

Abstract
Undifferentiated pleomorphic sarcomas, myxofibrosarcomas, and malignant peripheral nerve sheath tumors are characterized by complex genomic characteristics and aggressive clinical behavior. Recent advances in the understanding of the pathogenesis of these tumors may allow for the development of more-effective innovative therapeutic strategies, including immunotherapies. This review describes the current knowledge of the epidemiology, clinical presentation, treatment, and pathogenesis of these tumors and highlights ongoing and future research.
INTRODUCTION
Soft tissue sarcoma (STS) can be classified into two main genetic types: sarcomas with simple genomics associated with a simple genomic alteration and sarcomas with complex genomics characterized by complex karyotypes and genomic profiles. The latter type mainly comprises leiomyosarcomas, malignant peripheral nerve sheath tumors (MPNSTs), and pleiomorphic tumors previously classified as malignant fibrous histiocytoma (MFH), which in fact correspond to myxofibrosarcomas (MFSs), pleiomorphic liposarcomas/rhabdomyosarcomas, and true undifferentiated pleiomorphic sarcomas (UPSs).1 This review provides an update of the current research related to UPS, MFS, and MPNST, with particular emphasis on emerging mechanisms of tumorigenesis and their potential therapeutic impact.
UPS
Definition and Epidemiologic and Clinical Features
First described in 1964, MFH represented a group of STSs considered to be of probable fibrohistiocytic or fibroblastic lineage.1 MFH was considered as the most common STS of late adult life and was associated with a metastatic rate of 30% to 35% and a 5-year overall survival rate of 65% to 70%.2 The exact origin of MFH was a matter of debate for years. Despite extensive immunohistochemical and ultrastructural studies, a true histiocytic origin was never proved. Moreover, several studies that carefully reanalyzed tumors initially diagnosed as MFH have shown that a significant subset of these tumors show a specific line of differentiation (lipogenic, neurogenic, myogenic, or nonsarcomatous),3-6 which led to a general consensus that MFH represents a wastebasket of many tumors that share some morphologic similarities. For this reason, MFH now is considered obsolete terminology and has been replaced by the term UPS.7 UPSs account for 10% of adult STSs and represent one of the most common STSs of older adults, with most occurring in patients between the ages of 50 and 70 years.8 Pediatric UPSs are rare. The etiology is unknown, although some of these tumors occur in a previously irradiated field. Clinical features of UPS are not specific. Most are deep-seated lesions that enlarge rapidly and painlessly. The most frequent location are the limbs followed by the trunk. Superficial lesions (subcutaneous) are rare.
Histologic Diagnosis
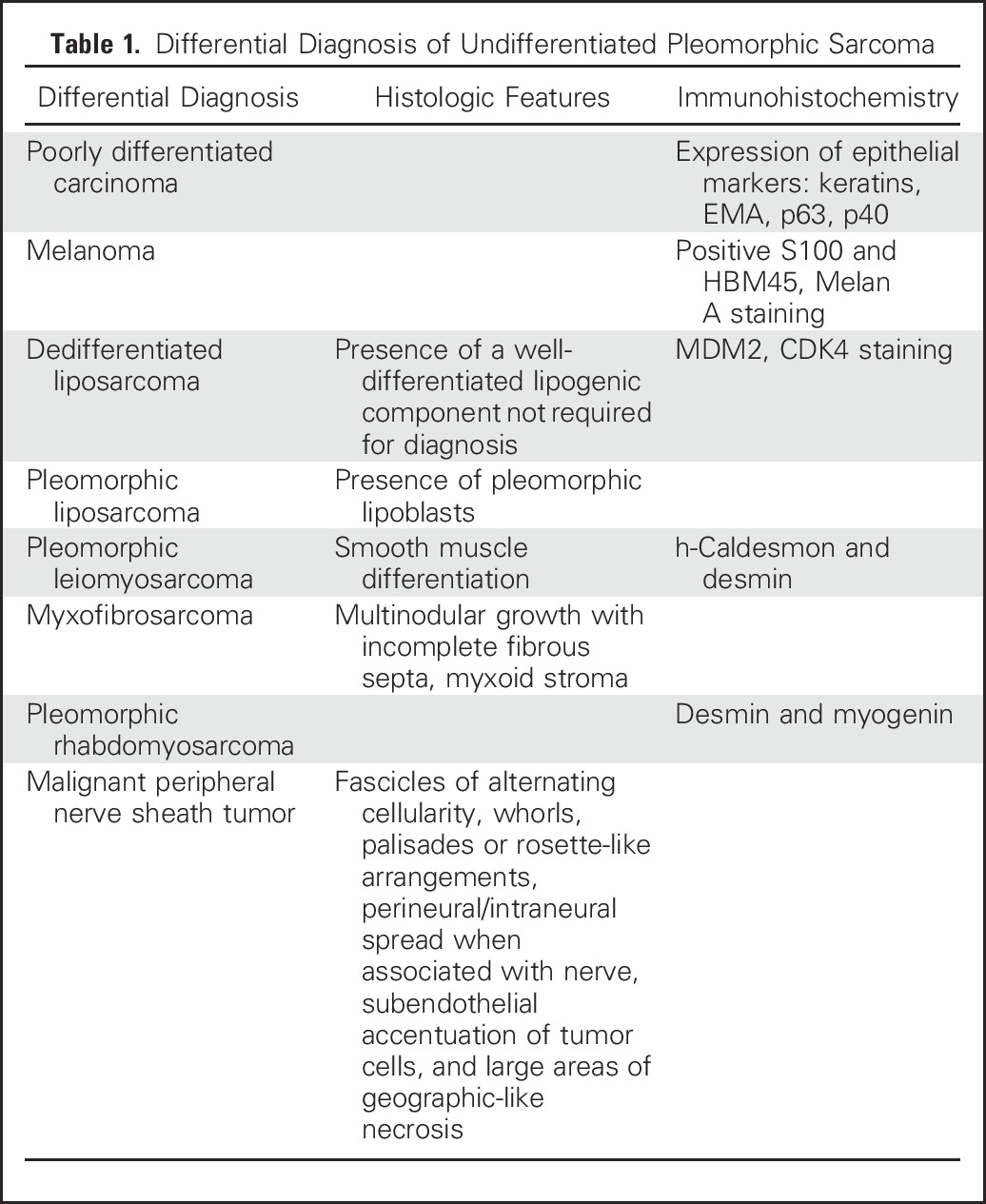
UPSs represent a diagnosis of exclusion on the basis of the absence of a specific line of differentiation after careful histologic examination and judicious use of ancillary techniques. The main differential diagnoses that must be considered and excluded before rendering the diagnosis of UPS are listed in Table 1.
Table 1.
Differential Diagnosis of Undifferentiated Pleomorphic Sarcoma
Molecular Features
Few studies have shed light on the genomics of MFH, and mechanisms of the oncogenesis in true UPS are unknown.9-13 The main series on MFH/UPS genomics has focused on comparative genomic hybridization or transcriptome analysis. MFH has been associated with inactivation of the RB1 gene or frequent loss of p53 function.14 However, the UPS mutational landscape has not been fully characterized. Gene expression profiling has been disappointing in detecting recurrent alterations in MFH. Baird et al11 identified two distinct groups of MFH: The first group carries a muscle profile that belongs to the ontologic family of motor activity, and the second group reveals an abundance of genes that belong to the ontologic family of immune activity and cell adhesion.
On the other hand, UPSs are the closest sarcoma state to mesenchymal stem cells.15 Activation of the Hedgehog and Notch signaling pathways have been described in cells with stem-like tumor-initiating potential from UPS.15 We and others have described overexpression of DKK1, an inhibitor of the Wnt canonical pathway, in UPS.16 Of note, activation of the β-catenin signaling has been found to be involved in preventing antitumor immunity in melanoma.17 However, correlation between these different pathways of interest and immunity involvement in tumor has not been assessed in UPS. Our group recently has shown that a small subset of UPS harbors fusions that involve the TRIO gene. The role of such a fusion gene in tumorigenesis remains to be defined, but these data suggest that aberrant regulation of Rho GTPase activities by guanine-nucleotide exchange factors may play an important role in UPS tumorigenesis.18 Recently, the VGLL3 and YAP1 genes which are TEAD cofactors in the Hippo signaling pathway have been shown to be overexpressed in a subset of UPS indicating a proportion of may be driven by the Hippo pathway.13
Management and Prognosis
As for other STSs, surgery performed by an experienced surgeon plus radiotherapy remain the cornerstone of treatment of nonmetastatic tumors. With the majority of these tumors being high grade, perioperative chemotherapy is an option, particularly with regard to the benefit in terms of overall survival recently demonstrated with this approach.19 In the metastatic setting, doxorubicin as a single agent or in combination with ifosfamide or olaratumab is likely to be the first choice of chemotherapy, which assumes that the findings with these treatments in other STSs also are applicable to UPS. In fact, despite being one the most common STS subtypes, few data are available in this specific subtype.20 Other drugs such as trabectedin, gemcitabine and docetaxel, and pazopanib also have shown activity in the advanced setting. Patients with advanced UPS have the worst outcome compared with other histologic STS subtypes.21
MFS
Definition and Epidemiologic and Clinical Features
MFS is one of the most common STSs in elderly patients, with the extremities and girdles being the most frequent sites.22 MFS affects men and women equally and usually presents as a slow-growing, often painless, deeply located tumor in 30% to 60% of patients. MFSs are characterized by a high risk of local recurrence related to a specific infiltrative growth pattern that leads deep-seated tumors to spread along vascular and fascial planes.23,24 Such patterns of tumor progression are well illustrated on magnetic resonance imaging (MRI), which frequently shows curvilinear extensions of high T2 signal with uptake of gadolinium-based contrast (Fig 1). As for other STSs, well-planned surgery is the cornerstone of treatment, and resection must be as large as possible because of the potential to spread over a considerable distance beyond the gross tumor margins. Insufficient tumor-free margins are associated with a high risk of local recurrence and, hence, a poor prognosis. Adjuvant radiotherapy usually is indicated to improve locoregional outcome.
Fig 1.

Magnetic resonance imaging (MRI) and histologic features of myxofibrosarcoma (MFS). (A) MFS of the left-side axillary region. Sagittal T2-weighted MRI shows three components: myxoid (red asterisk), fibrotic (blue arrow), and necrotic (black asterisk). (B) MFS of the right thigh. Axial T1-weighted MRI shows tumor spread along the fascial plane (green arrow, tail sign). (C) Macroscopic features of MFS. (D to F) Grossly, multinodular growth pattern and gelatinous, myxoid cut surface are shown. Microscopic features of MFS show the presence of three components: myxoid (red asterisk), fibrotic (arrow), and necrotic (black asterisk).
Molecular Features
Only a few studies of the molecular mechanisms involved in the tumorigenesis of MFS explain the specific patterns of progression. Molecular cytogenetic studies have identified a high level of genomic complexity with a recurrent amplification of the chromosome 5p region, the biologic significance of which is unknown.25 Okada et al26 recently analyzed the gene expression profiles of 64 primary high-grade MFSs and found that expression of ITGA10, which encodes integrin-α10, was significantly associated with worse outcomes. Knockdown of integrin-α10 specifically inhibited growth and survival and decreased RAC and AKT activation in MFS cells but not normal mesenchymal cells, which suggests that integrin-α10 signals through RAC and AKT in a tumor-specific manner. Given that the chromosome 5p amplicon contains TRIO, which encodes a guanine nucleotide exchange factor that activates RAC, and RICTOR, which encodes an essential subunit of mammalian target of rapamycin (mTOR) complex 2 that activates AKT, the authors hypothesized that integrin-α10 signals through TRIO and RICTOR to drive MFS tumorigenesis. Consistent with this hypothesis, integrin-α10 was found to physically interact with TRIO and RICTOR, and MFS cells require TRIO-dependent activation of RAC and RICTOR-dependent activation of AKT. Of note, pharmacologic inhibition of either RAC or mTOR reduce MFS growth in vivo, and simultaneous targeting of both RAC and mTOR lead to synergistic antitumor activity. These results indicate that high-grade MFS depends on integrin-α10 signaling through TRIO and RICTOR and provide insight into the etiology of this complex genomic tumor type and a rationale for clinical testing of inhibitors of such pathways in this disease.
MPNST
Definition and Epidemiologic and Clinical Features
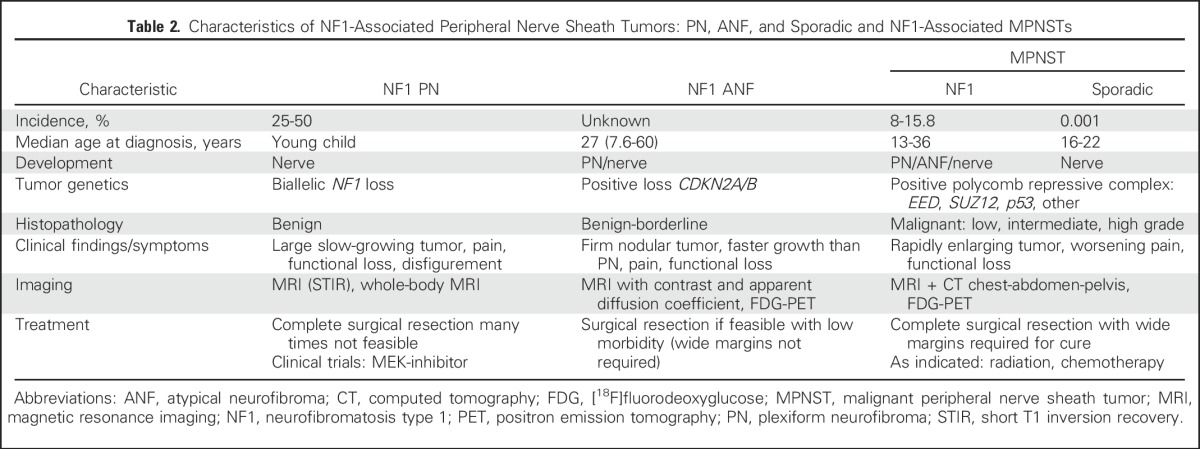
MPNSTs account for 4% of STSs and are characterized by a high risk of recurrence and poor outcome.27-29 One half of MPNSTs arise in individuals with neurofibromatosis type 1 (NF1), an autosomal disorder with an incidence of 1:3,000. The lifetime incidence of MPNST in NF1 is 15.8%,30 and MPNST is the leading cause of death in NF1. Complete surgical resection with wide negative margins is the only potentially curative therapy for MPNST.29 Advances in the understanding of the natural history of peripheral nerve sheath tumors in NF1 and the molecular pathogenesis of MPNST (Table 2) have resulted in the development of consensus research priorities and treatment strategies poised to accelerate the development of effective therapies for and prevention of MPNST.31,32
Table 2.
Characteristics of NF1-Associated Peripheral Nerve Sheath Tumors: PN, ANF, and Sporadic and NF1-Associated MPNSTs
Natural History of Peripheral Nerve Sheath Tumors and Importance of Atypical Neurofibromas as MPNST Precursor Lesions
Many MPNSTs in NF1 arise in preexisting, histologically benign, inoperable plexiform neurofibromas (PNs),33 which occur in up to 50% of individuals with NF1.34 PNs typically are diagnosed in young children and demonstrate the most rapid growth at an early age.35 Progressive growth of PNs can result in the development of substantial morbidities, such as disfigurement, pain, and functional impairment.36,37 The diagnosis of MPNST in this setting can be challenging because clinical findings and symptoms can overlap between PN and MPNST. The recent identification of atypical neurofibromas (ANFs) as precursor lesions to MPNST on the basis of loss of CDKN2A/B in ANF and MPNST but not in PN is a critical advance in the understanding of the pathogenesis of MPNST.38 This combined with the subsequent clinical and imaging characterization of ANFs have allowed for the development of strategies to prevent MPNST in NF1 (Higham et al, manuscript submitted for publication).39 Longitudinal use of whole-body short T1 inversion recovery MRI with volumetric MRI analysis in patients with NF1 and PN has allowed for the detection of distinct nodular lesions, many of which are ANFs on biopsy analysis (Fig 2). Unlike PNs, distinct nodular lesions and ANFs appear after early childhood, demonstrate a growth rate independent of age, and frequently grow faster than the surrounding typical PN.40 In addition, on imaging with [18F]fluorodeoxyglucose positron emission tomography, most ANFs are [18F]fluorodeoxyglucose avid in contrast to background uptake only in PN40 (Fig 2). The incidence of ANF in NF1 is not known, but the transformation of ANF to MPNST has been described, and in 63 patients with pathologically confirmed ANF, 19 (30%) had a history of MPNST; no regrowth of 57 completely resected ANFs was observed in this series (Higham et al, manuscript submitted for publication). Similar findings from Bernthal et al41 confirmed that recurrence of ANF or low-grade MPNST after resection with microscopically positive margins is an infrequent event. Resection of ANF without wide negative margins when feasible and with a low potential for morbidity, therefore, is recommended.32 Additional studies are needed to identify the incidence of ANF in NF1 and to develop better predictors (biomarkers) for the potential and timing of malignant transformation of ANF to MPNST, but given the absence of effective therapies for high-grade MPNST other than complete surgical resection with wide negative margins, the focal resection of ANFs may play a critical role in the prevention of MPNST. The definition of histopathologic features of nerve sheath tumors with regard to risk for malignant transformation becomes important in this setting, and a recent pathology consensus meeting proposed the name atypical neurofibromatous neoplasm of uncertain biologic potential for such tumors that require careful monitoring and, potentially, resection.42
Fig 2.

MRI and metabolic features of neurofibromas and malignant peripheral nerve sheath tumors (MPNSTs). (A) Axial (top panel) and coronal (bottom panel) short T1 inversion recovery magnetic resonance imaging of neck and chest plexiform neurofibroma in a female teenager with neurofibromatosis type 1. Neck pain for several months was attributed to stress and a heavy backpack. Development and progressive enlargement of a distinct nodular lesion were found in the neck and upper chest (arrows). In addition, growth of an anterior neck nodular lesion was found (arrowhead). [18F]fluorodeoxyglucose positron emission tomography at age 18 years demonstrated [18F]fluorodeoxyglucose avidity of the two nodular lesions. Biopsy of the deep nodular lesion (arrow) at age 18 years confirmed intermediate-grade MPNST. Surgical resection of MPNST and the anterior neck nodular lesion confirmed atypical neurofibroma (ANF). (B) Volume measurements of the deep and anterior neck nodular lesions over time showed parallel lesion growth from ages 13 to 15 years. Accelerated growth of the deep nodular lesion subsequent to this is concerning for malignant transformation.
Pathogenesis of MPNST
Mutations in TP53 and other genomic changes in addition to NF1 loss had been previously described in sporadic and NF1-associated MPNST.29 The more recent identification of somatic mutations in SUZ12 and EED, which encode for components of the polycomb repressive complex 2 (PRC2), provides new directions for developing MPNST therapies that target epigenetic mechanisms and bromodomain inhibition. In addition, the complete loss of trimethylated histone 3 lysine 27 (H3K27me3) as a result of SUZ12 ablation can be detected by immunohistochemistry as a novel marker for sporadic and NF1 MPNST.32
Preclinical and Clinical Trials for MPNST
To date, no clinical trial with targeted agents for MPNST has demonstrated substantial tumor shrinkage or prolongation in progression-free survival.31 Neurofibromas and MPNSTs are rich in macrophages, which provide the opportunity for targeted therapy. On the basis of preclinical studies in MPNST models, an early clinical trial that combined PLX3397, an oral small-molecule inhibitor of CSF1 and KIT, in combination with the mTOR inhibitor sirolimus was developed and is ongoing (ClinicalTrials.gov identifier: NCT02584647).43
With the goal to rationally select and prioritize promising agents for clinical trials, genetically engineered mouse models (GEMMs) of NF1-related neurofibromas and MPNST have been used to conduct preclinical trials through the NF Therapeutic Consortium. In a GEMM of NF1 neurofibroma,44 MEK inhibitors were the first targeted agents to result in consistent neurofibroma shrinkage,45 and this finding also was made in a clinical trial of the MEK inhibitor selumetinib for children with PN, where 71% of patients experienced a partial response with PN volume reduction.46 Preclinical trials in an MPNST GEMM (Nf1−/+;Trp53−/+ cis mice) have identified the first active combinations of targeted agents, which are being translated to the clinic.47,48 These models have the advantage of allowing evaluation of the microenvironment and immune infiltrate in tumors and, thus, assessment of the potential utility of immunotherapy for MPNST. Additional models that may reflect the pathogenesis of MPNST more closely, such as loss of SUZ12, have been developed.
Progress made in the understanding of NF1-related tumors, including MPNST, is largely the result of effective preclinical-clinical collaborations and of committed support of research through the Department of Defense and other sources.31 An effort to prospectively collect larger numbers of MPNST samples, clinical and imaging data, and blood samples for biomarker studies for comprehensive analyses is ongoing and will accelerate progress.32
MOLECULAR SIGNATURE OF ANEUPLOID STS: PROGNOSTIC AND PREDICTIVE RELEVANCE
Genomic instability is a hallmark of all human tumors49,50 and can be related to gene mutations, gene copy number alterations, structural chromosomal abnormalities such as translocations, telomere dysfunction, and whole-chromosome aneuploidy that later results in chromosome instability.50 Several diagnostic and prognostic signatures that characterize specific sarcoma subgroups have been reported.51 A common conclusion of these studies is that a correlation exists between cell pleomorphism and genomic complexity. For example, the most common adult sarcomas have complex karyotypes and pleomorphic histology, whereas sarcomas with chromosomal translocations often display a nonpleomorphic histology. Our research group identified and validated a 67-gene signature of chromosome instability that predicts metastasis in individuals with no translocation-related STSs, including undifferentiated sarcomas, leiomyosarcomas, and dedifferentiated liposarcomas.52,53 This signature (named complexity index in sarcomas [CINSARC]) was more reliable than histologic grading to predict metastasis-free survival in these tumors. Many of the genes identified encode for proteins involved in mitosis, cytokinesis, mitotic checkpoint, the cell cycle, and DNA repair. We also have shown that the same signature could predict clinical outcome in synovial sarcomas, the most frequent translocation-related STS.5
The correlation between CINSARC scores and response to chemotherapy in STS remains to be investigated. Of note, all the genes of the CINSARC signature are involved in the same biologic process (ie, control of chromosome integrity). Besides its prognostic value, chromosomal instability is associated with altered cytotoxic response.54 With regard to anthracyclines, chromosomal instability has been associated with improved response in tumor models such as breast cancer.55,56 We have observed a similar positive correlation between drug sensitivity and karyotypic complexity and heterogeneity in our panel of 25 sarcomas cell lines (unpublished data), which suggests that the CINSARC signature also has a predictive value in terms of response to chemotherapy. An ongoing prospective study aims to assess this hypothesis (ClinicalTrials.gov identifier: NCT02789384).
IMMUNOTHERAPY OF SARCOMAS WITH COMPLEX GENOMICS
Historically, sarcomas were the first tumor model for which immunotherapy was suggested as a relevant therapeutic strategy.57-59 The higher incidence of sarcoma in patients who are immunocompromised also supports the relevance of targeting the immune system in this disease.60,61
Programmed death-1 (PD-1) normally is expressed on the surface of activated T cells and suppresses unwanted or excessive immune responses, including autoimmune reactions. Its ligand PD-L1 can be expressed by various cells, including macrophages and tumor cells. The PD-1/PD-L1 interaction is a major pathway hijacked by tumors to suppress immune control. Several studies have assessed the expression of PD-L1 in sarcomas. Contradictory results have been published because of the limited number of patients included in the series, the multiplicity of assay methods and the reagents used, and the type of tumor material (tissue microarray v whole-tissue section) analyzed.62-64 Altogether, these studies have shown a heterogenous level of tumor-infiltrating lymphocytes by histologic subtype and a relative low incidence of PD-L1 expression compared with other solid tumors not > 10%.64,65 Of note, T-cell infiltration and PD-L1 expression were found to be higher in sarcomas with complex genomics and particularly in UPS than in other STSs.66,67 A correlation with the CINSARC signature also has been observed, with a high level of PD-L1 expression being more frequent in tumors with a high level of expression of signature genes.68 PD-1 inhibition in STSs and GI stromal tumors has limited activity in STSs, with objective response rates between 3% and 14% in two recently reported studies.68,69 The majority of objective responses have been observed in PD-L1–expressing UPS. We have recently reported that a high proportion of STS tumors are prominently infiltrated by CD163+ macrophages, which favor the M2 phenotype known to play a role in immunosuppression69 (Fig 3). Of note, we have observed that these tumor-associated macrophages expressed IDO1,69 which suggests that this pathway could preferentially contribute to the immunosuppressive phenotype of these cells and be an important mechanism of the primary resistance to PD-1 inhibition besides limited effector T-cell infiltration into the tumor, lack of PD-1/PD-L1 expression by the tumor, and tumor-infiltrating immune cells observed in this study.
Fig 3.

Massive infiltration by CD63/CD168+ macrophages in undifferentiated pleomorphic sarcoma.
In conclusion, although the outcomes for UPS, MFS, and MPNSTs have not changed, substantial advances in the understanding of the natural history and pathogenesis of these tumors have been made. These advances are being translated into trials with targeted therapies and provide hope for the identification of active therapies for STSs with specific histologies. Future research that combines treatment modalities, including immunotherapy, are warranted.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Biology and Management of Undifferentiated Pleomorphic Sarcoma, Myxofibrosarcoma, and Malignant Peripheral Nerve Sheath Tumors: State of the Art and Perspectives
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Brigitte C. Widemann
No relationship to disclose
Antoine Italiano
Research Funding: PharmaMar, GlaxoSmithKline, Novartis, MSD, AstraZeneca
Travel, Accommodations, Expenses: Roche, Novartis, Bayer AG, PharmaMar
REFERENCES
- 1.O’Brien JE, Stout AP: Malignant fibrous xanthomas. Cancer 17:1445-1455, 1964 [DOI] [PubMed] [Google Scholar]
- 2.Le Doussal V, Coindre JM, Leroux A, et al. : Prognostic factors for patients with localized primary malignant fibrous histiocytoma: A multicenter study of 216 patients with multivariate analysis. Cancer 77:1823-1830, 1996 [DOI] [PubMed] [Google Scholar]
- 3.Coindre JM, Mariani O, Chibon F, et al. : Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: A review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod Pathol 16:256-262, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Fletcher CD: Pleomorphic malignant fibrous histiocytoma: Fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am J Surg Pathol 16:213-228, 1992 [PubMed] [Google Scholar]
- 5.Fletcher CD, Gustafson P, Rydholm A, et al. : Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: Prognostic relevance of subclassification. J Clin Oncol 19:3045-3050, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Oda Y, Tamiya S, Oshiro Y, et al. : Reassessment and clinicopathological prognostic factors of malignant fibrous histiocytoma of soft parts. Pathol Int 52:595-606, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Fletcher CDM, Bridge JA, Hogendoorn P, et al (eds): WHO Classification of Tumours of Soft Tissue and Bone (ed 4). Lyon, France, IARC Press, 2013. [Google Scholar]
- 8.Henderson MT, Hollmig ST: Malignant fibrous histiocytoma: Changing perceptions and management challenges. J Am Acad Dermatol 67:1335-1341, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Gibault L, Pérot G, Chibon F, et al. : New insights in sarcoma oncogenesis: A comprehensive analysis of a large series of 160 soft tissue sarcomas with complex genomics. J Pathol 223:64-71, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Carneiro A, Francis P, Bendahl PO, et al. : Indistinguishable genomic profiles and shared prognostic markers in undifferentiated pleomorphic sarcoma and leiomyosarcoma: Different sides of a single coin? Lab Invest 89:668-675, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Baird K, Davis S, Antonescu CR, et al. : Gene expression profiling of human sarcomas: Insights into sarcoma biology. Cancer Res 65:9226-9235, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Lee YF, John M, Edwards S, et al. : Molecular classification of synovial sarcomas, leiomyosarcomas and malignant fibrous histiocytomas by gene expression profiling. Br J Cancer 88:510-515, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abehouse A, Adebamowo C, Adebamowow SN, et al. : Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171:950-965.e28, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pérot G, Chibon F, Montero A, et al. : Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. Am J Pathol 177:2080-2090, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang CY, Wei Q, Han I, et al. : Hedgehog and Notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer Res 72:1013-1022, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Matushansky I, Hernando E, Socci ND, et al. : Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest 117:3248-3257, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spranger S, Bao R, Gajewski TF: Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523:231-235, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Delespaul L, Lesluyes T, Pérot G, et al. : Recurrent TRIO fusion in nontranslocation-related sarcomas. Clin Cancer Res 23:857-867, 2017 [DOI] [PubMed] [Google Scholar]
- 19.Gronchi A, Ferrari S, Quagliuolo V, et al. : Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): An international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet Oncol 18:812-822, 2017 [DOI] [PubMed] [Google Scholar]
- 20.Reichardt P: Soft tissue sarcomas, a look into the future: Different treatments for different subtypes. Future Oncol 10:s19-s27, 2014. (suppl 8) [DOI] [PubMed] [Google Scholar]
- 21.Savina M, Le Cesne A, Blay JY, et al. : Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: The METASARC observational study. BMC Med 15:78, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willems SM, Debiec-Rychter M, Szuhai K, et al. : Local recurrence of myxofibrosarcoma is associated with increase in tumour grade and cytogenetic aberrations, suggesting a multistep tumour progression model. Mod Pathol 19:407-416, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Kaya M, Wada T, Nagoya S, et al: MRI and histological evaluation of the infiltrative growth pattern of myxofibrosarcoma. Skelet Radiol 37:1085-1090, 2008. [DOI] [PubMed]
- 24. doi: 10.2214/AJR.05.1130. Waters B, Panicek DM, Lefkowitz RA, et al: Low-grade myxofibrosarcoma: CT and MRI patterns in recurrent disease. AJR Am J Roentgenol 188:W193-W198, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Idbaih A, Coindre JM, Derré J, et al. : Myxoid malignant fibrous histiocytoma and pleomorphic liposarcoma share very similar genomic imbalances. Lab Invest 85:176-181, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Okada T, Lee AY, Qin LX, et al. : Integrin-α10 dependency identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discov 6:1148-1165, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans DG, Baser ME, McGaughran J, et al. : Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39:311-314, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferner RE, Gutmann DH: International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res 62:1573-1577, 2002 [PubMed] [Google Scholar]
- 29.Farid M, Demicco EG, Garcia R, et al. : Malignant peripheral nerve sheath tumors. Oncologist 19:193-201, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uusitalo E, Rantanen M, Kallionpää RA, et al. : Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol 34:1978-1986, 2016 [DOI] [PubMed] [Google Scholar]
- 31. Kim A, Stewart DR, Reilly KM, et al: Malignant peripheral nerve sheath tumors state of the science: Leveraging clinical and biological insights into effective therapies. Sarcoma 2017:7429697, 2017. [DOI] [PMC free article] [PubMed]
- 32. doi: 10.1093/jnci/djx124. Reilly KM, Kim A, Blakeley J, et al: Neurofibromatosis type 1-associated MPNST state of the science: Outlining a research agenda for the future. J Natl Cancer Inst 109:djx124, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meany H, Widemann B, Ratner N: Malignant peripheral nerve sheath tumors: Prognostic and diagnostic markers and therapeutic targets, in Upadhyaya M, Cooper D (eds): Neurofibromatosis Type 1. Berlin, Germany, Springer, 2012, pp 445-467. [Google Scholar]
- 34.Mautner VF, Asuagbor FA, Dombi E, et al. : Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro-oncol 10:593-598, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dombi E, Solomon J, Gillespie AJ, et al. : NF1 plexiform neurofibroma growth rate by volumetric MRI: Relationship to age and body weight. Neurology 68:643-647, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Kim A, Gillespie A, Dombi E, et al. : Characteristics of children enrolled in treatment trials for NF1-related plexiform neurofibromas. Neurology 73:1273-1279, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen R, Kluwe L, Fuensterer C, et al: Plexiform neurofibromas in children with neurofibromatosis type 1: Frequency and associated clinical deficits. J Pediatr 159:652-655.e2, 2011. [DOI] [PubMed]
- 38.Beert E, Brems H, Daniëls B, et al. : Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 50:1021-1032, 2011 [DOI] [PubMed] [Google Scholar]
- 39.Meany H, Dombi E, Reynolds J, et al. : 18-Fluorodeoxyglucose-positron emission tomography (FDG-PET) evaluation of nodular lesions in patients with neurofibromatosis type 1 and plexiform neurofibromas (PN) or malignant peripheral nerve sheath tumors (MPNST). Pediatr Blood Cancer 60:59-64, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Akshintala S, Bhaumik S, Venkatesan A, et al: Identification of lesions concerning for transformation to malignant peripheral nerve sheath tumors (MPNST) in neurofibromatosis 1 (NF1). Radiological Society of North America, 2014. [Google Scholar]
- 41.Bernthal NM, Putnam A, Jones KB, et al. : The effect of surgical margins on outcomes for low grade MPNSTs and atypical neurofibroma. J Surg Oncol 110:813-816, 2014 [DOI] [PubMed] [Google Scholar]
- 42. doi: 10.1016/j.humpath.2017.05.010. Miettinen M, Antonescu C, Fletcher C, et al: Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol 67:1-10, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patwardhan PP, Surriga O, Beckman MJ, et al. : Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res 20:3146-3158, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu J, Williams JP, Rizvi TA, et al. : Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 13:105-116, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jessen WJ, Miller SJ, Jousma E, et al. : MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest 123:340-347, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dombi E, Baldwin A, Marcus LJ, et al. : Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med 375:2550-2560, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Raedt T, Walton Z, Yecies JL, et al. : Exploiting cancer cell vulnerabilities to develop a combination therapy for Ras-driven tumors. Cancer Cell 20:400-413, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malone CF, Fromm JA, Maertens O, et al. : Defining key signaling nodes and therapeutic biomarkers in NF1-mutant cancers. Cancer Discov 4:1062-1073, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon DJ, Resio B, Pellman D: Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13:189-203, 2012 [DOI] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA: Hallmarks of cancer: The next generation. Cell 144:646-674, 2011 [DOI] [PubMed] [Google Scholar]
- 51.Nielsen TO, West RB: Translating gene expression into clinical care: Sarcomas as a paradigm. J Clin Oncol 28:1796-1805, 2010 [DOI] [PubMed] [Google Scholar]
- 52.Chibon F, Lagarde P, Salas S, et al. : Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med 16:781-787, 2010 [DOI] [PubMed] [Google Scholar]
- 53.Lagarde P, Przybyl J, Brulard C, et al. : Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31:608-615, 2013 [DOI] [PubMed] [Google Scholar]
- 54.Roschke AV, Lababidi S, Tonon G, et al. : Karyotypic “state” as a potential determinant for anticancer drug discovery. Proc Natl Acad Sci U S A 102:2964-2969, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munro AF, Twelves C, Thomas JS, et al. : Chromosome instability and benefit from adjuvant anthracyclines in breast cancer. Br J Cancer 107:71-74, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ignatiadis M, Singhal SK, Desmedt C, et al. : Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: A pooled analysis. J Clin Oncol 30:1996-2004, 2012 [DOI] [PubMed] [Google Scholar]
- 57. Curiel TJ: Historical perspectives and current trends in cancer immunotherapy, in Curiel TJ (ed): Cancer Immunotherapy, Paradigms, Practice and Promise. New York, NY, Springer, 2013, pp 3-16. [Google Scholar]
- 58.Coley WB: II. Contribution to the knowledge of sarcoma. Ann Surg 14:199-220, 1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiemann B, Starnes CO: Coley’s toxins, tumor necrosis factor and cancer research: A historical perspective. Pharmacol Ther 64:529-564, 1994 [DOI] [PubMed] [Google Scholar]
- 60.Gatti RA, Good RA: Occurrence of malignancy in immunodeficiency diseases. A literature review. Cancer 28:89-98, 1971 [DOI] [PubMed] [Google Scholar]
- 61.Penn I: Sarcomas in organ allograft recipients. Transplantation 60:1485-1491, 1995 [DOI] [PubMed] [Google Scholar]
- 62.Kim C, Kim EK, Jung H, et al. : Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer 16:434, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim JR, Moon YJ, Kwon KS, et al: Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One 8:e82870, 2013. [DOI] [PMC free article] [PubMed]
- 64.D’Angelo SP, Shoushtari AN, Agaram NP, et al. : Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol 46:357-365, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Toulmonde M, Adam J, Bessede A, et al: Integrative assessment of expression and prognostic value of PDL1, IDO, and kynurenine in 371 primary soft tissue sarcomas with genomic complexity. J Clin Oncol 34, 2016 (suppl; abstr 11008)
- 66. Bertucci F, Finetti P, Perrot D, et al: PDL1 expression is a poor-prognosis factor in soft-tissue sarcomas. Oncoimmunology 6:e1278100, 2017. [DOI] [PMC free article] [PubMed]
- 67. doi: 10.1002/cncr.30726. Pollack SM, He Q, Yearley JH, et al: T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 123:3291-3304, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tawbi HA, Burgess M, Bolejack V, et al: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18:1493-1501, 2017. [DOI] [PMC free article] [PubMed]
- 69. doi: 10.1001/jamaoncol.2017.1617. Toulmonde M, Penel N, Adam J, et al: Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: A phase 2 clinical trial. JAMA Oncol 10.1001/jamaoncol.2017 [epub ahead of print on June 29, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]