


Abstract
Biological processes are usually associated with genome-wide remodeling of transcription driven by transcription factors (TFs). Identifying key TFs and their spatiotemporal binding patterns are indispensable to understanding how dynamic processes are programmed. However, most methods are designed to predict TF binding sites only. We present a computational method, dynamic motif occupancy analysis (DynaMO), to infer important TFs and their spatiotemporal binding activities in dynamic biological processes using chromatin profiling data from multiple biological conditions such as time-course histone modification ChIP-seq data. In the first step, DynaMO predicts TF binding sites with a random forests approach. Next and uniquely, DynaMO infers dynamic TF binding activities at predicted binding sites using their local chromatin profiles from multiple biological conditions. Another landmark of DynaMO is to identify key TFs in a dynamic process using a clustering and enrichment analysis of dynamic TF binding patterns. Application of DynaMO to the yeast ultradian cycle, mouse circadian clock and human neural differentiation exhibits its accuracy and versatility. We anticipate DynaMO will be generally useful for elucidating transcriptional programs in dynamic processes.
INTRODUCTION
Transcription factors (TFs) bind to functional regulatory DNA sequences and regulate the expression of target genes. Hundreds of TFs have been identified from yeast to mammals (1,2). They play critical roles in ensuring the accuracy and specificity of transcription, not only under homeostatic conditions but also in various dynamic processes, such as cell cycle, development, differentiation and stress response (3–9). TFs do not merely establish appropriate levels of transcription, they also drive the progression of these dynamic processes. The functions of TFs are mostly context dependent (10). Mutations in TFs may dramatically affect gene expression under certain specific conditions or perturbations, though effects in the steady state may be minimal (11,12). Therefore, examining the dynamics and context of TF binding and activity is important for understanding their physiological functions. Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) has been widely used to examine genome-wide binding of TFs (13). However, each ChIP-seq experiment can only study one TF in one biological condition, and most recent studies were able to examine only a few TFs and conditions (14,15). A high-throughput ChIP approach still only examines limited TFs and conditions (16). An obvious challenge is how to examine genome-wide binding of hundreds of TFs simultaneously in a dynamic process consisting of multiple conditions (e.g. multiple time points), and how to identify TFs important for the dynamic process and prioritize them for subsequent functional studies. Directly applying ChIP-seq to all TFs and conditions would be laborious and costly, and often impossible due to limitations in materials, antibodies and reagents.
TFs usually recognize specific patterns of DNA sequences, known as TF binding motifs (4), characterized by consensus sequences or position-specific frequency matrices (PSFMs). The motif base readout and shape readout could determine protein–DNA recognition (17). Recent studies have shown that TF binding activities can be predicted by integrating static motif information with condition-dependent information on chromatin states and accessibility from high-throughput chromatin profiling data such as histone modification ChIP-seq and DNase I hypersensitivity sequencing (DNase-seq) data (18–26). This approach can predict binding sites of many TFs simultaneously. However, currently the approach is primarily used to predict TF binding sites (TFBSs) in a single condition or differential binding between pairs of conditions. Most existing methods do not provide a systematic solution to analyzing global regulatory programs in a dynamic process such as a time-course experiment with multiple conditions, which requires one to answer important questions such as which TFs are important for controlling the dynamic process, where and when these TFs bind, how their binding activities change across conditions, what major dynamic occupancy patterns of cis-regulatory elements exist, and which TFs are responsible for these patterns.
Here we introduce dynamic motif occupancy analysis (DynaMO), a computational pipeline that integrates TFBS prediction by random decision forests, binding site dynamic occupancy pattern discovery by unsupervised clustering, and DNA motif enrichment analysis to systematically address the above issues. DynaMO takes global chromatin profiling data from a dynamic process (e.g. time-course histone modification ChIP-seq or DNase-seq data) and TF binding motifs as input. It is designed to help users to achieve two goals: (i) predict important TFs responsible for dynamic processes; (ii) predict binding sites of these TFs and evaluate dynamic changes in their binding.
We applied DynaMO to study yeast metabolic cycle (YMC; also referred to as the ultradian cycle) (27), mammalian circadian cycle (28), and human neural differentiation (29). The first two systems exhibit coordinated oscillations of the transcriptome, epigenome and metabolome (30–32), and neural differentiation also shows dynamics of transcription and histone modification (33). Our analyses demonstrate that DynaMO is capable of unsupervised discovery of important TFs and their binding patterns for dynamic processes. Furthermore, it also allows one to more accurately predict TF binding sites.
MATERIALS AND METHODS
DynaMO overview
DynaMO is developed using the statistical programming language R (34). The objective of DynaMO is to integrate TF binding motif information with dynamic genome-wide chromatin profiling data to predict important TFs responsible for dynamic processes. It also predicts binding sites recognized by these TFs and dynamic changes of TF binding activities. We assume that chromatin profiling data collected from multiple biological conditions (e.g. multiple time points of a biological process) are available. The data may involve only one data type (e.g. ChIP-seq for one histone mark) or multiple data types (e.g. ChIP-seq for multiple histone marks) capable of serving as surrogates for TF binding activities. For each data type, data from all conditions are assumed to be available. DynaMO has three general components: (i) predicting TF binding sites; (ii) grouping binding sites with similar dynamic binding patterns into clusters; (iii) identifying TFs associated with each binding site cluster. For time course data, DynaMO can also describe temporal activities of predicted binding sites by fitting smooth temporal curves and extracting temporal characteristics from the fitted curves including the estimated time points corresponding to the maximal or minimal activity or the fastest changes in activity.
Data preprocessing
DynaMO takes locations of motif sites and aligned sequence reads from global chromatin profiling experiments as input (e.g. histone modification ChIP-seq data, DNase-seq data, etc.). For a given chromatin data type, each aligned read is shifted L/2 base pairs (bp) toward its 3′ end where L represents the expected DNA fragment size. This is because in experiments such as ChIP-seq, DNA is fragmented into small size smears and reads are typically generated from the ends of the DNA fragments through sequencing-by-synthesis. DNAs are synthesized in 5′-to-3′ direction. The L/2 bp shift toward the 3′ end of the read is equivalent to extending each read by L bp from its 5′ end to reconstruct its original DNA fragment and then extracting the center of the DNA fragment. For example, in the YMC analysis below, the DNA fragment size for the H3K9ac ChIP-seq data was expected to be ∼150 bp based on the size selection step in the ChIP-seq protocol. Therefore, L was set to 150 (i.e. reads were shifted 75 bp toward 3′ ends). The shifted reads are used for all subsequent analyses.
In our study, CisGenome (35) was used to map TF binding motifs to genomes. Motif sites with likelihood ratio ≥500 (i.e. the default cutoff value of CisGenome) were reported, and their locations were used for subsequent analyses. DynaMO first extends each motif site by W bp on both sides from the motif center. This creates a 2W bp long window centered at each motif site. The window is then divided into N consecutive bins. The length of each bin is B bp. Here B*N = 2W. For each data type and each biological condition, the number of shifted reads in each bin is counted, yielding an N-dimensional count vector for each motif site. The bin read counts are normalized across samples by the total read count of each sample. When there are D different data types, the above procedure will produce D vectors, and each vector is N-dimensional and contains read counts from N bins. They will be used as features by the random forests (RF) model below for TFBS prediction.
The procedure above involves two user-specified parameters W and B. The other two parameters D and L are determined by the experimental design. It is recommended that the window size parameter W should reflect the signal span (i.e. peak width) in the chromatin profiling data. Given W, the choice of bin size B represents a tradeoff between the features’ spatial resolution (i.e. bin size) and their dimension (i.e. bin number). Typically we choose B so that the bin size is ∼20–100 bp and the bin number is below 40–50 (i.e. N < 40–50) to avoid extremely high dimensionality. Take the YMC data as an example. After ChIP-seq peak calling described below, the mean width of H3K9ac peaks was 298 bp. Considering that TFs typically bind to nucleosome free regions and histone modification peaks are on nucleosomes adjacent to TFBSs, we set W = 300 and B = 20. This means that the 600 bp window surrounding each motif site was divided into 30 consecutive bins and each bin was 20 bp long. Read counts from these 30 bins were used by RF as features for TFBS prediction. In the human neural differentiation data, the mean width of the H3K27ac and H3K4me3 ChIP-seq peaks was 976 and 1166 bp respectively. In the mammalian circadian clock data, the mean peak width for H3K9ac and H3K4me1 was 896 and 984 bp respectively. Thus, the average span of chromatin signals in these two mammalian datasets was ∼1000 bp. Based on this, we set W = 1000 and B = 50. This implies that the 2000 bp window surrounding each motif site was divided into 40 consecutive bins and each bin was 50 bp long. In Supplementary Materials, we evaluated the impact of different values of W and B on TFBS prediction. It was found that DynaMO is relatively robust to different choices of W and B unless they deviate substantially from our recommended values (see details in Supplementary Materials).
TFBS prediction
DynaMO uses a two-pass algorithm to predict TFBSs.
Initial peak calling (First pass TFBS prediction): In this step, a conventional ChIP-seq peak caller is used to analyze global chromatin profiling data (e.g. histone modification ChIP-seq data, DNase-seq data, etc.). For each data type and each biological condition (e.g. each time point), genomic regions with enriched chromatin signals are identified. In principle, any peak caller compatible with the data type at hand can be used here. For instance, when one deals with a ChIP-seq dataset with both ChIP and input control samples, a ChIP-seq peak caller capable of handling control samples may be used. For a DNase-seq dataset without any input control sample, one should use a peak caller that does not require control samples. For convenience to integrate with the DynaMO R scripts, we used a peak caller developed in R, BayesPeak (36), to analyze data in this study. Peaks reported by BayesPeak with default parameters were the initial peaks.
RF (Second pass TFBS prediction): for each biological condition (e.g. each time point), all motif sites are separated into two classes: motif sites overlapping with an initial peak identified from the previous step in this condition, and motif sites that do not overlap with the initial peaks. From these two classes of motif sites, a positive training set and a negative training set are constructed for each TF for subsequent supervised learning. More precisely, suppose there are D data types. To construct the positive training set for each TF and biological condition, motif sites overlapping with the initial peaks identified from any data type are identified. These motif sites are ranked by the number of overlapping peaks (i.e. how many data types have peaks that overlap with each motif site) first and then in case of ties, by the sum of normalized peak signals (i.e. normalized read counts in the 2W bp window centered at motif sites) from all data types. The positive training set consists of 250 motif sites chosen from the top ranked sites. If a TF has fewer than 250 motif sites that overlap with initial peaks, all motif sites overlapping with the initial peaks are used as the positive training set. To construct the negative training set for each TF and biological condition, 250 motif sites are randomly sampled from the TF’s motif sites that do not overlap with any initial peak in this biological condition. In addition to the negative training set, we also randomly sample 500 motif sites and use them as background motif sites to derive the null distribution for P-value calculation. Next, the positive and negative training sets are used for constructing supervised TFBS prediction as follows:
For each TF and biological condition (e.g. each time point), DynaMO first constructs an RF model using the corresponding positive and negative training sets. This is done using the randomForest function in R (the number of trees is set to 500, the default value of the randomForest function). The features used by RF are the vectors of bin read counts surrounding each motif site described above. This RF model is TF- and condition-specific. In other words, each (TF, condition) pair has its own model. The trained model is then applied to all motif sites of the TF to predict whether each motif site is bound or not. Each motif site receives a vote value from the RF. The same RF model is also applied to all background motif sites to generate the null distribution of the vote values for P-value calculation.
For each biological condition, DynaMO also combines the training data from all TFs (e.g. all 175 motifs in YMC) together. Using the combined positive training set and negative training set, another RF model is trained. This model is not TF-specific. It is common to all TFs. However, it is still condition-specific. Applying the model to all motif sites and all background sites, DynaMO will generate another set of vote values for each motif site.
For each TF and condition, the two sets of vote values (one from the TF- and condition-specific RF, and the other one from the TF-invariant but condition-specific RF) are averaged at each motif site. Similarly, the two sets of vote values are also averaged for each background site to derive the null distribution. Based on the empirical cumulative distribution function of the null distribution (computed using the R function ‘ecdf’), a p-value is calculated for each motif site. The p-values are then converted into false discovery rate (FDR) (using the R function ‘fdr’) to account for multiple testing.
Clustering binding sites
Motif sites with FDR < 0.01 in any of the analyzed biological conditions (e.g. at any of the 16 time points in the YMC) are combined. These predicted binding sites from all TFs are pooled together. For each data type and biological condition, the normalized read count in the 2W bp long window at each binding site are log2 transformed after adding a pseudo-count of 1. For each data type, log-transformed read counts are organized into a matrix. Rows of the matrix correspond to motif sites, and columns correspond to biological conditions. Each row is standardized to have zero mean and unit standard deviation. When there are multiple data types, the matrix is first constructed and row-standardized for each data type. Then matrices from all data types are concatenated together. For C biological conditions and D data types, the combined matrix will have C*D columns in total. K-means clustering with Hartigan-Wong algorithm (‘kmeans’ function in R) is then applied to the combined matrix to cluster rows.
The number of clusters, K, is determined using the piecewise linear elbow method in (37). Briefly, K is set to different values. For each K, the rows of data matrix (i.e. binding sites) are clustered, and the proportion of total data variance unexplained by the cluster structure is computed. To do so, let 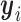 denote the row-standardized data for row i. Let M(i) be the cluster membership of the ith row. Let
denote the row-standardized data for row i. Let M(i) be the cluster membership of the ith row. Let 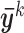 denote the mean of the kth cluster, and let
denote the mean of the kth cluster, and let 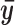 be the mean of all rows. The total data variance is
be the mean of all rows. The total data variance is 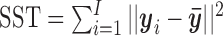 where
where  represents
represents 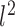 norm and I is the total number of rows. The variance unexplained by the cluster structure is
norm and I is the total number of rows. The variance unexplained by the cluster structure is 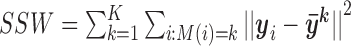 . The proportion of total data variance unexplained by the cluster structure is
. The proportion of total data variance unexplained by the cluster structure is 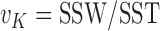 . As the cluster number K increases, the proportion of unexplained data variance decreases. Therefore,
. As the cluster number K increases, the proportion of unexplained data variance decreases. Therefore, 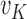 is a decreasing function of K. One can approximate this function using a continuous piecewise linear model
is a decreasing function of K. One can approximate this function using a continuous piecewise linear model 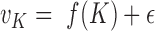 where
where 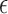 represents noise and f(K) consists of two regression lines (Supplementary Figure S1):
represents noise and f(K) consists of two regression lines (Supplementary Figure S1):
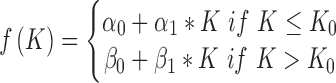 |
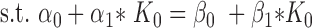 |
For a given junction point 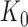 , this model can be fitted using the least squares approach. As
, this model can be fitted using the least squares approach. As 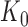 changes, the fitted model also changes. The
changes, the fitted model also changes. The 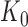 that produces the smallest squared error
that produces the smallest squared error 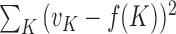 will be used as the optimal cluster number. For instance, in the YMC data the optimal cluster number obtained using this approach was 3. This result was consistent with the three known phases of the YMC (27).
will be used as the optimal cluster number. For instance, in the YMC data the optimal cluster number obtained using this approach was 3. This result was consistent with the three known phases of the YMC (27).
Identifying TFs associated with each cluster
After clustering, the number of predicted binding sites for each TF motif in each binding site cluster is counted. These counts are organized into a matrix. For instance, for YMC, the matrix had 175 rows (corresponding to 175 motifs) and 3 columns (corresponding to 3 clusters). To test whether a motif is enriched in a cluster, Fisher’s exact tests are conducted for each cell of the matrix (corresponding to a TF and cluster pair) using the following four numbers: the number of binding sites for the TF in the cluster (x1), the number of sites for the TF but not in the cluster (x0), the number of sites for other TFs in the cluster (z1), and the number of sites for other TFs and not in the cluster (z0). P-values from the tests are adjusted by the ‘p.adjust’ function with the Bonferroni method to account for multiple comparisons. For each TF motif, a fold enrichment in each cluster is also computed using [x1/(x1+x0)]/[(x1+z1)/(x1+z1+x0+z0)]. TFs are first ranked by adjusted P-values and then in the case of ties, by fold enrichment. TF motifs with adjusted P-values < 10−5 are shown in Figure 2.
Figure 2.

DynaMO predicts important TFs and temporal activities of TFs in YMC. (A) Clustering of predicted motif sites based on temporal H3K9ac signals. TFs with their motif sites enriched in clusters are listed to the right of the heat map. TFs whose expression is correlated with corresponding clusters are marked in red. A similar plot is presented in the companion paper by Kuang et al. (52) Figure 1B for the completeness of that study. (B) Phenotypic validation of TF mutants in YMC. The left panel shows the percentages of examined TFs with defect YMC. The right panel is a table summarizing the phenotypes. ‘+’ represents normal oscillation and ‘–’ represents abnormal oscillation. ‘+/–’ represents abnormal oscillation when the TF is mutated together with a mutation of another TF. ‘D’ means dampened oscillation; ‘A’ means changed amplitude; ‘F’ means a flat line of oxygen; ‘L-RC’ or ‘S-RC’ means longer or shorter periods of RC phase. (C) Examples of WT and abnormal oscillation in TF mutants.
DynaMO output
The enriched TFs identified above are reported. For each reported TF, DynaMO also reports the predicted binding sites in each biological condition. For each TF and condition, motif sites are ranked by the FDR obtained from TFBS prediction first. When multiple motif sites are tied because of identical FDR, their ranks are resolved using their ‘binding intensity’. The binding intensity of a motif site in a biological condition is computed using a projection approach that takes into account the characteristic shapes of read distributions around TFBSs. First, for a given data type, let the vector  be the normalized bin read counts for the N neighboring bins of motif site i. Let the vector
be the normalized bin read counts for the N neighboring bins of motif site i. Let the vector 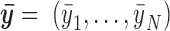 be the average of
be the average of  from the top 100 predicted binding sites for the TF in question. Here
from the top 100 predicted binding sites for the TF in question. Here 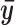 characterizes the average chromatin profile surrounding TFBSs. We first scale
characterizes the average chromatin profile surrounding TFBSs. We first scale 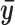 by
by 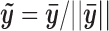 so that it has unit length. The binding intensity of each motif site i is then calculated as the inner product of
so that it has unit length. The binding intensity of each motif site i is then calculated as the inner product of 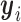 and
and 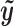 :
: 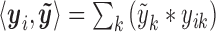 . This can be viewed as a weighted average of bin read counts surrounding motif site i where the weights are given by elements in
. This can be viewed as a weighted average of bin read counts surrounding motif site i where the weights are given by elements in 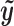 . Intuitively, given the same number of total bin read counts, a read distribution more similar to the characteristic chromatin profile determined by
. Intuitively, given the same number of total bin read counts, a read distribution more similar to the characteristic chromatin profile determined by 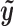 will yield a higher binding intensity. When there are multiple data types, the binding intensity is first computed for each data type separately. Then the average of binding intensities from all data types is computed as the predicted binding intensity. After computing the binding intensity of each motif site, the motif sites tied according to FDR are then ranked based on the binding intensity.
will yield a higher binding intensity. When there are multiple data types, the binding intensity is first computed for each data type separately. Then the average of binding intensities from all data types is computed as the predicted binding intensity. After computing the binding intensity of each motif site, the motif sites tied according to FDR are then ranked based on the binding intensity.
Characterization of temporal activities
The analyses described up to this point do not assume that the biological conditions have an intrinsic order. Thus, one may apply them to any dataset with multiple conditions. For data from time-course experiments, biological conditions will have a temporal order. For such data, DynaMO can further fit smooth temporal curves to describe binding intensities at predicted binding sites as functions of time. For each binding site, a smooth curve is fitted to the binding intensities from different time points using locally weighted scatterplot smoothing (LOESS) (38). The fitted curve and its derivative can be used to study temporal characteristics of the dynamic biological process. For example, one can estimate the time point at which the activity achieves its maximum or minimum (i.e. the derivative is zero), or when the activity increases or decreases at its fastest rate (i.e. the derivative achieves its maximum or minimum) (Supplementary Figure S2).
DynaMO analyses of YMC, neural differentiation and circadian clock
Details of DynaMO analysis of YMC, neural differentiation and circadian clock data, and validation experiments of DynaMO analysis in YMC can be found in Supplementary Methods. RNA-seq and ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE72263. The time for DynaMO to analyze the YMC data (175 motifs and ChIP-seq from 16 time points) was 2.4 h using 24 CPU cores (2.2 GHz) and 64 GB memory. On the same computer system, the running time for human neural differentiation and mammalian circadian clock data was 22.4 (525 motifs, 4 time points) and 0.97 h (1 motif, 6 time points) respectively.
RESULTS
DynaMO: an algorithm for predicting dynamic activities of TFs based on chromatin profiling
DynaMO is designed to couple TF binding motifs with global chromatin dynamic profiling experiments (e.g. time-course ChIP-seq data for histone modifications) to achieve two goals: (i) predict important TFs responsible for the dynamic processes; (ii) predict binding sites of these TFs and dynamic changes of their binding activities.
The DynaMO pipeline consists of three major steps (Figure 1A). In the first step, mapped sequence reads from chromatin profiling experiments are combined with computationally mapped DNA motif sites to predict TFBSs. In the second step, predicted binding sites for all TFs are pooled, and binding sites with similar dynamic binding patterns are grouped into clusters by automatic K-means clustering. In the third step, TFs important for each dynamic binding pattern (i.e. each binding site cluster) are identified through an enrichment analysis. For each TF and dynamic binding pattern pair, the enrichment analysis evaluates whether motif sites of the TF are enriched in the binding site cluster with the dynamic binding pattern as opposed to randomly distributed across different clusters. The rationale is that a TF acting in a specific time window of a process is more likely to be involved in driving dynamic changes in transcription than a TF functioning in a constitutive manner. After the analysis, DynaMO reports all identified dynamic binding patterns, identifies the TFs associated with each dynamic binding pattern and the predicted binding sites of each TF.
Figure 1.

Schematic view of DynaMO pipeline (A) and YMC (B). (A) DynaMO uses TF motifs and histone modification ChIP-seq reads as inputs. Motif sites are obtained by mapping PSFMs or consensus sequences to the genome. Histone modification peaks are detected by peak callers. Motif sites (red) overlapping peaks (blue) are selected as training data to construct RF models, which are used to predict potential TF binding sites. When multiple biological conditions (e.g. multiple time points) exist, TF binding sites in each condition are predicted and pooled across TFs and conditions. Pooled binding sites are clustered based on dynamic patterns of histone modification signals at these sites. TFs with associated binding sites enriched in clusters are predicted as important TFs and dynamic or temporal patterns of histone modification are predicted as dynamic or temporal activities of TFs. (B) Respiratory oscillations (dissolved O2 concentration) of prototrophic yeast strain under continuous nutrient limited condition with a period of 4–5 h. Three transcriptional and metabolic different phases are defined: oxidative (OX), reductive/building (RB) and reductive/charging (RC), which are marked by red, green and blue curves. The color scheme is used for labeling the three phases throughout the paper.
For the TFBS prediction in the first step, we employ a two-pass algorithm. In the first pass, a conventional ChIP-seq peak caller is used to perform initial peak calling to identify enriched ChIP signals. In the second pass, motif sites with and without enrichment signals are used as positive and negative training data, respectively, to train an RF model—a supervised prediction model—to learn the characteristic spatial distribution of chromatin signals surrounding putative TF binding sites. The shape, which may vary for different datasets, carries information that can help one to better discriminate true binding signals from noise. When there are multiple chromatin data types, the RF also allows one to conveniently integrate information from different data types. The trained model is then applied to all motif sites in the genome to reanalyze the chromatin profiling data and finally determine whether each motif site is bound or not by taking the signal shape information into account.
DynaMO can be used for both simple and complex experimental designs. When users only need to predict binding sites for one TF in one biological condition (e.g. one time point), they can choose to run only the first step of DynaMO pipeline. If users have one TF and multiple conditions to analyze, they can use DynaMO to predict the genome-wide dynamic binding pattern of the TF. When users need to screen hundreds of TFs to look for important ones in a dynamic process, DynaMO can provide complete prediction of which TFs are likely to be functional in the process, when, and where. Here we focus on the latter scenario.
DynaMO analysis of the yeast ultradian cycle
We first demonstrate the performance of DynaMO through a study of the yeast metabolic cycle (27,39). In the YMC, yeast cells are synchronized. Under a continuous, glucose-limited condition these synchronized cells exhibit respiratory oscillations resulting in oscillations of O2 consumption (Figure 1B). In parallel with the respiratory oscillations, more than half of the yeast genome is periodically expressed, peaking at three different phases, Oxidative (OX), Reductive/Building (RB) and Reductive/Charging (RC) (27,39). Genes encoding ribosome, amino acid metabolism and translation are expressed in the OX phase, indicating that OX is a growth phase. Mitochondrial and cell division cycle genes are induced in the RB phase. RC phase genes include those encoding stress response, protein degradation and non-respiratory modes of metabolism, such as glycolysis and fatty acid oxidation. Different cellular processes are temporally coordinated to maintain the metabolic oscillation of yeast, suggesting that multiple TFs control the specificity and accurate timing of gene expression. Here, we attempted to identify important TFs for this dynamic process using DynaMO and characterize their spatiotemporal binding activities.
We computationally mapped motif sites of 175 TFs to the yeast genome. Using DynaMO, we coupled these motif sites with time-course ChIP-seq data for histone H3K9ac to predict TFBSs at 16 different time points across one YMC. The H3K9ac ChIP-seq data derive from a previous study where we generated 16 time point ChIP-seq data for seven types of histone acetylation and methylation (32). Here, we focused on H3K9ac for predicting dynamic TF binding activities because this histone modification marks active promoters and is correlated with transcription and TF binding (18,40), and it shows the highest dynamics and temporal correlation with transcription (32). Moreover, our previous study has shown that active TFBSs are associated with an increased level of H3K9ac and changes of this histone mark can be used to predict differential TF binding activities between two biological conditions (21). After pooling all predicted TFBSs, DynaMO grouped TFBSs with similar temporally dynamic H3K9ac patterns into three clusters, and the temporal patterns of H3K9ac in these three clusters were highly consistent with the three known phases of the YMC (Figure 2A).
Since the data were from a time-course experiment, we further used DynaMO to fit smooth curves to describe temporal changes in H3K9ac at predicted binding sites. Based on the curves, one can extract important temporal features to characterize this dynamic process (Supplementary Figure S2A–C), a function not usually provided by other commonly used tools for analyzing chromatin profiling data. For instance, DynaMO computed the time corresponding to the maximal activity or the maximal activity change (i.e. maximal derivative) at each binding site. Supplementary Figure S2D shows the distribution of the peak (max) time and the time of the fastest H3K9ac increase for the predicted binding sites in each cluster. Taking clusters 1 and 2 as an example, the difference in the mean peak time of the maximal binding site activity between cluster 1 (which was active in the OX phase) and cluster 2 (which was active in the RB phase) was estimated to be 0.57 hr. The difference was statistically significant (two-sample t-test P-value < 2.2e-16, sample sizes for clusters 1 and 2 are 93662 and 44802 respectively). As another example, the difference in the time associated with the fastest increase rate between cluster 1 (OX) and cluster 2 (RB) was estimated to have a mean of 0.88 hour (two sample t-test P-value < 2.2e-16, sample size n1 = 93662, n2 = 44802). This analysis provides basic quantitative information about the YMC.
For each cluster, DynaMO identified its associated TFs based on analyzing which motifs were enriched. A total of 41 enriched TFs were identified with adjusted p-value<10−5 (Figure 2A and Supplementary Table S1). Sixteen TFs were enriched in cluster 1, in which H3K9ac peaks in the OX phase. Many of these TFs are involved in ribosome biogenesis, such as Tod6, Dot6, Rap1, Sfp1 and Stb3 (41–43), consistent with functions of genes expressed in the OX phase. Similarly, four TFs were enriched in cluster two, in which H3K9ac peaks in the RB phase. Among them, TFs regulating cell cycle genes, such as Mbp1, Swi6 and Xbp1 (44),were found. Twenty-one TFs were identified in cluster 3, including a number of TFs known to be involved in stress response such as Gis1, Mig1/2/3 and Msn2/4 (45–47). TFs regulating certain metabolic pathways, such as Adr1 (48), Rgm1 (49) and Tda9 (50) were observed in cluster 3 also. Overall, these results indicate that TFs enriched in a cluster are likely to be associated with specific biological processes. For each TF, we also predicted its binding sites. In total, 28 678 binding sites and their temporal patterns were predicted for the 41 enriched TFs (Supplementary Table S2), providing a resource not previously available for the study of YMC.
DynaMO identifies important TFs for regulating metabolic oscillations in the YMC
We asked whether TFs identified by DynaMO are actually important in maintaining normal metabolic cycles. From the top enriched TFs (ranked first by adjusted P-values and in the case of ties by fold enrichment), we randomly selected five TFs from the three clusters (Figure 2B). Five randomly selected non-enriched TFs were used as controls. For each of these 10 TFs, we attempted to disrupt its function in two different ways by constructing a deletion mutant and a C-terminus tag mutant respectively. We examined the O2 oscillation phenotypes of these mutants, a simple indicator of the ultradian cycle. As summarized in Figure 2B, enriched TFs identified by DynaMO show a much higher percentage of disrupted oscillations when mutated than do randomly selected non-enriched TFs (Supplementary Figure S3) (P-value < 0.01 by Fisher’s exact test).
The disrupted phenotypes observed in the mutants are characterized in Figure 2C. The first phenotype is a ‘flat line’ (F), as seen in the XBP1::3HA strain bearing only a C-terminal tag and similar to the behavior of gcn5Δ in a previous study (51), suggesting a complete loss of respiratory oscillation. Surprisingly, the xbp1Δ strain cycles normally. We hypothesize that the C-terminus tag of Xbp1 disrupts its interaction with other chromatin proteins or DNA. If this is true, the heterozygous diploid of the C-terminus tag strain should also show defects, i.e. the mutation is expected to be dominant or codominant. Indeed, XBP1::3HA/+ shows defective oscillations, although it performs better than the haploid (Supplementary Figure S3C). The second phenotype is damped amplitude (D), as seen in arg80Δ and XBP1::3HA/+. gcr1Δ exhibits yet another defective phenotype, namely an amplitude (A) defect; it is ever-increasing at first and then diminishes later. Yet another phenotype is observed in msn2Δ single mutant. The RC phase is longer than the WT strain in these mutants (L-RC) and it gets longer in each successive cycle/growth burst (Figure 2C). The extended RC phase was also observed in the msn2Δmsn4Δ double mutant (Supplementary Figure S3C, see the related paper by Kuang et al. (52) for detailed follow-up investigation of msn2Δmsn4Δ).
Together, our results show that DynaMO was able to identify important TFs in a dynamic process. This demonstrates the usefulness of DynaMO for investigators who need to select a few candidates from hundreds of TFs for functional study in a poorly studied system.
DynaMO predicts relevant TF binding sites and their temporal occupancy patterns
In order to evaluate the performance of DynaMO for predicting TF binding sites, we generated ChIP-seq data for Msn2 and Msn4 at six different time points across one metabolic cycle (Figure 3A). Msn2 and Msn4 are two stress-responsive TFs (45). Both were identified by DynaMO as regulators of metabolic oscillations. Using binding peaks identified from the Msn2 and Msn4 ChIP-seq data as a gold standard, we evaluated DynaMO’s ability to predict TFBSs.
Figure 3.

DynaMO predicts where and when TFs bind in YMC. (A) Six time-point WT cycling cells in YMC, including 1 OX, 2 RB and 3 RC time points, were collected for ChIP-seq of interesting TFs. The results were used for prediction validation. Red = OX, Green = RB, Blue = RC. (B) Curves of sensitivity versus rank for Msn2 binding sites prediction at each time point. Red curves are the performance of DynaMO and blue curves are the performance of CENTIPEDE. (C) ChIP-seq signals of Msn2 and H3K9ac at predicted Msn2 motif sites at T1 and T4. Each row represents a 500 bp genomic region centered on the predicted Msn2 binding site. Rows are ordered by Msn2 binding strength. (D) Curves of sensitivity versus nominal FDR for Msn2 and Msn4 binding sites prediction at each time point. (E) The averaged temporal patterns of Msn2 or Msn4 binding and H3K9ac at predicted motif sites in cluster 3. (F) The distribution of correlation between TF binding and H3K9ac at each motif site. Red/green/blue curves represent predicted binding sites in OX/RB/RC clusters. Black curves represent non-bound motif sites of Msn2 or Msn4 and gray curves represent random motif sites of other TFs.
Figure 3B shows the sensitivity as a function of the number of predicted Msn2 binding sites at each time point. As a reference, we compared DynaMO with CENTIPEDE, a widely used software package for binding site prediction (19). DynaMO showed higher predictive power than CENTIPEDE. Similar results were observed by examining Msn4 (Supplementary Figure S4A). This sensitivity-rank comparison shows that when the number of predicted binding sites is kept the same, DynaMO has higher sensitivity. In other words, DynaMO produced more true positives and fewer false positives than CENTIPEDE when these two methods were asked to produce the same number of predicted binding sites. This implies that DynaMO achieved a higher sensitivity ( = No. of true positives/total no. of TF-bound motif sites) at a lower false positive rate ( = No. of false positives/Total no. of motif sites not bound by TF = 1-specificity). Thus, the better sensitivity-rank performance also means that DynaMO has better receiver operating characteristic (ROC) (i.e. the sensitivity versus false positive rate curve), which is confirmed in Supplementary Figure S4B and C.
Figure 3C shows the spatial distribution of Msn2 and H3K9ac ChIP-seq reads at the predicted Msn2 binding sites in two different phases of the YMC, OX and RC. A ‘bimodal peak with U shape in the middle’ (bimodal U shape) pattern of the histone modification signals was found surrounding the predicted TFBSs, consistent with the known nucleosome occupancy pattern around many TFBSs (19). However, the bimodal U shape was not discovered in motif sites predicted not to be bound by TFs (Supplementary Figure S5A). Additionally, DynaMO further differentiated predicted binding sites from other motif sites by the intensities of H3K9ac. In other words, DynaMO predicts motif sites with very small and flat H3K9ac intensities as the least likely binding sites. This is consistent with the previously reported correlation between differential H3K9ac intensity and TF binding activity (21). By contrast, the unsupervised CENTIPEDE did not capture the ‘bimodal U shape’ well for the predicted binding sites, and it predicted motif sites with the reversed H3K9ac shape as the least likely binding sites. Also the H3K9ac intensities at the motif sites predicted to be bound by CENTIPEDE were not very different from the intensities at the predicted unbound motif sites (Supplementary Figure S5A). This demonstrates that the RF model used by DynaMO was able to capture the characteristic signal shapes and intensities when making predictions, potentially explaining the increased prediction accuracy of DynaMO.
In practical applications, the cutoff for reporting significant binding sites should be determined by FDR rather than by specificity (or ROC) due to multiple hypothesis testing. This is because when the majority of motif sites actually consist of noise (because they are in fact unbound in vivo), high specificity does not imply low FDR. Sensitivity-rank curves or ROC curves may be used to determine the best performing method. However, they do not reveal the actual sensitivity achievable by that method in practice when FDR is controlled at the desired level. Thus, we further examined the relationships between sensitivity and FDR. The performance varied across different time points and different TFs (Figure 3D). At the nominal FDR level of 25%, sensitivities range from 0.4 to 0.8 for Msn2 and 0.4 to 0.65 for Msn4. The sensitivity was lower for time points in OX and RB phases and higher for time points in RC phase. Of note, holding FDR equal, there was also substantial variation in the number of predicted TFBSs across different time points. The variation in the number of predicted TFBSs was consistent with the variation in the number of binding sites determined by Msn2/4 ChIP-seq. Overall, there were more binding sites in the RC phase than in the OX phase (Supplementary Figure S5B). This observation, along with the observed enrichment of Msn2/4 motif sites in cluster 3 (RC phase) in Figure 2A suggests that Msn2 and Msn4 binding might be more active in the RC phase. The smaller number of binding sites in OX and RB phases suggest that many more Msn2/4 motif sites at those time points correspond to noise. With the increased noise level, the statistical power for detecting signals is expected to decrease. This is consistent with the decreased sensitivity observed for OX and RB time points. Together, the above analyses show that one should not expect DynaMO to find all binding sites in practice. However, DynaMO can recover a substantial proportion of true binding sites, which will provide valuable information not otherwise available for guiding downstream functional studies when one does not have ChIP-seq data for the TFs themselves. A systematic investigation of the biology behind the variation in the number of Msn2/Msn4 binding sites is beyond the scope of this article. However, it would be interesting to investigate in the future whether such variation is linked to TF-TF cooperativity (e.g. presence or absence of cofactors in different time period for TF function) or TF residence time during the binding.
We further evaluated whether the TF binding dynamics can be predicted. We compared the temporal patterns of H3K9ac and Msn2/4 ChIP-seq signals at predicted Msn2/4 binding sites. Interestingly, in cluster 3, in which Msn2/4 was enriched, the trends of H3K9ac and Msn2/4 were very similar (Figure 3E and Supplementary Figure S5C), suggesting that the temporal pattern of H3K9ac can indicate the dynamics of TF binding. By contrast, in the other two clusters where Msn2/4 were not enriched, the temporal patterns of H3K9ac and Msn2/4 did not show strong correlation at the predicted Msn2/4 binding sites (Figure 3F and Supplementary Figure S5C).
Collectively, our analyses demonstrate how DynaMO can be used to identify important TFs and predict their binding sites. DynaMO predictions have helped us to obtain a deeper understanding of gene regulation in the YMC. Guided by these predictions, we conducted further biological studies and found Msn2/4 regulate yeast glycolysis genes. MSN2/4 deletion decreased the accumulation rate of acetyl-CoA, a key metabolite that drives cell growth and delayed the re-entry of quiescent cells into growth (52).
A comparison with other methods in the YMC analysis
Next, we systematically compared DynaMO with several existing methods including ZINBA (53) and ChromHMM (54) in addition to CENTIPEDE (Figure 4A, Supplementary Methods). We also compared DynaMO with a modified version of DynaMO in which the RF step was removed but all other procedures were kept the same. This comparison was used to evaluate whether RF helped with improving the analysis. CENTIPEDE, ZINBA and ChromHMM represent three different types of existing methods for analyzing chromatin profiling data. The objective of CENTIPEDE is to predict TFBSs using chromatin profiles and DNA motif information. Unlike DynaMO, CENTIPEDE does not cluster predicted binding sites based on their dynamic binding patterns in multiple biological conditions, nor does it identify enriched TFs associated with each binding pattern. ZINBA is a peak calling method that detects enrichment signals in chromatin profiling data. Unlike DynaMO and CENTIPEDE, ZINBA is a general-purpose peak caller rather than a method for predicting TFBSs. It does not predict TFBSs by combining the enrichment peaks in chromatin profiling data with DNA motif information. ZINBA also does not cluster peaks based on their dynamic binding patterns across multiple conditions, nor does it identify TFs associated with each binding pattern. ChromHMM is a genome segmentation method that divides a genome into non-overlapping windows based on analyzing multiple chromatin profiling datasets. The genome is segmented such that genomic positions within the same window have similar chromatin signal patterns whereas neighboring windows have different signal patterns. In addition to segmentation, all genomic segments are clustered based on their chromatin signal patterns across datasets. The segmentation and clustering are jointly solved using a Hidden Markov Model (HMM). After the analysis, biological functions of each cluster may be inferred by comparing its signal patterns with existing biological knowledge. Similar to DynaMO, ChromHMM allows one to identify different dynamic signal patterns through the clustering of genomic segments. However, unlike DynaMO, ChromHMM does not further predict TFBSs using DNA motif information, nor does it identify enriched TFs associated with each cluster. Genomic segments generated by ChromHMM can be very long and contain many TFBSs. For this reason, ChromHMM analysis is not designed to provide the resolution for identifying TFBSs.
Figure 4.

Performance comparison of DynaMO with other methods. (A) This table summarizes the methods and the analyses to be compared. A ‘√’ means the method is designed for this analysis and a ‘+’ means the method is customized by coupling a procedure in DynaMO for the analysis. (B) Percentages of cycling TFs among the top predicted TFs versus the number of predicted TFs are shown for different methods. TFs are ranked by the smallest enrichment P-values and biggest fold enrichment across clusters. (C) Curves of sensitivity versus rank for Msn2 and Msn4 binding sites prediction at each time point. (D) AUCs for curves in (C) are presented in heat maps. Bar plots show average performance scores across time points. The best method at each time point is given a score of 4 and the worst method is given a score of 1.
Among the compared methods, CENTIPEDE, ZINBA and ChromHMM do not provide functions to identify important TFs. We asked whether one can couple these methods with a motif enrichment analysis procedure similar to DynaMO to identify important TFs. To this end, we clustered CENTIPEDE predicted TFBSs and then identified enriched TF motifs for each cluster using a procedure similar to DynaMO. For ZINBA, we used motif sites covered by peaks to predict TFBSs. The predicted TFBSs were then clustered and enriched motifs were identified. For ChromHMM, we analyzed its genome segmentation results and identified enriched motifs for each cluster of genomic segments. In all cases, the cluster number was set to three, corresponding to the known number of cycling gene clusters in YMC. Additionally, DynaMO without RF was also run by setting the cluster number to three. In order to compare the ability of different methods to identify important TFs, one cannot use the 10 experimentally tested TFs in Figure 2B because they were selected based on the DynaMO analysis and may introduce bias in the comparison. For this reason, we obtained a list of TFs periodically expressed in the YMC based on independent gene expression time-course data in YMC (32) and used these cycling TFs to benchmark different methods. We reasoned that many TFs which drive YMC may also be periodically expressed in the cycles. For each method, all TFs identified from the enrichment analysis were ranked (first by adjusted P-values and in the case of ties by fold enrichment), and the number of cycling TFs among the top ranked TFs was computed. This analysis shows that by holding the number of reported TFs the same, DynaMO captured more cycling TFs among the predicted TFs than the other methods (Figure 4B), suggesting that DynaMO is better able to identify TFs relevant to this dynamic process. It should be pointed out that this comparison was only possible because we implemented additional procedures for clustering and identifying enriched motifs for CENTIPEDE, ZINBA and ChromHMM. Without writing additional computer programs, these existing methods cannot be directly run to conduct such analyses.
We further compared the ability of different methods to predict TFBSs. Here the compared methods include DynaMO, DynaMO without RF, CENTIPEDE and ZINBA. ChromHMM was not compared here since its genome segmentation was not designed for analyses at binding site resolution. For ZINBA, motif sites covered by peaks were used to predict TFBSs. Figure 4C shows the sensitivity of each method as a function of the number of predicted TFBSs. For each curve, we computed the area under the curve (AUC), and the AUCs are compared in Figure 4D. For each time point, methods were ranked based on their AUC. For each method, the average rank across the six time points is shown as a bar plot below the heat map in Figure 4D (the bigger the rank score the better). Since a method’s sensitivity-rank performance implies its ROC performance, the ROC comparison is skipped here. Figure 4C and D show that DynaMO outperformed the other three methods. The improvement of DynaMO over CENTIPEDE and ZINBA was substantial. DynaMO without RF also performed worse than DynaMO, and the improvement of DynaMO was sometimes substantial (e.g. Msn4 t3). Overall, DynaMO robustly performed the best. The bimodal U shape of histone mark signals at the binding sites and the difference in the H3K9ac intensity between the bound and unbound motif sites were also captured by ZINBA and DynaMO without RF but slightly weaker (Supplementary Figure S5A).
Collectively, our analyses demonstrate that DynaMO performed better than the other methods both in terms of the identification of important TFs and in terms of TFBS prediction.
DynaMO analyses of human neural differentiation and mammalian circadian clock
DynaMO can also be applied to genomes more complex than yeast. To demonstrate the versatility of DynaMO, we first applied it to predict important TFs in human neural differentiation, which is also coupled with dramatic transition of transcriptome and epigenome. A previous study (33) presented a time-course dataset of transcriptome and epigenome in early human neural differentiation, including stages of embryonic stem (ES) cells, neuroepithelial (NE) cells, early radial glial (ERG) cells and mid radial glial (MRG) cells. The study identified 244 transcriptional regulators differentially or highly expressed during this in vitro differentiation time course as potential key regulators. A total of 110 of these 244 regulators were subsequently validated through the short hairpin RNA (shRNA) knockdown screening (33). This provides a good benchmark dataset to evaluate the performance of DynaMO in predicting important TFs.
We mapped 525 TF motifs from TRANSFAC (55) to the human genome and ran DynaMO using four time point ChIP-seq data for H3K4me3 and H3K27ac. Both these histone marks have been shown to be associated with active TF binding (21). TFBSs predicted by DynaMO were automatically partitioned into three clusters based on the temporal H3K27ac and H3K4me3 signal patterns (Figure 5A). From the 525 motifs, we identified those that were tested in the shRNA knockdown experiment. These motifs were ranked by DynaMO based on their statistical significance and enrichment level as before (‘Materials and Methods’ section). Among the top 20 TFs predicted by DynaMO and included in the shRNA library, 75% exhibited significant activating or repressing functions upon shRNA knockdown (Figure 5B). This validation rate was much higher than the ∼30% validation rate of the original 244 regulators identified based on gene expression analysis alone. We compared DynaMO with CENTIPEDE, ZINBA, ChromHMM and DynaMO without RF for identifying important TFs (Supplementary Methods). The validation rates of these methods were consistently lower than DynaMO (Figure 5B). We examined the top TFs predicted by DynaMO in each cluster. Among the top 20 TFs predicted by DynaMO in each cluster and included in the shRNA library, 60–80% exhibited significant activating or repressing functions upon shRNA knockdown (Figure 5C), and TFs known to be involved in neural differentiation were captured such as the FOX protein families and POU domain proteins (Figure 5D and Supplementary Table S3). Together, these data suggest that DynaMO can greatly help with predicting important TFs across very different organisms and distinct dynamic systems.
Figure 5.

DynaMO analysis of TFs in human neural differentiation. (A) Clustering of predicted motif sites from 525 human-mouse motifs based on temporal H3K27ac and H3K4me3 signals at motif sites during neural differentiation. (B) Validation rates of predicted important TFs versus the number of predicted TFs in neural differentiation by different methods. A square marks the validation rate by gene expression only reported in the original paper (33) (C) Validation rate of important TFs predicted by DynaMO in three clusters. The plot shows percentage of the top 20 TFs in each cluster examined in the shRNA screen showing significant impact by shRNA knockdown. (D) Representative TFs from the top 20 of the total or examined TFs in each cluster.
Due to a lack of TF ChIP-seq data, the analysis of neural differentiation above could not examine the accuracy of DynaMO for predicting TFBSs. We therefore applied DynaMO to another dataset originally collected for studying the mammalian circadian clock. Transcriptional activators BMAL1, CLOCK and NPAS2 and repressors PER1, PER2, CRY1 and CRY2 form an auto-regulatory feedback loop, which regulates 24-h circadian oscillations of thousands of transcripts (28,31). A previous study by Koike et al. (31) systematically characterized the dynamics of genome-wide transcription and chromatin states every 4 h of a circadian cycle. Using the H3K9ac and H3K4me1 ChIP-seq data generated there, we predicted the spatiotemporal binding of BMAL1 at Ebox motif sites. Both H3K9ac and H3K4me1 have been shown to be associated with active TF binding (21). We evaluated the TFBS prediction performance of DynaMO using the BMAL1 ChIP-seq data.
We compared DynaMO with and without RF, CENTIPEDE and ZINBA. All methods used the same H3K9ac and H3K4me1 data and Ebox motif sites. Each method was applied to rank motif sites by jointly using H3K9ac and H3K4me1. To help evaluate the gain of integrating two data types, we also applied each method to rank motif sites using H3K9ac or H3K4me1 alone. For jointly analyzing H3K9ac and H3K4me1, DynaMO and CENTIPEDE were directly applied since both methods can handle multiple histone marks. DynaMO without RF cannot directly combine signals from multiple different data types. Therefore, in order to combine H3K9ac and H3K4me1, we first applied it to each histone mark separately to rank motif sites and then computed the average rank of each motif site. The average rank was then used to sort the motif sites again (denoted as ‘DynaMO w/o RF H3K4me1/H3K9ac’). Similarly, ZINBA does not provide a way to combine peak signals from multiple different histone marks. We also used the average rank approach above to combine H3K9ac and H3K4me1 (‘ZINBA H3K4me1/H3K9ac’). In DynaMO with RF, the information from different data types is integrated through RF (‘DynaMO H3K4me1+H3K9ac’). The role of RF is 2-fold. It automatically models the peak shape at motif sites within each data type, and it also provides a way to automatically integrate different data types. To help understand the role of RF in data integration, we also ran DynaMO on H3K9ac and H3K4me1 separately and then used the average rank approach instead of the RF to combine these two data types to rank motif sites (‘DynaMO H3K4me1/H3K9ac’). Note that both ‘DynaMO w/o RF H3K4me1/H3K9ac’ and ‘DynaMO H3K4me1/H3K9ac’ were based on first ranking the motif sites using each data type and then computing the average rank. The difference between these two methods is that when analyzing each data type, ‘DynaMO H3K4me1/H3K9ac’ used RF whereas ‘DynaMO w/o RF H3K4me1/H3K9ac’ used the peak signal obtained from the initial peak caller to rank motif sites. The difference between ‘DynaMO H3K4me1/H3K9ac’ and DynaMO (i.e. ‘DynaMO H3K4me1+H3K9ac’) is that the former only applied RF to each data type separately and then combined different data types using average rank, whereas the latter used H3K4me1 and H3K9ac jointly as features for RF and hence the two data types were automatically integrated through RF. In summary, the comparison between ‘DynaMO w/o RF H3K4me1/H3K9ac’ and ‘DynaMO H3K4me1+H3K9ac’ will reveal the overall contribution of RF in DynaMO. The comparison between ‘DynaMO w/o RF H3K4me1/H3K9ac’ and ‘DynaMO H3K4me1/H3K9ac’ will more specifically reveal the contribution of RF in terms of utilizing peak shape information within each data type. The comparison between ‘DynaMO H3K4me1/H3K9ac’ and ‘DynaMO H3K4me1+H3K9ac’ will more specifically reveal the contribution of RF in terms of combining different data types.
Figure 6A shows the sensitivity-rank curve of each method in all six time points for predicting BMAL1 binding sites. Figure 6B compares the AUC of different methods in a heat map. Again, for each time point, methods are ranked based on their AUC. Then for each method, the average rank across the six time points is shown in the bar plot below the heat map (the bigger the rank score the better). Comparisons of the same method running under different modes (i.e. H3K9ac only, H3K4me1 only or H3K9ac and H3K4me1 jointly) show that using both histone marks jointly allows one to more accurately predict TFBSs compared to using only one histone mark. Comparisons of different methods show that DynaMO offered higher TFBS prediction accuracy than the other methods. DynaMO with RF outperformed DynaMO without RF both when each histone mark was analyzed separately and when the two histone marks were analyzed jointly. Moreover, DynaMO (‘DynaMO H3K4me1+H3K9ac’) performed better than ‘DynaMO H3K4me1/H3K9ac’, and ‘DynaMO H3K4me1/H3K9ac’ performed better than ‘DynaMO w/o H3K4me1/H3K9ac’, indicating that RF is useful both for utilizing the peak shape information and for combining different data types. Collectively, these analyses demonstrate the advantage of using RF in DynaMO and further show the ability of DynaMO to produce better TFBS predictions than the other methods.
Figure 6.

DynaMO analysis of TFs in mammalian circadian clock. (A) Curves of sensitivity versus rank for BMAL1 binding sites prediction by different methods at each time point. H3K9ac+H3K4me1 means the two markers are integrated by the original methods to rank the motif sites. H3K9ac/H3K4me1 means the two markers are individually analyzed by the methods and the ranks are averaged to order the motif sites. (B) AUCs for curves in (A) are presented in a heat map. The bar plot shows average performance scores across all time points. The best method at each time point is given a score of 13 and the worst method is given a score of 1. (C) Left panel shows the average temporal patterns of BMAL1 and combined H3K9ac+H3K4me1 histone modification signal (i.e. the average of H3K9ac and H3K4me1 inner products) at predicted BMAL1 binding sites. Right panels show the distributions of correlation between BMAL1 and combined histone modification signal at all BMAL1 motif sites or predicted binding sites.
Supplementary Figure S6A shows the average read distribution surrounding the top and bottom motif sites ranked by DynaMO, CENTIPEDE, DynaMO without RF and ZINBA. Top sites identified by DynaMO and CENTIPEDE both exhibited the bimodal U shape of histone modification signals, whereas the signal shape from ZINBA was irregular. The read distribution from DynaMO without RF was similar to DynaMO, but the bimodal U shape in DynaMO without RF was slightly weaker. CENTIPEDE is based on a two-component mixture model. It predicted motif sites with a spatial pattern opposite to the U shape (i.e. a peak in the middle surrounded by relatively low intensity on both sides) as the least likely binding sites in this example. Also, CENTIPEDE did not clearly differentiate bound and unbound motif sites by the histone modification intensities. Compared to CENTIPEDE, DynaMO further distinguished top and bottom motif sites based on their differences in signal intensity (Supplementary Figure S6A). Thus, the RF used by DynaMO captured both the characteristic signal shapes and also the intensity information for making predictions.
We also examined how well DynaMO predicted temporal patterns of TF binding. The predicted temporal activities of the predicted TFBSs (‘Materials and Methods’ section) were consistent with the temporal patterns of BMAL1 binding determined by ChIP-seq (Figure 6C). Compared to random motif sites, the subset of motif sites predicted to be bound using DynaMO exhibited increased correlation between the predicted activity and BMAL1 ChIP-seq binding signals (Figure 6C). This further demonstrates the ability of DynaMO to filter out noisy motif sites and identify functional TFBSs and their dynamic binding patterns in mammalian genome.
Additionally, DynaMO analysis of the three CRY2 motifs exhibited dramatic differences of sequence specificity among the CRY2 binding site types (Supplementary Figure S6B). Ebox sites (recognized by BMAL1) and exd sites (recognized by CRY1/2) are largely excluded from each other in the same CRY2 peaks whereas NR sites usually co-exist with either Ebox or exd sites. NR sites also define another large group of CRY2 peaks which contain neither Ebox nor exd sites. This is consistent with the previous ChIP-seq analysis (31) showing that CRY2 peaks are divided into those overlapping with BMAL1 and CRY1 peaks, those overlapping with only CRY1 or BMAL1 peaks and a large proportion of CRY2 unique peaks. CRY1/2 peaks that are independent of BMAL1 may be due to lack of Ebox sites. Therefore, multiple mechanisms may exist for CRY2 recruitment.
DISCUSSION
In summary, we have introduced a method to identify important TFs and their binding sites, and to predict their recruitment/activity during dynamic processes. DynaMO is generally applicable to chromatin profiling data from multiple conditions. For data from time-course experiments, DynaMO can also be used to extract temporal characteristics of the dynamic process via curve fitting. Application of DynaMO to the yeast metabolic cycle, the mammalian circadian clock and neural differentiation demonstrates that it accurately and efficiently predicted when and where TFs bind, and also identified important TFs that regulate these processes. DynaMO is publicly available as an R package at https://github.com/spo111/DynaMO.
For constructing features used by random forests, DynaMO requires users to specify a window size W and a bin size B. Our analysis in Supplementary Materials and Supplementary Figure S7 suggests that DynaMO is relatively robust to different values of W and B chosen around our recommended values. DynaMO identifies TFs enriched in specific dynamic binding patterns through motif enrichment analysis. Empirically, our analyses have shown that this is a powerful approach for identifying truly important TFs for dynamic processes compared to random expectation. However, users still need to exercise caution in interpretation because enrichment does not necessarily equate with importance. Using other information may help one to better utilize the DynaMO results. For instance, when examining functions of candidate TFs, one may consider redundancy because the more important a TF is the more likely it has homologs (56–58). Furthermore, TFs often function in a cooperative fashion (e.g. through protein–protein interactions). Thus, co-enrichment of TFs with similar functions, acting in the same process or in the same protein complex may also point to which TFs are important in a system.
In this study, DynaMO was demonstrated using time-course histone modification data. In principle, DynaMO can also be used for other chromatin data types such as DNase-seq and ATAC-seq (59). An important open question for future research is to systematically understand the correlation between different chromatin data types with dynamic TF binding. Different chromatin data types may exhibit different temporal patterns relative to transcription. Certain chromatin data types may only mark active or repressive regions but do not change much in correspondence with transcription. Building a systematic catalog of chromatin data types most informative and uninformative for analyzing dynamic changes of global regulatory programs is therefore an important future goal.
DATA AVAILABILITY
RNA-seq and ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE72263. DynaMO is publicly available as an R package at https://github.com/spo111/DynaMO.
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. S. Takahashi for providing circadian TF motifs. We thank Y. Zhou for suggestions on neural differentiation. We thank W. Zhou, B. He for DNase hypersensitive regions and other statistical advice. We thank Adriana Heguy and her staff at the New York University School of Medicine Genome Technology Center for expert assistance with ChIP-Seq.
Footnotes
Present address: Zheng Kuang, Department of Immunology, The University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [U54GM103520, R01HG006841, R01HG006282]. Funding for open access charge: NIH [R01HG006282].
Conflict of interest statement. None declared.
REFERENCES
- 1. Heinemeyer T., Wingender E., Reuter I., Hermjakob H., Kel A.E., Kel O.V., Ignatieva E.V., Ananko E.A., Podkolodnaya O.A., Kolpakov F.A. et al. . Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998; 26:362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sandelin A., Alkema W., Engstrom P., Wasserman W.W., Lenhard B.. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004; 32:D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lemon B., Tjian R.. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000; 14:2551–2569. [DOI] [PubMed] [Google Scholar]
- 4. Hughes T.R., de Boer C.G.. Mapping yeast transcriptional networks. Genetics. 2013; 195:9–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kovacs L.A.S., Orlando D.A., Haase S.B.. Transcription network and cyclin/CDKs: The yin and yang of cell cycle oscillators. Cell Cycle. 2008; 7:2626–2629. [DOI] [PubMed] [Google Scholar]
- 6. Cao Y., Yao Z., Sarkar D., Lawrence M., Sanchez G.J., Parker M.H., MacQuarrie K.L., Davison J., Morgan M.T., Ruzzo W.L.. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell. 2010; 18:662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sandmann T., Girardot C., Brehme M., Tongprasit W., Stolc V., Furlong E.E.. A core transcriptional network for early mesoderm development in drosophila melanogaster. Genes Dev. 2007; 21:436–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jakobsen J.S., Braun M., Astorga J., Gustafson E.H., Sandmann T., Karzynski M., Carlsson P., Furlong E.E.. Temporal ChIP-on-chip reveals biniou as a universal regulator of the visceral muscle transcriptional network. Genes Dev. 2007; 21:2448–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Junion G., Spivakov M., Girardot C., Braun M., Gustafson E.H., Birney E., Furlong E.E.. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012; 148:473–486. [DOI] [PubMed] [Google Scholar]
- 10. Chua G., Robinson M.D., Morris Q., Hughes T.R.. Transcriptional networks: Reverse-engineering gene regulation on a global scale. Curr. Opin. Microbiol. 2004; 7:638–646. [DOI] [PubMed] [Google Scholar]
- 11. Chua G., Morris Q.D., Sopko R., Robinson M.D., Ryan O., Chan E.T., Frey B.J., Andrews B.J., Boone C., Hughes T.R.. Identifying transcription factor functions and targets by phenotypic activation. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:12045–12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aragon A.D., Rodriguez A.L., Meirelles O., Roy S., Davidson G.S., Tapia P.H., Allen C., Joe R., Benn D., Werner-Washburne M.. Characterization of differentiated quiescent and nonquiescent cells in yeast stationary-phase cultures. Mol. Biol. Cell. 2008; 19:1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valouev A., Johnson D.S., Sundquist A., Medina C., Anton E., Batzoglou S., Myers R.M., Sidow A.. Genome-wide analysis of transcription factor binding sites based on ChIP-seq data. Nat. Methods. 2008; 5:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D.. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng Y., Ma Z., Kim B., Wu W., Cayting P., Boyle A.P., Sundaram V., Xing X., Dogan N., Li J.. Principles of regulatory information conservation between mouse and human. Nature. 2014; 515:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garber M., Yosef N., Goren A., Raychowdhury R., Thielke A., Guttman M., Robinson J., Minie B., Chevrier N., Itzhaki Z.. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell. 2012; 47:810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abe N., Dror I., Yang L., Slattery M., Zhou T., Bussemaker H.J., Rohs R., Mann R.S.. Deconvolving the recognition of DNA shape from sequence. Cell. 2015; 161:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng C., Shou C., Yip K.Y., Gerstein M.B.. Genome-wide analysis of chromatin features identifies histone modification sensitive and insensitive yeast transcription factors. Genome Biol. 2011; 12:R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pique-Regi R., Degner J.F., Pai A.A., Gaffney D.J., Gilad Y., Pritchard J.K.. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011; 21:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei Y., Wu G., Ji H.. Global mapping of transcription factor binding sites by sequencing chromatin surrogates: A perspective on experimental design, data analysis, and open problems. Stat. Biosci. 2013; 5:156–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ji H., Li X., Wang Q.F., Ning Y.. Differential principal component analysis of ChIP-seq. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:6789–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blatti C., Kazemian M., Wolfe S., Brodsky M., Sinha S.. Integrating motif, DNA accessibility and gene expression data to build regulatory maps in an organism. Nucleic Acids Res. 2015; 43:3998–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo K., Hartemink A.J.. Using DNase digestion data to accurately identify transcription factor binding sites. Pac. Symp. Biocomput. 2013; 80–91. [PMC free article] [PubMed] [Google Scholar]
- 24. Kahara J., Lahdesmaki H.. BinDNase: A discriminatory approach for transcription factor binding prediction using DNase I hypersensitivity data. Bioinformatics. 2015; 31:2852–2859. [DOI] [PubMed] [Google Scholar]
- 25. Piper J., Elze M.C., Cauchy P., Cockerill P.N., Bonifer C., Ott S.. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 2013; 41:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jankowski A., Tiuryn J., Prabhakar S.. Romulus: Robust multi-state identification of transcription factor binding sites from DNase-seq data. Bioinformatics. 2016; 32:2419–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tu B.P., Kudlicki A., Rowicka M., McKnight S.L.. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science. 2005; 310:1152–1158. [DOI] [PubMed] [Google Scholar]
- 28. Feng D., Lazar M.A.. Clocks, metabolism, and the epigenome. Mol. Cell. 2012; 47:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirabayashi Y., Gotoh Y.. Epigenetic control of neural precursor cell fate during development. Natu. Rev. Neurosci. 2010; 11:377–388. [DOI] [PubMed] [Google Scholar]
- 30. Tu B.P., McKnight S.L.. Metabolic cycles as an underlying basis of biological oscillations. Nat. Rev. Mol. Cell Biol. 2006; 7:696–701. [DOI] [PubMed] [Google Scholar]
- 31. Koike N., Yoo S.H., Huang H.C., Kumar V., Lee C., Kim T.K., Takahashi J.S.. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012; 338:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuang Z., Cai L., Zhang X., Ji H., Tu B.P., Boeke J.D.. High-temporal-resolution view of transcription and chromatin states across distinct metabolic states in budding yeast. Nat. Struct. Mol. Biol. 2014; 21:854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziller M.J., Edri R., Yaffe Y., Donaghey J., Pop R., Mallard W., Issner R., Gifford C.A., Goren A., Xing J.. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature. 2015; 518:355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. R Development Core Team R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2015; Vienna: http://www.R-project.org/. [Google Scholar]
- 35. Ji H., Jiang H., Ma W., Johnson D.S., Myers R.M., Wong W.H.. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 2008; 26:1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cairns J., Spyrou C., Stark R., Smith M.L., Lynch A.G., Tavare S.. BayesPeak—an R package for analysing ChIP-seq data. Bioinformatics. 2011; 27:713–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ji Z., Ji H.. TSCAN: pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016; 44:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cleveland W.S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 1979; 74:829–836. [Google Scholar]
- 39. Tu B.P., Mohler R.E., Liu J.C., Dombek K.M., Young E.T., Synovec R.E., McKnight S.L.. Cyclic changes in metabolic state during the life of a yeast cell. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:16886–16891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pokholok D.K., Harbison C.T., Levine S., Cole M., Hannett N.M., Lee T.I., Bell G.W., Walker K., Rolfe P.A., Herbolsheimer E.. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005; 122:517–527. [DOI] [PubMed] [Google Scholar]
- 41. Huber A., French S.L., Tekotte H., Yerlikaya S., Stahl M., Perepelkina M.P., Tyers M., Rougemont J., Beyer A.L., Loewith R.. Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J. 2011; 30:3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lieb J.D., Liu X., Botstein D., Brown P.O.. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein–DNA association. Nat. Genet. 2001; 28:327–334. [DOI] [PubMed] [Google Scholar]
- 43. Marion R.M., Regev A., Segal E., Barash Y., Koller D., Friedman N., O'Shea E.K.. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:14315–14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mai B., Breeden L.. Xbp1, a stress-induced transcriptional repressor of the saccharomyces cerevisiae Swi4/Mbp1 family. Mol. Cell. Biol. 1997; 17:6491–6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Estruch F., Carlson M.. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of saccharomyces cerevisiae. Mol. Cell. Biol. 1993; 13:3872–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang N., Wu J., Oliver S.G.. Gis1 is required for transcriptional reprogramming of carbon metabolism and the stress response during transition into stationary phase in yeast. Microbiology. 2009; 155:1690–1698. [DOI] [PubMed] [Google Scholar]
- 47. Lutfiyya L.L., Iyer V.R., DeRisi J., DeVit M.J., Brown P.O., Johnston M.. Characterization of three related glucose repressors and genes they regulate in saccharomyces cerevisiae. Genetics. 1998; 150:1377–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Denis C.L., Young E.T.. Isolation and characterization of the positive regulatory gene ADR1 from saccharomyces cerevisiae. Mol. Cell. Biol. 1983; 3:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Estruch F. The yeast putative transcriptional repressor RGM1 is a proline-rich zinc finger protein. Nucleic Acids Res. 1991; 19:4873–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walkey C.J., Luo Z., Madilao L.L., van Vuuren H.J.. The fermentation stress response protein Aaf1p/Yml081Wp regulates acetate production in saccharomyces cerevisiae. PLoS One. 2012; 7:e51551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cai L., Sutter B.M., Li B., Tu B.P.. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell. 2011; 42:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuang Z., Pinglay S., Ji H., Boeke J.D.. Msn2/4 regulate expression of glycolytic enzymes and control transition from quiescence to growth. Elife. 2017; 6:e29938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rashid N.U., Giresi P.G., Ibrahim J.G., Sun W., Lieb J.D.. ZINBA integrates local covariates with DNA-seq data to identify broad and narrow regions of enrichment, even within amplified genomic regions. Genome Biol. 2011; 12:R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wingender E., Dietze P., Karas H., Knuppel R.. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996; 24:238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu Z., Killion P.J., Iyer V.R.. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007; 39:683–687. [DOI] [PubMed] [Google Scholar]
- 57. Gitter A., Siegfried Z., Klutstein M., Fornes O., Oliva B., Simon I., Bar-Joseph Z.. Backup in gene regulatory networks explains differences between binding and knockout results. Mol. Syst. Biol. 2009; 5:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y., Schnegelsberg P.N., Dausman J., Jaenisch R.. Functional redundancy of the muscle-specific transcription factors Myf5 and myogenin. Nature. 1996; 379:823–825. [DOI] [PubMed] [Google Scholar]
- 59. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE72263. DynaMO is publicly available as an R package at https://github.com/spo111/DynaMO.