

Abstract
Natural endogenously occurring peptides exhibit desirable medicinal properties, but are often limited in application by rapid proteolysis and inadequate membrane permeability. However, editing naturally occurring peptide sequences to develop peptidomimetic analogs created a promising class of therapeutics that can augment or inhibit molecular interactions. Here, we discuss a variety of chemical modifications, including L to D isomerization, cyclization, and unnatural amino acid substitution, as well as design strategies, such as attachment to cell-penetrating peptides, which are used to develop peptidomimetics. We also provide examples of approved peptidomimetics and discuss several compounds in clinical trials.
Introduction
The first treatment of a child with diabetes with insulin purified from bovine pancreas during the early 1920s proved the concept that human diseases can be treated by endogenously occurring peptides [1]. Initially, treating human disease with peptides was particularly difficult because of the challenges of peptide synthesis, which included a reduced yield and purity, low stability, and complicated delivery methods. Nevertheless, various approaches were developed to address these challenges. Two notable milestones include the advent of solid-phase peptide synthesis during the 1960s and progress in technological developments for peptide purification, such as HPLC. These accomplishments in peptide chemistry resulted in the ability to synthesize synthetic peptides more feasibly and to purify them more cost-effectively. Furthermore, strategies to optimize lead compounds from an endogenous peptide have become also more widely available [2].
An estimated 60 peptides are approved for human use worldwide, with an additional 140 peptidic therapeutics in different stages of clinical development, many of which are based upon natural endogenous sequences [3–5]. In 2010, four peptide drugs each produced global annual sales greater than US$1 billion [6]. The market share for peptide therapeutics continues to grow at an exponential rate, and is predicted to increase to an estimated US$25.4 billion by 2018 [5]. Yet, there remain significant challenges associated with natural endogenous peptide sequences as therapeutics [7]. These molecules are generally metabolically unstable and, consequently, have low oral bioavailability and membrane permeability. Linear peptides often have short half-lives (minutes to several hours), which might not be sufficient to deliver effective drug concentrations to the target tissue. A short plasma half-life is mainly the result of enzymatic degradation in the blood, liver, and kidney, as well as rapid renal clearance. Given that oral administration leads to degradation by the digestive system, many peptide therapeutics are administered via intraperitoneal or intravenous injection.
The limitations associated with therapeutic peptides can be overcome by modifying existing peptide sequences to create peptidomimetics, that is, peptides containing modified amino acids or chemical alterations [8]. Targeted changes in peptide sequences using various strategies improve therapeutic potential and reduce the adverse effects of peptides currently used. For example, desmopressin is a result of a one amino acid modification to vasopressin; this alteration eliminates the effect of raising blood pressure. A comprehensive list of peptides and peptidomimetics used in the clinic was previously reviewed [9]. Although some novel therapeutic peptides have been approved since, such as teduglutide and lixisenatide, in this review, we focus on how the modification of natural linear peptides, such as incorporation of nonproteinogenic amino acids or cyclization, can help overcome the hurdles associated with therapeutic peptides for treating human diseases. We selected examples for this review to demonstrate these concepts. We discuss optimization of peptides in nature and in the laboratory, as well as how these approaches have been leveraged to produce peptidomimetic therapeutics.
Sources of peptidomimetics
Peptides are often ligands for endogenous receptors, making peptidomimetics based on endogenous peptides promising lead compounds compared with antibodies or small molecules. Typically, endogenous precursor peptides are cleaved to generate biologically active metabolites. This natural processing can be taken advantage of to develop therapeutics based primarily on the active form of endogenous peptides. For example, octreotide is a truncated analog of somatostatin, with one disulfide bridge in which the D-Trp stabilizes a β-turn conformation around the D-Trp-Lys region, producing a similar effect to the parent compound [10,11]. Moreover, a truncated form of parathyroid hormone, teriparatide, retains only the anabolic effects of the parent compound. This is particularly useful because the catabolic effects of the endogenous peptide can lead to osteoporosis [10].
Although peptides are traditionally considered to regulate cellular responses as receptor ligands, recent evidence indicates that natural peptides also can act on intracellular targets to control cellular events [12]. Peptides are formed continuously inside cells as a result of protein degradation and turnover during cellular homeostasis. Most of these degradation products are further hydrolyzed into individual amino acids and reused for protein synthesis or intermediate metabolism. However, some intracellular peptides in mammalian cells escape complete degradation and instead regulate cellular protein kinases and phosphatases [12]. Identifying more biologically active intracellular peptides could reveal important normal and pathological cellular processes, and suggest new therapeutic candidates.
Peptides from non-mammalian species are also a valuable source of peptidomimetics. With less homology, these peptides are often unrecognized or less effectively degraded by human peptidases and proteases. Exenatide, the homolog of glucagon-like peptide-1 (GLP-1), was discovered in the venomous Gila monster lizard and shares approximately 50% homology with human GLP-1. The differences in the amino acid sequences of exenatide and GLP-1 allow the drug to be more metabolically stable, resulting in a longer half-life (2.5 hours) compared with human GLP-1 (4–5 min). Recent efforts have led to the introduction of an implantable depot to allow for extended administration of the drug [13].
L to D amino acid substitution
Most amino acids in nature exist in the L-amino acid configuration. The D-amino acid configuration, although infrequent for humans, is present in other species, such as cone snails, frogs, and bacteria [14]. Incorporation of D-amino acids into biologically active peptides can improve metabolic stability compared with L-amino acids given that few human enzymes hydrolyze peptide bonds with D-amino acids [14]. Here, we provide several examples of L to D amino acid substitution in the design of therapeutic peptides.
Vasopressin, or antidiuretic hormone (ADH), regulates plasma osmolality and volume to control water retention and blood vessel constriction, while also acting as a neurotransmitter with effects in the brain (Table 2) [15]. Several synthetic derivatives of vasopressin are available, including terlipressin, arginine vasopressin (argipressin), and desmopressin. In particular, desmopressin (1-desamino-8-D-arginine vasopressin) is a synthetic replacement for vasopressin, where the first amino acid has been deaminated and L-Arg has been substituted with D-Arg at the eighth position (Fig. 1). However, the modifications in desmopressin altered the receptor subtype specificity of the peptide, which affected the clinical indication. Desmopressin is mainly used to treat diabetes insipidus, nocturnal enuresis (bedwetting), and bleeding abnormalities. The antidiuretic activity of desmopressin is tenfold greater than vasopressin, but its activity as a vasoconstrictor is 1500-fold less [16]. As a result, desmopressin is slowly metabolized compared with vasopressin, and can be administered nasally, intravenously, or as an oral or sublingual tablet [16].
TABLE 2.
Peptidomimetic therapeutics mentioned in this review; primary function and target organ are indicated for each compound
| Target organ (indicated by number) | Peptidomimetic | Function |
|---|---|---|
| 1 | Pramlintide | Analog of amylin to treat T1DM and T2DM |
| 2 | Saralasin | Angiotensin II receptor agonist to reduce hypertension |
| 3 | Icatibant | Bradykinin 2 receptor antagonist to treat acute attacks of hereditary angioedema |
| 4 | Ziconotide | Blocks N-type voltage-gated calcium channels to treat chronic pain |
| 5 | Cyclosporine | Interferes with T cell activity to induce immunosuppression |
| 6 | Eptifibatide | Inhibits glycoprotein IIb/IIIa inhibitor to prevent blood clots |
| 7 | Vasopressin/desmopressin/terlipressin | Regulates plasma osmolality, cardiovascular, and brain functions |
| 8 | Linaclotide | Agonist that mimics guanylin and uroguanylin to treat chronic idiopathic constipation and constipation-predominant irritable bowel syndrome |
| 9 | Carbetocin | Agonist of peripheral oxytocin receptors to treat postpartum hemorrhage and bleeding |
| 10 | Daptomycin | Binds to Gram-positive bacterial membranes to inhibit growth |
| 11 | Afamelanotide | Analog of α-MSH to treat erythropoietic protoporphyria |
FIGURE 1.
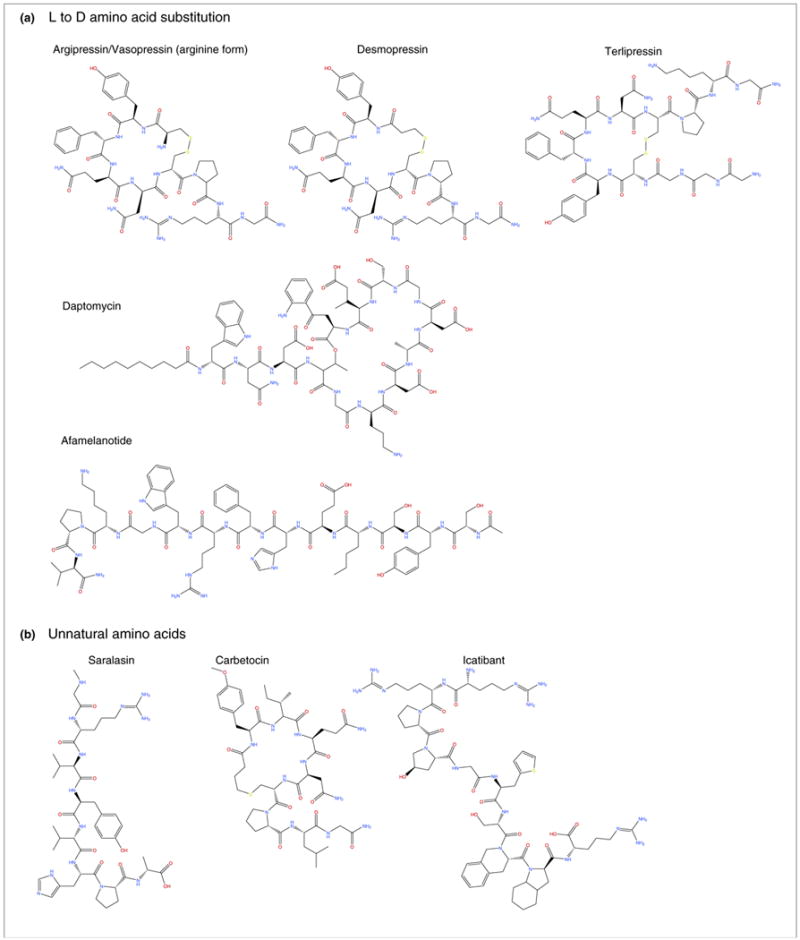
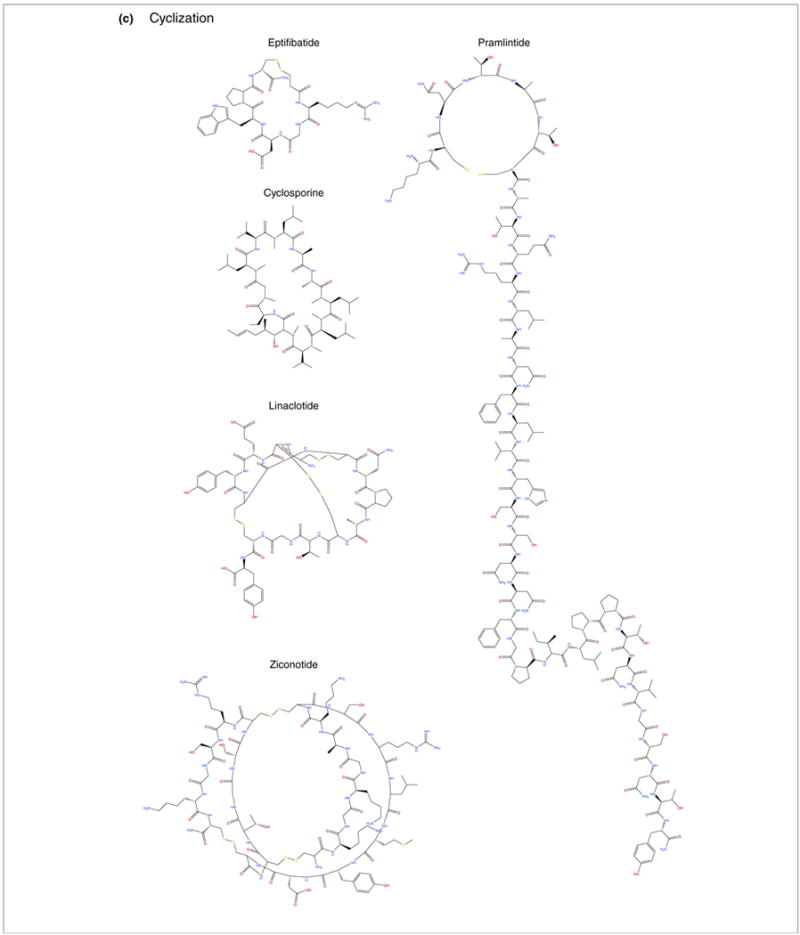
Peptidomimetic therapeutics approved for American, European, and/or Japanese pharmaceutical markets that were mentioned in this study (also see Table 1, main text). Compounds are organized by corresponding modifications: (a) L to D amino acid substitution; (b) incorporation of unnatural amino acids; and (c) and cyclization.
Another example of L to D amino acid substitution can be found in the development of antibiotic agents. Daptomycin is an antibacterial lipopeptide used to treat systemic infections caused by Gram-positive bacteria via a mechanism of action distinct from that of any other antibiotic and that is effective at any phase of bacterial growth. The compound was discovered during the late 1980s and was approved by the US Food and Drug Administration (FDA) in September 2003. The drug comprises 13 amino acids, ten of which are arranged in a cyclic fashion, and three on an exocyclic tail. Daptomycin contains several unnatural amino acids, such as D-alanine and D-serine, as well as the unusual amino acids L-kynurenine and L-3-methylglutamic acid. In addition, the N terminus tryptophan residue is coupled to decanoic acid, a medium-chain (C10) fatty acid. The peptide binds to the bacterial cell membrane, causing rapid disruption because of calcium efflux and inhibiting the synthesis of DNA, RNA, protein, and lipoteichoic acid. This results in rapid concentration-dependent bacterial cell death with a prolonged postantibiotic effect (Table 2) [17].
Afamelanotide is a synthetic analog of the naturally occurring melanocortin peptide alpha-melanocyte stimulating hormone (α-MSH), which can lead to skin tanning. The compound contains an L to D amino acid substitution of phenylalanine at position seven. Afamelanotide induces the production of darkening dermal pigment through melanogenesis, reducing sun damage to light-exposed skin. The drug was approved in Europe in October of 2014 to treat erythropoietic protoporphyria (EPP), a rare disease that causes intolerance to light. The plasma half-life can be as long as 100 min, which is much greater than the 20-min half-life of the natural peptide [18].
Unnatural amino acids
Incorporation of unnatural amino acids into peptide analogs or peptidomimetics can improve metabolic stability, while maintaining selectivity and potency of therapeutic peptides. In particular, unnatural amino acids support resistance to proteolysis, while also providing increased oral bioavailability [14]. Synthetic unnatural amino acids or modified noncanonical amino acids from other organisms can be substituted for canonical amino acids (the 20 proteinogenic amino acids encoded in the human body) to augment peptide sequences. For alternative applications, site-specific incorporation of an immunogenic unnatural amino acid into an antigenic peptide can overcome physiological mechanisms of tolerance to elicit an immune response analogous to vaccination (e.g. to combat self-tolerance in cancer or neurodegenerative disease) [19,20]. Unnatural amino acids are also valuable tools in chemical biology to support bio-orthogonal chemistry (reactions within organisms that do not affect their native biological processes) to accomplish selective cross-linking [21], and to assemble antibody–drug conjugates [22].
Unnatural amino acid-containing peptidomimetics can be developed by single-residue scanning of a parent sequence or scaffold [14], computational and rational design, or phenotype and/or genotype-linked display from phage [23] or yeast [24]. N-Alkyl amino acids (e.g. N-methylation) are useful for preventing proteolysis, or to scan for amino acid positions of functional interest [25,26]. Their position determines the extent of proteolytic protection, and affects the conformational freedom of the peptide backbone as well as adjacent side-chain residues [14]. Moreover, N-alkyl amino acids can be used in structure–activity relation (SAR) studies to perform N-alkyl scans. Each backbone NH can be sequentially and systematically alkylated, after which evaluating the biological consequence of each variant can help identify pharmacologically relevant residues. Another approach is illustrated by the Forcefield_NCAA platform (NCAA, noncanonical amino acid), which utilizes assisted model building with energy refinement (AMBER) force-field charge parameters derived by restrained electrostatic potential (RESP) fitting [27]. This platform was introduced in a study to help inform rational incorporation of unnatural amino acids and to predict pharmacological activity for derivatives of compstatin, which is a cyclic tridecapeptide inhibitor of complement component C3 originally derived from phage display libraries [27]. Using advances in molecular evolution and display, investigators rapidly selected high-affinity candidates from their library of 147 noncanonical and unnatural amino acids. Phage display is well established, and there are also other techniques, such as yeast display, based on combinatorial molecular display or in vitro compartmentalization, which are amenable to the display of unnatural amino acids [23].
Many therapeutic peptides containing unnatural amino acids and/or noncanonical amino acids target an aspect of cardiovascular homeostasis. Saralasin is an angiotensin II analog and partial agonist of the angiotensin II receptor, and is used to treat hypertension [28]. Saralasin contains substitutions to the angiotensin II sequence, including sarcosine at position 1, valine at position 5, and alanine at position 8 [28]. Substitution of the unnatural amino acids sarcosine supported resistance to aminopeptidase degradation, which improved the bioactivity of the compound. Icatibant is another peptidomimetic therapeutic where resistance to degradation is supported by incorporation of unnatural amino acids, including hydroxyproline, L-2-thienylalanine, tetrahydroisoquinolinecarboxylic acid, and octahydroindolecarboxylic acid [29]. The drug is a competitive bradykinin 2 receptor antagonist, used to treat acute attacks of hereditary angioedema in patients with C1-esterase inhibitor deficiency [30].
Unnatural amino acids are also utilized in carbetocin, which is a cyclic eight-amino acid analog of oxytocin used to limit postpartum hemorrhage and bleeding (particularly during a cesarean section), primarily through targeting peripheral oxytocin receptors. The carbetocin peptidomimetic contains methyltyrosine at position 2 and a thioether in place of a disulfide bond, which enhances its metabolic stability compared with earlier-generation lead compounds [31]. Overall, therapeutic peptides benefit widely from the incorporation of unnatural amino acids, especially to preserve metabolic stability.
Turn mimetics
The β-turn mimetic structure comprises i through i + 4 amino acid residues where Cαi and Cαi + 3 are equal to or less than 7 Å apart [32]. Importantly, the β-turn is one of three main recognition motifs (in addition to the α-helix and the β-strand) that govern protein–protein interactions (PPIs) and one of two main recognition motifs (in addition to the γ-turn) that mediate protein–G-protein-coupled receptor (GPCR) interactions [32]. More than 100 GPCRs (a common drug target) that recognize peptides are activated by β-turn motifs [33]. Taking advantage of this paradigm, Whitby and colleagues designed a β-turn mimetic library including compounds with canonical amino acids (Ala, Val, Leu, Ile, Ser, Thr, Met, Glu, Gln, Lys, His, Phe, Tyr, and Trp) as well as unnatural amino acids [4-chlorophenylalanine/Phe(4Cl), O-methyl tyrosine/TyrMe, homophenylalanine/HoPhe, homotyrosine/HoTyr, 4-chlorohomophenylalanine/HoPhe(4Cl), and naphthyl/Nap] to identify molecules that target opioid receptors [33]. The authors found a three-amino acid region in their β-turn mimetic scaffold that was crucial for target interactions, which informed high-affinity hits that bound the κ-opioid receptor (KOR) and included a basic molecule (Ki = 23 nM) and a nonbasic molecule (Ki = 390 nM) [33]. Other studies have also utilized unnatural amino acids in β-turn mimetics to target opioid receptors and identify by ‘turn can’ both agonists and antagonists for potential therapeutic applications [34].
Cyclization
Cyclic peptides are polypeptide chains with a cyclic ring structure. Depending on the desired cyclization site, there are several methods to arrange cyclic peptides, including head-to-tail, side chain-to-side chain, head-to-side chain, and side chain-to-tail cyclization. Several approaches have been used to generate cyclic peptides, such as backbone cyclization [35], peptide stapling [36], and native chemical ligation [37]. These structures can be formed with chemically stable bonds, such as an amide, lactone, ether, thioether, or disulfide bonds. Cyclization is often used to enhance conformational stability and improve biological activity compared to linear analogs. The resultant molecules are resistant to hydrolysis by peptidases because of conformational constraint and/or lack of amino and carboxyl termini. Cyclic peptides can support enhanced bioactivity as a result of their conformational rigidity, which decreases the entropy term of the Gibbs free energy to allow tighter binding of target molecules and receptor selectivity [38]. Several cyclic peptides are used commonly in the clinic, including gramicidin (a component of polysporin), the antibiotic vancomycin, and the immunosuppressant cyclosporine A. Here, we present several important examples that span various clinical indications.
Eptifibatide is a synthetic peptide derived from rattlesnake venom. The compound contains six amino acids, including a mercaptopropionyl unnatural amino acid located at the C terminus. The structure is a cyclic peptide with a bond between the cysteine and mercaptopropionyl residue, which supports greater stability than the parent peptide. Eptifibatide is an antiplatelet drug that functions as a glycoprotein IIb/IIIa inhibitor by preventing blood platelets from aggregating and forming blood clots. The drug is used to treat unstable angina (chest pain) and certain types of heart attack by intravenous injection (Tables 1 and 2) [39].
TABLE 1.
Peptidomimetic therapeutics mentioned in this review and approved for American, European, and/or Japanese pharmaceutical marketsa
| INNs | Brand names | Length | Sequences | Companies | Indications | Half-life |
|---|---|---|---|---|---|---|
| Daptomycin | Cubicin | 13 aa |
N-Decanoyl-Trp-Asp-Asp- threonylglycyl-ornithyl-Asp-D-alanyl- aspartylglycyl-D-seryl-threo-3-methyl- glutamyl-3-anthraniloyl-alanine[egr]1- lactone |
Cubist Pharmaceuticals | Antibiotic used to treat systemic and life-threatening infections caused by Gram-positive organisms | 8–9 hours |
| Afamelanotide | Scenesse | 13 aa | Ac-Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg- Trp-Gly-Lys-Pro-Val-NH2 |
Clinuvel Pharmaceuticals | Erythropoietic porphyries (EMEA and FDA orphan drug status, Phase 3) | 100 min |
| Eptifibatide | Integrilin | 7 aa | c[Mpa-homoArg-Gly-Asp-Trp-Pro-Cys]- NH2 |
Millennium Pharms, GSK, Schering-Plough | Acute coronary syndrome, unstable angina undergoing PCI | 2.5 hours |
| Cyclosporine | Neoral, Gengraf, Sandimmune, Arpimune Me, Graftin, Imusporin, Panimun Bioral | 11 aa | (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)- 30-Ethyl-33-[(1R,2R,4E)-1-hydroxy-2- methyl-4-hexen-1-yl]-6,9,18,24- tetraisobutyl-3,21-diisopropyl- 1,4,7,10,12,15,19,25,28-nonamethyl- 1,4,7,10,13,16,19,22,25,28,31- undecaazacyclotritriacontane- 2,5,8,11,14,17,20,23,26,29,32-undecone |
RGP life sciences, Ranbaxy laboratories, Cipla limited, Ecolity, Allergan, Novartis | Transplant (kidney, liver, and heart) rejection, rheumatoid arthritis, severe psoriasis | 5–18 hours |
| Linaclotide | Linzess and Constella | 16 aa | Cys-Cys-Glu-Tyr-Cys-Cys-Asp-Pro-Ala- Cys-Threonylglycyl-Cys-Tyr cyclo(1- 6),(2-10),(5-13)-tris(disulfide) |
Ironwood Pharmaceuticals | Irritable bowel syndrome (IBS) | 1.5 hours |
| Pramlintide | Symlin | 37 aa | Cyclo H-Lys-c[Cys-Asn-Thr-Ala-Thr-Cys]- Ala-Thr-Gln-Arg-Leu-Ala-Asn-Phe-Leu- Val-His-Ser-Ser-Asn-Asn-Phe-Gly-Pro-Ile- Leu-Pro-Pro-Thr-Asn-Val-Gly-Ser-Asn- Thr-Tyr]-NH2 |
Amylin Pharmaceuticals | T1DM, T2DM | 48 min |
| Ziconotide | Prialt | 25 aa | [Cys1-Cys16, Cys8-Cys20, Cys15-Cys25]- tricyclo H-[Cys1-Lys-Gly-Lys-Gly-Ala-Lys- Cys8-Ser-Arg-Leu-Met-Tyr-Asp-Cys15- Cys16-Thr-Gly-Ser-Cys20-Arg-Ser-Gly- Lys-Cys25]-NH2, acetate |
Elan Pharms | Severe chronic pain | 2.9–6.5 hours |
| Desmopressin acetate | DDAVP, Defirin, Desmopressin Acetate, Minirin, Minirinmelt, Octim, Stimate | 9 aa | c[Mpa-Tyr-Phe-Gln-Asn-Cys]-Pro-D-Arg- Gly-NH2, monoacetate trihydrate |
Apotex, Bausch & Lomb Pharms, Barr Labs, Behring, Ferring Pharms, Hospira, Pharmaceutique Noroit, Sanofi-Aventis, Teva | Central diabetes insipidus, nocturnal enuresis, nocturia, and stoppage of bleeding or hemorrhage in patients with hemophilia A | 158 min |
| Terlipressin acetate | Glypressin | 12 aa | H-Gly-Gly-Gly-c[Cys-Tyr-Phe-Gln-Asn- Cys]-Pro-Lys-Gly-NH2, acetate |
Ferring Pharmaceuticals | Bleeding esophageal varices | 50 min, 6 hours effective |
| Icatibant acetate | Firazyr | 10 aa | H-D-Arg-Arg-Pro-Hyp-Gly-Thi-Ser-D-Tic- Oic-Arg-OH, acetate |
Jerini AG | Hereditary angioedema | 1–2 hours |
| Carbetocin acetate | Duratocin, Lonactene, Pabal | 8 aa | c[Tyr(Me)-Ile-Gln-Asn-Cys((CH2)3CO2-)]- Pro-Leu-Gly-NH2, acetate |
Ferring Pharms | Prevention of uterine atony, control of postpartum bleeding or hemorrhage | 85–100 min |
Also see Fig. 1, main text.
Cyclosporine is an 11-amino acid cyclic peptide originally synthesized from Tolypocladium inflatum, and often used to prevent organ rejection in patients after liver, kidney, or heart transplants. The compound is hydrophobic and must be suspended in an emulsion because of its poor solubility in water, which results in a 3:1 equivalent oral:intravenous dose. Oral administration can be enabled by cyclization and encasement in a gelatin capsule to slow degradation, which is the preferred delivery route because of a higher incidence of anaphylactic reactions when given intravenously (Table 1 and Fig. 1) [40].
Linaclotide is a cyclic peptide comprising 14 amino acids, with three disulfide bonds. Linaclotide is an agonist that mimics the actions of endogenous guanylin (15-amino acid peptide) and uroguanylin (16-amino acid peptide), both of which activate the cell surface receptor for guanylate cyclase C (GC-C) and regulate electrolyte and water transport in intestinal and renal epithelia. The drug was approved by the FDA in August of 2012 for orally administered treatment of chronic idiopathic constipation and constipation-predominant irritable bowel syndrome (Tables 1 and 2) [41].
Pramlintide is a 37-amino acid cyclic polypeptide that differs structurally from human amylin by the replacement of alanine at position 25 as well as serine at positions 28 and 29, each with proline. The compound is an amylinomimetic, a functional analog of the naturally occurring pancreatic hormone amylin, which is released into the bloodstream in a similar pattern to insulin after eating. In addition to insulin, patients with diabetes are often deficient in amylin, which is normally active in the gastrointestinal and glucodynamic systems. Mimicking the activity of amylin, pramlintide can improve glycemic control by modulating the rate of gastric emptying, preventing postprandial rises in glucagon levels, and increasing satiety, thereby reducing caloric intake and potentiating weight loss. Pramlintide is used to treat both type 1 and 2 diabetes mellitus (T1DM and T2DM) via an injectable formulation (Table 1) [42].
Ziconotide is a 25-amino acid cyclic polypeptide with three disulfide bonds, which was derived from Conus magus (the cone snail). The compound is an analgesic agent used to treat severe and chronic pain. Ziconotide inhibits the release of neurochemicals associated with pain, including glutamate, calcitonin gene-related peptide, and substance P in the brain and spinal cord, by selectively inhibiting the N-type voltage-gated calcium channel (Tables 1 and 2) [43].
A rapidly emerging class of cyclic peptides is the stapled peptide, or hydrocarbon-stapled alpha-helical peptide. Staple peptides are locked into the bioactive conformation through a site-specific introduction of a chemical brace. The first stapled peptide was developed using ring-closing metathesis (RCM) on peptide antibiotics containing two alkenyl side chains. Since then, more stapled peptides have been developed [36]. In 2013, Aileron Therapeutics announced the successful completion of the first-in-human clinical trial of the stapled peptide drug ALRN-5281 for treatment of rare endocrine diseases, such as adult growth hormone deficiency and HIV lipodystrophy. A comprehensive review of the design of stapled peptides was published recently [44] and another review provides an excellent summary of stapled peptides and their applications to various human diseases, such as cancer, infection, and neurological disorders [45].
Peptide delivery
Peptides usually do not cross cellular membranes because the lipid bilayer comprises proteins and glycoproteins outwardly hydrophobic in nature. As a result, it is challenging for hydrophilic compounds, such as peptides, to permeate this barrier. One approach to overcome this limitation is to use cell-penetrating peptides (CPPs), also known as protein transduction domains, which can cross the cell membrane and deliver cargos with limited toxicity and without specific receptor recognition. CPPs were initially derived from hydrophilic proteins that can spontaneously cross cell membranes, first reported during the late 1980s. Currently, there are a variety of CPPs, most of which are positively charged short peptides (generally up to 30 residues), leading to over 25 clinical trials predominantly using a TAT-derived sequence [46].
Another important approach to deliver peptides and to extend their half-life is conjugation to fatty acids, as in the extended half-life of liraglutide compared with exenatide or native GLP-1. This modification allows the drug to bind albumin, thus protecting the peptide from degradation and slowing the release from albumin [47]. A longer-acting formulation of exenatide placed in a biomaterial matrix of poly-(D,L-lactide-co-glycolide) microspheres supports even slower release, thus enabling the agent to be injected subcutaneously only once per week (rather than twice daily) [48].
Additional promising peptide delivery methods include nasal, transmucosal, sublingual, transdermal, and topical administration [49,50]. These routes were initially complicated by low peptide permeability. However, for example, the transdermal delivery of peptides using electroporation of the outer skin can improve the delivery of some peptides [51]. This transient break in the skin layer allows the efficient delivery of peptides through a patch or pump [51]. Intranasal administration is effective for peptides <2000 kDa that can be absorbed through the nasal vascular bed, although intranasal delivery can be contraindicated for trans-sphenoidal hypophysectomy, or ineffective in the case of nasal congestion or mucosal atrophy.
Peptide drugs in development
Peptide and peptidomimetic therapeutics continue to attract increasing interest from academia and pharmaceutical industry. Peptides represent a family of potent and specific yet safe candidates for drug discovery and development. As more endogenous peptides are identified, characterized, and sequenced, additional therapeutic targets will emerge for clinical development. Many peptides recently discovered and characterized are presently being modified and developed for therapeutic use. We believe that many future peptidomimetic drugs will be derived from naturally occurring peptides. Modifications, such as local constraints, amino acid substitution, and cyclization, will help overcome many limitations. Moreover, alternative administration routes beyond injections will help broaden therapeutic applications.
There are several hundred novel therapeutic peptides in preclinical and clinical development, a few of which we cover here. Cenderitide is a peptide developed by combining native mammalian C-type natriuretic peptide and the C terminus of the dendroaspis natriuretic peptide isolated from venom of the green mamba. Cenderitide is the only dual natriuretic peptide receptor agonist that can act on the multiple disease processes with a role in negative outcomes associated with heart failure [52]. Cenderitide is being developed for outpatient treatment following hospitalization for acute heart failure, to be delivered continuously for up to 90 days using a subcutaneous infusion pump (presently in clinical trials).
Cilengitide is a cyclized penta-peptidomimetic designed to compete with the endogenous arginine-glycine-aspartic acid (RGD) peptide sequence that regulates integrin–ligand binding. Cilengitide selectively and potently blocks ligation of the αvβ3 and αvβ5 integrins, which are important for angiogenesis (forming new blood vessels) and other aspects of tumor development [53,54]. This compound is being developed for the treatment of glioblastoma and gliomas, where it could inhibit angiogenesis, tumor invasion, and proliferation (presently being evaluated in over 20 clinical trials). Although cilengitide had some promising results in a preliminary Phase 2 trial study based on efficacy and tumor delivery in patients with recurrent glioblastoma [55], several other studies, such as a Phase 2 trial investigating the efficacy and safety of combining cilengitide with temozolomide and procarbazine [56], did not yield positive results.
Szeto-Schiller (SS) peptides are cell permeable and highly charged tetrapeptides with a 3+ net charge at physiological pH. This charge results in selectivity for the inner mitochondrial membrane, independently of mitochondrial potential and without causing mitochondrial depolarization [57]. In isolated mitochondria, cultured cells and animal models, SS-31 (or MTP-131) selectively scavenges mitochondrial reactive oxygen species (ROS) and preserves mitochondrial function [58,59]. By decreasing mitochondrial ROS production, this compound has broad potential applications for the treatment of oxidative stress-related diseases, such as diabetic retinopathy, and is presently being evaluated in more than ten clinical trials [60].
Concluding remarks
Defining the human and nonhuman peptidome (all of the naturally occurring peptides) will allow investigators to identify new agents that regulate disease-mediating processes. Given that many PPIs are required for signal transduction, the generation of peptide and peptidomimetic inhibitors for these interactions can also provide novel drugs. Peptides were once undesirable pharmacologic agents because of their short half-life, inability to penetrate biological membranes, and generally limited bioavailability. Methods to increase peptide stability through the use of unnatural amino acids and to improve bioavailability through cyclization or cross-linking to membrane permeable peptides can overcome these limitations. Peptides produced endogenously by protein proteolysis and rationally designed peptide or peptidomimetic inhibitors of PPIs are likely to represent novel therapeutic leads for numerous unmet clinical needs.
Acknowledgments
This review was supported by NIH HL52141 (D.M.R.), NIH HL109212 (E.R.G.), Stanford University SPARK (N.Q.), and Stanford Graduate Fellowship (S.J.S.R.).
References
- 1.Banting FG, et al. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12:141–146. [PMC free article] [PubMed] [Google Scholar]
- 2.White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nat Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 3.Craik DJ, et al. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 4.Uhlig T, et al. The emergence of peptides in the pharmaceutical business: from exploration to exploitation. EuPA Open Proteom. 2014;4:58–69. [Google Scholar]
- 5.Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Sachdeva S. Peptides as ‘drugs’: the journey so far. Int J Pept Res Ther. 2016:1–12. [Google Scholar]
- 7.Otvos L, Jr, Wade JD. Current challenges in peptide-based drug discovery. Front Chem. 2014;2:62. doi: 10.3389/fchem.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bock JE, et al. Getting in shape: controlling peptide bioactivity and bioavailability using conformational constraints. ACS Chem Biol. 2013;8:488–499. doi: 10.1021/cb300515u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18:807–817. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Anthony L, Freda PU. From somatostatin to octreotide LAR: evolution of a somatostatin analogue. Curr Med Res Opin. 2009;25:2989–2999. doi: 10.1185/03007990903328959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ovadia O, et al. Improvement of drug-like properties of peptides: the somatostatin paradigm. Expert Opin Drug Discov. 2010;5:655–671. doi: 10.1517/17460441.2010.493935. [DOI] [PubMed] [Google Scholar]
- 12.Ferro ES, et al. Intracellullar peptides as putative natural regulators of protein interactions. J Neurochem. 2004;91:769–777. doi: 10.1111/j.1471-4159.2004.02757.x. [DOI] [PubMed] [Google Scholar]
- 13.Amiram M, et al. Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control. Proc Natl Acad Sci U S A. 2013;110:2792–2797. doi: 10.1073/pnas.1214518110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gentilucci L, et al. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr Pharm Des. 2010;16:3185–3203. doi: 10.2174/138161210793292555. [DOI] [PubMed] [Google Scholar]
- 15.Stoop R, et al. New opportunities in vasopressin and oxytocin research: a perspective from the amygdala. Annu Rev Neurosci. 2015;38:369–388. doi: 10.1146/annurev-neuro-071714-033904. [DOI] [PubMed] [Google Scholar]
- 16.Ozgonenel B, et al. How do you treat bleeding disorders with desmopressin? Postgrad Med J. 2007;83:159–163. doi: 10.1136/pgmj.2006.052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Ruiz A, et al. Daptomycin: an evidence-based review of its role in the treatment of Gram-positive infections. Infect Drug Resist. 2016;9:47–58. doi: 10.2147/IDR.S99046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim ES, Garnock-Jones KP. Afamelanotide: a review in erythropoietic protoporphyria. Am J Clin Dermatol. 2016;17:179–185. doi: 10.1007/s40257-016-0184-6. [DOI] [PubMed] [Google Scholar]
- 19.Grunewald J, et al. Mechanistic studies of the immunochemical termination of self-tolerance with unnatural amino acids. Proc Natl Acad Sci U S A. 2009;106:4337–4342. doi: 10.1073/pnas.0900507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roxin A, Zheng G. Flexible or fixed: a comparative review of linear and cyclic cancer-targeting peptides. Future Med Chem. 2012;4:1601–1618. doi: 10.4155/fmc.12.75. [DOI] [PubMed] [Google Scholar]
- 21.Furman JL, et al. A genetically encoded aza-Michael acceptor for covalent cross-linking of protein-receptor complexes. J Am Chem Soc. 2014;136:8411–8417. doi: 10.1021/ja502851h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hallam TJ, et al. Antibody conjugates with unnatural amino acids. Mol Pharm. 2015;12:1848–1862. doi: 10.1021/acs.molpharmaceut.5b00082. [DOI] [PubMed] [Google Scholar]
- 23.Galan A, et al. Library-based display technologies: where do we stand? Mol Biosyst. 2016;12:2342–2358. doi: 10.1039/c6mb00219f. [DOI] [PubMed] [Google Scholar]
- 24.Gietz RD. Yeast two-hybrid system screening. Methods Mol Biol. 2006;313:345–371. doi: 10.1385/1-59259-958-3:345. [DOI] [PubMed] [Google Scholar]
- 25.Kessler H. Peptide conformations 19. Conformation and biological-activity of cyclic-peptides. Angew Chem Int Ed Engl. 1982;21:512–523. [Google Scholar]
- 26.Chatterjee J, et al. N-methylation of peptides and proteins: an important element for modulating biological functions. Angew Chem Int Ed Engl. 2013;52:254–269. doi: 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]
- 27.Khoury GA, et al. Forcefield_NCAA: ab initio charge parameters to aid in the discovery and design of therapeutic proteins and peptides with unnatural amino acids and their application to complement inhibitors of the compstatin family. ACS Synth Biol. 2014;3:855–869. doi: 10.1021/sb400168u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavras H, Brunner HR. Role of angiotensin and its inhibition in hypertension, ischemic heart disease, and heart failure. Hypertension. 2001;37:342. doi: 10.1161/01.hyp.37.2.342. [DOI] [PubMed] [Google Scholar]
- 29.Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol. 2010;6:23. doi: 10.1186/1710-1492-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bork K, et al. Icatibant. Nat Rev Drug Discov. 2008;7:801–802. [Google Scholar]
- 31.Gruber CW, et al. Exploring bioactive peptides from natural sources for oxytocin and vasopressin drug discovery. Future Med Chem. 2012;4:1791–1798. doi: 10.4155/fmc.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitby LR, et al. Design, synthesis, and validation of a beta-turn mimetic library targeting protein-protein and peptide-receptor interactions. J Am Chem Soc. 2011;133:10184–10194. doi: 10.1021/ja201878v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz-Gomez G, et al. Update 1 of: over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem Rev. 2010;110:PR1–PR41. doi: 10.1021/cr900344w. [DOI] [PubMed] [Google Scholar]
- 34.Becker JA, et al. Ligands for kappa-opioid and ORL1 receptors identified from a conformationally constrained peptide combinatorial library. J Biol Chem. 1999;274:27513–27522. doi: 10.1074/jbc.274.39.27513. [DOI] [PubMed] [Google Scholar]
- 35.Gilon C, et al. Backbone cyclization – a new method for conferring conformational constraint on peptides. Biopolymers. 1991;31:745–750. doi: 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]
- 36.Schafmeister CE, et al. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 37.Dawson PE, et al. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 38.Moore SJ, et al. Knottins: disulfide-bonded therapeutic and diagnostic peptides. Drug Discov Today Technol. 2012;9:e1–e70. doi: 10.1016/j.ddtec.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 39.Saab F, et al. Bleeding risk and safety profile related to the use of eptifibatide: a current review. Expert Opin Drug Saf. 2012;11:315–324. doi: 10.1517/14740338.2012.650164. [DOI] [PubMed] [Google Scholar]
- 40.Survase SA, et al. Cyclosporin A: a review on fermentative production, downstream processing and pharmacological applications. Biotechnol Adv. 2011;29:418–435. doi: 10.1016/j.biotechadv.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 41.Love BL, et al. Linaclotide: a novel agent for chronic constipation and irritable bowel syndrome. Am J Health Syst Pharm. 2014;71:1081–1091. doi: 10.2146/ajhp130575. [DOI] [PubMed] [Google Scholar]
- 42.Jones MC. Therapies for diabetes: pramlintide and exenatide. Am Fam Physician. 2007;75:1831–1835. [PubMed] [Google Scholar]
- 43.Pope JE, Deer TR. Ziconotide: a clinical update and pharmacologic review. Expert Opin Pharmacother. 2013;14:957–966. doi: 10.1517/14656566.2013.784269. [DOI] [PubMed] [Google Scholar]
- 44.Tan YS, et al. Stapled peptide design: principles and roles of computation. Drug Discov Today. 2016 doi: 10.1016/j.drudis.2016.06.012. http://dx.doi.org/10.1016/j.drudis.2016.06.012. Published online June 18, 2016. [DOI] [PubMed]
- 45.Walensky LD, Bird GH. Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem. 2014;57:6275–6288. doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lonn P, Dowdy SF. Cationic PTD/CPP-mediated macromolecular delivery: charging into the cell. Expert Opin Drug Deliv. 2015;12:1627–1636. doi: 10.1517/17425247.2015.1046431. [DOI] [PubMed] [Google Scholar]
- 47.Madsbad S. Exenatide and liraglutide: different approaches to develop GLP-1 receptor agonists (incretin mimetics)-preclinical and clinical results. Best Pract Res Clin Endocrinol Metab. 2009;23:463–477. doi: 10.1016/j.beem.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 48.Kim D, et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Care Diabetes. 2007;30:1487–1493. doi: 10.2337/dc06-2375. [DOI] [PubMed] [Google Scholar]
- 49.Veuillez F, et al. Factors and strategies for improving buccal absorption of peptides. Eur J Pharm Biopharm. 2001;51:93–109. doi: 10.1016/s0939-6411(00)00144-2. [DOI] [PubMed] [Google Scholar]
- 50.Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11:905–910. doi: 10.1016/j.drudis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Prausnitz MR, et al. Current status and future potential of transdermal drug delivery. Nat Rev Drug Discov. 2004;3:115–124. doi: 10.1038/nrd1304. [DOI] [PubMed] [Google Scholar]
- 52.von Lueder TG, Krum H. New medical therapies for heart failure. Nat Rev Cardiol. 2015;12:730–740. doi: 10.1038/nrcardio.2015.137. [DOI] [PubMed] [Google Scholar]
- 53.Mas-Moruno C, et al. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med Chem. 2010;10:753–768. doi: 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becker A, et al. Metabolism and disposition of the alphav-integrin b3/b5 receptor antagonist cilengitide, a cyclic polypeptide, in humans. J Clin Pharmacol. 2015;55:815–824. doi: 10.1002/jcph.482. [DOI] [PubMed] [Google Scholar]
- 55.Gilbert MR, et al. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol. 2012;106:147–153. doi: 10.1007/s11060-011-0650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khasraw M, et al. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J Neurooncol. 2016;128:163–171. doi: 10.1007/s11060-016-2094-0. [DOI] [PubMed] [Google Scholar]
- 57.Zhao K, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 58.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 59.Chen M, et al. Mitochondria-targeted peptide MTP-131 alleviates mitochondrial dysfunction and oxidative damage in human trabecular meshwork cells. Investig Ophthalmol Vis Sci. 2011;52:7027–7037. doi: 10.1167/iovs.11-7524. [DOI] [PubMed] [Google Scholar]
- 60.Li J, et al. Mitochondria-targeted antioxidant peptide SS31 attenuates high glucose-induced injury on human retinal endothelial cells. Biochem Biophys Res Commun. 2011;404:349–356. doi: 10.1016/j.bbrc.2010.11.122. [DOI] [PubMed] [Google Scholar]