

Abstract
Human-driven global environmental changes have considerably increased the risk of biological invasions, especially the spread of human parasites and their vectors. Among exotic species that have major impacts on public health, the dengue fever mosquito Aedes aegypti originating from Africa has spread worldwide during the last three centuries. Although considerable progress has been recently made in understanding the history of this invasion, the respective roles of human and abiotic factors in shaping patterns of genetic diversity remain largely unexplored. Using a genome-wide sample of genetic variants (3,530 ddRAD SNPs), we analyzed the genetic structure of Ae. aegypti populations in the Caribbean, the first introduced territories in the Americas. Fourteen populations were sampled in Guyane and in four islands of the Antilles that differ in climatic conditions, intensity of urbanization, and vector control history. The genetic diversity in the Caribbean was low (He = 0.14–0.17), as compared with a single African collection from Benin (He = 0.26) and site-frequency spectrum analysis detected an ancient bottleneck dating back ∼300 years ago, supporting a founder event during the introduction of Ae. aegypti. Evidence for a more recent bottleneck may be related to the eradication program undertaken on the American continent in the 1950s. Among 12 loci detected as FST-outliers, two were located in candidate genes for insecticide resistance (cytochrome P450 and voltage-gated sodium channel). Genome–environment association tests identified additional loci associated with human density and/or deltamethrin resistance. Our results highlight the high impact of human pressures on the demographic history and genetic variation of Ae. aegypti Caribbean populations.
Keywords: invasive species, human impact, adaptation, bottleneck, insecticide resistance, mosquito
Introduction
Human activities at the worldwide scale have considerably increased the risk of biological invasions in the last decades. The introduction and establishment of exotic species are driving native biodiversity decline and ecosystems degradation (Simberloff et al. 2013). Assessing current and future risks as well as improving management interventions to limit the rates and impacts of invasions requires knowledge on the evolutionary processes promoting invasion (Lee 2002; Allendorf and Lundquist 2003; Wilson et al. 2009; Estoup and Guillemaud 2010; Prentis and Pavasovic 2013). Since the 1980s, the field of invasion science has contributed to theoretical understanding of biological invasions (Sax et al. 2005; McGeoch et al. 2012; Spear et al. 2013) but fundamental questions have not been fully answered (Facon et al. 2006; Colautti and Lau 2015; Packer et al. 2017). A general framework regarding the mechanisms involved at each stage of an invasion has been adopted (Blackburn et al. 2011), and the relevance of genetic and genomic approaches to decipher colonization pathways and to understand the role of local adaptation and preadaptation in determining invasion success is now well-recognized (Lawson Handley et al. 2011; Chown et al. 2015). The response of nonnative species to ongoing environmental changes and the role of human past control strategies on pest evolutionary trajectories are timely research questions (Darling and Côté 2008; Bradley et al. 2010; Treasure et al. 2014).
In the recent history, biological invasions have become a major public health concern due to the expansion of disease vectors and the emergence of vector-borne diseases (Lounibos 2002; Tatem et al. 2006). Among the most threatening nonnative species, mosquitoes are vectors of parasitic and viral diseases, and their expansion has often coincided with important outbreaks of human diseases such as malaria, yellow and dengue fevers, and chikungunya and zika viruses. The typically anthropophilic species Aedes aegypti, native to African countries, comprises two endemic forms (forest vs. domestic) supported by genetic, morphological, ecological, and behavioral differences (Brown et al. 2011, 2014; Powell and Tabachnick 2013). Although the details of the shift from a forest to a domestic form in the native range are still debated, all populations outside Africa appear to derive from the same ancestral domestic African populations (Bennett et al. 2016; Gloria-Soria et al. 2016). The species has spread worldwide as a result of human transportations but mainly throughout tropical and subtropical regions with year-round breeding confined to latitudes <33°N (Brown et al. 2014). Unlike its close relative species Aedes albopictus, Ae. aegypti has not invaded temperate regions, suggesting that climate might be a limiting ecological factor for this species range expansion. However, the respective roles of human and climate in shaping patterns of genetic diversity and local adaptation remain largely unexplored.
Sources of spatially varying selection pressures for mosquito populations encompass climatic variables such as temperature and precipitation (Fouque et al. 2006; Canyon et al. 1999; Costa et al. 2010; Brady et al. 2013; Goindin et al. 2015), and habitat descriptors such as human density or insecticide treatment intensity (Paris et al. 2010; Barbosa et al. 2011; Marcombe et al. 2013; Nkya et al. 2014). The Caribbean represents an outstanding natural laboratory for evaluating how exotic populations rapidly respond to newly encountered environments. Here, we focused on the French Antilles archipelago (Martinique, Guadeloupe, St-Barthélemy, St-Martin) and French Guiana (Guyane). These areas share the same history of introduction, giving populations about the same time period to evolve, but differ in climatic conditions (tropical vs. equatorial), intensity of urbanization, and of insecticide use. Ae. aegypti was presumably introduced during the African slave trade (17th–18th century) first to the Antilles and then to the American continent (Bryant et al. 2007). In the 1950s, the Pan American Health Organization (PAHO) conducted an eradication campaign on the American continent with the massive use of insecticides (DDT, dieldrin; Soper 1963; Camargo 1967). The species was declared eradicated in the American continent, but not in the Antilles from which the recolonization of American countries presumably took place in the 1970s (Wallis et al. 1983; Monteiro et al. 2014). Dengue outbreaks became more and more frequent since the 1990s with increasing number of cases of severe hemorrhagic forms (Gubler and Clark 1994), and the evolution of resistance to DDT in populations have triggered the massive use of deltamethrin, thereby selecting highly resistant populations (Faucon et al. 2015; Dusfour et al. 2015; Goindin et al. 2017).
Here, we analyzed the distribution of genetic variation in populations in the Caribbean using genome-wide distributed molecular markers. More specifically we aimed at i) determining the genetic structure of Ae. aegypti at global and local scales, ii) inferring the demographic history of these bridgehead invasive populations, and iii) detecting genes showing signatures of local adaptation to climatic and human-induced selective pressures in the Caribbean.
Materials and Methods
Sample Collection
Immature stages of mosquitoes were collected in 14 localities in two regions in the Caribbean: 2 localities in Guyane and 12 in the Antilles distributed across four islands: St-Barthélemy, St-Martin, Guadeloupe (including the island La Désirade), and Martinique (including the Îlet Long islet) (table 1 and fig. 1). Three individuals from Benin (laboratory strain SBE) were genotyped as outgroup. Collection sites were domestic breeding habitats and in each locality, collections constituted 10–20 larvae. Field-collected larvae were either directly preserved in ethanol 75%, or pooled and reared in standard laboratory conditions and the obtained F1 progeny reared to four-instar larvae.
Table 1.
Sampling Sites Characteristics
| Region | Population Name | Code | Longitude | Latitude | Human Density (inh/km2) | Total Precipitation (mm/year) | Mean Temperature (°C) | N | Deltamethrin RR50a | Collector |
|---|---|---|---|---|---|---|---|---|---|---|
| Guadeloupe | Anse Bertrand | AB | −61.50 | 16.46 | 78.6 | 1,310 | 26.73 | 3 | 28.1 | D. Goindin |
| Guadeloupe | Baie-Mahault | BM | −61.58 | 16.27 | 656.5 | 1,535 | 26.58 | 3 | 13.6 | D. Goindin |
| Guadeloupe | La Désirade | DES | −61.04 | 16.32 | 73.6 | 1,083 | 26.38 | 3 | NA | D. Goindin |
| Guadeloupe | St-François | SF | −61.29 | 16.26 | 245.1 | 1,321 | 26.51 | 1 | 13.7 | D. Goindin |
| North islands | St-Barthélemy | SBH | −62.82 | 17.89 | 430.2 | 995 | 26.68 | 2 | 9.3 | D. Goindin |
| North islands | Sandy Ground | SMO | −63.10 | 18.05 | 528 | 1,032 | 26.77 | 3 | 12.4 | D. Goindin |
| North islands | Oyster Pond | SME | −63.08 | 18.06 | 528 | 1,051 | 26.35 | 3 | 12.4 | D. Goindin |
| Guyane | Cayenne | CAY | −52.32 | 4.92 | 2,425 | 3,105 | 26.04 | 3 | 750 | I. Dusfour |
| Guyane | St-Georges | GEO | −51.80 | 3.89 | 1.7 | 3,400 | 26.02 | 3 | 750 | I. Dusfour |
| Martinique | Îlet Long | ILO | −60.86 | 14.61 | 0 | 1,932 | 26.79 | 3 | 5.1 | S. Marcombe |
| Martinique | Lamentin | LAM | −61.00 | 14.62 | 236 | 1,992 | 26.68 | 3 | 5.6 | S. Marcombe |
| Martinique | Rivière salée | RSAL | −60.96 | 14.52 | 326 | 1,964 | 26.74 | 3 | 3.7 | S. Marcombe |
| Martinique | St-Anne | SAN | −60.85 | 14.43 | 121 | 1,880 | 26.82 | 3 | 6.7 | S. Marcombe |
| Martinique | St-Pierre | SPIER | −61.16 | 14.76 | 113 | 1,832 | 26.01 | 3 | 4 | S. Marcombe |
| Benin | SBE | 2.36 | 6.38 | 1,227 | 27.42 | 3 | 1 | S. Marcombe |
Resistance ratio as compared with the Bora-Bora laboratory strain (Marcombe et al. 2012, 2013; Faucon et al. 2015; Goindin et al. 2017).
Fig. 1.

—Locations of the 15 collection sites assigned to six biogeographical regions based on the presence of natural barriers to dispersion (geographical distance and sea) and administrative borders: Benin (Africa), Guyane, Martinique, Guadeloupe, St-Barthélemy, and St-Martin.
DNA Extraction and ddRADseq Library Preparation
DNA was extracted from each individual larva (3 larvae per location) using cetyl trimethyl ammonium bromide (CTAB) chloroform/isoamyl alcohol protocol (Doyle and Doyle 1987). A double-digested RAD (Restriction site Associated DNA) experiment was conducted on 45 individuals using a modified version of the protocol previously described (Peterson et al. 2012; Capblancq et al. 2015). Briefly, 250 ng of DNA template from each individual was double-digested with ten units each of SbfI–HF and MspI (New England Biolabs Inc.) at 37 °C for 1 h using the CutSmart buffer provided with the enzymes. Digestion was further continued together with the ligation of P1 (individually indexed) and P2 adapters in a thermocycler (60 cycles of 2-min digestion at 37 °C and 2-min ligation at 16 °C, followed by heat inactivation of the enzymes at 65 °C for 10 min). An equal volume of all the digested-ligated individuals was pooled and purified with Agencourt AMPure XP beads (Beckman Coulter, France). After migration on 1.6% agarose gel, fragments between 250 and 500 bp were excised and purified with QIAquick Gel Extraction Kit (Qiagen, Germany). The ddRAD library was amplified in ten independent replicates of 15 PCR cycles (initial denaturation 10 min. 98 °C; 15 cycles of 98 °C for 10 s, 66 °C for 30 s and 72 °C for 1 min; followed by a final 10-min extension period at 72 °C) in a final volume of 20 µl with 1 µl of DNA template, 10 mM of dNTPs, 10 µM of each PCR primers (Peterson et al. 2012) and 2 U/µl of Taq Phusion-HF (New England Biolabs Inc.). The low number of amplification cycles conducted in ten independent tubes guarantees that errors during amplification will not generate artificial SNPs in the final ddRAD library. The ten PCR products were pooled, purified with QIAgen MinElute PCR Purification Kit (Qiagen, Germany) and sequenced on an Illumina Hi-Seq 2500 Illumina sequencer (1/10 lane, paired-end 2×125 bp, Fasteris SA, Switzerland).
SNPs Genotyping
The ∼31 million DNA reads obtained were used to call SNP genotypes with the STACKS pipeline (Catchen et al. 2013) as follows: the process_radtags function was first run to demultiplex the data and filter the reads on their quality. Reads with low quality (Illumina filter) or uncalled bases were removed (options -c, -q, and –r). Only SbfI reads that were paired with MspI reads were retained. We then mapped the reads of each individual to the reference Aedes aegypti genome (AaegL3, https://www.vectorbase.org/; bwa mem), and then filtered on alignment quality >35 (samtools view, –q 35), and aligned bam files were used to build loci from reference (pstacks). A catalog of the loci from all the individuals was built using the cstacks function, with a maximum of five mismatches for merging two individual loci into the catalogue (-n). Loci within each individual were searched against the catalog (sstacks function) and a SNP data set was produced with the genotype of each individual for every polymorphic position with a minimum read depth m = 3 (populations function). For genetic analyses, only SNPs present in >60% of the whole sample were retained.
Phylogenetic Relationships and Population Structure
Phylogenetic relationships among individuals were inferred based on the concatenated sequences of individuals using variable and invariant sites as recommended (Leache et al. 2015) and UIPAC codes for polymorphic sites. All polymorphic positions present in at least 24 individuals were used. A Maximum Likelihood (ML) tree was generated using the GTR model with a Gamma distribution parameter allowing rate variation across sites (Γ = 0.02). We performed 700 rapid heuristic bootstrap followed by a thorough ML search implemented in the program RAxML v.8 (Stamatakis 2014). The number of bootstrap replicates was determined using the automatic BS convergence criterion (Pattengale et al. 2010).
In order to detect population genetic structure, we first performed a Discriminant Analysis of Principal Components (Jombart et al. 2010) using the R package adegenet v1.4.2 (Jombart 2008) with the number of populations as the number of clusters. Then we used the “structure-like” algorithm sNMF for inferring admixture coefficients for all individuals as implemented in the R package LEA (Frichot et al. 2014; Frichot and François 2015). The number of genetic clusters, K, was varied from 1 to 10. We performed 100 runs for each value of K, and retained the best run for each K based on the cross-entropy criterion. We surveyed the results for all K and stopped increasing this value when at least one genetic cluster contained less than two individuals.
Genetic Diversity and Geographical Differentiation
Genetic diversity by individual (individual heterozygosity: observed heterozygosity), within each sampled locality (population genetic diversity: expected heterozygosity), and between all population pairs was assessed using GENEPOP4.7 software (Raymond and Rousset 1995). FIS and FST were estimated according to Weir and Cockerham (1984). Genetic variation partitioning among and within populations and geographical groups was tested in hierarchical analyses of molecular variance (AMOVA) using nonparametric permutational procedures (1,023 iterations) as implemented in Arlequin 3.5 (Excoffier et al. 2005). We first tested for the effect of the three sampling regions (Benin/Guyane/Antilles) on genetic diversity. We then focused on the Caribbean populations to test for genetic continuity between Guyane and Antilles, and for genetic similarities among the four islands of the Antilles.
In order to assess the respective roles of geographical distance and the presence of sea in population genetic differentiation of Ae. aegypti, multiple correlations between genetic (FST/[1−FST]), straight-line geographic distance (square-root transformed), and distance based on the presence/absence of sea (coded as 0/1) were tested by performing a multiple regression on distance matrix (MRM in the R package ecodist, Goslee and Urban 2007) using permutation tests of significance (999 permutations) for regression coefficients and R-squared.
Demographic History Inference
To infer the demographic history of the Caribbean mosquito populations, we used the stairway plot method, which infers effective population size changes over time using the SNP site frequency spectrum (SFS) (Liu and Fu 2015). The SFS was computed after imputation of missing genotypes following the method of Chi et al. (2013), using the ancestry and allele frequency matrices (Q-matrix and F-matrix) estimated from the sNMF model with K = 6. The mutation rate was set to 1.6×10−8 base pair per generation, and a generation time of 1 month was used (Bennett et al. 2016). The total sequence length was 847 Mb (3,530×240×100bp). We assessed the statistical errors of effective population size estimates by using 50 bootstrap replicates of the stairway plot algorithm.
To measure the intensity of genetic drift (population-specific effective size and immigration rate), we calculated population-specific FST by using the hierarchical Bayesian algorithm BayeScan v2.1 (Foll and Gaggiotti 2008). This approach is based on a continent-island model in which subpopulation allele frequencies are correlated through a common migrant gene pool, thereby reflecting the well documented recent history of colonization of the Caribbean from an African ancestor (Brown et al. 2014; Gloria-Soria et al. 2016). Population-specific FST coefficients are estimated by the difference in allele frequency between the common gene pool and each population.
Locus-Specific Departure from Neutrality
To search for loci more differentiated than expected under the neutrality assumption, we used the hierarchical Bayesian BayeScan package v2.1 (Foll and Gaggiotti 2008), which incorporates the uncertainty in allele frequencies due to small population sample sizes. Selection is introduced by decomposing each locus FST into a population-specific component, shared by all loci, and a locus-specific component (α) shared by all the populations using a logistic regression. Departure from neutrality at a locus is assumed when the locus-specific component is necessary to explain the observed pattern of diversity (α significantly different from 0). The estimation of model parameters was automatically tuned on the basis of short pilot runs (ten pilot runs, length 2,000). The sample size was set to 10,000 and the thinning interval to 50 as suggested by the authors of BayeScan (Foll and Gaggiotti 2008), resulting in a total chain length of 500,000 iterations, performed four times independently. The loci were ranked according to their false discovery rate controlling for multiple testing (q value), and all loci with a q value <0.05 were retained as outliers.
Environmental Variables and Genome–Environment Association Tests
For each sampled locality, we extracted all the 19 bioclimatic variables averaged on 30 years (1960–1990) from the WorldClim database (http://www.worldclim.org; Hijmans et al. 2005) and we used a principal component analysis (PCA) to reduce these variables to two uncorrelated climatic variables: mean temperature and total precipitation (supplementary fig. S1, Supplementary Material online). In addition, two human-induced local pressure proxies were considered: human density and resistance to deltamethrin. Human density (inhabitants per km2) from the 2014 census was extracted from the INSEE database (https://www.insee.fr). Resistance to deltamethrin, the main insecticide currently used against adult mosquitoes, was used as a proxy for the local insecticide selective pressure (Marcombe et al. 2012, 2013; Faucon et al. 2015; Goindin et al. 2017) (table 1). These two anthropogenic variables are not correlated (Pearson’s R = 0.25, P = 0.36).
In order to determine which environmental (climatic or anthropogenic) variables have the most important impact on genetic variation in Ae. aegypti populations, we used two different correlative methods: redundancy analysis (RDA, Legendre and Legendre 1998), and latent factor mixed models (LFMM, Frichot et al. 2013). RDA is a constrained version of principal component analysis that summarizes the component of genetic variation explained by a set of environmental variables. We used RDA as implemented in the R package vegan (Oksanen et al. 2017). To remove the effect of geography on genetic variation, we used a combination of each sampled locality coordinates (latitude + longitude) as a conditional factor in the RDA (Nadeau et al. 2016). To determine how much of the genetic variation is uniquely explained by each environmental factor, how much is uniquely explained by geography, and how much is explained by some joint effect of these factors, a series of RDA and partial RDA (conditioned on geography) models was constructed: a full model with all climate and anthropic variables as explanatory variables, all the four models with one environmental factor, or with anthropic factors (deltamethrin + human density), or with climatic factors (temperature + precipitation) as explanatory variables. The loadings of RDA (one-factor models) were used as test statistics for testing association with each environmental variable (Forester et al. 2016). In LFMM, population structure and other confounding variables are modeled by unobserved (latent) factors, and the statistical tests are adjusted for those latent factors. The latent factor models were run with K = 6 factors, following the results of population structure analyses. We evaluated association between each SNP and each environmental variable by running 30 independent replicates of LFMM. Assuming a Gaussian distribution under the null hypothesis, Z-scores from all runs were combined using the Stouffer method and the P values were adjusted by using a genomic inflation factor correction (Frichot and François 2015; François et al. 2016). The Storey–Tibshirani FDR control algorithm was applied to detect the most interesting loci with an expected FDR level of 10% (Storey and Tibshirani 2003). RDA and LFMM tests were applied on a subset of the SNP data set without missing genotypes (578 SNPs), and we extrapolated the level of resistance to deltamethrin in the island La Désirade (DES) to the average level observed in Guadeloupe. The effects of each environmental variable on the SNP data set were tested separately with the default settings of the RDA and LFMM algorithms.
Results
SNP Genomic Distribution
Approximately 31 million high-quality reads were obtained with an average 700,000 reads per individual. Individuals with <300,000 reads were removed from the analysis. Thirty-nine individuals were kept for genetic analysis (table 1), and mapped to the reference genome (1,376 Mb consisting of 4,758 supercontigs 0.06–5 Mb available at http://aaegypti.vectorbase.org). For each individual, about half the total reads mapped to the genome (mapping quality > 35) at a unique position. An average of 12,000 “RADtags” (a pair of 120-bp sequences flanking a genomic region 0–400 bp) were obtained per individual. A total of 15,128 loci (RADtags) were identified across the 39 individuals, distributed over 1,355 supercontigs representing a total length of 1,280 Mb (93% of the 1,376 Mb Ae. aegypti genome, supplementary table S1, Supplementary Material online). A total of 15,989 SNPs was called of which 3,530 SNPs present in >60% of the individuals were retained (median missing data per SNP= 5%). These SNPs were distributed over 2,535 RADtags distributed throughout 719 supercontigs (supplementary table S2, Supplementary Material online), with 1 up to 19 RADtags per supercontig. The number of RADtags on a supercontig was correlated with the length of the supercontig (Pearson’s R = 0.64, P < 0.001). These 719 supercontigs represented a total length of 1,009 Mb (73.3% of Ae. aegypti genome), and the density of RADtags was 3.5 per Mb. Assuming an estimated average recombination rate in Ae. aegypti c = 0.3 cM/Mb, or 3.3 Mb/cM, we expect to have on an average eleven RADtags every 1% recombination event which is enough to identify genomic regions undergoing selective sweep, even if none of our RADtag are directly under selection (Wilfert et al. 2007).
Phylogenetic Relationships and Population Structure
The ML phylogenetic tree inferred from all sites (794,070 sites, including 11,763 variable sites) showed three genetic groups supported by high bootstrap values (fig. 2). The first group corresponds to individuals from St-Barthélemy. The second included all individuals from St-Martin. The third group clustered populations from Martinique, Guadeloupe, and Guyane. Within this group, the two populations from Guyane, GEO, and CAY, clustered together into two distinct subgroups, and all populations from Guadeloupe clustered together into a distinct subgroup. For non-African samples, the sNMF analysis provided strong evidence for geographic population structure and only weak evidence for genetic admixture (fig. 2). For K = 3–6, each genetic cluster corresponded to a well-defined geographic region (supplementary fig. S2, Supplementary Material online). The best model for K = 6 identified six main regions (North Islands, Guadeloupe, Martinique, populations BM, GEO, and CAY) as differentiated genetic groups. A single individual from St-Barthélemy was misassigned to the Guadeloupean cluster, and the unique individual from SF failed to be assigned to a genetic cluster. Models for K greater than six further partitioned the Guadeloupe and Martinique Islands into smaller regions that matched with sampling localities (supplementary fig. S2, Supplementary Material online). In the DAPC, most individuals were correctly assigned to their geographical region, except for two out of the three samples from BM (Guadeloupe), which were assigned to Martinique (fig. 3A and B).
Fig. 2.

—Phylogenetic relationships and genetic clustering among Ae. aegypti Caribbean samples. (A) The Maximum Likelihood (ML) tree was rooted with the sequences from Benin (N = 39; 794,070 SNPs). Colors indicate the geographic region: Nord islands (dark blue), Martinique (light blue), Guadeloupe (green), and Guyane (orange). Bootstrap support values ≥50% and≥95% are shown in italic and in bold respectively. Scale represents the mean number of nucleotide substitutions per site. Sequence labels are given as population code and individual number. (B) Genetic clustering (sNMF) for American samples (N = 36) for K = 6 (see supplementary fig. S2, Supplementary Material online, for K = 3–8).
Fig. 3.

—Results of the Discriminant Analysis in Principal Component performed with populations as prior clusters. (A) DAPC scatterplot of LD1 versus LD2 for the 14 localities studied; (B) assignment probabilities of each individual to each of the four geographical regions: Guyane, Martinique, Guadeloupe, North islands.
Genetic Diversity and Geographical Differentiation
Individual heterozygosity ranged from 0.1347 (in CAY) to 0.1754 (in SBE; table 2) and was not significantly different among regions (Adjusted R2 = 0.13, F5,8= 1.403, P = 0.32). Population diversity (expected heterozygosity) ranged from 0.1431 to 0.2593 and was significantly higher only in SBE (adjusted R2 = 0.85, F5,8= 16.69, P = 0.0004). FIS ranged from 0.01 to 0.32 and was again higher in SBE (adjusted R2 = 0.69, F5,8= 6.954, P = 0.008). Therefore, the five different regions studied (Guadeloupe, Martinique, St-Martin, St-Barthélemy, and Guyane) shared comparable ranges of lower genetic diversity indices as compared with the only African population sampled. The number of singletons, defined as variants uniquely represented in the total sample, was much higher in SBE (mean value = 108) than in the Caribbean populations (mean value = 9.6, two-sample t-test P = 0.03, table 2). Pairwise population differentiation (Weir and Cockerham FST) ranged from 0 (between SPIER and SAN, two localities from Martinique) to 0.25 between African and Caribbean populations (supplementary table S3, Supplementary Material online). Average FST across all loci (excluding the 5% most differentiated loci) was 0.07 across the 14 American populations. The presence of sea and the geographic distance were both predictors of population genetic differentiation (supplementary table S4, Supplementary Material online, R2 = 0.47, P = 0.001). Most of the genetic variation was found within individuals (>70%, see table 3), and ∼4% of the variance was found among populations within groups, whatever the tested geographical clustering. The genetic differentiation between the sampling regions was high (13% of total variance when considering Benin, Guyane, and the Antilles) to moderate (4% of total variance when considering Guyane and the Antilles).
Table 2.
Genetic Indices Per Sampled Locality
| Population Name | Code | Region | He | Ho | FIS | FSTa | Singletons (sd) |
|---|---|---|---|---|---|---|---|
| Anse Bertrand | AB | Guadeloupe | 0.1670 | 0.1492 | 0.107 | 0.1055 | 9.7 (1.5) |
| Baie-Mahault | BM | Guadeloupe | 0.1658 | 0.1469 | 0.114 | 0.1204 | 8.0 (4.4) |
| La Désirade | DES | Guadeloupe | 0.1618 | 0.1428 | 0.117 | 0.1403 | 9.7 (4.7) |
| St-François | SF | Guadeloupe | NA | NA | NA | 0.1304 | 15 (NA) |
| St-Barthélemy | SBH | North islands | 0.1761 | 0.1591 | 0.096 | 0.1307 | 16 (14) |
| Oyster Pond | SME | North islands | 0.1777 | 0.1558 | 0.124 | 0.1078 | 11 (2.9) |
| Sandy Ground | SMO | North islands | 0.1557 | 0.1393 | 0.105 | 0.1889 | 6.7 (1.5) |
| Cayenne | CAY | Guyane | 0.1431 | 0.1347 | 0.059 | 0.2518 | 6.7 (2.3) |
| St-Georges | GEO | Guyane | 0.1737 | 0.1566 | 0.098 | 0.1361 | 10 (4.4) |
| Îlet Long | ILO | Martinique | 0.1637 | 0.1513 | 0.076 | 0.1735 | 10 (0.7) |
| Lamentin | LAM | Martinique | 0.1714 | 0.1477 | 0.138 | 0.0833 | 10 (10) |
| Rivière Salée | RSAL | Martinique | 0.1651 | 0.1448 | 0.123 | 0.0929 | 13 (1.5) |
| St-Anne | SAN | Martinique | 0.1640 | 0.1436 | 0.124 | 0.0785 | 4.6 (4.1) |
| St-Pierre | SPIER | Martinique | 0.1725 | 0.1702 | 0.013 | 0.0443 | 10 (1.4) |
| SBE | SBE | Benin | 0.2593 | 0.1744 | 0.328 | 108 (31) |
He, expected heterozygosity; Ho, observed heterozygosity; NA, not available.
Population-specific FST calculated by BayeScan. High values indicate genetic drift (small effective size, low migration) from a common ancestral population.
Table 3.
Hierarchical Analysis of Molecular Variance (AMOVA) for the Three Different Genetic Partitioning Hypotheses
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variance |
|---|---|---|---|---|
| Benin/Guyane/Antilles | ||||
| Among groups | 2 | 887 | 18.75 | 13.06*** |
| Among populations within groups | 12 | 1,931 | 6.95 | 4.84* |
| Among individuals within populations | 24 | 3,019 | 7.88 | 5.48*** |
| Within individuals | 39 | 4,292 | 110.05 | 76.62** |
| Total | 77 | 10,130 | 143.63 | |
| Guyane/Antilles | ||||
| Among groups | 4 | 917 | 6.08 | 4.87*** |
| Among populations within groups | 9 | 1,327 | 5.96 | 4.77*** |
| Among individuals within populations | 22 | 2,576 | 4.21 | 3.37 |
| Within individuals | 36 | 3,912 | 108.67 | 86.99*** |
| Total | 71 | 8,732 | 124.92 | |
| Antilles | ||||
| Among islands | 3 | 604 | 4.63 | 3.81*** |
| Among populations within islands | 8 | 1,117 | 4.72 | 3.88*** |
| Among individuals within populations | 18 | 2,090 | 3.93 | 3.23 |
| Within individuals | 30 | 3,248 | 108.27 | 89.08*** |
| Total | 59 | 7,059 | 121.54 | |
P < 0.001; **P < 0.01; *P < 0.05.
Demographic History of Populations
The SFS computed from the imputed SNP data set revealed an excess of common variants compared with the neutral expectations (fig. 4A). The stairway plot method used to infer demographic events revealed a strong decrease in effective population size that occurred ∼300 years ago (fig. 4B). In addition, the stairway curve exhibited a strong decrease in population size (down to near extinction) ∼100 years ago, followed by a rapid recovery and population expansion during the latest century. The population-specific FST coefficients ranged from 0.04 in St-Pierre (Martinique) up to 0.25 in CAY (Guyane) (table 2). As expected for populations undergoing genetic drift effect, populations with high specific FST also had low genetic diversity (Pearson’s R= −0.74, P < 0.01).
Fig. 4.
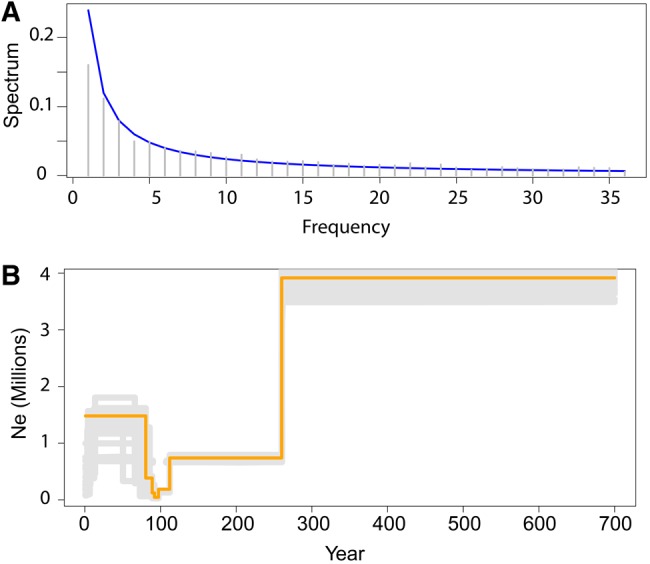
—Demographic history of Caribbean populations of Aedes aegypti. (A) SNP frequency spectrum with a deficit in low frequency alleles as compared with expectation under a neutral model (blue line) and (B) estimates of effective population size obtained from the stairway plot method applied to imputed genotypes.
Genetic Variation under Selection and Association with Environmental Variables
BayeScan detected 12 loci more differentiated than the background variants and potentially under selection, all with α positive, which indicates divergent ongoing selection (table 4). Among the 12 outliers detected by BayeScan, five were located in (or close to) genes that are involved in transcription regulation (nucleic acid binding or ribosomal protein), or are known to be potentially involved in insecticide resistance, such as cytochrome P450 or voltage-gated sodium channel genes (table 5).
Table 4.
Summary of the 12 SNPs Identified as Outliers by BayeScan (qval > 0.05)
| Locus ID | Posterior Prob | Log10(PO) | qval | α | FST |
|---|---|---|---|---|---|
| 2619_48 | 1.0 | 1,000 | 0 | 2.2 | 0.5 |
| 1272_118 | 1.0 | 1,000 | 0 | 2.0 | 0.5 |
| 4396_57 | 1.0 | 2.7 | 0.001 | 2.0 | 0.5 |
| 9098_14 | 1.0 | 2.3 | 0.002 | 1.8 | 0.4 |
| 4107_83 | 1.0 | 1.6 | 0.006 | 1.6 | 0.4 |
| 14191_81 | 1.0 | 1.5 | 0.013 | 1.7 | 0.4 |
| 454_74 | 1.0 | 1.5 | 0.013 | 1.6 | 0.4 |
| 4204_34 | 1.0 | 1.4 | 0.016 | 1.4 | 0.4 |
| 13604_97 | 0.9 | 1.1 | 0.023 | 1.8 | 0.5 |
| 10476_44 | 0.9 | 1.0 | 0.029 | 1.4 | 0.4 |
| 4205_13 | 0.9 | 0.9 | 0.036 | 1.2 | 0.3 |
| 3085_26 | 0.9 | 0.8 | 0.045 | 1.5 | 0.4 |
Note.—See supplementary table S5, Supplementary Material online, for all 3,530 loci characteristics.
PO, Posterior odds.
Table 5.
Within-Gene or Near-Gene SNPs among the 12 Outliers Identified by BayeScan Analysis
| Locus ID | Locus Position | Gene ID | Gene Descriptiona | Molecular Functionb | Expression Factorc |
|---|---|---|---|---|---|
| 4396_57 | 1.221_433624 | * AAEL006802 | Cytochrome P450 | Monooxygenase/oxidoreductase activity | Insecticides (2, 4, 6, 8, 9); dengue infection (1, 5); Bacterial infection (3, 7, 10) |
| 9098_14 | 1.47_801966 | # AAEL002013 | Nucleic acid binding; Zinc ion binding | West Nile virus and dengue infection (1, 5); Bacterial infection (3, 7, 11); Permethrin exposure (8) | |
| * AAEL001996 | Zinc finger protein | Nucleic acid binding; Zinc ion binding | Dengue infection (1, 5); Permethrin exposure (8); Bacterial infection (3, 7, 13); Habitat (12) | ||
| 4107_83 | 1.212_658124 | # AAEL006596 | West Nile virus infection (1); dengue infection (1, 5); Habitat (12) | ||
| 13604_97 | 1.920_113831 | * AAEL013788 | Sodium/calcium exchanger | Calcium: sodium antiporter activity | Dengue infection (1); Bacterial infection (3, 13); Insecticides (9); Habitat (12) |
| 3085_26 | 1.179_1472374 | * AAEL005901 | RpS3a | Ribonucleoprotein, Ribosomal protein | Yellow fever virus infection (1); Bacterial infection (3); Habitat (12) |
Note.—Locus position: chromosome(supercontig)_position.
VectorBase annotation.
Near coding region (<20 kb).
In coding region.
UniProt annotation.
Significantly altered expression under conditions listed as experimental factors.
(1) (Colpitts et al. 2011); (2) (Bariami et al. 2012); (3) (Choi et al. 2012); (4) (Riaz et al. 2009); (5) (Behura et al. 2011); (6) (Strode et al. 2008); (7) (Rancès et al. 2012); (8) (Poupardin et al. 2010); (9) (Riaz et al. 2013); (10) (Ye et al. 2013); (11) (Canton et al. 2015); (12) (McBride et al. 2014); (13) (Desjardins et al. 2015).
Local adaptation to environmental conditions can generate correlations between environmental variables and allele frequency (Frichot et al. 2013). RDA showed that 18.2% of genome-wide variation (578 SNPs) can be explained by environmental variation (fig. 5 and table 6). Geography accounted for 12.8% of total genetic variation, in line with the AMOVA results (3,530 SNPs). When correcting for geography, the environmental variables still explained >13% of total variation, with anthropic and climatic factors explaining 8% and 6% of total genetic variation respectively (table 6). LFMM and RDA produced congruent results and detected together eight loci associated with deltamethrin resistance. All except one loci detected by RDA were also detected by LFMM (table 7). The candidate loci displayed a common pattern of polymorphism in which the derived allele was present at high frequency in Guyane, and at low frequency elsewhere. For human density, LFMM detected seven loci associated with this variable (RDA found six loci, three loci were discovered by both methods). No polymorphism was associated to temperature and only one locus was associated with annual precipitation variation. None of the SNPs discovered by LFMM or RDA were detected as outliers by BayeScan.
Fig. 5.

—Redundancy analysis plot, summarizing the result of multiple regressions of genotypes (578 SNPs excluding missing data) against four environmental variables: temperature, precipitation, human density, and deltamethrin resistance (in blue), not constrained by geography. The four environmental variables explained 18% of total genetic variation. Anthropic factors (deltamethrin + human density) explained 9% of total variation, and climate (temperature + precipitation) explained 10% of total variation.
Table 6.
Redundancy Analyses (RDAs) Partitioning Genetic Variation into Geography and Environmental Variables
| Variables | Proportion of Variance Constrained (%) | Conditioned with Geographyb |
|---|---|---|
| Geographya | 12.78 | |
| Human density | 4.75 | 4.18 |
| Deltamethrin | 5.23 | 4.42 |
| Precipitation | 5.17 | 3.57 |
| Temperature | 5.51 | 2.86 |
| Anthropic factorsa | 9.05 | 7.94 |
| Climatic factorsa | 10.01 | 5.96 |
| All environmental variables | 18.21 | 13.33 |
Geography: (Latitude + Longitude); Anthropic factors: (Human density + Deltamethrin); Climatic factors: (Precipitation + Temperature).
Proportion in percentage of genetic variation constrained by environmental variables removing the 12.78% explained by geography.
Table 7.
Significance Values for LFMM and RDA Tests
| Locus ID | Deltamethrina |
Pearson’s R | Human Densitya |
Pearson’s R | ||
|---|---|---|---|---|---|---|
| LFMM | RDA | LFMM | RDA | |||
| 2396_47 | 3.20 | 0.65 | ||||
| 3036_41 | 2.95 | −0.53 | ||||
| 3036_120 | 2.79 | 3.24 | 0.52 | |||
| 3037_48 | 3.24 | −0.49 | ||||
| 3038_100 | 3.12 | 3.36 | −0.54 | 3.29 | −0.47 | |
| 3039_81 | 3.24 | 3.36 | 0.54 | 3.28 | 0.47 | |
| 3053_67 | 4.39 | 3.00 | −0.88 | |||
| 3228_68 | 2.99 | −0.49 | ||||
| 4122_74 | 3.65 | 2.71 | −0.58 | |||
| 4256_106 | 3.18 | 3.14 | −0.54 | |||
| 5855_28 | 2.80 | −0.41 | ||||
| 6505_59 | 3.59 | 2.99 | 0.68 | |||
| 7409_33 | 4.73 | 3.70 | −0.71 | |||
| 7546_102 | 3.16 | 0.54 | ||||
| 9844_61 | 3.35 | 2.82 | 0.65 | |||
| 13059_16 | 2.96 | −0.64 | ||||
Minus log10 P values for LFMM and RDA tests.
Discussion
Colonization History and Demographic Bottlenecks
Several previous studies based on molecular markers have found evidence for the ancestral status of African populations of Aedes aegypti for all pantropical populations (Powell and Tabachnick 2013; Brown et al. 2014; Gloria-Soria et al. 2016). Our results support the single out-of-Africa origin as previously reported, with genetic differentiation between the Caribbean and African populations (SBE) higher (average FST 0.18) than between Caribbean populations (average FST 0.07). Furthermore, the African individuals had a high number of singletons, and the genetic diversity was much lower in all Caribbean populations than in the African one.
The reduction of genetic diversity in invasive populations could indicate a founder effect during the introduction of the species in the Caribbean. Demographic changes in populations, such as bottlenecks, are not only expected to reduce population genetic diversity but also to change the allele frequency spectrum (Maruyama and Fuerst 1985). Here, the SFS also supports a founder event in Caribbean populations, as shown by the low number of rare variants. The strongest evidence for a bottleneck during the introduction of Ae. aegypti is the severe decrease in effective population size ∼300 years ago. The first suspected dengue-like epidemics were reported in 1635 in Martinique and Guadeloupe suggesting that its vector had been introduced in the Caribbean during the 17th century (Tabachnick 1991; Urdaneta-Marquez and Failloux 2011; San Martín et al. 2012; Powell and Tabachnick 2013; Weaver and Lecuit 2015). However, it is difficult to attribute these outbreaks to dengue without a detailed clinical picture. Our results suggest that Ae. aegypti populations established in the Caribbean at the beginning of the 18th century. This establishment could be related to the intensification of the triangular trade between France, West Africa (Senegal, Benin), and French Antilles after the creation of the “Compagnie du Sénégal” in 1673. This date of establishment of the species in the Caribbean is for the first time inferred from molecular markers, and needs to be confirmed by a larger sampling in this region.
The stairway plot method applied to infer the demographic history of populations also detected a more recent bottleneck. Although the precise dates remain uncertain, the pattern of variation in effective population size coincides with the discovery of the role of Ae. aegypti in yellow fever immediately followed by vector control measures that took place at the turn of the 20th century during the construction of the Panama canal, and with an extensive use of insecticides by humans together with the rise of resistance mutations in Aedes populations during the second part of 20th century. The lowest genetic diversity was recorded in CAY (Cayenne, Guyane), the most urbanized locality sampled. This low diversity was not expected as Cayenne offers many breeding opportunities for Ae. aegypti and experiences recurrent dengue epidemics sustained by huge mosquito populations. This locality also exhibits the highest specific FST, which is indicative of a recent bottleneck in this population (reduced effective population size). This bottleneck could be related to the eradication campaign against Ae. aegypti in the American continent, back in the 1950s (PAHO 1997) or to the contemporary intense insecticide applications in this city, resulting in highly resistant populations to the most used insecticide, deltamethrin. However in GEO (St-Georges, Guyane), populations are also highly resistant to deltamethrin and still maintain a higher genetic diversity than in CAY. Furthermore, these two Guyane localities are genetically distinct (ML tree and cluster analyses). The road linking Cayenne and St-Georges is recent, and St-Georges is geographically much closer and mainly connected to Brazil, where Ae. aegypti populations are large and genetically diversified (Monteiro et al. 2014). Recurrent migration from Brazil might mitigate the genetic diversity lost due to intense insecticide spraying.
The reappearance of Ae. aegypti in areas declared free of the species occurred as soon as control had been relaxed in the 1970s (Wallis et al. 1983). The establishment of new populations in Guyane is probably due to the recolonization from neighboring countries. The present phylogenetic relationships show that the two populations sampled in Guyane diverged from Caribbean populations. Because eradication has failed in the Antilles (PAHO 1997), these populations could have acted as bridgehead populations for the recolonization of Guyane, as already suggested for southern Brazil (Linss et al. 2014; Monteiro et al. 2014). Unraveling the origin of recently established populations in Guyane however requires comparing genetic similarities with samples from other countries of South America, for example, from Brazil and Venezuela (Monteiro et al. 2014), and from other locations worldwide.
Patterns of Genetic Diversity and Geographical Scale of Differentiation in the Antilles
The level of genetic differentiation among the four islands sampled in the Lesser Antilles estimated by AMOVA was low but significant, and spatial patterns of variation indicate a high genetic structure. Bayesian clustering of individuals revealed that North islands, Martinique, and Guadeloupe formed three distinct clusters, with a significant pattern of isolation by distance, indicating gene flow between neighboring populations. Furthermore, the ML phylogram shows that St-Martin and St-Barthélemy (North islands) are separated in two different groups. This geographical differentiation is linked to the presence of the sea that restricts gene flow between regions, subjecting within-region groups of populations to genetic drift. The genetic diversity in all the Antilles was comparable. However, at small spatial scale (within islands), we found evidence for the role of human movement and habitat conditions in shaping genetic diversity and the level of genetic differentiation between populations.
The two localities sampled in St-Martin present contrasting patterns of genetic diversity, Oyster Pond population being genetically more diversified than Sandy Ground population. Oyster Pond is a residential area, with a high-income socio-economic neighborhood as compared with Sandy Ground, the suburb of the largest city of the island, Marigot. The contrasted socio-economic status of the two sampled localities could influence habitat availability, with different population dynamics in distinct socio-economic urban environments (LaDeau et al. 2015).
Populations from Martinique and Guadeloupe have comparable levels of genetic diversity and appear to be connected as shown by the poorly resolved phylogeny for samples from these two islands. These two highly touristic destinations are daily connected by flight and boat, and have been connected for a long time, being the first lands discovered by Christopher Columbus as early as 1493 and were both heavily involved in the slave trade (17th and 18th centuries). Furthermore evidence for contemporary exchange of migrants was found as two samples from Baie-Mahault, the only industrial locality in Guadeloupe with the international airport and port, were genetically close to Martinique samples (DAPC results). In Martinique, the inhabited islet Îlet Long, 10 km from the main island, is less diversified (with higher specific FST) than any other populations in Martinique, reflecting limited connectivity with other populations, and allelic loss through genetic drift associated to small effective population size. The genetic diversity pattern observed in Martinique is consistent with previous work based on 6 microsatellites and 319 transcriptomic SNPs (Marcombe et al. 2013) conducted in the same localities. In Guadeloupe, the island La Désirade, 30 km from the main island, is the least diversified population, with the highest specific FST, and all samples cluster together in the phylogenetic tree, indicating genetic distinctiveness from the main island. Although La Désirade is nowadays daily connected with the main island by boat, this was not the case back in the 1990s, where the lack of infrastructure (no water supply) prevented the development of tourism.
Signatures of Selection in the Genome of Ae. aegypti
In this study, we conducted two different local adaptation analyses. First, we focused on populations from the Antilles and Guyane and we used FST based outlier tests (BayeScan) to detect loci more differentiated than expected by chance across populations. These loci are putatively under ongoing divergent selection, but this method gives no indication of which selective pressure is at play. Second, we conducted genome–environment association tests (LFMM and multivariate RDA) on the whole data set (including the African population) to decipher which environmental factor has the main impact on local adaptation in Caribbean populations. BayeScan identified twelve loci more differentiated than expected under neutral distribution. Five of them were within or close to annotated genes, coding for one cytochrome P450, one voltage-gated sodium channel and three transcription factors. The cytochrome P450 gene family is well known to be involved in insecticide metabolism in mosquitoes (David et al. 2013). Here, the cytochrome P450 identified (CYP9J22) has been shown to be upregulated in two pyrethroid-resistant Caribbean populations (Cuba and Grand Cayman) (Bariami et al. 2012), suggesting it is involved in metabolic resistance to pyrethroids (including deltamethrin). Mutations in the voltage-gated sodium channel (gene AAEL006019 on supercontig 1.186) cause a resistance phenotype to pyrethroid insecticides (including deltamethrin) known as knockdown resistance or kdr, that have been extensively studied, including in Caribbean populations that show high frequency for the kdr mutation (>80%, Marcombe et al. 2012). Among the ddRAD markers used in this study, four were located on supercontig 1.186, including one marker <6,000 bp from AAEL06019 gene (marker 3328, supplementary table S2, Supplementary Material online). However, BayeScan identified none of these loci as outlier. One possible explanation is a nearly fixed kdr mutation in all invasive populations studied. Indeed, the marker 3328 was polymorphic only in the African samples (not included in the BayeScan analysis); loss of polymorphism is consistent with a selective sweep in the kdr genomic region in the Caribbean populations. Interestingly, another voltage-gated sodium channel gene (AAEL013788) was detected as outlier in the present study. This gene was shown to be down-regulated in imidacloprid-resistant strains (Riaz et al. 2013), and up-regulated upon bacterial (Colpitts et al. 2011) or viral infection (Choi et al. 2012), in domestic versus forest populations, and in human versus animal fed mosquitoes (McBride et al. 2014). This gene involved in the nervous influx appears to be central in many aspects of the mosquito biology and represents a good putative candidate for being under human-induced selective pressure. Among SNPs detected as FST-outliers (BayeScan), most are probably not directly involved in adaptation, since genomic variation rather than transcriptomic functional variation was screened. Our primary objective was not to identify precisely the genes under selection, but to identify the main selective factors (climatic vs. human-induced selective pressures) driving local adaptation in these anthropophilic invasive populations.
Overall association tests, geography had a prominent influence on genetic variation as compared with environmental factors. Geography explained ∼12% of total genetic variation (AMOVA and RDA results). However, when geography was controlled for (partial RDA), anthropic and climatic factors explained 8% and 6% of the residual variance respectively. Both LFMM and RDA identified seven SNPs associated with deltamethrin resistance and three loci associated with human density, but only one locus associated to precipitation, and none to temperature, suggesting that anthropic factors are more important than climatic factors in selecting specific regions of the genome. At the study scale, the mean annual temperature varied little, ranging from 26.0 °C (in St-Pierre, Martinique) to 26.8 °C (in Sandy Ground, St-Martin), but even small changes in larval breeding temperature or in adult rearing conditions were shown to induce changes in the physiology, immune condition, development time, fecundity, and survival of mosquitoes (Beserra et al. 2009; Goindin et al. 2015; Yee et al. 2017). Total precipitation was more variable than temperature across sites, ranging from 959 mm (in St-Barthélemy, tropical climate) up to 3,505 mm (in St-Georges, Guyane, equatorial climate). However, these climatic conditions do not appear to induce strong adaptive differentiation across sampled localities. All the samples were collected in domestic breeding sites and the presence of water containers suitable for breeding are presumably available all year round independently of the local precipitation level. This mitigating effect of man on mosquito habitat may explain why more loci were associated with human-induced pressures than with climatic factors, together with the fact that the two anthropogenic variables analyzed were more variable across sites than the two climatic variables.
Combining the results from different methods can reduce false discovery rates (de Villemereuil et al. 2014) and help to detect loci under strong selection (Lotterhos and Whitlock 2015; Nadeau et al. 2016). Although LFMM and RDA were partly concordant, they detected loci that were not identified as outliers by BayeScan. There are several explanations to the absence of overlap between these methods. First, the geographical scale was different as BayeScan was conducted only on Caribbean populations (14 populations and 3,530 SNPs) while RDA and LFMM also included the African population but was conducted on a SNP subset excluding missing data (15 populations and 578 SNPs). Second, the four environmental variables tested only accounted for 18.2% of total genetic variation (RDA analysis), and other nonstudied factors might be involved in shaping patterns of genetic variability among populations that were detected by BayeScan. For exemple, Temephos and Bti (Bacillus thuringiensis israelensis) are two insecticides currently used against mosquitoes in the Caribbean region that were shown to act as selective agents (Paris et al. 2010; Marcombe et al. 2013). Crop pesticides and various pollutants were also shown to induce metabolic responses in mosquito and represent other potential selective factors (Marcombe et al. 2012; Poupardin et al. 2012; Nkya et al. 2014). Third, inconstancies between different methods have already been observed (de Villemereuil et al. 2014; Lotterhos and Whitlock 2015; Nadeau et al. 2016) and could be attributed to confounding variation of demographic history and adaptive patterns (Lotterhos and Whitlock 2015), which makes difficult to separate neutral from selected loci. Furthermore, Nadeau et al. (2016) showed small overlap between methods depending on the neutral population structure correction. Indeed, overcorrection and undercorrection could lead to increase the rate of false negatives and false positives respectively. Here, we applied a correction for population genetic structure with K = 6, representing the geographical areas considered in this study, but using other K values (K = 5 or 7) had no impact on the results (not shown). SNPs that are not shared across methods should not be discarded entirely as simulations showed that loci under weak selection are often detected by only one method (Lotterhos and Whitlock 2015). Altogether, our results highlight the difficulty to accurately detect signatures of local adaptation using genetic-environment association tests when there is collinearity between environmental variation and neutral population structure (Frichot et al. 2015). Indeed, populations from Guyane present distinct genetic patterns at some loci, which may be due to drift (accentuated by the recent history of insecticide treatments) or to selection by deltamethrin intensive use, but most likely the two factors acting concomitantly.
Conclusion
The evolutionary history of Ae. aegypti since its introduction in the Caribbean is a complex mixture of population contractions and expansions in link with human movement and spatio-temporal changes in insecticides use. Our study showed that the establishment of Ae. aegypti in the Caribbean is recent and has been subjected to several bottlenecks since its introduction. Even slight changes in insecticide doses could induce changes in the strength and direction of selection, and selective pressures other than the insecticides used for mosquito control can affect the resistance allele dynamics (Milesi et al. 2016). We attempted to disentangle signatures of local adaptation in Caribbean populations that have different demographic histories. We identified a set of SNPs that could be involved in local adaptation in response to human impact. The adaptive potential of these bridgehead Caribbean populations is further illustrated by their evolutionary success in invading all the continents but Antarctica in the last decades, adapting to the large variety of human-shaped habitats encountered throughout the Americas and Eurasia. Further investigations are needed to evaluate the role of insecticide treatments and climatic conditions in shaping adaptive patterns, especially with a greater number of populations analyzed to maximize the range of environmental variation (Meirmans 2012).
Supplementary Material
Supplementary figures S1-S2, tables S1-S5 are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Sebastien Marcombe for collecting all samples from Martinique and for providing the SBE strain, and Isabelle Dusfour (Institut Pasteur Guyane) for providing the samples from Guyane. We thank Julien Renaud for help with the bioclimatic variables analyses. This work was funded by Centre National de la Recherche Scientifique, Universite Grenoble Alpes and Institut Pasteur Guadeloupe.
Literature Cited
- Allendorf FW, Lundquist LL.. 2003. Introduction: population biology, evolution, and control of invasive species. Conserv Biol. 17(1):24–30.http://dx.doi.org/10.1046/j.1523-1739.2003.02365.x [Google Scholar]
- Barbosa S, Black WC, Hastings I.. 2011. Challenges in estimating insecticide selection pressures from mosquito field data. PLoS Negl Trop Dis. 5(11):e1387.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariami V, Jones CM, Poupardin R, Vontas J, Ranson H.. 2012. Gene amplification, ABC transporters and Cytochrome P450s: unraveling the molecular basis of pyrethroid resistance in the dengue vector, Aedes aegypti. PLoS Negl Trop Dis. 6(6):e1692.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura SK, Gomez-Machorro C, Harker BW, et al. 2011. Global cross-talk of genes of the mosquito Aedes aegypti in response to dengue virus infection. PLoS Negl Trop Dis. 5(11):e1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett KL, Shija F, Linton Y-M, et al. 2016. Historical environmental change in Africa drives divergence and admixture of Aedes aegypti mosquitoes: a precursor to successful worldwide colonization? Mol Ecol. 25(17):4337–4354. [DOI] [PubMed] [Google Scholar]
- Beserra EB, Fernandes CRM, Silva SAD, et al. 2009. Effects of temperature on life cycle, thermal exigency and number of generations per year estimation of Aedes aegypti (Diptera, Culicidae). Iheringia Serie Zool. 99(2):142–148. [Google Scholar]
- Blackburn TM, Pyšek P, Bacher S, et al. 2011. A proposed unified framework for biological invasions. Trends Ecol Evol. 26(7):333–339. [DOI] [PubMed] [Google Scholar]
- Bradley BA, Blumenthal DM, Wilcove DS, Ziska LH.. 2010. Predicting plant invasions in an era of global change. Trends Ecol Evol. 25(5):310–318. [DOI] [PubMed] [Google Scholar]
- Brady OJ, Johansson MA, Guerra CA, et al. 2013. Modelling adult Aedes aegypti and Aedes albopictus survival at different temperatures in laboratory and field settings. Parasites Vectors 6:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JE, Evans BR, Zheng W, et al. 2014. Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito: evolutionary genetic history of Aedes aegypti. Evolution 68(2):514–525.http://dx.doi.org/10.1111/evo.12281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JE, McBride CS, Johnson P, et al. 2011. Worldwide patterns of genetic differentiation imply multiple “domestications” of Aedes aegypti, a major vector of human diseases. Proc R Soc B Biol Sci. 278(1717):2446–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant JE, Holmes EC, Barrett ADT.. 2007. Out of Africa: a molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 3(5):e75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo S. 1967. History of Aedes aegypti eradication in the Americas. Bull World Health Organ. 36(4):602–603. [PMC free article] [PubMed] [Google Scholar]
- Canton PE, Cancino-Rodezno A, Gill SS, et al. 2015. Transcriptional cellular responses in midgut tissue of Aedes aegypti larvae following intoxication with Cry11Aa toxin from Bacillus thuringiensis. BMC Genomics 16(1):1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canyon DV, Hii JLK, Müller R.. 1999. Adaptation of Aedes aegypti (Diptera: Culicidae) oviposition behavior in response to humidity and diet. J Insect Physiol. 45(10):959–964. [DOI] [PubMed] [Google Scholar]
- Capblancq T, Després L, Rioux D, et al. 2015. Hybridization promotes speciation in Coenonympha butterflies. Mol Ecol. 24(24):6209–6222. [DOI] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, et al. 2013. Stacks: an analysis tool set for population genomics. Mol Ecol. 22(11):3124–3140.http://dx.doi.org/10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi EC, Zhou H, Chen GK, et al. 2013. Genotype imputation via matrix completion. Genome Res. 23(3):509–518.http://dx.doi.org/10.1101/gr.145821.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y-J, Fuchs JF, Mayhew GF, et al. 2012. Tissue-enriched expression profiles in Aedes aegypti identify hemocyte-specific transcriptome responses to infection. Insect Biochem Mol Biol. 42(10):729–738.http://dx.doi.org/10.1016/j.ibmb.2012.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chown SL, Hodgins KA, Griffin PC, et al. 2015. Biological invasions, climate change and genomics. Evol Appl. 8(1):23–46.http://dx.doi.org/10.1111/eva.12234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colautti RI, Lau JA.. 2015. Contemporary evolution during invasion: evidence for differentiation, natural selection, and local adaptation. Mol Ecol. 24(9):1999–2017.http://dx.doi.org/10.1111/mec.13162 [DOI] [PubMed] [Google Scholar]
- Colpitts TM, Cox J, Vanlandingham DL, et al. 2011. Alterations in the Aedes aegypti transcriptome during infection with West Nile, dengue and yellow fever viruses. PLoS Pathog. 7(9):e1002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa EAP, Santos EM, Correia JC, et al. 2010. Impact of small variations in temperature and humidity on the reproductive activity and survival of Aedes aegypti (Diptera, Culicidae). Rev Bras Entomol. 54(3):488–493.http://dx.doi.org/10.1590/S0085-56262010000300021 [Google Scholar]
- Darling ES, Côté IM.. 2008. Quantifying the evidence for ecological synergies. Ecol Lett. 11(12):1278–1286. [DOI] [PubMed] [Google Scholar]
- David J-P, Ismail HM, Chandor-Proust A, et al. 2013. Role of cytochrome P450s in insecticide resistance: impact on the control of mosquito-borne diseases and use of insecticides on Earth. Philos Trans R Soc B Biol Sci. 368:20120429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villemereuil P, Frichot É, Bazin É, François O, Gaggiotti OE.. 2014. Genome scan methods against more complex models: when and how much should we trust them? Mol Ecol. 23(8):2006–2019. [DOI] [PubMed] [Google Scholar]
- Desjardins CA, Sanscrainte ND, Goldberg JM, et al. 2015. Contrasting host–pathogen interactions and genome evolution in two generalist and specialist microsporidian pathogens of mosquitoes. Nat Commun. 6:7121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle J, Doyle J.. 1987. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull. 19:11–15. [Google Scholar]
- Dusfour I, Zorrilla P, Guidez A, et al. 2015. Deltamethrin resistance mechanisms in Aedes aegypti populations from three french overseas territories worldwide. PloS Negl Trop Dis. 9(11):e0004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoup A, Guillemaud T.. 2010. Reconstructing routes of invasion using genetic data: why, how and so what?: Reconstructing routes of invasion. Mol Ecol. 19(19):4113–4130.http://dx.doi.org/10.1111/j.1365-294X.2010.04773.x [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S.. 2005. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online 1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Facon B, Genton B, Shykoff J, et al. 2006. A general eco-evolutionary framework for understanding bioinvasions. Trends Ecol Evol. 21(3):130–135.http://dx.doi.org/10.1016/j.tree.2005.10.012 [DOI] [PubMed] [Google Scholar]
- Faucon F, Dusfour I, Gaude T, et al. 2015. Identifying genomic changes associated with insecticide resistance in the dengue mosquito Aedes aegypti by deep targeted sequencing. Genome Res. 25(9):1347–1359.http://dx.doi.org/10.1101/gr.189225.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Gaggiotti O.. 2008. A Genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180(2):977–993.http://dx.doi.org/10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forester BR, Jones MR, Joost S, et al. 2016. Detecting spatial genetic signatures of local adaptation in heterogeneous landscapes. Mol Ecol. 25(1):104–120.http://dx.doi.org/10.1111/mec.13476 [DOI] [PubMed] [Google Scholar]
- Fouque F, Carinci R, Gaborit P, et al. 2006. Aedes aegypti survival and dengue transmission patterns in Guyane. J Vector Ecol. 31:390–399.http://dx.doi.org/10.3376/1081-1710(2006)31[390:AASADT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- François O, Martins H, Caye K, Schoville SD.. 2016. Controlling false discoveries in genome scans for selection. Mol Ecol. 25(2):454–469. [DOI] [PubMed] [Google Scholar]
- Frichot E, François O, O’Meara B.. 2015. LEA : an R package for landscape and ecological association studies. Methods Ecol Evol. 6(8):925–929. [Google Scholar]
- Frichot E, Mathieu F, Trouillon T, Bouchard G, François O.. 2014. Fast and efficient estimation of individual ancestry coefficients. Genetics 196(4):973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot E, Schoville SD, Bouchard G, François O.. 2013. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol. 30(7):1687–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot E, Schoville SD, de Villemereuil P, et al. 2015. Detecting adaptive evolution based on association with ecological gradients: orientation matters! Heredity 115(1):22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloria-Soria A, et al. 2016. Global genetic diversity of Aedes aegypti. Mol Ecol. 25(21):5377–5395.,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goindin D, Delannay C, Gelasse A, et al. 2017. Levels of insecticide resistance to deltamethrin, malathion, and temephos, and associated mechanisms in Aedes aegypti mosquitoes from the Guadeloupe and St Martin islands (French West Indies). Infect Dis Poverty 6(1):38.http://dx.doi.org/10.1186/s40249-017-0254-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goindin D, Delannay C, Ramdini C, et al. 2015. Parity and longevity of Aedes aegypti according to temperatures in controlled conditions and consequences on dengue transmission risks. PLoS One 10(8):e0135489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslee SC, Urban DL.. 2007. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Software 22(7):1–19. [Google Scholar]
- Gubler DJ, Clark GG.. 1994. Community-based integrated control of Aedes aegypti : a brief overview of current programs. Am J Trop Med Hyg. 50(6 Suppl):50–60. [DOI] [PubMed] [Google Scholar]
- Hijmans RJ, Cameron SE, Parra JL, et al. 2005. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 25(15):1965–1978.http://dx.doi.org/10.1002/joc.1276 [Google Scholar]
- Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405.http://dx.doi.org/10.1093/bioinformatics/btn129 [DOI] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F.. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDeau SL, Allan BF, Leisnham PT, et al. 2015. The ecological foundations of transmission potential and vector-borne disease in urban landscapes. Funct Ecol. 29:889–901.http://dx.doi.org/10.1111/1365-2435.12487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson Handley L-J, Estoup A, Evans DM, et al. 2011. Ecological genetics of invasive alien species. BioControl 56(4):409–428.http://dx.doi.org/10.1007/s10526-011-9386-2 [Google Scholar]
- Leache AD, Banbury BL, Felsenstein J, et al. 2015. Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst Biol. 64(6):1032–1047.http://dx.doi.org/10.1093/sysbio/syv053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CE. 2002. Evolutionary genetics of invasive species. Trends Ecol Evol. 17(8):386–391.http://dx.doi.org/10.1016/S0169-5347(02)02554-5 [Google Scholar]
- Legendre P, Legendre L.. 1998. Numerical ecology. 2nd English ed Amsterdam (The Netherlands: ): Elsevier. [Google Scholar]
- Linss JG, Brito L, Garcia G, et al. 2014. Distribution and dissemination of the Val1016Ile and Phe1534Cys Kdr mutations in Aedes aegypti Brazilian natural populations. Parasites Vectors 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Fu Y-X.. 2015. Exploring population size changes using SNP frequency spectra. Nat Genet. 47(5):555–559.http://dx.doi.org/10.1038/ng.3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotterhos KE, Whitlock MC.. 2015. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol Ecol. 24(5):1031–1046.http://dx.doi.org/10.1111/mec.13100 [DOI] [PubMed] [Google Scholar]
- Lounibos LP. 2002. Invasions by insect vectors of human disease. Annu Rev Entomol. 47:233–266.http://dx.doi.org/10.1146/annurev.ento.47.091201.145206 [DOI] [PubMed] [Google Scholar]
- Marcombe S, Mathieu RB, Pocquet N, et al. 2012. Insecticide resistance in the dengue vector Aedes aegypti from Martinique: distribution, mechanisms and relations with environmental factors. PLoS One 7(2):e30989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcombe S, Paris M, Paupy C, et al. 2013. Insecticide-driven patterns of genetic variation in the dengue vector Aedes aegypti in Martinique Island. PLoS One 8(10):e77857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Fuerst PA.. 1985. Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics 111:675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride CS, Baier F, Omondi AB, et al. 2014. Evolution of mosquito preference for humans linked to an odorant receptor. Nature 515(7526):222–227.http://dx.doi.org/10.1038/nature13964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch MA, Spear D, Kleynhans EJ, et al. 2012. Uncertainty in invasive alien species listing. Ecol Appl. 22(3):959–971.http://dx.doi.org/10.1890/11-1252.1 [DOI] [PubMed] [Google Scholar]
- Meirmans PG. 2012. The trouble with isolation by distance. Mol Ecol. 21(12):2839–2846.http://dx.doi.org/10.1111/j.1365-294X.2012.05578.x [DOI] [PubMed] [Google Scholar]
- Milesi P, Lenormand T, Lagneau C, et al. 2016. Relating fitness to long-term environmental variations in natura. Mol Ecol. 25(21):5483–5499.http://dx.doi.org/10.1111/mec.13855 [DOI] [PubMed] [Google Scholar]
- Monteiro FA, Shama R, Martins AJ, et al. 2014. Genetic diversity of Brazilian Aedes aegypti: patterns following an eradication program. PLoS Negl Trop Dis. 8(9):e3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Meirmans PG, Aitken SN, Ritland K, Isabel N. 2016. The challenge of separating signatures of local adaptation from those of isolation by distance and colonization history: The case of two white pines. Ecol Evol. 6(24):8649–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkya TE, Akhouayri I, Poupardin R, et al. 2014. Insecticide resistance mechanisms associated with different environments in the malaria vector Anopheles gambiae: a case study in Tanzania. Malaria J. 13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet GF, Friendly M, et al. 2017. vegan: community ecology package. R package version 2.4-3. https://CRAN.R-project.org/package=vegan
- Packer JG, Meyerson LA, Richardson DM, et al. 2017. Global networks for invasion science: benefits, challenges and guidelines. Biol Invasions 19(4):1081–1096.http://dx.doi.org/10.1007/s10530-016-1302-3 [Google Scholar]
- Pan American Health Organization (PAHO). 1997. Cooperation of the Pan American Health Organization in the health sector reform processes. Washington, DC.
- Paris M, Boyer S, Bonin A, et al. 2010. Genome scan in the mosquito Aedes rusticus: population structure and detection of positive selection after insecticide treatment: population structure at regional scale. Mol Ecol. 19(2):325–337.http://dx.doi.org/10.1111/j.1365-294X.2009.04437.x [DOI] [PubMed] [Google Scholar]
- Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A.. 2010. How many bootstrap replicates are necessary? J Comput Biol. 17(3):337–354. [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, et al. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7(5):e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupardin R, Riaz MA, Jones CM, et al. 2012. Do pollutants affect insecticide-driven gene selection in mosquitoes? Experimental evidence from transcriptomics. Aquat Toxicol. 114-115:49–57. [DOI] [PubMed] [Google Scholar]
- Poupardin R, Riaz MA, Vontas J, et al. 2010. Transcription profiling of eleven cytochrome P450s potentially involved in xenobiotic metabolism in the mosquito Aedes aegypti. Insect Mol Biol. 19(2):185–193.http://dx.doi.org/10.1111/j.1365-2583.2009.00967.x [DOI] [PubMed] [Google Scholar]
- Powell JR, Tabachnick WJ.. 2013. History of domestication and spread of Aedes aegypti – a review. Mem Inst Oswaldo Cruz 108(Suppl 1):11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentis PJ, Pavasovic A.. 2013. Understanding the genetic basis of invasiveness. Mol Ecol. 22(9):2366–2368.http://dx.doi.org/10.1111/mec.12277 [DOI] [PubMed] [Google Scholar]
- Rancès E, Ye YH, Woolfit M, et al. 2012. The relative importance of innate immune priming in Wolbachia-mediated dengue interference. PLoS Pathog. 8(2):e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F.. 1995. GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. J Hered. 86(3):248–249.http://dx.doi.org/10.1093/oxfordjournals.jhered.a111573 [Google Scholar]
- Riaz MA, Chandor-Proust A, Dauphin-Villemant C, et al. 2013. Molecular mechanisms associated with increased tolerance to the neonicotinoid insecticide imidacloprid in the dengue vector Aedes aegypti. Aquat Toxicol. 126:326–337. [DOI] [PubMed] [Google Scholar]
- Riaz MA, Poupardin R, Reynaud S, et al. 2009. Impact of glyphosate and benzo[a]pyrene on the tolerance of mosquito larvae to chemical insecticides. Role of detoxification genes in response to xenobiotics. Aquat Toxicol. 93(1):61–69.http://dx.doi.org/10.1016/j.aquatox.2009.03.005 [DOI] [PubMed] [Google Scholar]
- San Martín JL, Brathwaite Dick O, del Diego J, et al. 2012. The history of dengue outbreaks in the Americas. Am J Trop Med Hyg. 87(4):584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sax DF, Gaines, Steven D, et al. 2005. Species invasions: insights into ecology, evolution, and biogeography. Sunderland, MA, USA: Sinauer Associates. [Google Scholar]
- Simberloff D, Martin J-L, Genovesi P, et al. 2013. Impacts of biological invasions: what’s what and the way forward. Trends Ecol Evol. 28(1):58–66. [DOI] [PubMed] [Google Scholar]
- Soper FL. 1963. The elimination of urban yellow fever in the Americas through the eradication of Aedes aegypti. Am J Public Health Nations Health 53:7–16.http://dx.doi.org/10.2105/AJPH.53.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear D, Foxcroft LC, Bezuidenhout H, et al. 2013. Human population density explains alien species richness in protected areas. Biol Conserv. 159:137–147.http://dx.doi.org/10.1016/j.biocon.2012.11.022 [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313.http://dx.doi.org/10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R.. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 100(16):9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strode C, Wondji CS, David J-P, et al. 2008. Genomic analysis of detoxification genes in the mosquito Aedes aegypti. Insect Biochem Mol Biol. 38(1):113–123.http://dx.doi.org/10.1016/j.ibmb.2007.09.007 [DOI] [PubMed] [Google Scholar]
- Tabachnick WJ. 1991. Evolutionary genetics and arthropod-borne disease: the yellow fever mosquito. Am Entomol. 37:14–26.http://dx.doi.org/10.1093/ae/37.1.14 [Google Scholar]
- Tatem AJ, Hay SI, Rogers DJ.. 2006. Global traffic and disease vector dispersal. Proc Natl Acad Sci U S A. 103(16):6242–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treasure AM, Chown SL, Diez J.. 2014. Antagonistic effects of biological invasion and temperature change on body size of island ectotherms. Divers Distributions 20(2):202–213.http://dx.doi.org/10.1111/ddi.12153 [Google Scholar]
- Urdaneta-Marquez L, Failloux A-B.. 2011. Population genetic structure of Aedes aegypti, the principal vector of dengue viruses. Infect Genet Evol. 11(2):253–261.http://dx.doi.org/10.1016/j.meegid.2010.11.020 [DOI] [PubMed] [Google Scholar]
- Wallis GP, Tabachnick WJ, Powell JR.. 1983. Macrogeographic genetic variation in a human commensal: Aedes aegypti, the yellow fever mosquito. Genet Res. 41(03):241–258.http://dx.doi.org/10.1017/S0016672300021315 [DOI] [PubMed] [Google Scholar]
- Weaver SC, Lecuit M.. 2015. Chikungunya virus and the global spread of a mosquito-borne disease. N Engl J Med. 372(13):1231–1239. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC.. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370. [DOI] [PubMed] [Google Scholar]
- Wilfert L, Gadau J, Schmid-Hempel P.. 2007. Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity 98(4):189–197.http://dx.doi.org/10.1038/sj.hdy.6800950 [DOI] [PubMed] [Google Scholar]
- Wilson JRU, Dormontt EE, Prentis PJ, Lowe AJ, Richardson DM.. 2009. Something in the way you move: dispersal pathways affect invasion success. Trends Ecol Evol. 24(3):136–144. [DOI] [PubMed] [Google Scholar]
- Ye YH, Woolfit M, Rancès E, O’Neill SL, McGraw EA.. 2013. Wolbachia-associated bacterial protection in the mosquito Aedes aegypti. PLoS Negl Trop Dis. 7(8):e2362.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee DA, Ezeakacha NF, Abbott KC.. 2017. The interactive effects of photoperiod and future climate change may have negative consequences for a wide-spread invasive insect. Oikos 126(1):40–51.http://dx.doi.org/10.1111/oik.03635 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.