1. Note that comparable yields are obtained for deuteration and hydrogenation of the same substrate although a longer reaction time is needed for deuteration due to kinetic isotope effect (Table 1). Both the electron-rich and -deficient iodinated substrates can be deuterated by D2O (Table 1, 2a–2n). The deuterium can be specifically installed at different positions on the aryl ring without negative impact on efficiency (Table 1, 2a–2c). Owing to the mild reaction conditions, sensitive groups such as cyan, ester, amino, hydroxyl, aldehyde, and ketone are not affected (Table 1, 2h–2m), in sharp contrast to current C–H/C–D exchange and early-stage C–X deuteration processes involving strong bases. The latter requires expensive deuterium sources (CD3OD38, DCOONa39), harsh reaction conditions (e.g., BuLi at −78 °C)40 and is disadvantaged by very narrow substrate scopes. Although the H/D exchange of -NH2 and -OH groups happens in D2O inevitably, the influence on the deuteration of C–I bond is negligible (Table 1, 2h and 2i). Heterocyclic aryl iodides, as well as iodinated substrates containing a long aliphatic chain and naphthyl are good candidates, giving rise to the deuterated products in high yields (Table 1, 2o–2r). The unactivated or activated alkyl iodides are also amenable to our strategy, albeit the former gives a slightly lower yield (Table 1, 2s–2v). In addition, the number of deuterium on aryl ring can be controlled by halogen chemistry to give mono-, bi-, or multi-deuterated products, which is extremely challenging in C–H/C–D exchange (Table 1, 2w–2ab). In addition, C–Br, C–Cl, and C–F bonds which are usually non-labile using other photocatalytic methods can be deuterated in the presence of some activated functional groups (Table 1, 2ac–2ai).
In addition to the direct deuterium incorporation on target molecules, the heavy water D2O-splitting strategy proposed here can be developed into a series of useful D-containing tool kits bearing both deuterium atom and linkage moieties. For example, we have successfully incorporated deuterium on aryl halogen, boric acid, alkyne, and alkene moieties-containing molecules (Table 2, 2aj–2ar), which are often fragile under harsh conditions as well as noble metal catalytic system. The elegance of the approach is that deuterium can be readily incorporated on pharmaceutical molecules or advanced materials through subsequent Suzuki coupling, Click reaction, Heck reaction, or C–H bond insertion reaction (Table 2, eq. 1 ~ 4), which is challenging by current methods. Potentially useful deuterated molecules can be synthesized by our strategy. For example, using the kinetic isotope effect, a deuterium atom at the ortho-position of an amide group is helpful for probing reaction mechanisms in metal-catalyzed transformations, or for determining the reaction sites of a reaction (Table 2, 2ap). By converting C–Br bond to C–D bond, the deuterium labeled pharmaceutical precursor 2aq is synthesized, which can undergo further ester hydrolysis to form D-containing nicotinic acid. Nicotinic acid is a benchmarked anti-hypercholesterolemia drug and among the top 200 pharmaceutical products produced in 200941. The halogen-containing amino acids can also be deuterated via the C–X transformation (2ar). Through polymerase chain reaction, deuterium can be integrated into polypeptides, protein etc. for investigating their property owing to the isotope effect. The utility of this deuterated strategy is further demonstrated by a tandem deiodination-cyclization-deuteration of 2-(allyloxy)-1-iodobenzene, from which a radical pathway for the deuteration of halides may be operational (Table 2, eq. 5)26. The success in the preparation of deuterated benzofuran scaffold opens up the window on the follow-up synthesis of more complex deuterium-bearing molecules with new stereocentres through radical addition or coupling. Therefore, C–X to C–D transformation can provide a powerful tool kit for deuterium labeling in synthesis and drug investigation.
To illustrate the robustness and scalability of our approach, a gram-scale hydrodehalogenation of 1w (1.53 g) was carried out with a yield of 71% after irradiation for 16 h at r.t. (Supplementary Fig. 7). Moreover, porous CdSe nanosheets can be recycled and maintain a reaction efficiency of >90% after 3 runs. A slight reduction of the reactivity was observed in the fourth run (Supplementary Fig. 8), which may be due to mild photo-corrosion and/or loss of catalyst during recovery and washing process42. The excellent arylamine group tolerance (Tables 1, 2h) can be attributed to the competition with the sacrificial agent (Na2SO3). As Na2SO3 has an apparently lower oxidation potential than the valence band potential of CdSe and the oxidation potential of arylamine43, the photo-generated holes will be preferentially transferred to the sacrificial agent, which is in excess, and not to the arylamine. For alkenes and alkynes (Table 2, 2am & 2an), the super high selectivity is due to the lack of noble metal catalysts for activation44, as well as the kinetically unfavorable radical addition compared to the fast radical coupling.
Reaction mechanism for photocatalytic C–X to C–D transformations
The photocatalytic C–X bond deuteration by D2O involves a radical pathway and a possible mechanism is proposed in Fig. 4a. The reaction begins with the absorption of substrates on the photocatalysts45. Upon irradiation, electron-hole pairs are generated on porous CdSe29,30,46. Owing to the defective sites, besides electron-hole recombination, electrons are trapped and transferred to D2O and/or organic halides to form the corresponding deuterium radicals47 or the aryl radical anion (ArX·-)26–28. Following, the coupling of deuterium radical and ArX·- leads to the formation of deuterated product. The D radicals can also self-couple to release D242,47. The catalyst then recovers to its ground state after accepting an electron from the sacrificial agent (Na2SO3), whereupon SO32− converts to SO42− owing to oxidation48. The electron transfer from the CdSe to iodobenzene is evidenced by PL and femtosecond transient absorption (TA) spectroscopy. We observe a significant quenching of PL from CdSe, together with a faster decay in the bleaching band at 455 nm in TA spectra upon the addition of iodobenzene (Fig. 4b and Supplementary Fig. 15–18). A shorter electron transfer time constant was obtained from single-wavelength dynamics at 450 nm in the presence of iodobenzene, as compared to when it was absent.
Fig. 4.

Mechanism of the photocatalytic deuteration reaction. a possible radical pathway for C–X to C–D transformation; b comparison of femtosecond transient absorption kinetics of porous CdSe with/without the addition of iodobenzene in CHCl3. Pump: 400 nm, probe: 450 nm; c EPR measurements of 1 mM CdSe nanosheets with 4 mM DMPO and 0.13 M Na2SO3 in (1:1 v/v) CH3CN/H2O under UV (>280 nm) irradiation: (1) with 2 mM p-iodoanisole in the dark, (2) with 2 mM p-iodoanisole after 30 s irradiation, (3) without p-iodoanisole after 35 min irradiation, and (4) only with 1 mM CdSe and 4 mM DMPO in iodobenzene after 150 s irradiation; * and # represent peaks from DMPO-OH and DMPO-C radicals
The radical pathway for photocatalytic deuteration is confirmed by electron paramagnetic resonance (EPR) measurements in Fig. 4c and Supplementary Fig. 11–14. By trapping radicals with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) molecules, which forms stable radical adducts, the characteristic EPR signals of DMPO-OH adducts (quartet peaks, aN = aH = 14.9 G, marked by *) can be detected as water/hydroxides are oxidized to hydroxyl radicals by photo-generated holes49. Although we could not detect the presence of hydrogen radicals due to trace amounts of oxygen in the system, hydroperoxyl adducts (DMPO-OOH, marked by △) could be detected50, which might be generated from the combination of hydrogen radicals with trace amount of oxygen in the system. Three new peaks that can be assigned to the reduction product of DMPO-C adducts (aN = aH = 13.9 G, marked by #) are clearly seen after the addition of iodides51. The presence of free radicals in this system is further confirmed by the exponential decay of the EPR peak area with time (Supplementary Fig. 11) after a radical scavenger, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) is added. Supplementary Fig. 14 compares the EPR results for porous and non-porous CdSe nanosheets. The absence of EPR signals from DMPO-OH and DMPO-C adducts in non-porous CdSe nanosheets points to the much lower photocatalytic activity of this material compared to its porous counterpart.
In conclusion, using porous 2D-CdSe nanosheets as photocatalysts, selective and efficient C–X bond deuteration can be achieved with D2O. A wide range of organic iodides including aryl-, alkyl-, and alkynyl iodides can be effectively deuterated in excellent yields and with good functional groups tolerance. This method is also powerful enough to deuterate less reductively labile C–Br, C–Cl and even C–F bonds in the presence of activating groups on the ring. The good performance is due to high reductive potential of porous CdSe nanosheets and its high density of catalytic sites. This strategy introduces spatially precise and controllable number of deuterium atoms on the substrates compared to current C–H/C–D exchange. Importantly, we can further develop a D-labeled tool kit including deuterated aryl halogen, boric acid, alkyne, alkene, etc., which are useful D-containing building blocks for drugs or advanced materials. The radical coupling strategy can be further exploited to rapidly generate molecular complexity via radical cascade cyclization, for example, for ring closure and the generation of new stereo centers, or in the total synthesis of complex molecules.
Methods
Synthesis of porous CdSe nanosheets
We used a modified method from Ref. 33 to introduce nanopores on CdSe nanosheets. In brief, 1.5 mmol of CdCl2 was dissolved in 5 mL of oleylamine and 5 mL of octylamine at 120 °C for 2 h under Ar. The solution was mixed with 4.5 mmol of selenide dispersed in 2.5 mL of oleylamine and 2.5 mL of octylamine at r.t. The resulting mixture was heated to 100 °C for 16 h under Ar. After precipitation in ethanol and re-dispersing in CHCl3, as-formed CdSe nanosheets were purified using a silica column. The acidic environment of silica column causes the corrosion of CdSe to form nanopores. The porous CdSe nanosheets were then recovered by rotary evaporation.
Photocatalytic C–X to C–D transformation
Typically, 5 mg of porous CdSe nanosheets, 0.1 mmol of 4-iodoanisole (1c) and Na2SO3 (0.25 M) were dispersed in a CH3CN/D2O (2.5 mL/1.5 mL) mixture and sonicated for 30 min. The reaction mixture was then irradiated with a Xenon lamp visible light (150 W, λ > 420 nm) for 8 to 10 h at r.t. under Ar. After reaction, the mixture was centrifuged to remove photocatalyst. The supernatant was extracted by adding 5 mL of CH2Cl2 and the organic phase was analyzed by GC-MS. The conversions and yields were calculated from standard calibration curves. The yield was calculated by dividing the amount of the obtained desired product by the theoretical yield. Hydrogenated products were prepared by using CH3CN/H2O mixture under identical conditions. Details on reaction setup and synthesis of substrates can be found in Supplementary Fig. 19 ~ 76 and Supplementary Methods.
Characterization equipment
STEM and EELS (Nion UltraSTEM-100 with aberration- correction, 60 kV), TEM/EDS (FEI Titan, 80 kV), AFM (Dimension Fast Scan), XPS (AXIS UltraDLD, monochromatic Al Kα), XRD (Bruker D8), NMR (Bruker AV300), EPR (JEOL FA200), GC (Agilent 7890 A), GC-MS (Agilent 5975 C inert MSD with triple-axis detector), UV-Vis (Shimadzu UV-3600), FT-IR (Varian 3100), PL (Horiba Fluorolog-3), TA & Pump-Probe (Spectra Physics, Ti:sapphire femtosecond laser), Electrochemistry (Autolab PGSTAT30 with a 3-electrodes cell).
Data availability
All data are available from the authors upon reasonable request.
Electronic supplementary material
Acknowledgements
K.P. Loh thanks the National Research Foundation, Prime’s Minister Office, for support under the mid-sized research centre (CA2DM), and also MOE Tier 2 grant “Porous, Conjugated Molecular Framework for Energy Storage (MOE2016-T2-1-003).” C. Su thanks NNSFC (51502174), Shenzhen Peacock Plan (Grant No. 827-000113, KQTD2016053112042971), and Science and Technology Planning Project of Guangdong Province (2016B050501005) for financial support. We acknowledge Dr. Ji’en Wu for his contributions on EPR measurements, Dr. Tobias Lünskens, Reinhard Haselberges, Dr. Dong Shi, Dr. Bo Liu, and Mr. Xing Li for PL measurements, Ms. Qianqian Hu for XPS measurements, Prof. Bin Zhang and Dr. Wei Meng for insightful discussions. This work was supported in part by the U.S. Department of Energy, Office of Science, Basic Energy Science, Materials Sciences and Engineering Division, and through a user project at ORNL’s Center for Nanophase Materials Sciences (CNMS), which is a DOE Office of Science User Facility. C.L. appreciates funding support from Shenzhen University and the China Postdoctoral Science Foundation (2016M592519). Z.C. thanks the NGS Scholarship for support.
Author contributions
C.L., Z.C. conceived the research and wrote the draft. C.L. and Z.C. synthesized the materials and C.L. performed the photocatalytic reactions. X.Z., Z.C., and W.Z. conducted TEM characterization and data analysis. G.N., C.L., and Z.C. conducted EPR measurements. H.Z., C.L., Z.C. and Q.X. performed the transient measurements. K.L. performed AFM measurement. Q.G., W.T., W.F., B.T., X.P., and J. L. assisted with materials characterization and data analysis. C.S. and K.P.L supervised the research. All authors discussed and commented on the manuscript.
Competing interests
The authors declare no competing financial interests.
Footnotes
Cuibo Liu and Zhongxin Chen contributed equally to this work.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-017-02551-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chenliang Su, Email: chmsuc@szu.edu.cn.
Kian Ping Loh, Email: chmlohkp@nus.edu.sg.
References
- 1.Simmons EM, Hartwig JF. On the interpretation of deuterium kinetic isotope effects in C–H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed. 2012;51:3066–3072. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 2.Miyashita M, Sasaki M, Hattori I, Sakai M, Tanino K. Total synthesis of Norzoanthamine. Science. 2004;305:495–499. doi: 10.1126/science.1098851. [DOI] [PubMed] [Google Scholar]
- 3.Vardeny ZV, et al. Isotope effect in spin response of π-conjugated polymer films and devices. Nat. Mater. 2010;9:345–352. doi: 10.1038/nmat2633. [DOI] [PubMed] [Google Scholar]
- 4.Yu RP, Hesk D, Rivera N, Pelczer I, Chirik PJ. Iron-catalysed tritiation of pharmaceuticals. Nature. 2016;529:195–199. doi: 10.1038/nature16464. [DOI] [PubMed] [Google Scholar]
- 5.Katsnelson A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 2013;19:656. doi: 10.1038/nm0613-656. [DOI] [PubMed] [Google Scholar]
- 6.Isin EM, Elmore CS, Nilsson GN, Thompson RA, Weidolf L. Use of adiolabeled compounds in drug metabolism and pharmacokinetic studies. Chem. Res. Toxicol. 2012;25:532–542. doi: 10.1021/tx2005212. [DOI] [PubMed] [Google Scholar]
- 7.Belleau B, Burba J, Pindell M, Reiffenstein J. Effect of deuterium substitution in sympathomimetic amines on adrenergic responses. Science. 1961;133:102. doi: 10.1126/science.133.3446.102. [DOI] [PubMed] [Google Scholar]
- 8.Magano J, Dunetz JR. Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals. Chem. Rev. 2011;111:2177–2250. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt C. First deuterated drug approved. Nat. Biotechnol. 2017;35:493–494. doi: 10.1038/nbt0617-493. [DOI] [PubMed] [Google Scholar]
- 10.Atzrodt J, Derdau V, Fey T, Zimmermann J. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 2007;46:7744–7765. doi: 10.1002/anie.200700039. [DOI] [PubMed] [Google Scholar]
- 11.Taglang C, et al. Enantiospecific C–H activation using ruthenium nanocatalysts. Angew. Chem. Int. Ed. 2015;54:10474–10477. doi: 10.1002/anie.201504554. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee B, Krishnakumar V, Gunanathan C. Selective α‑deuteration of amines and amino acids using D2O. Org. Lett. 2016;18:5892–5895. doi: 10.1021/acs.orglett.6b02978. [DOI] [PubMed] [Google Scholar]
- 13.Liu K, et al. Solid-state deuterium NMR of imidazole ligands in Cytochrome c peroxidase. J. Am. Chem. Soc. 1998;120:10199–10202. doi: 10.1021/ja9732385. [DOI] [Google Scholar]
- 14.Crabtree R, Felkin H, Morris G. Cationic iridium diolefin complexes as alkene hydrogenation catalysts and isolation of some related hydrido complexes. J. Organomet. Chem. 1977;141:205–215. doi: 10.1016/S0022-328X(00)92273-3. [DOI] [Google Scholar]
- 15.Nilsson GN, Kerr WJ. The development and use of novel iridium complexes as catalysts for ortho-directed hydrogen isotope exchange reactions. J. Label. Comp. Radiopharm. 2010;53:662–667. doi: 10.1002/jlcr.1817. [DOI] [Google Scholar]
- 16.Ma S, Villa G, Thuy-Boun PS, Homs A, Yu JQ. Palladium-catalyzed ortho-selective C–H deuteration of arenes: evidence for superior reactivity of weakly coordinated palladacycles. Angew. Chem. Int. Ed. 2014;53:734–737. doi: 10.1002/anie.201305388. [DOI] [PubMed] [Google Scholar]
- 17.Emmert MH, Gary JB, Villalobos JM, Sanford MS. Platinum and palladium complexes containing cationic ligands as catalysts for arene H/D exchange and oxidation. Angew. Chem. Int. Ed. 2010;49:5884–5886. doi: 10.1002/anie.201002351. [DOI] [PubMed] [Google Scholar]
- 18.Hale LVA, Szymczak NK. Stereoretentive deuteration of α‑chiral amines with D2O. J. Am. Chem. Soc. 2016;138:13489–13492. doi: 10.1021/jacs.6b07879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, Moore BS. Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chem. Rev. 2017;117:5619–5674. doi: 10.1021/acs.chemrev.6b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrone DA, Ye J, Lautens M. Modern transition-metal-catalyzed carbon-halogen bond formation. Chem. Rev. 2016;116:8003–8104. doi: 10.1021/acs.chemrev.6b00089. [DOI] [PubMed] [Google Scholar]
- 21.Kuriyama M, et al. Deuterodechlorination of aryl/heteroaryl chlorides catalyzed by a palladium/unsymmetrical NHC system. J. Org. Chem. 2016;81:8934–8946. doi: 10.1021/acs.joc.6b01609. [DOI] [PubMed] [Google Scholar]
- 22.Qiu GYS, Li YW, Wu J. Recent developments for the photoinduced Ar–X bond dissociation reaction. Org. Chem. Front. 2016;3:1011–1027. doi: 10.1039/C6QO00103C. [DOI] [Google Scholar]
- 23.Corrigan N, Shanmugam S, Xu JT, Boyer C. Photocatalysis in organic and polymer synthesis. Chem. Soc. Rev. 2016;45:6165–6212. doi: 10.1039/C6CS00185H. [DOI] [PubMed] [Google Scholar]
- 24.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staveness D, Bosque I, Stephenson CRJ. Free radical chemistry enabled by visible light-induced electron transfer. Acc. Chem. Res. 2016;49:2295–2306. doi: 10.1021/acs.accounts.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh I, Ghosh T, Bardagi JI, König B. Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science. 2014;346:725–728. doi: 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen JD, D’Amato EM, Narayanam JMR, Stephenson CRJ. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nat. Chem. 2012;4:854–859. doi: 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]
- 28.Senaweera SM, Singh A, Weaver JD. Photocatalytic hydrodefluorination: facile access to partially fluorinated aromatics. J. Am. Chem. Soc. 2014;136:3002–3005. doi: 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y, Huang Y, Zhang B. Rational design of semiconductor-based photocatalysts for advanced photocatalytic hydrogen production: the case of cadmium chalcogenides. Inorg. Chem. Front. 2016;3:591–615. doi: 10.1039/C5QI00217F. [DOI] [Google Scholar]
- 30.Jiao Y, Zheng Y, Jaroniecb M, Qiao SZ. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015;44:2060–2086. doi: 10.1039/C4CS00470A. [DOI] [PubMed] [Google Scholar]
- 31.Su C, et al. Probing the catalytic activity of porous graphene oxide and the origin of this behaviour. Nat. Commun. 2012;3:1298. doi: 10.1038/ncomms2315. [DOI] [PubMed] [Google Scholar]
- 32.Su C, et al. Tandem catalysis of amines using porous graphene oxide. J. Am. Chem. Soc. 2015;137:685–690. doi: 10.1021/ja512470t. [DOI] [PubMed] [Google Scholar]
- 33.Son JS, et al. Large-scale soft colloidal template synthesis of 1.4 nm thick CdSe nanosheets. Angew. Chem. Int. Ed. 2009;48:6861–6864. doi: 10.1002/anie.200902791. [DOI] [PubMed] [Google Scholar]
- 34.Deng D, et al. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016;11:218–230. doi: 10.1038/nnano.2015.340. [DOI] [PubMed] [Google Scholar]
- 35.Jensen SC, Homan SB, Weiss EA. Photocatalytic conversion of nitrobenzene to aniline through sequential proton-coupled one-electron transfers from a cadmium sulfide quantum dot. J. Am. Chem. Soc. 2016;138:1591–1600. doi: 10.1021/jacs.5b11353. [DOI] [PubMed] [Google Scholar]
- 36.Voiry D, Yang J, Chhowalla M. Recent strategies for improving the catalytic activity of 2D TMD nanosheets toward the hydrogen evolution reaction. Adv. Mater. 2016;28:6197–6206. doi: 10.1002/adma.201505597. [DOI] [PubMed] [Google Scholar]
- 37.Meites, L., Zuman, P., Scott, W. J. & Lampugani, L. CRC handbook series in organic electrochemistry. CRC press (1977);
- 38.Janni M, Peruncheralathan S. Catalytic selective deuteration of halo(hetero) arenes. Org. Biomol. Chem. 2016;14:3091–3097. doi: 10.1039/C6OB00193A. [DOI] [PubMed] [Google Scholar]
- 39.Zhang HH, Bonnesen PV, Hong K. Palladium-catalyzed Br/D exchange of arenes: selective deuterium incorporation with versatile functional group tolerance and high efficiency. Org. Chem. Front. 2015;2:1071–1075. doi: 10.1039/C5QO00181A. [DOI] [Google Scholar]
- 40.Alonso F, Beletskaya IP, Yus M. Metal-mediated reductive hydrodehalogenation of organic halides. Chem. Rev. 2002;102:4009–4091. doi: 10.1021/cr0102967. [DOI] [PubMed] [Google Scholar]
- 41.McGrath N, A. Brichacek M, Njardarson JT. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ. 2010;87:1348–1349. doi: 10.1021/ed1003806. [DOI] [Google Scholar]
- 42.Li XB, et al. Mechanistic insights into the interface-directed transformation of thiols into disulfides and molecular hydrogen by visible-light irradiation of quantum dots. Angew. Chem. Int. Ed. 2014;53:2085–2089. doi: 10.1002/anie.201310249. [DOI] [PubMed] [Google Scholar]
- 43.Neta P, Huie RE. Free-radical chemistry of Sulfite. Environ. Health Perspect. 1985;64:209–217. doi: 10.1289/ehp.8564209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong JJ, et al. Combining visible light catalysis and transfer hydrogenation for in situ efficient and selective semihydrogenation of alkynes under ambient conditions. Chem. Commun. 2016;52:1800–1803. doi: 10.1039/C5CC08697C. [DOI] [PubMed] [Google Scholar]
- 45.Kisch H. Semiconductor photocatalysis for chemoselective radical coupling reactions. Acc. Chem. Res. 2017;50:1002–1010. doi: 10.1021/acs.accounts.7b00023. [DOI] [PubMed] [Google Scholar]
- 46.Wang JJ, et al. Photocatalytic hydrogen evolution from glycerol and water over nickel-hybrid cadmium sulfide quantum dots under visible-light irradiation. ChemSusChem. 2014;7:1468–1475. doi: 10.1002/cssc.201400028. [DOI] [PubMed] [Google Scholar]
- 47.Meng QY, et al. A cascade cross-coupling hydrogen evolution reaction by visible light catalysis. J. Am. Chem. Soc. 2013;135:19052–19055. doi: 10.1021/ja408486v. [DOI] [PubMed] [Google Scholar]
- 48.Zhao J, Holmes MA, Osterloh FE. Quantum confinement controls photocatalysis: a free energy analysis for photocatalytic proton reduction at CdSe nanocrystals. ACS Nano. 2013;7:4316–4325. doi: 10.1021/nn400826h. [DOI] [PubMed] [Google Scholar]
- 49.Ipe BI, Lehnig M, Niemeyer CM. On the generation of free radical species from quantum dots. Small. 2005;1:706–709. doi: 10.1002/smll.200500105. [DOI] [PubMed] [Google Scholar]
- 50.Clément JL, et al. Assignment of the EPR spectrum of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) superoxide spin adduct. J. Org. Chem. 2005;70:1198–1203. doi: 10.1021/jo048518z. [DOI] [PubMed] [Google Scholar]
- 51.Michail K, Siraki AG. Post-trapping derivatization of radical-derived EPR-silent adducts: application to free radical detection by HPLC/UV in chemical, biochemical, and biological systems and comparison with EPR spectroscopy. Anal. Chem. 2012;84:6739–6746. doi: 10.1021/ac301142c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon reasonable request.