


Abstract
Human Pumilio proteins, PUM1 and PUM2, are sequence specific RNA-binding proteins that regulate protein expression. We used RNA-seq, rigorous statistical testing and an experimentally derived fold change cut-off to identify nearly 1000 target RNAs—including mRNAs and non-coding RNAs—that are functionally regulated by PUMs. Bioinformatic analysis defined a PUM Response Element (PRE) that was significantly enriched in transcripts that increased in abundance and matches the PUM RNA-binding consensus. We created a computational model that incorporates PRE position and frequency within an RNA relative to the magnitude of regulation. The model reveals significant correlation of PUM regulation with PREs in 3′ untranslated regions (UTRs), coding sequences and non-coding RNAs, but not 5′ UTRs. To define direct, high confidence PUM targets, we cross-referenced PUM-regulated RNAs with all PRE-containing RNAs and experimentally defined PUM-bound RNAs. The results define nearly 300 direct targets that include both PUM-repressed and, surprisingly, PUM-activated target RNAs. Annotation enrichment analysis reveal that PUMs regulate genes from multiple signaling pathways and developmental and neurological processes. Moreover, PUM target mRNAs impinge on human disease genes linked to cancer, neurological disorders and cardiovascular disease. These discoveries pave the way for determining how the PUM-dependent regulatory network impacts biological functions and disease states.
INTRODUCTION
Post-transcriptional mechanisms play key roles in regulation of gene expression, acting to control mRNA processing, localization, translation and stability. The richness and diversity of post-transcriptional control is alluded to by the discovery of a multitude of RNA-binding regulatory factors, including RNA-binding proteins (RBPs) and small RNAs (1,2). Moreover, the application of genomic technologies to study post-transcriptional regulation has provided a panoramic view of the expansive role of RBPs in gene regulation (3). A crucial remaining challenge is to identify the RNAs that are bound by each RBP, and to measure the functional impact of that RBP on RNAs. Accomplishing this goal can provide important insights into the regulatory activity and biological roles of the RBP, leading to a better understanding of the mechanisms that control gene expression networks.
In this study, we sought to identify RNAs that are functionally regulated by the human RBPs, PUMILIO1 (PUM1) and PUMILIO2 (PUM2). These RBPs share a conserved RNA-binding domain, the Pumilio Homology Domain (Pum-HD), which defines the eukaryotic PUF family (named after Drosophila melanogaster Pumilio and Caenorhabditis elegans Fem-3 Binding Factor) (4–6). The Pum-HD is composed of eight repeats of a three α-helical module that form a crescent-shaped molecule (7–9). Each repeat can recognize a ribonucleotide base, mediated by a three amino acid–RNA recognition motif (TRM). In the cases of human PUM1 and PUM2, each protein binds with high affinity and specificity to the Pumilio Response Element (PRE) with the consensus 5′-UGUANAUA (where N is A, C, G or U) (7,10–14).
The indistinguishable RNA-binding specificities of PUM1 and PUM2 suggested the potential for functional overlap. Indeed, several lines of evidence indicate redundant functions. First, the PUMs are highly similar; their Pum-HD domains are 94% identical with highly similar three dimensional structures and their TRMs are identical (10,15,16). Second, both PUMs are coincidently expressed widely throughout tissues and cell types (15). Third, each PUM can act as a repressor; when directed to a reporter mRNA, either PUM inhibits protein expression and reduces the mRNA level with similar magnitude (17). Moreover, in cellular repression assays, the two PUMs are functionally redundant; when one is depleted, the other can repress an mRNA bearing PREs in its 3′ untranslated region (UTR) (17). There is even evidence of compensatory regulation of the two PUMs; each PUM binds the others mRNA via PREs (11,12) and knockdown of PUM1 results in a >30% increase of PUM2 mRNA expression (ENCODE datasets ENCSR745WVZ and ENCSR620PUP) and increased PUM2 protein expression (18). It has also been directly demonstrated that both PUMs must be depleted to fully alleviate repression mediated by PRE elements (17,18). In light of the overlapping function of PUM1 and PUM2, it is important to consider both PUMs when investigating the RNAs they regulate.
The repressive activity of human PUMs is consistent with the inhibitory activities of PUF family members from model organisms including mouse, Xenopus, Drosophila, C. elegans and Saccharomyces cerevisiae (4,19,20). Though the mechanism of repression by PUM proteins is not fully understood, key principles have emerged. PUM proteins repress protein expression from a target mRNA through multiple mechanisms including inhibition of translation and acceleration of mRNA degradation (20). Protein expression is reduced in part by PUM-mediated antagonism of the translation factor poly(A)-binding protein (21). PUMs also repress by recruiting the CCR4-NOT deadenylase complex and consequently promote shortening of the poly(A) tail, which further reduces translation and leads to mRNA degradation (17,21,22). Multiple repression domains in the N-terminus of PUM proteins contribute to degradation of PRE-containing reporter mRNAs (21,23). As a result of these mechanisms, PUM-mediated repression is manifested in the substantial decrease in the level of PRE-containing RNAs, as we and others have previously documented (12,17,24,25).
In model organisms, several strategies have been utilized to identify the functions of PUF proteins and their target mRNAs. Genetic analysis revealed phenotypes of PUF mutants including defects in development, fertility and neurological functions (20,26) and, in certain cases, regulation of specific mRNAs related to these phenotypes have been reported. For example, D. melanogaster Pumilio represses the hunchback mRNA to control embryonic development (27–30). In the germline, D. melanogaster Pumilio controls stem cell proliferation and fertility by repressing Cyclin B and Mei-P26 mRNAs (31–35). In the nervous system, D. melanogaster Pumilio affects long term memory formation and was shown to repress the dlg1 mRNA in the mushroom body—a site of learning and memory formation (36,37). Moreover, D. melanogaster Pumilio regulates development and morphology of neurons (e.g. by regulation of hid mRNA (38,39)) and motoneuron function (e.g. by regulation of paralytic and eIF-4E mRNAs (40–42)). Biochemical approaches identified hundreds of additional mRNAs bound by D. melanogaster Pumilio, indicating a broader regulatory role (43,44). In C. elegans, PUFs also regulate specific mRNAs to control germline stem cells and formation of gametes (5,45,46).
In mammals, an important gap in our understanding of the biological roles and regulatory functions of mammalian PUMs arises from the lack of information regarding their functional impact on gene expression. Important insights are emerging from genetic analysis in mice, where PUM1 and PUM2 genes have been individually disrupted, revealing several phenotypes (47,48). In a few instances, target mRNAs related to the mutant phenotypes have been identified, summarized in Supplementary Table S1. PUM1 or PUM2 knockout mice have reduced body mass (48,49). Simultaneous knockout of both PUM genes was recently reported to be lethal (50). Interestingly, the relationships of PUMs to fertility and neurological processes appear to be conserved in mammals. Disruption of either PUM has negative effects on fertility of mice, though the severity of the phenotypes varies. PUM1 knockout caused reduced ovarian follicle formation and meiotic development of oocytes, resulting in infertility in female mice (48). The target mRNAs underlying this phenotype remain unknown. In male mice, knockout of PUM1 reduced fertility substantially and increased apoptosis of developing sperm as a result of increases in p53 pathway components, some of which are bound and regulated by PUM1 (25) (Supplementary Table S1). PUM2 is also implicated in the male germline, as shown by smaller testes in a PUM2 gene trap mutant mouse, yet these mice remained fertile (47). The mRNAs regulated by PUM2 in the germline remain unknown. Though binding of PUM2 to several germline mRNAs has been reported (51–53), functional effects on PUM2 on these transcripts remains undocumented.
Several lines of evidence also support regulatory roles of mammalian PUMs in the nervous system. Knockout of PUM2 causes a number of neurological abnormalities in mice, including deficiencies in spatial and object memory, and impaired behavior and reduced seizure threshold (49). Mouse and human PUM1 repress the Ataxin1 (ATXN1) mRNA, encoded by the Spinocerebellar Ataxia Type 1 (SCA-1)-associated gene in neurons and cultured cells (54). Further, PUM1 knockout mice exhibit defective motor function and their brains exhibit signs of neurodegeneration (54). In cultured neurons, depletion of PUM2 altered neuronal morphology and electrophysiology, and alleviated repression of the translation factor eIF4E (55). PUM2 was also shown to control axon potential in rat cortical pyramidal neurons by repressing the sodium channel Nav1.6/SCN8A, an ortholog of the D. melanogaster Pumilio target paralytic (56). Interestingly, PUM2 is present in dendritic mRNA–protein particles that may mediate repression and localization of mRNAs to synapses (55,57), but the identity of these transcripts remains unknown.
In cell culture studies, depletion of PUMs was shown to promote cell proliferation and PUM1 repressed expression of the CDKN1B/p27 tumor suppressor (18). Over-expression of PUM2, in combination with human Nanos proteins, was reported to inhibit the mRNA encoding the transcription factor E2F3 in bladder cancer cells (58). Furthermore, over-expression of PUM2 was shown to repress ERK2 and p38α reporter genes in human embryonic stem cells (59). More recently, PUMs were reported to control genome stability in cultured cells and over-expression of PUM1 or PUM2 reduced levels of 21 mRNAs related to genome stability (Supplementary Table S1) (24). While illuminating, these studies analyzed the PUMs individually, though both PUMs are present in the cells and can bind and repress the same PRE-containing mRNAs. Several studies identified thousands of RNAs bound by PUM1 and PUM2 in cultured human cells using PAR-CLIP (photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation) or RIP-Chip (RNP immunoprecipitation–microarray) approaches (11–13). Moreover, depletion of PUM1 was found to stabilize three PUM1-bound mRNAs, consistent with PUM1-mediated mRNA degradation (Supplementary Table S1) (12). While these experiments implicate the potential far-reaching effects of PUMs, a remaining challenge is to identify the repertoire of functionally regulated mRNAs.
Here, we sought to identify RNAs that are functionally regulated by PUM1 and PUM2 by depleting both PUMs from human cells and measuring differential RNA expression using RNA-sequencing (RNA-seq) analysis. The advantages of RNA-seq analysis are numerous: most importantly, its dynamic range of detection is broad, quantitative and reproducible (60). By knocking down PUM1 and PUM2, we circumvent possible complications with the proteins’ overlapping functionality. We took a multi-dimensional approach to identify PUM-regulated target RNAs using the following criteria. First, the abundance of PUM-regulated RNAs should reproducibly change when PUMs are depleted. We identified nearly 1000 RNAs that were significantly, differentially expressed upon PUM knockdown. Second, to identify high confidence directly regulated PUM targets, we identified the subset of differentially expressed RNAs that contain a PRE motif. We found that PUM-repressed RNAs were highly enriched among the set with the PRE motif. Moreover, we integrated experimental data demonstrating physical interaction of the differentially expressed RNAs with the PUMs. Unexpectedly, we also identified a minor category of direct PUM-target RNAs that are stabilized rather than destabilized by PUMs. We then validated regulation of multiple PUM targets, including repressed and activated categories, by quantitative reverse transcription and polymerase chain reaction (qRT-PCR) analysis, quantitative western blot analysis and reporter gene assays. We interrogated enrichment of regulatory motifs and gene ontologies within the PUM-regulated mRNAs, providing important new insights into their regulatory roles of PUMs in diverse biological processes including cell–cell communication and adhesion, major developmental signaling pathways and disease associations. Together, our data and analyses provide new insights into the biological processes controlled by PUMs and their mechanism of regulation.
MATERIALS AND METHODS
Cell culture and siRNA transfections for RNA-seq
Human HEK293 cells were obtained from ATCC and cultured at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium with glucose and 1× Penicillin/Streptomycin/Glutamine and 10% Fetal Bovine Serum (FBS) (Gibco) as described previously (17). For RNAi, PUMs were knocked down in HEK293 cells using On-target Plus Smartpool siRNAs (GE Dharmacon) for PUM1 (L-014179-00), PUM2 (L-014031-02). The siRNA sequences are reported Supplementary Table S2. Non-targeting control siRNAs (D-001810-01)(GE Dharmacon) served as a negative control. For RNA-seq experiments, HEK293 cells (2 × 105 cells per well) were seeded into a 12-well culture plate in 1 ml of medium. Four replicates were performed for each condition. After 24 h, 500 μl culture medium was removed and cells were transfected with 10 fmoles of siRNAs in 500 μl of transfection mix (10 nM final concentration of siRNA per well) using Dharmafect-I (GE Dharmacon) following the manufacturer’s specifications. After 8 h of siRNA treatment, 500 μl culture medium was refreshed. After 24 h, 500 μl culture medium was removed and cells were transfected again with an additional 10 fmoles of siRNAs using Dharmafect-I. After 8 h of siRNA treatment, 650 μl culture medium was refreshed. Cells were harvested 48 h after last transfection for RNA purification and RNA-seq experiments using three of the replicates. Protein lysates were created from one replicate by lysing cells on ice in TNEMN150 (50 mM Tris pH 8, 0.5% Non-idet P40, 1 mM ethylenediaminetetraacetic acid (EDTA), 2 mM MgCl2, 150 mM NaCl) with 1× protease inhibitor cocktail (1 mM PMSF, 50 μg/ml of aprotinin, 50 μg/ml of pepstatin and 50 μg/ml leupeptin) as previously described (17).
RNA-seq
RNA-seq analysis was performed on RNA purified from HEK293 cells using the Maxwell simplyRNA cells kit (Promega) and a Maxwell 16 instrument. Purity and integrity of total RNA was determined using a Bioanalyzer (Agilent) before the sequencing preps: all total RNA samples had RIN values of 10, indicating high quality and integrity. Polyadenylated RNAs were then isolated from the total RNA using oligo-dT purification (Agencourt) and prepared for sequencing using TruSeq reagents and protocol (TruSeq RNA Sample preparation kit 48 (#RS-930-2001, 15008136, Rev A, Nov 2010, Illumina). Each sample’s library was barcoded according to the Illumina TruSeq methodology. Libraries were then analyzed via Bioanalyzer to verify proper size and concentration. Samples were submitted to the University of Michigan Sequencing Core for paired end, 100 base sequencing on an Illumina HiSeq instrument. Libraries were multiplexed in a single ‘lane’. Low quality sequence reads were filtered by Illumina’s CASAVA software. The resulting dataset was subjected to FASTQC analysis and trimmed to give 86 quality base reads. Sequencing yielded on average 33 million reads per sample.
The data analysis pipeline included analysis with the Tuxedo suite: (i) TopHat 2.0.9 and Bowtie 2.1 were used to map and align reads, Cufflinks was then used to quantify abundance of each transcript, and Cuffmerge and Cuffdiff (2.1.1) using a false discovery rate of ≤0.05 were used to assess differential abundance as described in (61–63); genes that could not be detected in the RNA-seq data were excluded from downstream analysis. Relative abundance was reported in fragments per kilobase of exon per million fragments (FPKM values). Fold change between the PUM knockdown and non-targeting control samples was calculated and the data was assessed using a 1.3-fold cutoff in addition to the False Discovery Rate (FDR) threshold described above. Confidence intervals for log fold change values were established by bootstrapping using sampled fragment abundances generated by Cufflinks during differential expression calling. Complete reporting of ENCODE guidelines for RNA-seq experiments is included in the Supplementary Data. Both the raw and processed data have been submitted to the Gene Expression Omnibus (Accession GSE95412).
Reverse transcriptase and quantitative PCR
A minimum of three replicate samples of HEK293 cells, grown in 6-well plates, were transfected with either non-targeting control siRNA or PUM1 and PUM2 siRNA smartpools as described above. Total RNA was purified from each sample using Maxwell simplyRNA cells kit (Promega), including on bead DNase treatment and then subjected to qRT-PCR analysis (64). cDNA was synthesized using GoScript reverse transcriptase (Promega) and random hexamers (IDT) as indicated by the manufacturer’s instructions and as described in (64).
Quantitative PCR was carried out using GoTaq qPCR master mix (Promega) and a CFX96 instrument (Bio-Rad). Cycling conditions were as follows: (i) 95°C for 3 min, (ii) 95°C for 10 s, (iii) 65°C for 30 s and (iv) 72°C for 40 s. Steps (ii) through (iv) were repeated for a total of 40 cycles. Negative control reactions were performed in the absence of template or reverse transcriptase. Cycle thresholds (Ct) were measured using the CFX Manager software and analyzed using the ΔΔCt method (65,66). ΔCt was calculated by normalizing to the 18S gene Ct values. We then calculated ΔΔCt as follows: ΔΔCt = ΔCt (target RNAi) − ΔCt (control RNAi). Fold change values were then calculated as fold change = 2−ΔΔCt. Knockdown of PUM1 and PUM2 mRNAs were verified in each experiment by qRT-PCR analysis. Primer sequences are provided in Supplementary Table S2. All primer set amplification efficiencies were optimized to 90–110% at 200 nM final concentration, as reported in Supplementary Table S2. Statistical analysis is described below, and log2 fold change, number of replicates and statistical values are reported in Supplementary Table S3. Complete reporting of MIQE guidelines for QPCR Experiments is included in Supplementary Data.
Plasmids
Renilla luciferase reporters (RnLuc) were created using the psiCheck1 plasmid (Promega), which has a minimal 3′ UTR with a multiple cloning site and synthetic cleavage and polyadenylation signal (64). To create 3′ UTR reporters for PUM target mRNAs, the target 3′ UTR sequence of the gene of interest was amplified from human genomic DNA (Promega), beginning just after the stop codon and ending just before the cleavage-polyadenylation signal, with primers that added flanking Xho1 and Not1 sites. Primer sequences are reported in Supplementary Table S2. The SMPDL3A 3′ UTR was generated as a gBlock (IDT). Restriction digested PCR products were then inserted into the Xho1 and Not1 sites of psiCheck1. To inactivate each PRE in psiCheck1 reporters, nucleotide positions 1–3 of the PRE 5′-UGUACAUA PREs within each 3′ UTR were mutated to ACA using either Quikchange or inverse PCR strategies. 3′ UTR PRE (1×, 2×, 3× and 4×) reporters were all cloned using inverse PCR with the primers listed in Supplementary Table S2. The firefly luciferase (FfLuc) plasmid, pGL4.13 (Promega) was used as a transfection efficiency control (64).
Luciferase assays
Renilla (75 ng) and firefly (25 ng) reporter plasmids were transfected into 20 000 HEK293 cells per well of 96-well plates using FuGENE HD (Promega). Forty-eight hours after transfection, luciferase activity was measured with Dual-Glo reagent using a Glomax Discover luminometer (Promega). To normalize for transfection efficiency, a relative response ratio was calculated by dividing the relative light unit values of RnLuc by those for FfLuc for each individual well (64). A minimum of three replicates were analyzed per condition and results were replicated in multiple independent experiments. Fold change values were then calculated relative to the control condition, as indicated in the figure legends. For minimal PRE reporters in Figure 1, fold change was calculated relative to the corresponding mutant PRE reporter. For target gene 3′ UTR reporters in Figures 3 and 4, fold change was calculated relative to the parental vector psiCheck1 with a minimal 3′ UTR containing no regulatory elements. Statistical analysis is described below and values and statistical data are reported in Supplementary Table S3.
Figure 1.

Transcriptome-wide analysis identifies PUM-regulated transcripts in HEK293 cells. (A) Western blot detection of PUM1 and PUM2 in HEK293 cells treated with PUM1 and PUM2 (PUM) or non-target control (NTC) siRNAs. Western blot detection of GAPDH served as a control for equivalent loading of the sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. (B) Density plot comparing the observed log2 fold change values from RNA-seq experiments to the overall abundance of transcripts for each corresponding gene in the NTC control case. (C) Effects of increasing numbers of wild-type (WT) PRE sites relative to mutated PRE (PREmt) sites in a minimal luciferase reporter construct. For this and subsequent figures showing luciferase or qRT-PCR-based assays, we analyzed the data using a hierarchical Bayesian model (see ‘Materials and Methods’ section for details) to make optimal use of the experimental information available; observed data are shown as points and the error bars define a 95% credible interval. We mark as significant (*) any case with a 95% posterior probability of having the observed sign, and as highly significant (**) any case with a 95% chance of showing at least a 1.3-fold change (that is, exceeding the magnitude of the significance threshold used in analyzing our RNA-seq data). N.b. the 1× PRE case shown in this panel does not reach the threshold for being highly significant, since its observed value was used to define that very threshold. (D) Volcano plot of log2 fold change (PUM knockdown relative to NTC) versus adjusted P-values for all gene expression levels. Several specific classes of genes are highlighted as described in the legend, including previously reported PUM targets from mouse and human and PUM repressed and activated targets that are validated in this study. (E) Density plot comparing log2 fold change values to expression levels (plotted as log2 FPKM in the NTC control samples) for only the genes that showed significant changes >1.3-fold in magnitude in response PUM1/2 knockdown.
Figure 3.

Validation of PUM-mediated repression of direct target mRNAs. (A) Change in levels of FZD8 mRNA or protein upon PUM1 and PUM2 knockdown, assessed using the indicated methods including RNA seq or qRT-PCR using three RNAi conditions, or by quantitative western blotting. PUM knockdown was achieved using individual siRNAs (PUM1-1 or PUM1-3; PUM2-2 or PUM2-4) or Smart Pools (SP) of four specific siRNAs to each target. Error bars indicate 95% confidence intervals (RNA-seq) or credible intervals (other assays) obtained as described in ‘Materials and Methods’ section. We mark values with a posterior probability of change in the indicated direction >95% with one asterisk (*), and those with a posterior probability >95% of passing the 1.3-fold change cut-off applied to our RNA-seq data with two asterisks (**). (B) Analysis of DEK mRNA and protein levels in response to PUM1 and PUM2 knockdown using RNA-seq, qRT-PCR and quantitative western blot assays. (C) Analysis of SMPDL3A mRNA levels in response to PUM1 and PUM2 knockdown using RNA-seq or qRT-PCR assays. (D) Analysis of RET mRNA levels in response to PUM1 and PUM2 knockdown using RNA-seq or qRT-PCR assays. (E) Analysis of ANO4 mRNA levels in response to PUM1 and PUM2 knockdown using RNA-seq or qRT-PCR assays. (F) Analysis of NOVA2 mRNA levels in response to PUM1 and PUM2 knockdown using RNA-seq or qRT-PCR assays. (G) Analysis of SCUBE1 mRNA levels in response to PUM1 and PUM2 knockdown using RNA-seq or qRT-PCR assays. (H) Analysis of L1CAM transcript levels in response to PUM1 and PUM2 knockdown using RNA-seq and qRT-PCR. (I) Analysis of LEFTY2 transcript and protein levels in response to PUM1 and PUM2 knockdown, assessed by RNA-seq and quantitative western blotting. (J) In vivo reporter assay testing the effects of a PRE mutation in the 3′ UTR of FZD8. Shown are log2 fold change values in RnLuc activity relative to an RnLuc control for cells transfected with RnLuc bearing the FZD8 3′ UTR WT or the same sequence with a PREmt. Significance stars follow the convention in panel A, and are shown relative to RnLuc itself (above each symbol) or between the WT and PREmt cases (crossbar). (K) Reporter gene analysis of PRE-mediated regulation by the DEK 3′ UTR, comparing the regulatory activities of the two WT and mutant PREs in the DEK 3′ UTR. (L) Reporter gene analysis of PRE-mediated regulation by the SMPDL3A 3′ UTR, comparing the regulatory activities of WT and mutant PRE SMPLDL3A 3′ UTRs. (M) Reporter gene analysis of PRE-mediated regulation by the RET 3′ UTR, comparing the regulatory activities of WT and mutant PRE RET 3′ UTRs.
Figure 4.
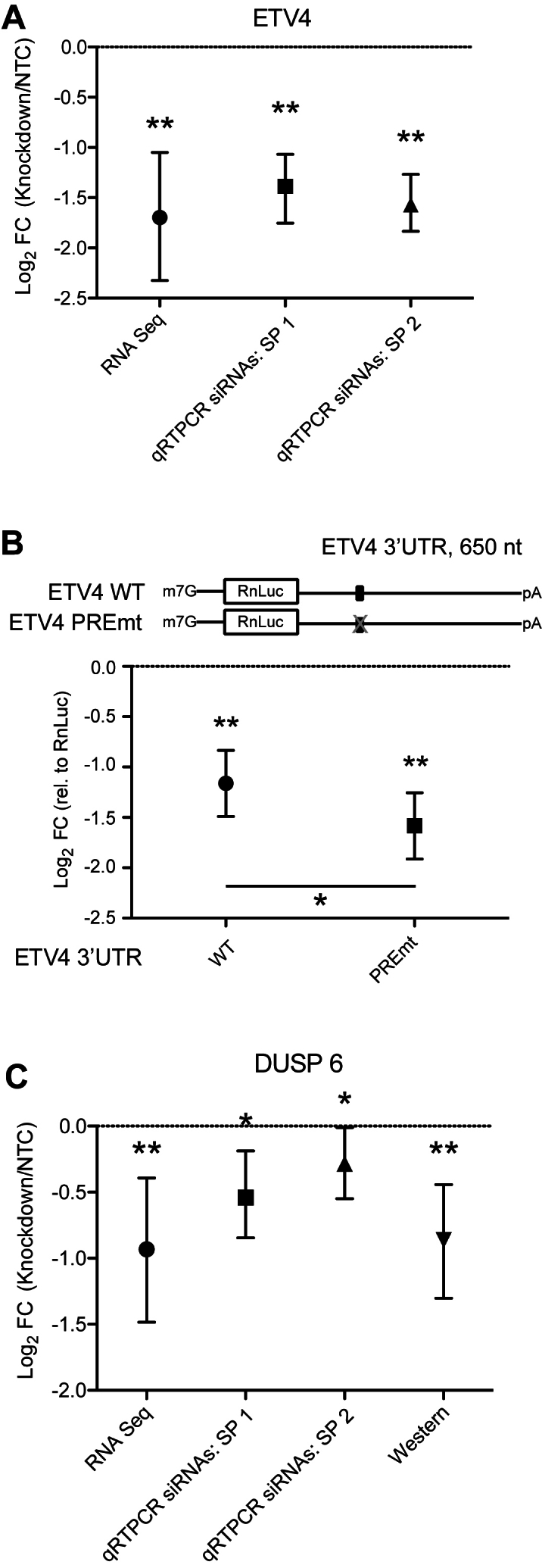
Validation of PUM-mediated activation of direct target mRNAs. (A) Comparison of ETV4 transcript levels in response to PUM knockdown assessed via RNA-seq or qRT-PCR assays with two independent experiments. Error bars indicate 95% confidence intervals (RNA-seq) or credible intervals (other assays) obtained as described in ‘Materials and Methods’ section. We mark values with a posterior probability of change in the indicated direction >95% with one asterisk (*), and those with a posterior probability >95% of passing the 1.3-fold change cut-off applied to our RNA-seq data with two asterisks (**). (B) Reporter gene analysis comparing the effect of WT or mutant PRE in the ETV4 3′ UTR on expression of the RnLuc reporter. Significance stars follow the convention in panel A, and are shown relative to RnLuc itself (above each symbol) or between the WT and PREmt cases (crossbar). (C) Comparison of the effects of PUM knockdown on DUSP6 transcript and protein levels assessed by RNA-seq, qRT-PCR and quantitative western blot.
Quantitative western blotting
For quantitative western blotting of PUM target genes, 4 × 105 HEK293 cells in 2 ml of medium were seeded per well of a 6-well plate. After 24 h, cells were transfected with On-target Plus Smartpool siRNAs for PUM1 (12.5 fmol) and PUM2 (12.5 fmol) or non-targeting control (25 fmol) siRNAs (25 nM final) using Dharmafect-1 reagent (GE Dharmacon). Twenty-four hours later, cells were transfected a second time with the siRNAs. Forty-eight hours later, cells were harvested and homogenized in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% Nonidet-P40, 1% sodium deoxycholate and 0.1% SDS) containing protease inhibitors (1 mM Phenylmethanesulfonyl fluoride (PMSF), 50 μg/ml of aprotinin, 50 μg/ml of pepstatin and 50 μg/ml leupeptin). Concentration of the whole cell lysate was measured using a DC Lowry Protein Assay (Bio-Rad). Samples of the cell lysates, containing the indicated protein mass, were then prepared in 1× SDS-PAGE loading dye, heated at 37°C for 10 min and then loaded into the wells of a 4–15% gradient SDS-PAGE gel (Bio-Rad). Electrophoresis was performed for 75 min at 110 V and then proteins were transferred at 70 V for 2 h at 4°C to Immobilon-P membrane (Millipore). Blots were blocked in blotto (5% W/V non-fat dried milk in 1× phosphate buffered saline (PBS) and 0.1% Tween-20) overnight at 4°C. Blots were incubated in primary antibody for 1 h at room temperature with gentle rocking, washed 3× for 10 min with blotto and incubated in secondary antibody-HRP conjugates (Pierce) for 1 h at room temperature with gentle rocking. Blots were washed 3× for 10 min with gentle rocking, developed with either Pierce ECL or Millipore Immobilon reagent and visualized with X-ray film. Primary antibodies are listed below. Specific bands were quantified using ImageJ densitometry software and signal from target proteins was normalized to the loading control GAPDH or β-Actin. Antibody signal response was assessed through the titration of control protein lysate and a standard curve with linear response range was determined for each antibody, as shown in Supplementary Figure S1, with data and statistics reported in Supplementary Table S3. Fold change in each protein level was determined by comparing western blot signal from PUM knockdown lysates relative to the non-targeting control samples from three western blot analyses and were analyzed using the Bayesian approach described below. Triplicate western blots, along with standard curves, are reported in Supplementary Figure S1 and values and statistics are reported in Supplementary Table S3.
Antibodies
| Anti-PUM1 | Custom antigen-affinity purified rabbit polyclonal antibody, raised against PUM1 unique region: amino acids 1–127. |
| Anti-PUM2 | Custom antigen-affinity purified rabbit polyclonal antibody, raised against PUM2 amino acids 100–150. |
| Anti-DEK | BD Transduction Laboratories (catalog no. 610948) |
| Anti-DUSP6 | Abcam (catalog no. EPR129Y) |
| Anti-FZD8 | Sigma Life Science (catalog no. HPA045025) |
| Anti-CDKN1B | BD Transduction Laboratories (catalog no. 610241) |
| Anti-LEFTY2 | Abcam (catalog no. EPR5444) |
| Anti-GAPDH | Life Technologies (catalog no. AM4300) |
| Anti-β−Actin | MP Biomedical (clone C4, catalog no. 08691001) |
Cell cycle analysis
For cell cycle analysis, reported in Supplementary Figure S2, 3 × 105 HEK293 cells were seeded in 6-well plates and transfected with either non-targeting control siRNA or PUM1 and PUM2 siRNA Smartpools as described above. This was performed for five replicates and all data and statistics are reported in Supplementary Table S3. After 72 h, cells were trypsinized and resuspended in PBS. Cells were then fixed with 100% ice-cold ethanol for 20 min. Fixed cells then were centrifuged and prepared in PBS containing 50 mg/ml propidium iodide and 100 mg/ml RNAse Type I-A solution. Cells were incubated at room temperature for 20 min and then over night at 4°C in the dark. Using a FACsAria III instrument, cells were sorted and analyzed with FlowJo software by the University of Michigan’s FACs core.
Data analysis
Quantitative RT-PCR, western blotting and luciferase assay data were analyzed using a multilevel Bayesian model to account properly for the multiple levels of uncertainty present in our experimental data (variations between biological samples and between technical replicates). Input data were ΔCt values relative to a reference housekeeping gene (for qRT-PCR), densitometry-based protein levels normalized to either GAPDH or actin (for western blots), or log ratios of RnLuc normalized to firefly luciferase luminescence (for luciferase assays). While exact details varied as appropriate for the experimental setup, in general we modeled the average for each biological replicate (performed on a separate day) using a t distribution centered on the mean for any particular sample type and variation between replicates on a given day as a t distribution centered on the mean for that day. We inferred separate variance parameters for each biological sample type, but inference for number of degrees of freedom in the t distributions at both levels were pooled across all biological samples for a given experiment type, as was the variance parameter for the technical replicates. In appropriate cases (e.g. luciferase data where all samples were compared to firefly luciferase), we added an additional term accounting for the shared day-to-day variation in the reference sample, which was shared across all experiments performed on a given day. All parameters began using non-informative priors. Models were fitted using JAGS (67), running four independent Monte Carlo chains and convergence was assessed by ensuring that the Gelman–Rubin shrinkage statistic for all reported parameters was <1.1 (68). We typically report the expected value for the parameter alongside 95% credible intervals using the highest posterior density approach, and also frequently base our inferences on the posterior probability that a given difference is biologically meaningful (>1.3-fold change, following the criteria used in our analysis of RNA-seq data); the exact quantities reported in each case are noted in the text. Our analysis is essentially an extension of the BEST method for analysis of continuous data (69), adapted to make use of the additional information present in our hierarchical experimental design, as we properly model co-variance between replicates performed on the same day or in the same experiment. Data, key statistics and the number of experimental data points obtained for each experiment are reported in Supplementary Table S3.
GO term enrichment analysis
We performed gene ontology (GO) term enrichment analysis using iPAGE (70) with default settings, using either the GO term database shipped with iPAGE or custom-built PANTHER and DAVID correlation tables (71–74). All enrichment analyses were performed on a set of eight bins constructed as three-bit binary numbers by separately considering whether each gene (i) showed significant differences in abundance in our RNA-seq data; (ii) contained at least one 5′-UGUANAUW PRE motif; and (iii) was identified as a PUM bound mRNA in RIP-CHIP and/or PAR-CLIP experiments (Supplementary Table S4) (11–13).
Motif detection
We identified sequence motifs that were significantly informative of the abundance profiles in our PUM-knockdown RNA-seq experiments using a development version of Finding Informative Regulator Elements (FIRE) (75). We applied FIRE to the 5′ promoter and 3′ UTR regions in the default human annotation shipped with the software, using as input the PUM-knockdown RNA-seq log fold change scores (discretized into 15 equally populated bins, and filtering out any transcripts with <5 FPKM under both conditions of comparison, as well as PUM1 and PUM2 themselves). We subsequently identified potential RBPs corresponding to each motif using the TOMTOM web server (76) with default settings, using the Ray et al. RNA Homo sapiens database (77).
Regression model fitting
For fitting the models of PUM effect on transcript levels as a function of PUM site number and location, we considered only the subset of transcripts with transcript levels of at least 5 FPKM in both conditions, in order to ensure that our fits were based on robust data. We used the nlsLM function of the minpack.lm R package to perform all non-linear least-squares fits. Confidence intervals were generated using bootstrapping with 1000 simulated replicates.
RESULTS
Identification of 930 mRNAs regulated by human PUMs
To globally identify mRNAs that are regulated by human PUMs, we performed RNA-seq to measure changes in mRNA levels in response to RNAi-mediated depletion of both PUM1 and PUM2. For this analysis, we used the human embryonic kidney cell line, HEK293, which has characteristics of kidney, adrenal, and neuronal tissues (78,79) and are highly amenable to transfection-based RNAi using siRNAs. PUMs are well-expressed in kidney, adrenal and neuronal cells as reported by the Illumina bodyMap2 transcriptome, and both PUMs were previously verified to be active in HEK293 cells (17). Four replicate samples were prepared for RNAi of PUMs and compared to four negative control samples treated with non-targeting control (NTC) siRNAs. Efficient depletion of PUM1 and PUM2 was verified from one replicate of each condition by western blot (Figure 1A), and RNA was extracted from the remaining three replicates for each condition and analyzed by Illumina RNA-seq. We also analyzed potential effects of PUM1 and PUM2 depletion on cell cycle; no significant effect was observed under these conditions (Supplementary Figure S2).
More than 26 million reads were obtained for each sample and mapped to >22 000 genes. Using this data, normalized expression values (in FPKM units) were determined for each gene (Supplementary Table S4). The expression values spanned a range of seven orders of magnitude, and were not strongly correlated with the fold change values (Figure 1B). From this data, we confirmed reproducible depletion of the PUM1 and PUM2 mRNAs, with a 64 and 73% reduction in the respective mRNA levels observed relative to NTC (Supplementary Table S4). The data were analyzed to detect statistically significant changes in abundance between the NTC and PUM1 and PUM2 knockdown samples, and significant fold changes (q < 0.05) in expression of mRNAs between the two conditions ranged from 3.81 to 0.287. Given the demonstrated partial redundancy of PUM1 and PUM2 (described in the Introduction), we used simultaneous knockdown of both PUMs to obtain the most comprehensive possible picture of PUM-dependent regulation. While there are no publicly available datasets with individual PUM1 or PUM2 knockdowns in HEK293 cells, we noted that the magnitude of changes in transcript levels for transcripts with identifiable PREs in their 3′ UTRs was generally greater than observed in an ENCODE dataset for individual PUM1 or PUM2 knockdowns in K562 cells (see Supplementary Figure S3 for details), providing further justification for our dual knockdown approach.
To establish an appropriate fold change cutoff parameter for differential expression, we measured the magnitude of repression caused by one or more wild-type (WT) PREs relative to mutant PREs located in the context of a minimal 3′ UTR of a RnLuc reporter gene. Previously, we used this strategy to demonstrate that three consensus PREs caused 3- to 4-fold repression of reporter protein and mRNA levels relative to mutant PREs wherein the UGU motif, which is essential for the PUM–RNA interaction, was changed to ACA to prevent PUM binding (17). With one PRE, we observed a 1.3-fold decrease in the reporter expression relative to the mutant version, thereby showing the magnitude of PUM/PRE dependent repression of a model mRNA (Figure 1C, 1× PRE). Additional PREs (up to four) caused a 2-fold increase in repression for the second and then third site, and 4-fold more repression for a fourth PRE site, as determined relative to corresponding reporters with an equal number of PREmt sites (Figure 1C, 2× PRE, 3× PRE and 4× PRE; Supplementary Table S3). Thus, PRE/PUM-mediated repression was proportional in this reporter mRNA context. Based on these observations, we imposed a cutoff of 1.3-fold change for biological significance in our RNA-seq analysis (matching the observed effect of a single PRE site), coupled with a q-value threshold of 0.05 for statistical significance. Using this combined test, we found that mRNAs from 930 unique genes were reproducibly, differentially expressed in the PUM knockdown samples; this will be called the ‘Response’ dataset (Figure 1D). The complete list of differentially expressed mRNAs is available in Supplementary Table S4.
Of the 930 Response genes, 634 increased in abundance in the absence of PUMs, indicating that they are negatively regulated (i.e. repressed) by PUM1 and PUM2 (Figure 1D). Conversely, 296 RNAs were reduced in abundance upon PUM knockdown (excluding the PUMs themselves), implicating PUMs in positive regulation (i.e. activation) of those genes, either directly or indirectly. The Response gene set contained mRNAs previously shown to be regulated in humans and other mammals (Supplementary Table S1). Indeed, eight previously reported PUM-regulated genes were differentially expressed, including PCNA, stem loop binding protein (SLBP), CKS2, CCNA1, UBA2, CDK1, DUSP6 and ACHE, representing a significant enrichment of known targets (odds ratio = 4.02, P = 4.74 × 10−4, χ2 test; note that throughout the text, all references to a χ2 test refer to the (n − 1)/n corrected variant of the χ2 test, first proposed by Pearson (80) and later recommended by Campbell (81)). These observations serve as a validation of our approach for identification of PUM targets.
This Response dataset provides an extensive list of mRNAs responding to PUM depletion. That said, the inventory is not exhaustive and false negatives are possible, including those genes that are not expressed in this cell type, that are regulated by mechanisms that supersede PUM regulation or that failed to meet our criteria for fold change and reproducibility. One example is CDKN1B, a previously reported PUM target (18). Though CDKN1B mRNA was detected and increased in abundance upon PUM depletion, the change did not meet our criteria for significance. In Supplementary Figure S4, we confirmed PUM and PRE mediated repression of CDKN1B mRNA using qRT-PCR and protein expression by quantitative western blotting and reporter gene assays. The results demonstrate that PUM1 and PUM2 directly repress CDKN1B expression through two PRE elements located in the 3′ UTR of its mRNA.
We also examined whether there is a correlation between PUM-regulated genes and their expression level. In our data, PUM Response mRNAs span a broad range of expression including repressed and activated mRNAs (Figure 1E). Therefore, PUMs do not appear to preferentially regulate genes in a manner that depends on their expression level. Together, our results identify a large set of novel PUM-regulated mRNAs including those that are repressed or activated by PUMs. This list is likely to contain a collection of direct PUM targets that are bound and regulated by PUMs, in addition to indirectly affected mRNAs.
Data-driven identification of PUM regulatory motifs
To identify sequence determinants related to differential expression of RNAs in our Response dataset, we applied the FIRE program (75). Motifs significantly enriched in the 3′ UTR of PUM-responsive transcripts are shown in Figure 2A and B. The strongest such element is Motif 1, 5′-UGUAHAUW, which is highly enriched in transcripts that increased in abundance upon PUM deletion (i.e. PUM repressed targets). Motif 1 very closely matches the PRE consensus 5′-UGUANAUA, accounting for flexibility in recognition that has previously been documented at nucleotide position 5 and, to a lesser degree, nucleotide position 8 (7,10–13,82,83). Drawing on both our analysis and existing literature, we use the functionally enriched motif, 5′-UGUANAUW, to define PREs for the remainder of this work. This modified Motif 1 is contained in 432 of 598 possible genes in the increased dataset; odds ratio 3.77, P < 10−5, χ2 test). The choice between the degenerate W and strict A at the last motif position reflects a sensitivity/specificity trade off: relative to the 5′-UGUANAUA motif, our PRE definition shows modestly higher sensitivity (0.722 for the degenerate motif versus 0.585 for the strict motif) and modestly lower specificity (0.591 for the degenerate motif and 0.723 for the strict) in identifying members of the increased dataset. The degenerate motif shows a slightly superior odds ratio (3.77 versus 3.68) and the classifiers show similar Matthews correlation coefficients (0.119 for the degenerate versus 0.127 for the strict), motivating our choice to use the more sensitive motif. The PRE motif is bound by PUMs with high affinity and is necessary and sufficient to confer PUM-mediated mRNA repression (Figure 1C) (7,10–13,17,82,83). Consistent with this, the majority of previously reported PUM-regulated mRNAs possess one or more PREs in their 3′ UTR (Supplementary Table S1). Thus, our functional analysis directly links the PRE/Motif 1 consensus to a significant number of mRNAs that are functionally regulated by PUMs. In summary, the motif analysis clearly demonstrates the dominance of a PRE-like motif emerging solely from our data, which is consistent with our understanding of PUM regulatory activity.
Figure 2.

The PRE is enriched in PUM-regulated transcripts. (A) Identification of RNA-sequence motifs significantly correlated with changes in transcript abundance using FIRE. The top of the panel shows the distribution of transcript log2 fold changes in each of a set of discretized bins, and below the enrichment or depletion of the four identified motifs in each bin is shown. Significant enrichment is observed for Motif 1 (which is highly identical to documented PUM1 and PUM2 binding sites, the PRE) in strongly PUM-repressed transcripts, and for Motif 2 in transcripts that decrease in abundance upon PUM knockdown. (B) Sequence logos for Motifs 1 and 2 identified by the analysis in panel A. (C) Predictions of putative PUM target transcripts. The distribution of the number and location of predicted PREs at various locations within all annotated transcripts in UCSC genome version hg19. Indicated locations are 5′ and 3′ UTRs and coding sequence (CDS) and non-protein coding RNAs (ncRNAs). Transcript numbers include all annotated transcript isoforms. Circles in the plot have areas proportional to the weight of that point in the distribution of all transcripts with at least one PRE in the corresponding location. (D) Number of genes that we observed to be activated by PUM1 and PUM2 that have at least one PRE in each indicated location. (E) Number of genes in the set that we observed to be repressed by PUM1 and PUM2 that have at least one PRE in each location. Stars here indicate cases with significantly enriched overlap between the presence of a PRE and membership in the Response dataset (P < 0.05, χ2 test). (F) Overlaps between our ‘Response’, ‘Predicted’ and ‘Bound’ datasets. Pairwise overlap comparisons show P-values based on χ2 tests; the three way overlap shows the Woolf test for the null hypothesis of homogeneous conditional probabilities (note that for the purposes of the analysis in this panel, we only considered genes that were part of the set with overlapping names between the genomic annotations used for analysis of the RNA-seq data and transcript boundaries, which is why only 867 Response genes are shown). ′None′ indicates the number of genes that were not included in any of the three categories.
FIRE identified one additional motif, Motif 2 (5′-BYCSWCSC), which is over-represented in the 3′ UTR of the PUM-activated genes (Figure 2A and B). Motif 2 occurs in the 3′ UTR of 139 out of 269 cases in the decreasing set but 41.3% of genes overall (odds ratio 1.53, P = 0.000627, χ2 test) and is even more strongly enriched across the entire transcripts of the decreasing set (235 out of 269 cases, odds ratio 1.66, P = 0.004886); note that for all analysis comparing our Response dataset with other datasets, we omit genes which could not be mapped properly between the datasets. The function of this motif is currently unknown, but it is recognized by the TOMTOM program (76) as having similarity to known binding motifs for multiple RBPs, including PCBP2 and HNRNPK; the details of all potential matches are summarized in Supplementary Table S5. As none of the candidate proteins show significant changes in expression upon PUM knockdown, either Motif 2 represents the activity of an as-yet unidentified RBP, or arises due to an interaction between PUMs and the Motif 2-binding protein that alters the effects of the PUM from inhibitory to activating. Some support for the latter possibility arises due to the fact that PREs and Motif 2 co-occur in the 3′UTRs of genes significantly more frequently than expected by chance (odds ratio 1.98, P < 0.000001).
Defining high confidence direct PUM target mRNAs
We next sought to identify high confidence, direct PUM target mRNAs within our Response gene set by applying two additional criteria. First, direct PUM targets should contain at least one PRE. Second, experimental evidence should support PUM binding to the direct target mRNA. To identify subsets of genes fitting into the first of those categories, we first surveyed all transcripts for the presence of the PRE motif 5′-UGUANAUW. In the human transcriptome (UCSC genome version Hg19 using TxDb.Hsapiens.UCSC.hg19 annotations), a total of 22 115 transcriptional units in 7822 unique genes contained one or more PRE motifs; these are referred to as the ‘Predicted’ PUM target dataset (Supplementary Tables S4 and 6).
Restricting our analysis to gene names that could be cross-referenced across all relevant databases, we found that 548 of 867 Response genes contained a PRE (odds ratio 2.49, P < 0.000001, χ2 test). Analysis of the location of PREs within each transcript shows that the majority of sites occur in 3′ UTRs or in non-coding RNAs, and furthermore, the number of sites per transcript is higher in those regions than in the 5′ UTR or CDS (Figure 2C; P < 10−12, asymptotic permutation test for each pairwise comparison). We also observed a strong and significant enrichment for PREs in the 3′ UTRs of genes in the Response dataset, indicating that sites in the 3′ UTR were more likely to be functional, both for positive and negative regulation (Figure 2D and E). Indeed, the presence of a PRE in the 3′ UTR substantially increased the odds of a gene being in our differentially regulated set (odds ratio 2.45, P < 10−10; χ2 test) whereas the effect was weaker in the CDS (odds ratio 1.53, P = 0.000092) and not statistically significant in the 5′ UTR (odds ratio 1.07, P = 0.827).
PUM regulatory effects are strongest for PREs located in the 3′ UTR and non-coding RNAs
To further analyze the regulatory role played by PREs in different numbers and locations in transcripts, we fitted a series of models to compare the numbers of PREs found in the 5′ UTR, coding sequence (CDS) and 3′ UTR to the observed log ratio (PUM RNAi/NTC RNAi) of expression in our RNA-seq datasets. Based on exploratory data analysis (described in Supplementary Data), we found that the effects of PUM binding could best be represented using a model where the effect of PRE was dependent on the probability of having at least one PRE in the 5′ UTR, CDS or 3′ UTR, with different strengths of effect from each binding location. We also developed a separate model for PREs in long non-coding RNAs (lncRNAs). The models were of the form:
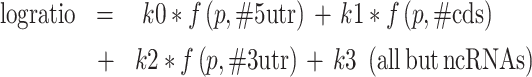 |
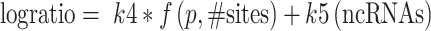 |
where, 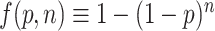 .
.
Here, logratio is the log2 of the observed expression ratio between PUM knockdown and control conditions (the ratio itself is inversely proportional, under reasonable assumptions, to the ratio of decay rates between the PUM knockdown and control conditions, as described in Supplementary Data), ‘#5utr’ denotes the number of PREs in the 5′ UTR and so on for the others ‘#’ variables. P should be interpreted as the probability that any particular PUM site is bound, and thus f(p,n) is the probability that a transcript region with n PREs is bound by at least one PUM molecule. As detailed in Supplementary Data, this biologically informed model proved superior to a family of purely statistical models relating the number of PUM sites to observed log ratios. The fitted coefficients are shown in Table 1; note that the model for lncRNAs was fitted separately from the others, with P constrained to the value for the main fit.
Table 1. Fitted parameters for the biologically informed model described in the text to link transcript abundance upon PUM depletion to the number and nature of PUM sites present in the transcript.
| PRE location | Fitted coefficient | P-value | 95% CI |
|---|---|---|---|
| k0 (5′ UTR) | 0.002 | 0.9652 | −0.111–0.101 |
| k1 (CDS) | 0.128 | 0.0023 | 0.023–0.201 |
| k2 (3′ UTR) | 0.260 | 5.21*10−10 | 0.143–0.319 |
| k4 (lncRNA) | 0.272 | <2*10−16 | 0.208–0.327 |
| P | 0.217 | 2.18*10−6 | 0.134–0.298 |
From these regressions we can derive several key insights. First, as suggested by Figure 2, the magnitude of effect of each PUM site is greatest for a PRE in the 3′ UTR, intermediate in strength for a PRE in the CDS and insignificant for a PRE in the 5′ UTR. Second, while the lncRNA model was fitted independently, the behavior of PUM sites on non-coding RNAs closely resembles those on the 3′ UTR of RNAs in our general pool. Third, as detailed in Supplementary Data, there appears to be anti-cooperativity in the presence of multiple PUM sites on a single transcript, as the effects of increasing numbers of PREs are less than additive. A logical interpretation is that once a transcript is bound by a PUM, there is no additional effect from having additional PUM molecules bind and thus the main effect of large numbers of PUM sites is to increase the probability of binding to at least one of those sites. Finally, while the fitted model parameters are statistically significant and provide a global description of the effects of an average PRE, the coefficient of determination of the fitted model (defined as the square of the Pearson correlation between the modeled and actual values) is only 0.068 and thus there is substantial variation in the effects of PUM sites in individual transcripts that is not explained by our global model, likely due to factors such as variations in the binding affinity of PUMs to different site contexts, involvement of PUM-regulated factors in decay rates and feedback regulation that violates the assumptions of our model interpretation (see Supplementary Data for details). As detailed in the Supplementary Data, we also observe that the efficacy of a single PRE appears to vary depending on the base at the eighth position, with an Adenosine at nucleotide position 8 having a higher probability of being bound (p) in the model above a Uridine, when the two parameters were allowed to vary independently.
PUM regulatory effects show strong correlations with in vivo RNA binding data
To complement the prediction of direct PUM target RNAs from the RNA-seq data described above, we considered PUM-associated RNAs identified by RIP-ChIP and PAR-CLIP methods from human cells reported in three studies. Morris et al. performed RIP-ChIP using rabbit polyclonal antibodies to identify human PUM1-associated RNAs in HeLa S3 cells: 726 associated RNAs were identified in that study (12). Galgano et al. performed RIP-ChIP using rabbit polyclonal antibodies to human PUM1 and PUM2 in HeLa S3 cells and identified 1040 PUM1 and 435 PUM2 associated RNAs (11). Hafner et al. performed PAR-CLIP by overexpressing and purifying FLAG-tagged PUM2 from HEK293 cells to identify some 3000 PUM2-bound RNAs (13). Together, these analyses provide a list of 4071 total PUM1 and PUM2 bound mRNAs, which we designated the ‘Bound’ dataset. Of these, 3368 could be mapped to genes in our analyzed datasets (Supplementary Table S4) and are considered in the analysis below. Comparison of the Bound dataset with the Predicted dataset identified 2519 genes (37% of Predicted dataset, 75% of Bound dataset; see Figure 2F) that met both criteria, demonstrating significant enrichment of the PRE (Motif 1) in the majority of PUM-bound mRNAs. The minority of Bound RNAs that do not have a consensus PRE may be the result of mismatch tolerance in the PUM recognition site not incorporated into our consensus search, alternate PUM–RNA contacts (though no such mechanism is known), false positives from PUM overexpression or immunoprecipitation, or PUM association with those RNAs mediated by RNA-binding partners.
To obtain information on how the known and predicted PUM-binding patterns affect transcript levels in HEK293 cells, we next compared the genes found in the Bound+Predicted datasets to the Response dataset, yielding 581 overlapping genes (67% of Response). Both the Bound-Response and Bound-Predicted associations showed significant enrichments of overlap (χ2 test, P < 0.000001; Figure 2F). We then applied all three criteria to identify the highest confidence direct PUM targets: 297 genes contain at least one PRE (Predicted), were shown to associate with one or both PUMs (Bound) and met our criteria for significant differential regulation by PUMs (Response) (Figure 2F). Members of this high confidence set are cataloged in Supplementary Table S4, and include the previously reported, PUM-regulated mRNAs UBA2, CKS2 and ACHE (Supplementary Table S1). The biological roles of members in this set are discussed in more detail below.
As noted above, our analysis used an experimentally derived 1.3-fold change cut-off criteria and significance calling for inclusion of genes in the Response dataset. To explore how the fold change cut-off affected the overlap of the number of genes differentially expressed in response to PUM depletion with the Bound or Predicted datasets, we re-examined our RNA-seq dataset without a fold change criteria or with a more stringent 1.5-fold change criteria, while maintaining a q-value threshold of 0.05 for statistical significance (Supplementary Table S4). Results are shown in Supplementary Figure S5. The number of differentially expressed genes scored by each fold change cut-off increased from 363 (stringent) to 965 (no fold change cut-off) but did not substantially alter the rate of overlap with the Bound and Predicted sets.
Validation of PUM-mediated repression of target mRNAs
We validated PUM-mediated regulation of several targets, chosen based on their fold change, statistical significance and biological importance (addressed in the ‘Discussion’ section) including the PUM-repressed mRNAs FZD8, DEK, SMPDL3A, RET, ANO4, NOVA2, SCUBE1, LEFTY2 and L1CAM. First, we measured changes between control and PUM knockdown samples using qRT-PCR. This analysis used total RNA purified from cells and random priming for reverse transcription; thus, the qRT-PCR also allows us to confirm that the poly(A) selection did not bias the RNA-seq results. Depletion of PUM1 and PUM2 mRNAs was accomplished using two different individual siRNAs for each PUM, or pools of four siRNAs and depletion of each PUM mRNA in these experiments was confirmed by qRT-PCR (Supplementary Table S3). For PUM repressed targets FZD8 (Figure 3A), DEK (Figure 3B), SMPLD3A (Figure 3C), RET (Figure 3D), ANO4 (Figure 3E), NOVA2 (Figure 3F), SCUBE1 (Figure 3G) and L1CAM (Figure 3H), the fold change of each target mRNA measured by qRT-PCR corresponds to the change observed in the RNA-seq data (Supplementary Figure S6), thereby verifying repression by PUMs.
We also performed quantitative western blotting to measure PUM-mediated repression of protein expression. This analysis was restricted to FZD8, DEK and LEFTY2 by the availability of high quality antibodies. Protein levels were measured in a minimum of three replicates and standard curves were generated to verify the linear response of the assay (Supplementary Figure S1 and Table S3). FZD8 protein levels reproducibly increased 5.56-fold upon PUM knockdown, which is almost a 3-fold increase over the mRNA fold change observed in RNA-seq and qRT-PCR experiments (Figure 3A). DEK protein levels increased 1.56-fold, which is approximately the same fold increase as the mRNA levels (Figure 3B). Interestingly, the LEFTY2 target, which is among the most strongly affected at the mRNA level, exhibited a significant but smaller magnitude change at the protein level (Figure 3I), perhaps reflecting other parameters that influence its protein levels, such as translation efficiency and/or protein turnover rate.
As an additional approach to verify PUM-mediated repression of FZD8, DEK, SMPDL3A and RET, we assessed the regulatory activities of their 3′ UTRs and the importance of PRE(s) using RnLuc reporter constructs. Reporter activity of each construct was compared to RnLuc with an unregulated minimal 3′ UTR, allowing us to measure the effect of the WT 3′ UTR and compare the impact of WT and mutant PREs. We observed that the 3′ UTR of FZD8 caused substantial repression of RnLuc activity, whereas a PRE mutation nearly eliminated repression (Figure 3J), consistent with PUM1 and PUM2 being dominant regulators of this 3′ UTR. The DEK 3′ UTR has a positive effect on RnLuc expression, and mutation of two PREs increased expression further (Figure 3K), indicative of a repressive role. Likewise, a PRE mutation in the SMPDL3A 3′ UTR (Figure 3L) and RET 3′ UTR (Figure 3M) reporters increased reporter activity relative to the corresponding WT 3′ UTRs. From these data, we conclude that PREs present in each target mRNA 3′ UTR confer PUM-mediated repression of reporter protein expression.
Validation of PUM-mediated activation of target mRNAs
We observed that a third of the Response RNAs decreased in abundance upon PUM depletion, indicating that PUMs positively affect their expression; we label these genes as PUM-activated (Supplementary Table S4). In the datasets that we considered here, 116 of the 269 PUM-activated genes for which comparison was possible contain one or more PREs, and 60 of 269 are also in the Bound dataset (Supplementary Table S4). A total of 46 PUM-activated genes match the high confidence criteria of being significantly regulated, containing a PRE and inclusion in the Bound dataset. We applied our validation strategies to two of these mRNAs: ETV4 and DUSP6. In the RNA-seq analysis, we detected a 3.2-fold decrease in ETV4 mRNA in response to PUM depletion (Figure 4A and Supplementary Table S3), which we confirmed in two separate qRT-PCR experiments by measuring ETV4 mRNA in total RNA isolated from cells treated with PUM or control siRNAs (Figure 4A). A suitable antibody was not available for ETV4 and thus we were not able measure the effect at the protein level. As an alternative approach, we verified a positive regulatory role of a PRE located in the ETV4 3′ UTR using RnLuc reporter assays (Figure 4B and Supplementary Table S3). The ETV4 3′ UTR has repressive activity relative to a minimal control 3′ UTR, but inactivation of the PRE made the ETV4 3′ UTR even more inhibitory, consistent with PUM-binding having a positive regulatory effect (Figure 4B). For DUSP6, we observed a 1.9-fold decrease in the mRNA level in response to PUM depletion, which was corroborated by qRT-PCR measurements (Figure 4C and Supplementary Table S3) and by quantitative western blotting using a specific antibody for DUSP6 (Figure 4C; Supplementary Figure S1 and Table S3). Again we note strong concordance between fold changes measured by RNA-seq and qRT-PCR (Supplementary Figure S6). Taken together, the data indicate that PUM-activation of ETV4 and DUSP6 mRNAs is manifested by binding of PUMs to the PREs resulting in increased abundance of the mRNA and encoded protein.
Indirect mechanisms also appear to contribute to the observed PUM-mediated activation. Though PREs are present in 116 of 269 PUM-activated genes, they are not significantly enriched (odds ratio = 1.05, P = 0.7566, χ2 test), nor is there an enrichment of Bound, Pum-activated transcripts (88 out of 269 are in the Bound set; odds ratio = 1.04, P = 0.8199, χ2 test). Therefore, indirect mechanisms mediated by PUM regulation of other regulatory factors, such as other RBPs, likely account for the majority of activated targets. As noted above, Motif 2 is enriched among PUM-activated transcripts and may play a role in stabilizing PUM-activated transcripts, although there are likely additional paths to indirect PUM-mediated activation that may be elucidated in future studies (see ‘Discussion’ section).
Biological significance of newly identified PUM target RNAs
We analyzed the transcripts regulated by PUM1 and PUM2 and found that they encode proteins involved in a variety of important biological, biochemical and disease related processes, providing new insights into the PUM regulon. We applied the iPAGE package (70) to identify pathways significantly affected by PUM1/2 knockdown by separately flagging each transcript with membership in the Response, Predicted PRE-containing and Bound datasets (Supplementary Table S4). The iPAGE package selects a non-redundant set of annotations with maximal mutual information to a given set of inputs; thus, this analysis allowed us to find gene categories, pathways and disease associations which are either over- or under-represented in the datasets. In our analysis, we considered annotations significantly enriched or depleted in GO terms (Figure 5A), PANTHER terms (Figure 5B) (73) and Genetic Association Database and Online Mendelian Inheritance in Man annotations from the DAVID database (Figure 5C)).
Figure 5.

Enrichment of ontology terms among subsets of PUM targets. (A) List of GO terms showing significant mutual information with membership in the Regulated, Bound and Predicted gene sets discussed in the text, calculated using iPAGE. The color of each cell shows the P-value for significance of the enrichment (red) or depletion (blue) at that cell, scale is shown at the bottom of the figure; black bordered cells individually show P-values < 0.05 after Bonferroni correction across their row. The plotting and highlighting conventions described here apply to all panels of the Figure. (B) As in panel A, for PANTHER terms. (C) DAVID disease annotations showing significant mutual information with membership in the Regulated, Bound and Predicted sets. (D) GO terms showing significant mutual information with the observed log2 fold changes upon PUM knockdown, measured in our RNA-seq dataset. The log2 fold changes were discretized into 15 equally populated bins, with expression levels shown above the plot. (E) As in panel D, for PANTHER terms. (F) As in panel D, for DAVID Disease terms.
We first focus on the high confidence target genes (Response+Bound+Predicted) (Figure 5A and B). Included in this category are genes involved in developmental signaling (sFRP: WNT signaling pathway; Notch signaling pathway), physical cell–cell interactions (integrins, extracellular matrix glycoproteins), hormonal signaling (PDGF signaling pathway, guanyl-nucleotide exchange factors), neurotransmitter/hormone secretion (Golgi vesicle transport, vesicle coat protein) and fatty acid metabolism (pantothenate kinase activity, 1-acylglycerol-3-phosphate O-acyltransferase activity, diacylglycerol lipase). In addition, signal transduction (guanyl-nucleotide exchange factor activity, protein serine/threonine phosphatase complex), DNA polymerase processivity factor and negative regulation of transcription classifications were significantly enriched in high confidence PUM targets.
Consideration of the individual categories (Response, Bound or Predicted) informs our understanding of the regulatory potential of PUMs. The Response dataset, inclusive of direct and indirect targets, contains enriched terms pertaining to cell–cell adhesion, intermediate filaments, nucleosomes, insulin IGF pathway, extracellular matrix glycoprotein and membrane metalloprotease and endopeptidases (Figure 5A and B). PUM Response genes are also enriched for neural functions (e.g. neurotransmitter binding, Slit/Robo axon guidance) that are prevalent in PUM-regulated mRNAs detected in HEK293 cells, consistent with the neuronal and adrenal cell characteristics of this cell line (78,79). The pattern of transcripts demonstrated to be PUM-bound indicates potential additional regulatory roles in the mitotic cell cycle, regulation of I-kappaB kinase and PDGF signaling pathways. In contrast to the Response and Bound datasets, the Predicted targets are not limited to transcripts detected in the studied cell lines and relate more broadly to the distribution of PREs across the transcriptome. Analysis of these Predicted PUM targets revealed significant enrichment for genes important for neural functions (e.g. members of the voltage-gated ion channel class), adenosine triphosphate (ATP)-binding cassette transporters, metalloendopeptidases and nuclear hormone receptors (Figure 5A and B).
Analysis of the genes over-represented in a subset of the three categories of PUM targets is also informative, and we observe both distinct patterns and substantial overlap in certain terms. In the Response+Bound dataset, enriched terms include ubiquitin conjugating enzyme, axon guidance by Slit/Robo, cyclin D, insulin receptor substrate family and 4-aminobutyrate aminotransferase activity (which catabolizes the neurotransmitter γ-aminobutyric acid, GABA) (Figure 5A and B). In the Response+Predicted category, enriched terms include homophilic cell adhesion, integrin complex, cadherins, extracellular matrix glycoprotein, TGF-β receptors, serine/threonine kinase activity and metalloendopeptidases (Figure 5A and B). Our analysis also showed significant under-representation of GO and PANTHER terms in some PUM target categories (Figure 5A and B), many of which correspond to core metabolic and cellular processes, indicating that PUMs are not involved in the regulation of core biological processes, but rather, regulate specific pathways and functions.
Our analysis also identified several disease associations for the various classes of PUM targets (Figure 5C). The three categories of PUM targets are variously enriched for terms associated with adiposity, obesity and insulin signaling, consistent with the identification above of key roles for PUM in regulating both lipid metabolism and hormonal signaling. Over-representation of several disease terms pertaining to cardiovascular, skeletal, liver and kidney diseases was also observed, and male infertility was also enriched in PUM targets (Figure 5C). Several types of neurological disorders were enriched in PUM target categories including GABA transaminase deficiency, Bosch-Boonstra-Schaaf syndrome and brain neoplasms (Figure 5C).
Several pathways and disease associations also reveal links to cancer (e.g. Notch and WNT signaling, cell cycle and cell death terms). Indeed, of the 565 cancer genes in the COSMIC cancer gene census (84,85), we find that 330 genes are predicted to be PUM targets on the basis of possessing one or more PREs (P < 0.000001), 209 are in the Bound category (P < 0.000001) and 45 were regulated by PUMs (Response dataset) in HEK293 cells (P = 0.000751; χ2 test) (Supplementary Table S7). Among the set of genes in the Response dataset were well-known cancer genes including both oncogenes (e.g. ETV4, RET and DEK) and tumor suppressors (e.g. CDKN1C, BTG1). Moreover, 16 cancer genes are high confidence PUM targets.
To gain insight into the directionality of PUM regulation, we used iPAGE to identify over- and under-represented GO and PANTHER and DAVID terms among repressed or activated transcripts in the Response dataset (Figure 5D–F). We found that several biological functions show patterns of repression by PUMs, including cell migration, cell adhesion, extracellular matrix glycoprotein, sodium ion binding, circulation, nucleosome components, p53 pathway and peptide hormone production. Other terms are over-represented in PUM activated transcripts, such as carbon–carbon lyase activity, proteasome and adenomatous polyposis coli. In contrast, other terms show significant depletion from the highly PUM-repressed bins (i.e. ribosomal components and mRNA processing factors), consistent with our observations above.
To visualize specific PUM-responsive pathways, we plotted differential expression of individual components in relationship to Response, Bound and Predicted classification (Figure 6). The graphs reveal two dominant patterns: PUMs either broadly repress members of a particular pathway, or cause a switch from the activity of one set of genes in a pathway to a different set. Examples of the former paradigm can be seen in Figure 6, which shows similar patterns for nearly all differentially expressed genes in the GO terms for guanyl-nucleotide exchange factor activity (Figure 6A), integrin complex (Figure 6B), homophilic cell adhesion (Figure 6C) and PDGF signaling pathway (Figure 6D). Notably, although most differentially expressed mRNAs were in the Predicted set, similar behaviors are observed for GO term members that lack PREs, suggesting indirect effects via changes in expression of proteins that are themselves PUM-regulated. A striking example of indirect PUM regulation of an enriched GO term is the case of nucleosome components (Figure 6E), where the majority of term members show increases in transcript levels upon PUM knockdown (many of them passing the threshold for membership in the Response set), but few are identified as direct PUM targets based on the presence of a PRE or prior binding data (see also Figure 5). At present there is insufficient information to definitively determine the path of information flow between PUM and the histone transcript levels. One likely contributor is indirect regulation via SLBP, a crucial factor in histone RNA processing, stability, translation and decay (86). SLBP itself contains a PRE and is significantly upregulated by PUM knockdown (Figure 1D). Other as-yet unrecognized factors likely play additional roles in modulating histone transcript levels in response to PUM activity.
Figure 6.

Overview of transcript level behavior for enriched PUM-regulated gene term members. In each panel, we show volcano plots for all members of select enriched Gene Ontology terms identified in our iPAGE analysis; membership in the Predicted, Response and Bound sets discussed in the text are indicated by fill state, point size and color, respectively, as shown in the legend at the bottom. We individually label each gene that either passes our RNA-seq significance thresholds, or which showed >2-fold change in transcript level or q-value < 0.01 regardless of the dual significance criteria. Panels A–F, in order, show results for: (A) guanyl-nucleotide exchange factor activity (GO:0005085), (B) integrin complex (GO:0008305), (C) homophilic cell adhesion (GO:0007156), (D) PDGF signaling pathway (PANTHER P00047), (E) nucleosome (GO:0000786) or (F) Notch signaling pathway (GO:0007219).
A simple example of the switch-like pattern of PUM-mediated regulation is the case of pantothenate kinase activity (GO:0004594), where PANK1 shows a log2 fold change of −0.528 and PANK3 shows a log2 fold change of +0.527 upon PUM knockdown (changes to PANK2 and PANK4 are smaller in magnitude and were not significant), suggesting that for this pathway PUM activity induces a shift to a profile of pantothenate kinase expression more resembling that of gluconeogenic tissues (87). Figure 6F shows a more complex example of the same phenomenon, where the differentially expressed members of the Notch signaling pathway are split between PUM-activated and PUM-repressed targets, likely reflecting a similar functional shift of Notch signaling driven by PUM activity in appropriate tissues. The majority of Notch pathway members showing PUM responsiveness appear to be direct targets, based on the presence of a predicted PUM motif and/or experimentally identified PUM binding, and includes several atypical PUM-activated targets (TFDP2, APH1B and MAML1).
DISCUSSION
The results of this research expand the number of mRNAs that are regulated by PUMs, illuminating their post-transcriptional regulatory networks, biological functions and roles in controlling expression of disease genes. In addition, our findings provide new insights into the activities and mechanisms of PUM regulation.
Human PUMs act through both direct and indirect mechanisms
Our analysis indicates that the PUM-regulated RNAs contain both direct targets bearing a PRE and are bound by PUMs, and indirectly affected transcripts. Some 63% of the PUM-regulated targets contained a PRE/Motif1 and 38% were also shown to be bound by PUMs (11–13), consistent with direct regulation of these mRNAs by PUMs. Extending this analysis to the entire transcriptome, ∼42% of annotated transcripts contain one or more PREs (32% have a PRE in the 3′ UTR), suggestive of broad regulatory potential for PUMs extending beyond the cell type analyzed in this study.
We identified several hundred high confidence PUM-regulated RNAs that contain a PRE and are bound by PUMs. We selected 11 of these direct targets, chosen to represent different physiological functions and verified PUM-dependent regulation with multiple assays. Several such targets belong to signaling and cell adhesion pathways that function in developmental processes and relate to human disease. For instance, multiple validated direct targets have specialized functions in neurodevelopment. The FZD8 mRNA encodes a homolog of frizzled, the WNT receptor protein (88) and is predominantly expressed in the brain where it controls brain development and size (89). PUMs also repress the neural cell adhesion molecule, L1CAM, which participates in nervous system development (90). L1CAM expression promotes migration of metastatic cancer cells and dysfunction of L1CAM causes X-linked spastic paraplegia and CRASH syndrome (corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraplegia and hydrocephalus). The PUM target SCUBE1 encodes a cell adhesion molecule with roles in endothelial cells, platelets and thrombosis (91). The LEFTY2 target mRNA produces a TGF-β family growth factor that participates in development of left–right body asymmetry (92) and differentiation of embryonic stem cells (93). We also validated PUM repression of the NOVA2 mRNA, which encodes a neuron specific RBP that regulates alternative splicing decisions to control neuronal development and axon pathfinding (94).
We validated direct PUM repression of multiple cancer genes, including two proto-oncogenes: RET and DEK. The RET protein is a receptor tyrosine kinase for glial cell derived neurotrophic factor (GDNF) ligands and normally controls cell proliferation, neuronal migration and cell differentiation. Mutations in RET cause thyroid and endocrine cancers and Hirschsprung’s disease, a childhood neurodevelopmental disease (95,96). DEK is a proto-oncogene that associates with chromatin and is linked to cancers, inflammation and arthritis (97,98). We also demonstrated PUM repression of the SMPDL3A mRNA, which produces an acid sphingomyelinase-like phosphodiesterase implicated in bladder cancer (99,100).
Our results revealed that PUMs regulate the ANO4 mRNA, which encodes Anoctamin-4, a regulator of aldosterone production (101,102). Aldosterone is a steroid hormone that modulates blood pressure, thus potentially linking PUMs to cardiovascular function. We also detected PUM repression of seven previously reported PUM target mRNAs encoding proteins involved in replication (SLBP, PCNA), cell cycle and proliferation (CDK1, CKS2 and Cyclin A1), protein sumoylation (UBA2) and neurotransmission (ACHE) (Supplementary Table S1). Taken together, these results provide robust confirmation of the PUM-mediated repression of target mRNAs and provide new insights into the multiple pathways modulated by PUMs.
The observed PUM-mediated regulation of the 37% of Response RNAs that lack a PRE consensus may be the result of indirect effects. For instance, these RNAs may be controlled by an intermediary factor that is directly regulated by PUMs (as we explore below). Identification of these RNAs is nonetheless important for understanding PUM impact on gene expression. Alternatively, PUMs might interact with these transcripts via alternative RNA-binding modes or via protein–protein interactions with another RBP. In support of the latter idea, we recently reported that the RNA-binding specificity and affinity of D. melanogaster Pumilio is cooperatively controlled by a partner, the RBP Nanos (103). Such combinatorial partners for human PUMs are at this time poorly characterized and future work will investigate the potential influence of combinatorial control.
Implications for PUM repression mechanism
Our results provide several insights into the mechanism of PUM-mediated repression. Previous work established that PUMs reduce translation and accelerate mRNA decay, manifested in a corresponding reduction in protein expression (12,17,21,23). These effects were characterized for a small number of mRNAs (Supplementary Table S1), and our data extends those observations to a broader set of natural target mRNAs. Interestingly, for some validated targets, we observe a larger magnitude of repression at the protein level, suggesting that translation of certain transcripts may be preferentially affected by PUMs relative to RNA decay (e.g. FZD8). The parameters that influence this effect remain unknown, but may involve regulated changes in poly(A) tail length and the translational activity of poly(A) binding protein, and their resulting influence on translation efficiency and RNA decay.
We find that location of the PRE is an important determinant for PUM-mediated repression, with significant correlation of PREs in 3′ UTRs and to a lesser degree, in CDSs; whereas, PREs in 5′ UTRs do not correlate. Consistent with this observation, analysis of our global datasets indicates that PREs are both more prevalent and most effective in the 3′ UTR of transcripts (see Supplementary Figure S7 for a comparison of predictions and observations). Our reporter data show that PUM-mediated repression increases with the number of 3′ UTR-bourne PREs, within the tested range of 1–4 PREs (Figure 1). Likewise, on a global level, the strength of repression generally increases with the number of PREs, but in a sub-linear fashion (Supplementary Figure S7). The effect of increasing numbers of PREs could be modeled most parsimoniously by considering the strength of repression to be directly related to the probability that a transcript was bound by at least one PUM molecule (Table 1), and thus the effect of multiple sites was primarily to raise the probability of binding so that transcript degradation could be initiated. Qualitatively equivalent conclusions are reached even if the fit is restricted to transcripts which rose in abundance upon PUM depletion (data not shown), removing the possibility that the uncommon PUM-stabilized transcripts are responsible for our conclusion. It is notable that whereas a global fit of our RNA-seq data suggest less-than-linear effects for increasing number of 3′ UTR PREs (Supplementary Figure S7), our direct in vivo data on a model transcript (Figure 1C) show at least linear response for 1× and 2× PREs and apparently synergistic effects for the 3× and 4× PREs. The discrepancy likely arises due to the un-natural context of the closely spaced 3× and 4× PREs in the minimal 3′ UTR of the RnLuc reporter that may promote cooperative binding of PUMs and co-repressors.
PRE occupancy will depend on the affinity of PUM for a PRE sequence and the intracellular concentration of the PUMs and their multitude of mRNA targets. PUM–PRE interactions have been biochemically analyzed extensively, with dissociation constants measured in the low to subnanomolar range (6,7,10,17,104). The intracellular concentration of PUM1 and PUM2 have been estimated to be ∼12 nM for PUM1 and ∼3 nM for PUM2 in HCT116 cells (24). In our experimental system, HEK293, we observe approximately equal levels of PUMs to equivalent amounts of HCT116 cell extract (data not shown), and thus PUM1 and PUM2 concentration is probably above the dissociation constant for the consensus PRE. Other factors that cannot be addressed at this time are likely to influence PRE occupancy, including competing RNA-binding factors, RNA structure and cooperativity with RNA-binding partners.
Despite these new relationships of PUM regulation relative to PRE number and location, our ability to predict the strength of repression caused by the PREs in a given transcript remains quite poor. One interpretation of this observation is that other factors and parameters influence the response of PRE-containing RNAs, which are not accounted for by our model and remain to be measured. These may include interspatial relationship of PREs, binding sites for other trans-acting factors, RNA structure, and the kinetics of RNA-binding, translational inhibition and mRNA degradation. It is also likely that for some PRE-containing mRNAs, other regulatory mechanisms supersede the effect of PUM regulation.
MicroRNAs are candidates for combinatorial control alongside PUMs, as we observe significantly enrichment of known microRNA targets, derived from miRTarBase (105), among our Bound, Response and Predicted sets (P < 10−10 in call cases, adjusted chi-squared test). Also, a previous study reported inter-spatial relationship of PUM and microRNA-binding sites (11). However, no single microRNA with significant overlap is observed to target >∼10% of the members of the PUM-activated or ∼20% of the members of the PUM-repressed gene sets in our experimental data. Whereas many microRNAs have individually small numbers of targets in these sets (data not shown), any regulatory overlap between PUMs and microRNAs likely involves a complex mixture of interactions that will need to be mapped out by future experiments.
Our analysis focused on the well-documented role of PUMs in regulation of RNA decay, we cannot exclude other possible regulatory mechanisms that may act on subsets of PUM target RNAs. For instance, though translation and RNA stability are intimately linked (106–109), it remains possible that PUMs could control translation of certain mRNAs without a corresponding effect on the transcript’s level. Such a mechanism could operate in specialized contexts, such in oocytes, where decay of maternal RNAs is often inactive (110). The future application of quantitative proteomics to measure PUM-mediated translational control will be necessary to explore this possibility. PUMs may also exclusively control intracellular localization of certain transcripts, which would not be detected in our Response dataset, though those RNAs would likely be included in the Predicted and/or Bound datasets.
It is important to acknowledge other limitations of our approach. As our primary goal was to identify high confidence PUM-regulated RNAs, we imposed strict criteria on our significance calling and fold change. As a result, false negatives are probable in our Response dataset. Indeed, the PUM target CDKN1B was just below the imposed threshold, though it is clearly regulated by PUMs (Supplementary Figure S4) (18). Moreover, the target RNAs must be reproducibly expressed and detected; transcripts with too much variability between replicates were ignored as they did not pass statistical cut-offs and many genes were not detected or were too low in expression in this cell population. Limitations of RNAi efficiency can also impose false negatives. To mitigate this issue, we optimized RNAi of PUM1 and PUM2 and verified substantial depletion by western blot, qRT-PCR and RNA-seq. RNA targets may have been missed due to alternative RNA processing that could produce RNA isoforms that lack PREs, such as by shortening of 3′ UTRs. While the resulting RNA-seq data do not define an exhaustive list, the new dataset substantially expands our understanding of PUM-responsive RNAs and provides important new insights into their impact on gene expression.
Evidence of PUM-mediated activation
While PUMs typically act as repressors, surprisingly, we find evidence of PUM-mediated activation, manifested by reduced transcript level upon depletion of PUMs. For many activated transcripts, these effects may be indirect; however, several pieces of evidence support direct PUM activation of specific RNAs. Of the 269 RNAs that decreased in abundance upon PUM depletion, 43% contain one or more PREs and 22% were reported to be bound by PUMs. Moreover, we find that 17% of PUM-activated mRNAs (47 total) are high confidence direct targets that are bound by PUMs and contain a PRE. This group includes the ETV4 and DUSP6 mRNAs for which we validated PUM dependent activation, manifested by significantly increased mRNA and protein expression. It is noteworthy that while activation of mRNAs by PUMs in mammals was unprecedented, previous reports suggest that PUF family members can enhance expression in certain contexts in other species (111–113). While this manuscript was under review, a new study reported that PUMs bind to and promote expression of the PRE-containing FOXP1 mRNA to control hematopoiesis (114).
The potential significance of PUM-mediated activation is illustrated by the importance of the affected genes. For example, the PUM-activated genes ETV1, 4 and 5 are members of the ETS translocation variant subfamily of oncogenic transcription factors that are linked to multiple cancers (115). All three ETV mRNAs contain a PRE and thus are likely direct targets. We validated PUM/PRE regulation of high confidence direct target ETV4, which is involved in development of the hippocampus (116), promotes cell proliferation, motility, invasion (115) and is over-expressed in a variety of cancers including prostate, breast and colorectal cancers (117,118) illustrating the importance of regulating its expression. We also validated PUM regulation of the DUSP6 mRNA, which encodes the dual specificity protein phosphatase 6, a negative regulator of specific MAP kinases including ERK (119). DUSP6 regulates heart development (120), and mutations contribute to congenital hypogonadotropic hypogonadism (121) and breast cancer (122). Moreover, upregulation of DUSP6 is associated with glioblastoma, tumor growth and chemo-resistance (123).
The mechanism(s) of direct PUM-mediated activation is currently unknown, and we suggest several means, some of which are not mutually exclusive: (i) PUM binding could displace a dominant repressor, resulting in stabilization/activation. (ii) PUM could act combinatorially with additional RNA-binding factors that alter its regulatory activity, switching from repression to stabilization. Precedent for combinatorial activation comes from analysis in Xenopus oocytes, where placement of a PRE proximal to a Cytoplasmic Polyadenylation Element (CPE) enhanced translation by stabilizing CPE-binding protein (CPEB) on that mRNA (124). However, we do not detect enrichment of CPEs in the PUM-activated target mRNAs, so it is unlikely that CPEB is responsible for the observed effects. Our analysis of sequence-response correlations identified enrichment of Motif 2 in the PUM-activated gene set, including some high confidence targets, raising the possibility of combinatorial control. (iii) PUM RNA-binding could alter the secondary structure of the target RNA, modulating the accessibility of a binding site for a stabilizing factor. (iv) PUM could directly activate by recruiting factors that stabilize the mRNA and/or promote translation. Precedent for this idea comes from C. elegans, where the PUF protein, fem-3 binding factor (FBF), switches from repressor to activator by changing protein partners (112). FBF represses mRNAs in the mitotic region of the nematode germline but can switch to activation in the meiotic region by pairing with the poly-adenosine polymerase GLD2, which extends the target mRNAs poly(A) tail to stabilize and enhance translation of the transcript (112). In a similar manner, the CPEB RBP has been shown to act as a bifunctional switch (125). To date, no interaction between human PUMs and GLD2 orthologs has been reported. Future research will be necessary to test each of these hypotheses and expand our understanding of the molecular mechanisms underlying PUM-mediated activation.
PUMs regulate expression of genes from multiple pathways and biological processes
Our findings reveal that PUMs regulate genes with diverse biological functions and here we highlight several themes. A prominent category of PUM targets includes genes involved in cell–cell and cell–extracellular matrix contacts, cell migration and cell–cell communication including integrins, cadherins, extracellular matrix glycoproteins and membrane metalloproteases. Signaling pathway categories are also prevalent PUM targets including Notch, PDGF, TGF-β receptor, insulin receptor binding, serine/threonine phosphatase, guanine nucleotide exchange factor and WNT/Frizzled signaling pathways. Neural pathways are enriched, including neural development, maturation and axon guidance pathway components such as the Slit/Robo pathway. Notch and WNT/Frizzled pathways overlap with this category (89,126). Neurotransmitter hormone secretion, binding and catabolism pathways (specifically GABA) were also enriched in PUM targets. Other categories include replication fork, negative regulation of transcription, fatty acid metabolism and ubiquitin ligase activity. While these findings are derived from one cell type, the information we gained can be extrapolated to the transcriptome through analysis of all PRE containing mRNAs, which revealed potential for PUMs to regulate additional functional categories including voltage gated ion channels, ATP-binding cassette transporters and nuclear hormone receptors. Interestingly, we note multiple functional relationships between the targets of human and Drosophila Pumilio proteins, including Notch, insulin receptor, WNT signaling pathways, voltage gated ion channels, axon guidance, cell adhesion and migration (44).
Many of the PUM targets have important roles in developmental processes and their dysfunction or dysregulation contribute to human diseases including cancer, cardiovascular and neuro-developmental diseases. Indeed, we observed enrichments among PUM-regulated genes of neurological disease categories including GABA-transaminase deficiency, muscular dystrophy and brain neoplasms. One disorder linked to PUM targets is Bosch–Boonstra–Schaaf optic atrophy (127), an autosomal dominant disorder characterized by delayed development, moderate intellectual disability and optic atrophy. Male infertility was also linked to the PUM-regulated RNAs, which is intriguing given the conserved function of PUF proteins in controlling fertility and spermatogenesis (25,47,48). We also observed enrichment of disease terms related to cardiovascular diseases, including blood pressure and cardiac output, and abdominal obesity and the bone disorder osteopetrosis, revealing unanticipated connections of PUMs to these conditions.
The connection of human PUMs to regulation of neuronal genes is intriguing given that mammalian PUMs are present in neurons, where they are thought to participate in localization and translational control of target mRNAs (55–57). Currently the identities of neuronal PUM target mRNAs in humans remain largely unknown and we anticipate that the targets identified in our analysis may be relevant, including L1CAM and FZD8. As discussed in the introduction, PUF proteins have documented roles in neurological processes (36–41,128,129), and studies in mice linked PUMs to neurodegeneration and behavioral defects (49,130). Several neuronal target mRNAs were reported including SCA1/ATXN1 mRNA (encoding Ataxin 1) (54) and the voltage-gated sodium ion channel SCN8A/Nav1.6 (56). Neither of these targets met our criteria for inclusion in the Response dataset, though they both contain PREs. We did detect direct PUM regulation of another voltage-gated sodium ion channel, SCN9A/Nav1.7, which is involved in pain sensation and is linked to extreme pain disorders (131). The relationship of mammalian PUMs to neuronal functions is further emphasized by a study published while this manuscript was in revision, which reported that mouse PUM1 and PUM2 bind and regulate a collection of mRNAs in the hippocampus (50). Moreover, the neural-specific knockout of both PUM1 and PUM2 resulted in defective neurogenesis and compromised learning and memory formation.
Our findings indicate that PUMs regulate or have the potential to control a plethora of cancer genes: 58% of cancer genes in the COSMIC database contain PREs, 37% were bound by PUMs and 8% were regulated by PUMs in the Response dataset (Supplementary Table S7). The PUM-regulated cancer genes include oncogenes, tumor suppressors and prevalent cancer-related pathways (e.g. WNT/Frizzled, PDGF, TGF-β and Notch signaling). Moreover, PUM bound transcripts are enriched in cell cycle and cell death pathways. These pathways need to be tightly regulated as their dysregulation contributes to cancer and developmental defects. For instance, the WNT pathway is documented to control a variety of processes including development, proliferation of stem cells, cell migration and cell fate. Mutation or overexpression of WNT proteins or Frizzled receptors, both of which are repressed by PUMs, contributes to a variety of cancer types (88,132). The effects of PUMs on the outputs of these pathways, including initiation and progression of cancer, remains to be explored.
Pathway analysis of PUM regulons raises interesting questions pertaining to the regulatory logic. PUMs may act as a molecular switch that turns a regulon off (or on). Alternately, PUMs may act as an overall antagonist of a pathway by reducing expression of multiple components simultaneously, thereby diminishing the magnitude of responses. Our data favor the latter scenario, as we observe that in general, the magnitude of PUM regulation is relatively small, though comparable to other post-transcriptional mechanisms (133,134). By shortening the lifespan of mRNAs within a pathway, PUMs may confer dynamic responsiveness of pathway components and downstream targets, which is often important for transient phenomenon such a signaling (135). The observed effects of PUMs on guanyl-nucleotide exchange factor, integrin complex, cell adhesion and PDGF signaling pathway components are consistent with this idea (Figure 6).
The effects of PUM regulation on the Notch pathway are more complex (Figure 6F). Notch signaling controls an array of functions in development (e.g. heart development), stem cell regulation and differentiation, with direct relevance to cancer and cardiomyopathy (136). PUMs appear to affect ligand and receptor components and downstream outputs. Some Notch components are directly PUM-repressed, including the jagged JAG1 and JAG2 ligands. The Delta/Notch-like epidermal growth factor-related receptor ligand is also repressed, but lacks a PRE. Likewise, the NOTCH3 receptor, which functions in the vascular system, is repressed by PUMs (137). PUMs also repress the TMEM100, a transmembrane Notch component that functions in angiogenesis. PUMs directly repress the Adam10 metalloproteinase that cleaves Notch receptors (138). In contrast, PUMs directly activate the APH1B subunit of the γ-secretase complex, which catalyzes the intramembrane cleavage of Notch receptor, releasing the Notch intracellular domain to migrate to the nucleus where it mediated a transcriptional response (136). Multiple players involved in the nuclear events of Notch signaling are repressed by PUMs including Transducin-like enhancer protein 1 (TLE 1), a negative transcription regulator and the HEY1 transcription repressor, which is involved in neuronal and cardiovascular development (139). Other nuclear factors are activated by PUMs, including direct effects on the DNA-binding Transcription Factor Dp-2 (TFDP2) and Mastermind-like protein 1 (MAML1) transcriptional coactivator protein (140). Cyclin D1 (CCND1) is directly regulated by Notch signaling, and we observe that it is indirectly affected by depletion of PUMs. Thus PUMs are likely to have broad effects on the function of Notch signaling. Overall, PUM-mediated repression of the Notch pathway illustrates a potential role for PUM in scaling the strength of lateral induction (141,142) through simultaneous, co-directional control of Notch ligands and receptors. Because Notch pathway outputs are complex, context specific and in part determined by the repertoire of transcriptional regulators present in a particular cell type (136), future work will be necessary to measure the effects of PUM regulation on Notch mediated processes.
PUM regulation of non-coding RNAs
Our transcriptome-wide analysis revealed many ncRNAs that contain PREs including several with the highest number of PREs (Supplementary Table S6). The HCG11 lncRNA has 19 PREs, whereas the recently described non-coding RNA Activated by DNA damage (NORAD/LINC00657) has 17 PREs (24,143). Both NORAD and HCG11 are intronless, poly-adenylated ncRNAs with nearly identical sequence. Their genes are located on different chromosomes and appear to be transcribed by promoters derived from endogenous retroviral long terminal repeats. NORAD is reported to be cytoplasmic (24,143).
We observe that PUMs reduce the levels of certain non-coding RNAs. Indeed, NORAD and HCG11 were stabilized ∼1.3-fold upon PUM depletion, and both changes were statistically significant (q = 0.033 for NORAD, 0.040 for HCG11) (Supplementary Table S4). The PUM-FIT model indicates significant correlation of PUM repression and PREs in non-coding RNAs, exhibiting as strong correlation as PREs in mRNA 3′ UTRs (Table 1 and Supplementary Figure S7). Moreover, published evidence demonstrates PUM1 and PUM2 binding to PREs in NORAD and HCG11 (13,24,143). Thus, NORAD and HCG11 match our criteria as direct PUM targets.
The observed regulation of ncRNAs by PUMs is intriguing but the significance is unclear due to our limited understanding of the affected RNAs. While the molecular function of HCG11 is unknown, NORAD was proposed to act as a competitive inhibitor of PUMs, mediated by its many PREs and its transcriptional induction by DNA-damage (24), much like miRNA sponges or competing endogenous RNAs (144–147). This model does not address how the lncRNAs themselves might escape PUM-mediated degradation. Instead, our data show that NORAD is degraded by a PUM-mediated mechanism. Synthesizing this information, we hypothesize that the PUM-lncRNA interaction forms a regulatory loop (148). In this model, PUMs regulate both mRNAs and lncRNAs (e.g. NORAD), reducing their stability. Induction of lncRNA expression by a stimulus, such as DNA damage mediated induction of NORAD, could lead to competitive inhibition of PUM activity, thereby transiently alleviating PUM repression of mRNAs. At the same time, PUMs would destabilize the lncRNA, diminishing its level back to the original state.
We interrogated our RNA-seq dataset for evidence of competitive inhibition in HEK293 cells by calculating the abundance of PREs in expressed ncRNAs and mRNAs, using the normalized expression levels (FPKM counts). We found >6-fold excess of PREs in protein-coding transcripts relative to ncRNAs. NORAD and HCG11 each account for <1% of all PREs in these cells. Moreover, we observe PUM-dependent regulation of protein-coding transcripts in the highest expression range (e.g. PCNA). Thus, in this cell type under these conditions, ncRNAs would not be expected to substantially, competitively inhibit PUM function. Consistent with this idea, computational modeling of mRNA versus competitor ncRNA indicates that a very large increase in competing PREs in ncRNA, exceeding the PRE abundance in mRNAs, would be necessary to effectivity inhibit PUMs (149).
CONCLUSION
The results of this research have illuminated the diversity of target mRNAs regulated by PUM1 and PUM2 and have broad implications for PUM-mediated control of important biological pathways and processes including cellular interactions, motility and cell signaling. Future research will be necessary to discern the effects of PUM regulation on these processes in cells and in animal model systems. In particular, the relationships of PUM targets to developmental and neurological processes compels further exploration. The ability of PUMs to repress or activate transcripts in specific contexts requires further analysis of the mechanism(s) of mRNA regulation and exploration of the cis- and trans-acting factors that determine these activities. Our results also document PUM-mediated regulation of a number of disease genes linked to neurological and cardiovascular disorders and cancer, suggesting that dysregulation may arise due to loss or gain of function of PUMs in disease states. The observed redundancy of PUM1 and PUM2 function and their potential for auto- and cross-regulation may act as buffers that mitigate the potential deleterious effects (11,12) (Supplementary Table S6). Thus, genetic analysis will need to interrogate the expression and mutations of both PUMs when exploring their biological functions and connections to diseases.
AVAILABILITY
Processed and raw data for all high throughput sequencing experiments described here are available from the Gene Expression Omnibus (accession GSE95412).
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Goldstrohm laboratory for helpful suggestions and May Tsoi for technical assistance.
Footnotes
Present addresses:
Jamie Van Etten, Masonic Cancer Center, University of Minnesota, MN 55455, USA.
Brittany Bowman, Lineberger Cancer Center, University of North Carolina, Chapel Hill, NC 27599, USA.
Contributor Information
Jennifer A Bohn, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Jamie L Van Etten, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Trista L Schagat, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Promega Corporation, Madison, WI 53711, USA.
Brittany M Bowman, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Richard C McEachin, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Lydia Freddolino, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Aaron C Goldstrohm, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN 55455, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Cancer Society Research Scholar Grant [RSG-13–080-01-RMC to A.C.G.]; National Institute of General Medical Sciences, National Institutes of Health [R01GM105707 to A.C.G., R00GM097033 to L.F.]; University of Michigan Center for Genetics and Genomics Pilot Feasibility Grant (to A.C.G., T.L.S.); Promega Corporation; Michigan Predoctoral Training Program in Cellular Biotechnology through NIH National Research Service Award [5T32GM008353 to J.A.B.]. Funding for open access charge: Institutional Funds from the University of Minnesota.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajewsky N. microRNA target predictions in animals. Nat. Genet. 2006; 38(Suppl):S8–S13. [DOI] [PubMed] [Google Scholar]
- 3. Marguerat S., Bahler J. RNA-seq: from technology to biology. Cell Mol. Life Sci. 2010; 67:569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wickens M., Bernstein D.S., Kimble J., Parker R. A PUF family portrait: 3΄UTR regulation as a way of life. Trends Genet. 2002; 18:150–157. [DOI] [PubMed] [Google Scholar]
- 5. Zhang B., Gallegos M., Puoti A., Durkin E., Fields S., Kimble J., Wickens M.P. A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line. Nature. 1997; 390:477–484. [DOI] [PubMed] [Google Scholar]
- 6. Zamore P.D., Williamson J.R., Lehmann R. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA. 1997; 3:1421–1433. [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X., McLachlan J., Zamore P.D., Hall T.M. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002; 110:501–512. [DOI] [PubMed] [Google Scholar]
- 8. Wang X., Zamore P.D., Hall T.M. Crystal structure of a Pumilio homology domain. Mol. Cell. 2001; 7:855–865. [DOI] [PubMed] [Google Scholar]
- 9. Edwards T.A., Pyle S.E., Wharton R.P., Aggarwal A.K. Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell. 2001; 105:281–289. [DOI] [PubMed] [Google Scholar]
- 10. Lu G., Hall T.M. Alternate modes of cognate RNA recognition by human PUMILIO proteins. Structure. 2011; 19:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galgano A., Forrer M., Jaskiewicz L., Kanitz A., Zavolan M., Gerber A.P. Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One. 2008; 3:e3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris A.R., Mukherjee N., Keene J.D. Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol. Cell. Biol. 2008; 28:4093–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr, Jungkamp A.C., Munschauer M.et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hall T.M. De-coding and re-coding RNA recognition by PUF and PPR repeat proteins. Curr. Opin. Struct. Biol. 2016; 36:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spassov D.S., Jurecic R. Cloning and comparative sequence analysis of PUM1 and PUM2 genes, human members of the Pumilio family of RNA-binding proteins. Gene. 2002; 299:195–204. [DOI] [PubMed] [Google Scholar]
- 16. Spassov D.S., Jurecic R. The PUF family of RNA-binding proteins: does evolutionarily conserved structure equal conserved function?. IUBMB Life. 2003; 55:359–366. [DOI] [PubMed] [Google Scholar]
- 17. Van Etten J., Schagat T.L., Hrit J., Weidmann C.A., Brumbaugh J., Coon J.J., Goldstrohm A.C. Human Pumilio proteins recruit multiple deadenylases to efficiently repress messenger RNAs. J. Biol. Chem. 2012; 287:36370–36383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kedde M., van Kouwenhove M., Zwart W., Oude Vrielink J.A., Elkon R., Agami R. A Pumilio-induced RNA structure switch in p27-3΄ UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010; 12:1014–1020. [DOI] [PubMed] [Google Scholar]
- 19. Quenault T., Lithgow T., Traven A. PUF proteins: repression, activation and mRNA localization. Trends Cell Biol. 2011; 21:104–112. [DOI] [PubMed] [Google Scholar]
- 20. Miller M.A., Olivas W.M. Roles of Puf proteins in mRNA degradation and translation. Wiley Interdiscip. Rev. RNA. 2011; 2:471–492. [DOI] [PubMed] [Google Scholar]
- 21. Weidmann C.A., Raynard N.A., Blewett N.H., Van Etten J., Goldstrohm A.C. The RNA binding domain of Pumilio antagonizes poly-adenosine binding protein and accelerates deadenylation. RNA. 2014; 20:1298–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldstrohm A.C., Hook B.A., Seay D.J., Wickens M. PUF proteins bind Pop2p to regulate messenger RNAs. Nat. Struct. Mol. Biol. 2006; 13:533–539. [DOI] [PubMed] [Google Scholar]
- 23. Weidmann C.A., Goldstrohm A.C. Drosophila Pumilio protein contains multiple autonomous repression domains that regulate mRNAs independently of Nanos and brain tumor. Mol. Cell. Biol. 2012; 32:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee S., Kopp F., Chang T.C., Sataluri A., Chen B., Sivakumar S., Yu H., Xie Y., Mendell J.T. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016; 164:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen D., Zheng W., Lin A., Uyhazi K., Zhao H., Lin H. Pumilio 1 suppresses multiple activators of p53 to safeguard spermatogenesis. Curr. Biol. 2012; 22:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arvola R.M., Weidmann C.A., Tanaka Hall T.M., Goldstrohm A.C. Combinatorial control of messenger RNAs by Pumilio, Nanos and brain tumor proteins. RNA Biol. 2017; 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lehmann R., Nusslein-Volhard C. Involvement of the pumilio gene in the transport of an abdominal signal in the Drosophila embyro. Nature. 1987; 329:167–170. [Google Scholar]
- 28. Wreden C., Verrotti A.C., Schisa J.A., Lieberfarb M.E., Strickland S. Nanos and pumilio establish embryonic polarity in Drosophila by promoting posterior deadenylation of hunchback mRNA. Development. 1997; 124:3015–3023. [DOI] [PubMed] [Google Scholar]
- 29. Wharton R.P., Sonoda J., Lee T., Patterson M., Murata Y. The Pumilio RNA-binding domain is also a translational regulator. Mol. Cell. 1998; 1:863–872. [DOI] [PubMed] [Google Scholar]
- 30. Barker D.D., Wang C., Moore J., Dickinson L.K., Lehmann R. Pumilio is essential for function but not for distribution of the Drosophila abdominal determinant Nanos. Genes Dev. 1992; 6:2312–2326. [DOI] [PubMed] [Google Scholar]
- 31. Lin H., Spradling A.C. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 1997; 124:2463–2476. [DOI] [PubMed] [Google Scholar]
- 32. Asaoka-Taguchi M., Yamada M., Nakamura A., Hanyu K., Kobayashi S. Maternal Pumilio acts together with Nanos in germline development in Drosophila embryos. Nat. Cell Biol. 1999; 1:431–437. [DOI] [PubMed] [Google Scholar]
- 33. Gilboa L., Lehmann R. Repression of primordial germ cell differentiation parallels germ line stem cell maintenance. Curr. Biol. 2004; 14:981–986. [DOI] [PubMed] [Google Scholar]
- 34. Parisi M., Lin H. The Drosophila pumilio gene encodes two functional protein isoforms that play multiple roles in germline development, gonadogenesis, oogenesis and embryogenesis. Genetics. 1999; 153:235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Joly W., Chartier A., Rojas-Rios P., Busseau I., Simonelig M. The CCR4 deadenylase acts with Nanos and Pumilio in the fine-tuning of Mei-P26 expression to promote germline stem cell self-renewal. Stem Cell Rep. 2013; 1:411–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen G., Li W., Zhang Q.S., Regulski M., Sinha N., Barditch J., Tully T., Krainer A.R., Zhang M.Q., Dubnau J. Identification of synaptic targets of Drosophila pumilio. PLoS Comput. Biol. 2008; 4:e1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubnau J., Chiang A.S., Grady L., Barditch J., Gossweiler S., McNeil J., Smith P., Buldoc F., Scott R., Certa U.et al. The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr. Biol. 2003; 13:286–296. [DOI] [PubMed] [Google Scholar]
- 38. Ye B., Petritsch C., Clark I.E., Gavis E.R., Jan L.Y., Jan Y.N. Nanos and Pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Curr. Biol. 2004; 14:314–321. [DOI] [PubMed] [Google Scholar]
- 39. Bhogal B., Plaza-Jennings A., Gavis E.R. Nanos-mediated repression of hid protects larval sensory neurons after a global switch in sensitivity to apoptotic signals. Development. 2016; 143:2147–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mee C.J., Pym E.C., Moffat K.G., Baines R.A. Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J. Neurosci. 2004; 24:8695–8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Menon K.P., Sanyal S., Habara Y., Sanchez R., Wharton R.P., Ramaswami M., Zinn K. The translational repressor Pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eIF-4E. Neuron. 2004; 44:663–676. [DOI] [PubMed] [Google Scholar]
- 42. Schweers B.A., Walters K.J., Stern M. The Drosophila melanogaster translational repressor pumilio regulates neuronal excitability. Genetics. 2002; 161:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gerber A.P., Luschnig S., Krasnow M.A., Brown P.O., Herschlag D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:4487–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laver J.D., Li X., Ray D., Cook K.B., Hahn N.A., Nabeel-Shah S., Kekis M., Luo H., Marsolais A.J., Fung K.Y.et al. Brain tumor is a sequence-specific RNA-binding protein that directs maternal mRNA clearance during the Drosophila maternal-to-zygotic transition. Genome Biol. 2015; 16:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crittenden S.L., Bernstein D.S., Bachorik J.L., Thompson B.E., Gallegos M., Petcherski A.G., Moulder G., Barstead R., Wickens M., Kimble J. A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature. 2002; 417:660–663. [DOI] [PubMed] [Google Scholar]
- 46. Crittenden S.L., Eckmann C.R., Wang L., Bernstein D.S., Wickens M., Kimble J. Regulation of the mitosis/meiosis decision in the Caenorhabditis elegans germline. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003; 358:1359–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu E.Y., Chang R., Salmon N.A., Reijo Pera R.A. A gene trap mutation of a murine homolog of the Drosophila stem cell factor Pumilio results in smaller testes but does not affect litter size or fertility. Mol. Reprod. Dev. 2007; 74:912–921. [DOI] [PubMed] [Google Scholar]
- 48. Mak W., Fang C., Holden T., Dratver M.B., Lin H. An important role of Pumilio 1 in regulating the development of the mammalian female germline. Biol. Reprod. 2016; 94:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Siemen H., Colas D., Heller H.C., Brustle O., Pera R.A. Pumilio-2 function in the mouse nervous system. PLoS One. 2011; 6:e25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang M., Chen D., Xia J., Han W., Cui X., Neuenkirchen N., Hermes G., Sestan N., Lin H. Post-transcriptional regulation of mouse neurogenesis by Pumilio proteins. Genes Dev. 2017; 1354–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fox M., Urano J., Reijo Pera R.A. Identification and characterization of RNA sequences to which human PUMILIO-2 (PUM2) and deleted in Azoospermia-like (DAZL) bind. Genomics. 2005; 85:92–105. [DOI] [PubMed] [Google Scholar]
- 52. Spik A., Oczkowski S., Olszak A., Formanowicz P., Blazewicz J., Jaruzelska J. Human fertility protein PUMILIO2 interacts in vitro with testis mRNA encoding Cdc42 effector 3 (CEP3). Reprod. Biol. 2006; 6:103–113. [PubMed] [Google Scholar]
- 53. Spik A., Oczkowski S., Olszak A., Kotecki M., Formanowicz P., Blazewicz J., Jaruzelska J. Candidate mRNAs interacting with fertility protein PUMILIO2 in the human germ line. Reprod. Biol. 2006; 6(Suppl. 1):37–42. [PubMed] [Google Scholar]
- 54. Gennarino V.A., Singh R.K., White J.J., De Maio A., Han K., Kim J.Y., Jafar-Nejad P., di Ronza A., Kang H., Sayegh L.S.et al. Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell. 2015; 160:1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vessey J.P., Schoderboeck L., Gingl E., Luzi E., Riefler J., Di Leva F., Karra D., Thomas S., Kiebler M.A., Macchi P. Mammalian Pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:3222–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Driscoll H.E., Muraro N.I., He M., Baines R.A. Pumilio-2 regulates translation of Nav1.6 to mediate homeostasis of membrane excitability. J. Neurosci. 2013; 33:9644–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vessey J.P., Vaccani A., Xie Y., Dahm R., Karra D., Kiebler M.A., Macchi P. Dendritic localization of the translational repressor Pumilio 2 and its contribution to dendritic stress granules. J. Neurosci. 2006; 26:6496–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miles W.O., Tschop K., Herr A., Ji J.Y., Dyson N.J. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012; 26:356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee M.H., Hook B., Pan G., Kershner A.M., Merritt C., Seydoux G., Thomson J.A., Wickens M., Kimble J. Conserved regulation of MAP kinase expression by PUF RNA-binding proteins. PLoS Genet. 2007; 3:e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5:621–628. [DOI] [PubMed] [Google Scholar]
- 61. Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013; 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Van Etten J., Schagat T.L., Goldstrohm A.C. A guide to design and optimization of reporter assays for 3΄ untranslated region mediated regulation of mammalian messenger RNAs. Methods. 2013; 63:110–118. [DOI] [PubMed] [Google Scholar]
- 65. Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 66. Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008; 3:1101–1108. [DOI] [PubMed] [Google Scholar]
- 67. Plummer M. JAGS: a program for analysis of Bayesian graphical models using gibbs sampling. Proceedings of the 3rd International Workshop on Distributed Statistical Computing. 2003; 1–10. [Google Scholar]
- 68. Gelman A., Rubin D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992; 7:457–472. [Google Scholar]
- 69. Kruschke J.K. Bayesian estimation supersedes the t test. J. Exp. Psychol. Gen. 2013; 142:573–603. [DOI] [PubMed] [Google Scholar]
- 70. Goodarzi H., Elemento O., Tavazoie S. Revealing global regulatory perturbations across human cancers. Mol. Cell. 2009; 36:900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gene Ontology, C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015; 43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mi H., Poudel S., Muruganujan A., Casagrande J.T., Thomas P.D. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016; 44:D336–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Elemento O., Slonim N., Tavazoie S. A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell. 2007; 28:337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A.et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013; 499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shaw G., Morse S., Ararat M., Graham F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002; 16:869–871. [DOI] [PubMed] [Google Scholar]
- 79. Lin Y.C., Boone M., Meuris L., Lemmens I., Van Roy N., Soete A., Reumers J., Moisse M., Plaisance S., Drmanac R.et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014; 5:4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pearson E.S. The choice of statistical tests illustrated on the interpretation of data classed in a 2 X 2 table. Biometrika. 1947; 34:139–169. [DOI] [PubMed] [Google Scholar]
- 81. Campbell I. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat. Med. 2007; 26:3661–3675. [DOI] [PubMed] [Google Scholar]
- 82. Cheong C.G., Hall T.M. Engineering RNA sequence specificity of Pumilio repeats. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13635–13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gupta Y.K., Nair D.T., Wharton R.P., Aggarwal A.K. Structures of human Pumilio with noncognate RNAs reveal molecular mechanisms for binding promiscuity. Structure. 2008; 16:549–557. [DOI] [PubMed] [Google Scholar]
- 84. Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A.et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011; 39:D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004; 4:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Marzluff W.F., Wagner E.J., Duronio R.J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 2008; 9:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dansie L.E., Reeves S., Miller K., Zano S.P., Frank M., Pate C., Wang J., Jackowski S. Physiological roles of the pantothenate kinases. Biochem. Soc. Trans. 2014; 42:1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Klaus A., Birchmeier W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer. 2008; 8:387–398. [DOI] [PubMed] [Google Scholar]
- 89. Boyd J.L., Skove S.L., Rouanet J.P., Pilaz L.J., Bepler T., Gordan R., Wray G.A., Silver D.L. Human-chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr. Biol. 2015; 25:772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kiefel H., Bondong S., Hazin J., Ridinger J., Schirmer U., Riedle S., Altevogt P. L1CAM: a major driver for tumor cell invasion and motility. Cell Adh. Migr. 2012; 6:374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wu M.Y., Lin Y.C., Liao W.J., Tu C.F., Chen M.H., Roffler S.R., Yang R.B. Inhibition of the plasma SCUBE1, a novel platelet adhesive protein, protects mice against thrombosis. Arterioscler Thromb. Vasc. Biol. 2014; 34:1390–1398. [DOI] [PubMed] [Google Scholar]
- 92. Shiratori H., Hamada H. TGFbeta signaling in establishing left-right asymmetry. Semin. Cell Dev. Biol. 2014; 32:80–84. [DOI] [PubMed] [Google Scholar]
- 93. Kim D.K., Cha Y., Ahn H.J., Kim G., Park K.S. Lefty1 and lefty2 control the balance between self-renewal and pluripotent differentiation of mouse embryonic stem cells. Stem Cells Dev. 2014; 23:457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Saito Y., Miranda-Rottmann S., Ruggiu M., Park C.Y., Fak J.J., Zhong R., Duncan J.S., Fabella B.A., Junge H.J., Chen Z.et al. NOVA2-mediated RNA regulation is required for axonal pathfinding during development. Elife. 2016; 5:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pacheco M.C. Multiple endocrine neoplasia: a genetically diverse group of familial tumor syndromes. J. Pediatr. Genet. 2016; 5:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McKeown S.J., Stamp L., Hao M.M., Young H.M. Hirschsprung disease: a developmental disorder of the enteric nervous system. Wiley Interdiscip. Rev. Dev. Biol. 2013; 2:113–129. [DOI] [PubMed] [Google Scholar]
- 97. Mor-Vaknin N., Saha A., Legendre M., Carmona-Rivera C., Amin M.A., Rabquer B.J., Gonzales-Hernandez M.J., Jorns J., Mohan S., Yalavarthi S.et al. DEK-targeting DNA aptamers as therapeutics for inflammatory arthritis. Nat. Commun. 2017; 8:14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sanden C., Gullberg U. The DEK oncoprotein and its emerging roles in gene regulation. Leukemia. 2015; 29:1632–1636. [DOI] [PubMed] [Google Scholar]
- 99. Wright K.O., Messing E.M., Reeder J.E. Increased expression of the acid sphingomyelinase-like protein ASML3a in bladder tumors. J. Urol. 2002; 168:2645–2649. [DOI] [PubMed] [Google Scholar]
- 100. Traini M., Quinn C.M., Sandoval C., Johansson E., Schroder K., Kockx M., Meikle P.J., Jessup W., Kritharides L. Sphingomyelin phosphodiesterase acid-like 3A (SMPDL3A) is a novel nucleotide phosphodiesterase regulated by cholesterol in human macrophages. J. Biol. Chem. 2014; 289:32895–32913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tian Y., Schreiber R., Kunzelmann K. Anoctamins are a family of Ca2+-activated Cl- channels. J. Cell Sci. 2012; 125:4991–4998. [DOI] [PubMed] [Google Scholar]
- 102. Maniero C., Zhou J., Shaikh L.H., Azizan E.A., McFarlane I., Neogi S., Scudieri P., Galietta L.J., Brown M.J. Role of ANO4 in regulation of aldosterone secretion in the zona glomerulosa of the human adrenal gland. Lancet. 2015; 385(Suppl. 1):S62. [DOI] [PubMed] [Google Scholar]
- 103. Weidmann C.A., Qiu C., Arvola R.M., Lou T.F., Killingsworth J., Campbell Z.T., Tanaka Hall T.M., Goldstrohm A.C. Drosophila Nanos acts as a molecular clamp that modulates the RNA-binding and repression activities of Pumilio. Elife. 2016; 5:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zamore P.D., Bartel D.P., Lehmann R., Williamson J.R. The PUMILIO-RNA interaction: a single RNA-binding domain monomer recognizes a bipartite target sequence. Biochemistry. 1999; 38:596–604. [DOI] [PubMed] [Google Scholar]
- 105. Chou C.H., Chang N.W., Shrestha S., Hsu S.D., Lin Y.L., Lee W.H., Yang C.D., Hong H.C., Wei T.Y., Tu S.J.et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44:D239–D247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen Y.H., Coller J. A universal code for mRNA stability. Trends Genet. 2016; 32:687–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hu W., Sweet T.J., Chamnongpol S., Baker K.E., Coller J. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature. 2009; 461:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K.E., Graveley B.R.et al. Codon optimality is a major determinant of mRNA stability. Cell. 2015; 160:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tat T.T., Maroney P.A., Chamnongpol S., Coller J., Nilsen T.W. Cotranslational microRNA mediated messenger RNA destabilization. Elife. 2016; 5:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Walser C.B., Lipshitz H.D. Transcript clearance during the maternal-to-zygotic transition. Curr. Opin. Genet. Dev. 2011; 21:431–443. [DOI] [PubMed] [Google Scholar]
- 111. Lee C.D., Tu B.P. Glucose-regulated phosphorylation of the PUF protein Puf3 regulates the translational fate of its bound mRNAs and association with RNA granules. Cell Rep. 2015; 11:1638–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Suh N., Crittenden S.L., Goldstrohm A., Hook B., Thompson B., Wickens M., Kimble J. FBF and its dual control of gld-1 expression in the Caenorhabditis elegans germline. Genetics. 2009; 181:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kaye J.A., Rose N.C., Goldsworthy B., Goga A., L’Etoile N.D. A 3΄UTR pumilio-binding element directs translational activation in olfactory sensory neurons. Neuron. 2009; 61:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Naudin C., Hattabi A., Michelet F., Miri-Nezhad A., Benyoucef A., Pflumio F., Guillonneau F., Fichelson S., Vigon I., Dusanter-Fourt I.et al. PUMILIO/FOXP1 signaling drives expansion of hematopoietic stem/progenitor and leukemia cells. Blood. 2017; 129:2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Oh S., Shin S., Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta. 2012; 1826:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fontanet P.A., Rios A.S., Alsina F.C., Paratcha G., Ledda F. Pea3 transcription factors, Etv4 and Etv5, are required for proper hippocampal dendrite development and plasticity. Cereb. Cortex. 2016; 1–14. [DOI] [PubMed] [Google Scholar]
- 117. Pellecchia A., Pescucci C., De Lorenzo E., Luceri C., Passaro N., Sica M., Notaro R., De Angioletti M. Overexpression of ETV4 is oncogenic in prostate cells through promotion of both cell proliferation and epithelial to mesenchymal transition. Oncogenesis. 2012; 1:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Aytes A., Mitrofanova A., Kinkade C.W., Lefebvre C., Lei M., Phelan V., LeKaye H.C., Koutcher J.A., Cardiff R.D., Califano A.et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E3506–E3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kidger A.M., Keyse S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016; 50:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Maillet M., Purcell N.H., Sargent M.A., York A.J., Bueno O.F., Molkentin J.D. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J. Biol. Chem. 2008; 283:31246–31255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Miraoui H., Dwyer A.A., Sykiotis G.P., Plummer L., Chung W., Feng B., Beenken A., Clarke J., Pers T.H., Dworzynski P.et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am. J. Hum. Genet. 2013; 92:725–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Boulding T., Wu F., McCuaig R., Dunn J., Sutton C.R., Hardy K., Tu W., Bullman A., Yip D., Dahlstrom J.E.et al. Differential roles for DUSP family members in epithelial-to-mesenchymal transition and cancer stem cell regulation in breast cancer. PLoS One. 2016; 11:e0148065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Messina S., Frati L., Leonetti C., Zuchegna C., Di Zazzo E., Calogero A., Porcellini A. Dual-specificity phosphatase DUSP6 has tumor-promoting properties in human glioblastomas. Oncogene. 2011; 30:3813–3820. [DOI] [PubMed] [Google Scholar]
- 124. Pique M., Lopez J.M., Foissac S., Guigo R., Mendez R. A combinatorial code for CPE-mediated translational control. Cell. 2008; 132:434–448. [DOI] [PubMed] [Google Scholar]
- 125. Ivshina M., Lasko P., Richter J.D. Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu. Rev. Cell Dev. Biol. 2014; 30:393–415. [DOI] [PubMed] [Google Scholar]
- 126. Louvi A., Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006; 7:93–102. [DOI] [PubMed] [Google Scholar]
- 127. Chen C.A., Bosch D.G., Cho M.T., Rosenfeld J.A., Shinawi M., Lewis R.A., Mann J., Jayakar P., Payne K., Walsh L.et al. The expanding clinical phenotype of Bosch-Boonstra-Schaaf optic atrophy syndrome: 20 new cases and possible genotype-phenotype correlations. Genet. Med. 2016; 18:1143–1150. [DOI] [PubMed] [Google Scholar]
- 128. Menon K.P., Andrews S., Murthy M., Gavis E.R., Zinn K. The translational repressors Nanos and Pumilio have divergent effects on presynaptic terminal growth and postsynaptic glutamate receptor subunit composition. J. Neurosci. 2009; 29:5558–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Muraro N.I., Weston A.J., Gerber A.P., Luschnig S., Moffat K.G., Baines R.A. Pumilio binds para mRNA and requires Nanos and Brat to regulate sodium current in Drosophila motoneurons. J. Neurosci. 2008; 28:2099–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rouhana L., Wang L., Buter N., Kwak J.E., Schiltz C.A., Gonzalez T., Kelley A.E., Landry C.F., Wickens M. Vertebrate GLD2 poly(A) polymerases in the germline and the brain. RNA. 2005; 11:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kapetis D., Sassone J., Yang Y., Galbardi B., Xenakis M.N., Westra R.L., Szklarczyk R., Lindsey P., Faber C.G., Gerrits M.et al. Network topology of NaV1.7 mutations in sodium channel-related painful disorders. BMC Syst. Biol. 2017; 11:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Anastas J.N., Moon R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013; 13:11–26. [DOI] [PubMed] [Google Scholar]
- 133. Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Selbach M., Schwanhausser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008; 455:58–63. [DOI] [PubMed] [Google Scholar]
- 135. Yang E., van Nimwegen, Zavolan E., Rajewsky M., Schroeder N., Magnasco M., Darnell J.E. Jr Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003; 13:1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bray S.J. Notch signalling in context. Nat. Rev. Mol. Cell. Biol. 2016; 17:722–735. [DOI] [PubMed] [Google Scholar]
- 137. Penton A.L., Leonard L.D., Spinner N.B. Notch signaling in human development and disease. Semin. Cell Dev. Biol. 2012; 23:450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Saftig P., Lichtenthaler S.F. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog. Neurobiol. 2015; 135:1–20. [DOI] [PubMed] [Google Scholar]
- 139. Weber D., Wiese C., Gessler M. Hey bHLH transcription factors. Curr. Top Dev. Biol. 2014; 110:285–315. [DOI] [PubMed] [Google Scholar]
- 140. Saint Just Ribeiro M., Wallberg A.E. Transcriptional mechanisms by the coregulator MAML1. Curr. Protein Pept. Sci. 2009; 10:570–576. [DOI] [PubMed] [Google Scholar]
- 141. Petrovic J., Formosa-Jordan P., Luna-Escalante J.C., Abello G., Ibanes M., Neves J., Giraldez F. Ligand-dependent Notch signaling strength orchestrates lateral induction and lateral inhibition in the developing inner ear. Development. 2014; 141:2313–2324. [DOI] [PubMed] [Google Scholar]
- 142. Hartman B.H., Reh T.A., Bermingham-McDonogh O. Notch signaling specifies prosensory domains via lateral induction in the developing mammalian inner ear. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:15792–15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tichon A., Gil N., Lubelsky Y., Havkin Solomon T., Lemze D., Itzkovitz S., Stern-Ginossar N., Ulitsky I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 2016; 7:12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010; 465:1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ebert M.S., Sharp P.A. Emerging roles for natural microRNA sponges. Curr. Biol. 2010; 20:R858–R861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ebert M.S., Sharp P.A. MicroRNA sponges: progress and possibilities. RNA. 2010; 16:2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Poliseno L., Salmena L., Riccardi L., Fornari A., Song M.S., Hobbs R.M., Sportoletti P., Varmeh S., Egia A., Fedele G.et al. Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010; 3:ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007; 8:450–461. [DOI] [PubMed] [Google Scholar]
- 149. Jens M., Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat. Rev. Genet. 2015; 16:113–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.