

Abstract
A core tenet of precision oncology is the rational choice of drugs to interact with patient-specific biological targets of interest, but it is currently difficult for researchers to obtain consistent and well-supported target information for pharmaceutical drugs. We review current drug target interaction resources and critically assess how supporting evidence is handled. We introduce the concept of a unified Cancer Targetome to aggregate drug target interactions within an evidence-based framework. We discuss current unmet needs and the implications for evidence based-clinical omics. The focus of this review is precision oncology but the discussion is highly relevant to targeted therapies of any area.
Keywords: Precision Medicine, Molecular Targeted Therapy, Evidence-based Medicine, Data Curation, Cancer Targetome
Precision Oncology Requires Rigorous Drug Target Information
The advent of precision oncology (see Glossary) is often hallmarked with the development of the targeted therapy imatinib to treat BCR/ABL1 positive chronic myeloid leukemia (CML) [1]. Over time, the term precision oncology has evolved to include the use of genetic biomarkers to guide treatment selection as well as refer to the emerging paradigm of treating cancer in a mutation-centric manner over a histology-centric manner [2–4]. However, the promise of precision oncology has been dimmed with the realization that only a small number of genetic variants in cancer are currently actionable with approved drugs [2,5]. Much of the work focused on expanding what is considered to be actionable in cancer genomics has focused on characterizing cancer-associated and driver genes and prioritization of these candidates for therapeutic intervention [6–9]. But any endeavor to expand the actionable space and thereby expand patient treatment options requires that we have a working knowledge of the interactions between drugs and their biological targets.
As illustrated in Figure 1 (Key Figure), drug-target interactions play an integral role in many different precision oncology applications. Clinical trials for cancer therapies are at the forefront of design and methodology development [10]. Newly emerging trial designs include umbrella trials, in which patients with the same type of cancer are assigned to different treatment arms according to key genetic variants [11,12], and basket trials, in which patients are assigned to treatment based on genetic variant but irrespective of cancer type [13,14]. Both of these trial designs rely on drug-target interaction information. Computational and predictive modeling approaches to predict drug response or anticipate adverse drug reactions require both primary and secondary target information for a complete picture [15–19]. Drug repurposing, or finding alternate uses for existing drugs often makes use of secondary or so-called “off-target” binding, where a drug binds to a target other than the one it was designed for [20,21]. Lastly, designing combinatorial drug treatment for a patient based on multiple genetic variants requires knowledge of drugs interacting with targets affected by each of those genetic variants [22]. Each of these examples requires knowledge of the biological targets that a drug may potentially interact with, but the specific context of a precision medicine application will dictate more or less rigorous requirements for the strength of supporting evidence for a drug-target interaction. Because this information can directly impact drug or target prioritization decisions and ultimately affect treatment options for patients, it is imperative that researchers have access to drug-target interaction information with clear literature and experimental evidence.
Figure 1. Precision Oncology Applications Rely on Drug-Target Interaction Information. A. Umbrella Clinical Trial with Multiple Treatment Arms.
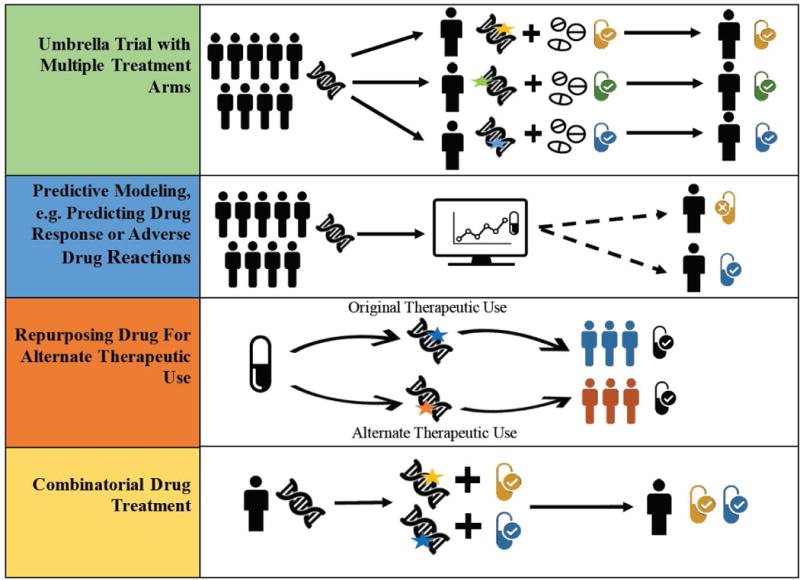
Patients are assigned to different treatment arms in clinical trial according to their genetic alterations. Drug treatments in each arm are determined according to interactions between drugs and priority genomic variants in tumor. B. Predictive Modeling e.g. Predicting Drug Response or Adverse Drug Reactions. Patient genomic data is used with in silico drug treatment simulation to predict which patients will respond beneficially (or adversely) to particular drug treatments. C. Repurposing Drug for Alternate Therapeutic Use. Drug binding information at additional or “secondary” targets can be used to repurpose a drug for a secondary therapeutic indication. D. Combinatorial Drug Treatment. A combined treatment of drugs is chosen for a patient using his or her genetic variant information and prioritization of variant-related targets according to known interactions with existing drugs.
Historically, the scope of the approved drug-target interaction space has been difficult to pin down precisely. Since the first characterization of the druggable genome [23,24] nearly twenty years ago, estimates for the number of biological targets for approved drugs has varied both with the definition of target and scope of data collection [25–30]. The realization that many currently approved drugs display polypharmacological or non-selective behavior [31] has added another layer of complexity to characterizing the drug-target interaction space.
Current public informatics resources for drug-target interaction information do not reflect a strong and consistent understanding of cancer drug binding across multiple targets. While the broader drug-target interaction space in the public domain faces the limitation of sparsity (only so many drug-target interaction pairs have been tested), there is a plethora of drug-target interaction and bioactivity information that is available but currently underutilized by the precision oncology research community. Hurdles to using this information include the need for aggregation across resources, unclear reference lineage, and differing types of supporting evidence. These challenges pose significant barriers to researchers looking to critically assess existing target annotations for a particular drug and this task quickly becomes intractable as the number of drugs of interest increases.
Current Resources for Drug-Target Interactions
Here, we briefly review resources and databases for drug-target interaction information (Table 1). Current resources for drug-target interaction data can be broadly categorized into two types, drug-centric and bioactivity-centric resources. Resources such as DrugBank, Therapeutic Targets Database, and KEGG Drug contain drug-target annotations supported by literature evidence and are subject to manual curation, but they currently do not incorporate experimental binding activity evidence [32–36]. Other resources, such as the International Union of Basic and Clinical Pharmacology/British Pharmacological Society (IUPHAR/BPS) Guide to Pharmacology, include manually curated experimental binding activities with drug-target annotations [37]. The Drug Gene Interaction Database aggregates drug-target annotation across multiple sources, allowing the user to see the parent sources and total literature reference count per drug-target interaction, but it does not currently include binding activity evidence [38]. Other resources that provide experimental binding evidence for target annotations for approved drugs and/or clinical trial drugs include DrugCentral, Pharos, SuperTarget, and STITCH [9,39–41]. The Open Targets is a recently released academic-industry collaborative resource that includes drug-target interaction information, but it is currently more focused on enabling target validation efforts [42]. While all of these resources allow for multiple targets per drug, differing standards for target inclusion can result in discrepant target annotation across resources [43].
Table 1. Databases for Drug-Target Interactions.
Database name and URL, Year Established, Current Version (Release Date, as available on August 4, 2017), Drugs/Compounds (A= approved drugs, Cl = clinical trial/investigational drugs, N = nutraceuticals/natural products, L= ligands, W= withdrawn), Target Inclusion Criteria (Manual = manual curation involved, Threshold= activity threshold applied, All= all targets listed (applicable to bioactivity databases), Supporting Evidence (PubMed ID, Bindingselected = selected binding values for drug-target interaction, Bindingall = all binding values for drug-target interaction reported, External DB= parent source database as applicable), and database license. License information reflects most recent information in publication or from database websites as of August 4, 2017.
| Database (URL) |
Description | Year Est. |
Version (Release Date) |
Drugs/ Compounds |
Target Inclusion Criteria |
Supporting Evidence |
License | Ref. |
|---|---|---|---|---|---|---|---|---|
| DrugBank https://www.drugbank.ca/ | Drug database including FDA-approved small molecules, FDA-approved biotech drugs, nutraceuticals, and experimental drugs. | 2006 | 5.0.7 (07/06/17) | A, Cl, N, W | Manual | PubMed | Public for non-commercial use1 | [32, 33] |
| Therapeutic Targets Database http://bidd.nus.edu.sg/BIDD-Databases/TTD | Includes therapeutic protein and nucleic acid targets, targeted disease condition, pathway information, and drug interactions. | 2002 | 4.3.02 (08/25/11) Site updated 09/10/15 | A, Cl | Manual | PubMed | No license indicated 2 | [34] |
| IUPHAR/BPS3 http://www.guidetopharmacology.org/ | Expert-curated resource for pharmacological, chemical, genetic, functional and pathophysiological data on targets of approved and experimental drugs. | 2013 | 2017.4 (05/23/17) | A, Cl | Manual | PubMed, US Patent, Bindingselected | Database ODbl4, contents are CC-BY SA 3.05 | [37] |
| DGIdb6 http://dgidb.genome.wustl.edu/ | Integrated from 13 primary sources for “druggable genome”, genes with known drug interactions or genes that are potentially druggable. | 2013 | 3.0 (06/30/17) | A, Cl | Manual | External DB, PubMed | GNU General Public License V3 | [38] |
| Open Targets https://www.targetvalidation.org/ | Informatics platform for target validation with extensive evidence associating targets and diseases. | 2016 | 3.2.0 (07/27/17) | A, Cl | All | External DB (ChEMBL) | Public | [42] |
| Pharos https://pharos.nih.gov/idg | Contains druggable human protein targets as part of the NIH Illuminating the Druggable Genome project, also includes target information for full human proteome. | 2016 | Uses 4.6.2 of Target Central Resource | A, Cl, L | Threshold | Bindingselected | CC BY SA-4.07 International | [9] |
| DrugCentral http://drugcentral.org/ | Drug compendium of structure, bioactivity, regulator, pharmacologic action and indication information for active pharmaceutical ingredients approved by FDA, EMA, and PMDA. | 2016 | 9.4 (04/25/17) | A8 | Threshold | Bindingselected | CC BY-SA 4.0 International | [39] |
| SuperTarget http://insilico.charite.de/supertarget/ | Web-based warehouse that integrates drug-related information with indication, adverse effect, metabolism, and gene ontology terms for target proteins | 2008 | NA | A, L | All | External DB, PubMed, Bindingall | CC BY-NC-SA 3.09 US | [40] |
| STITCH http://stitch.embl.de/ | Aggregates protein-chemical interactions from both experimental and manually curated evidence. | 2008 | 5.0 (2016) | A, L | All | External DB, PubMed, Bindingall | CC BY-NC-SA 4.010,11 | [41] |
| KEGG Drug http://www.genome.jp/kegg/drug/ | Drug information for approved drugs from Japan, US, and Europe. | 2010 | 04/03/17 | A8 | Manual | NA | KEGG Medicus publicly available12 | [35, 36] |
| BindingDB https://www.bindingdb.org/ | Experimentally determined binding affinities for protein-ligand complexes, scientific extracted from literature, selected databases, and US patents. | 2000 | 08/01/17 | A, Cl, L | All | External DB, PubMed, Bindingall | CC BY 3.013,14 US | [45] |
| ChEMBL https://www.ebi.ac.uk/chembl/ | Large-scale bioactivity database with information manually extracted from medicinal chemistry literature. | 2011 | 23 (05/17) | A, Cl, L | All | External DB, PubMed, Bindingall | CC BY-3.0 SA | [44] |
| PubChem BioAssay https://pubchem.ncbi.nlm.nih.go v/# | Repository for results of high throughput screening experiments for small molecules and RNA interference. | 2004 | NA | A, Cl, L | All | External DB, PubMed, Bindingall | Public domain15 | [46, 47] |
Abbreviations and Footnotes
DrugBank Open Data datasets (subset of full database) available under Creative Commons CCO 1.0 International License
All rights reserved
International Union of Basic and Clinical Pharmacology/British Pharmacological Society Guide to PHARMACOLOGY
Open Data Commons Open Database License
Creative Commons Attribution-Share Alike 3.0
Drug-Gene Interaction Database
Creative Commons Attribution-ShareAlike 4.0
Approved Drugs in Japan (PMDA), US (FDA), and Europe (EMA)
Creative Commons Attribution-NonCommercial-ShareAlike 3.0
Creative Commons Attribution-NonCommercial-ShareAlike 4.0
For chemical-protein interaction information
Academic users require subscription for FTP access, commercial users require subscription through Pathway Solutions
Creative Commons Attribution 3.0
Data sourced from ChEMBL is CC BY-SA 3.0
Data supplied by contractors or non-federal government entities or employees may be subject to copyright. However, PubChem listed as (ODbL) at https://www.healthdata.gov/dataset/pubchem
Bioactivity databases such as ChEMBL, BindingDB, and PubChem Bioassay aggregate chemical compound experimental binding activity information through manual extraction or text mining from the literature and other bioactivity databases [44–48]. These resources offer different coverage with respect to compounds, targets, and interactions due to differences in data scope, collection methods, and curation [49–51].
While bioactivity databases offer a wealth of potential compound-target information due to large scale collection of high throughput screening results [52], they do not directly provide drug-target interaction annotation, and it is therefore up to the user to determine an appropriate binding activity threshold when collecting and assessing experimental binding activity data. This presents its own challenge, as the choice of an appropriate activity threshold depends on the biological context of the problem. For determining bioactivity of compounds, the threshold of 10,000nM (10μM) is often used, but a much stricter threshold of 100nM or under is more appropriate when requiring interactions to be relevant to drug binding [31,53]. Paolini et al. required the best activity across assay types (IC50, EC50, Ki, and Kd) to be less than 10,000nM in their analysis of global pharmacological space [31]. Similarly, Koutsakas et al. used a bioactivity threshold of 10,000nM to obtain a balance between chemical space coverage and the inclusion of weakly active compounds [54]. This bioactivity threshold has been used by others in target prediction methods [55,56], or analysis of drug-target annotations [57], while other groups have used more conservative bioactivity thresholds across assay types (1,000nM) [58] in target prediction or used only single assay type (KD <3,000nM) in calculating selectivity measures [59]. Finan et al. used a threshold of 100nM on ChEMBL bioactivity data (across all assay types) to supplement target annotation found in company pipelines and the literature for approved and clinical trial drugs [60]. The Pharos platform, which presents data from the Target Central Resource Database (and uses a Target Development Level scheme to group targets based on level of study and association with small molecule bioactivity), uses bioactivity thresholds based on target family specific cut-offs [9].
The Need for a Unified Cancer Targetome
While there are many resources for drug-target interaction and compound bioactivity data, it is still an enormous task to collect, assess, potentially reconcile, and make informed decisions about putative drug-target interactions. This challenge is illustrated by a recent comprehensive analysis of all FDA-approved drugs, which curated all efficacy drug targets (as defined by Santos et al.) through an extensive search of both prescribing information and the scientific literature [30]. There is a critical need for aggregation of drug-target information in a framework that allows for assessment of the supporting evidence for each interaction.
We aggregate drug-target interaction and bioactivity data for FDA-approved antineoplastic drugs from four publicly available resources and introduce a framework for categorizing the type of evidence supporting each interaction to create a unified Cancer Targetome. Briefly, we selected these four resources in an effort to obtain representative coverage of the drug-target interaction space that is both publicly available and widely used by the research community. DrugBank is a popular resource for drug and drug-target data that is used widely by pharmacy and medicinal researchers, clinicians, educators, and the public [33]. Therapeutic Targets Database offers extended coverage for biological targets [61]. IUPHAR utilizes expert manual curation and rigorously requires experimental binding evidence from a primary source for all drug-target interactions [37]. However, IUPHAR typically provides only one experimental binding assay value for each drug-target interaction, so we also included an aggregated bioactivity database (BindingDB) in our collection efforts for the Cancer Targetome. BindingDB provides a wide coverage of binding assay data by aggregating across the scientific literature as well as from other bioactivity resources such as ChEMBL and PubChem [45]. Across four resources (DrugBank, Therapeutic Targets Database, IUPHAR, and BindingDB), we retrieved a total of 137 drugs and 658 targets participating in a total of 6385 unique drug-target relationships. We emphasize that the number of unique drug-target relationships should not be regarded as an estimate of actual drug-target binding space, as many of these relationships are supported experimental binding values that reflect very weak binding. DrugBank provided the highest coverage of drugs participating in drug-target relationships while BindingDB provided the highest coverage of targets participating in drug-target relationships (Supp. Figure 1). BindingDB also provided the highest coverage of unique drug-target relationships, which can be interpreted as experimentally tested drug-target interactions but not necessarily “true” drug-target binding events.
To assess the strength of supporting evidence for collected drug-target interactions, we develop a three level evidence scale. Evidence levels I, II, or III are assigned to drug target relationships retrieved from a database with no additional supporting information, with supporting literature information, or with supporting literature information and at least one reported experimental binding value, respectively. Experimental binding values may be reported as Kd, Ki, IC50, or EC50 assay values. Because drug-target information is aggregated across multiple databases, each unique drug-target relationship may have different types of supporting evidence reported across all four databases and therefore can be associated with multiple evidence levels. As we require increasing levels of supporting evidence for drug-target relationships, we see an overall decrease in coverage of drugs, targets, and unique relationships as expected (Figure 2). Within the Level III evidence tier, we can further threshold according to the numeric value of reported experimental binding activities. This allows us to triage experimentally tested drug-target relationships to those that have been reported with a binding value that is potentially relevant for drug and target binding having clinical impact.
Figure 2. Cancer Targetome Aggregated Counts for Drugs, Targets, and Unique Interactions by Evidence Level.
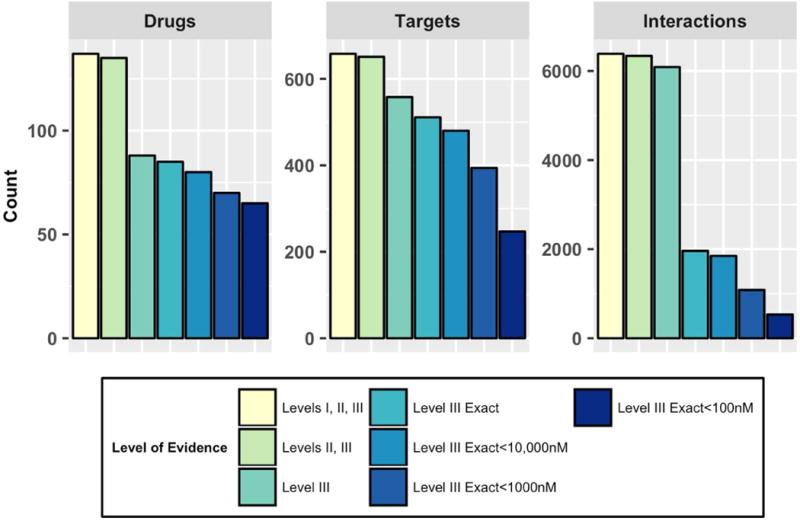
Drugs are FDA-approved antineoplastic drugs (total 137), targets are unique human UniProt Accession IDs (total 658) and interactions are unique relationships (total 6385) between one drug and one target. Counts are colored by supporting evidence level: Level I indicates database annotation only, Level II indicates database and literature reference annotation, Level III indicates database, literature, and experimental binding value annotation. Level III Exact refers to annotation of a binding value with an exact (“=”) binding value reported, rather than “<” or “>”. Thresholds on Level III binding activities were applied at 10,000nM, 1,000nM, and 100nM.
We demonstrate how the Cancer Targetome framework allows for filtering of aggregated drug-target relationships to those meeting particular evidence criteria. For instance, to obtain an estimate of drug-target interaction space for which there is strong experimental evidence to support nanomolar binding interactions, e.g. relevant to clinically achievable doses for a given drug, we can require Level III evidence and further threshold to reported binding affinities less than 100nM, which produces a total of 529 unique drug-target interactions. Interestingly, of these 529 putative drug-target binding interactions, the majority are reported by only one database, with only a quarter of these putative binding interactions reported by two or more databases (Supp. Figure 2A). Within this set of putative interactions, we can also examine the “best” or minimum experimental binding affinity value reported for each unique drug-target interaction and the database that is responsible for contributing this value. While the majority of such minimum assay values are contributed by BindingDB, IUPHAR contributes the minimum assay value for approximately 50 interactions (over 10%) (Supp. Figure 2B). This example highlights the benefit of aggregation across multiple sources to provide the research community with a more comprehensive resource for precision oncology.
Protein Kinase Inhibitors Are Highly Experimentally Tested Against Targets
The majority of antineoplastic drugs have been experimentally tested against less than twenty protein targets. This sparsity of the publicly available drug-target interaction space has been discussed by others [62] and presents a key limitation for efforts by the research community to assess drug promiscuity, or binding to “secondary” targets. However, a small set of drugs (all protein kinase inhibitors) have been experimentally tested with more than three hundred targets (Supp. Figure 3), providing us with several examples of drugs with extensive binding data with which we may assess potential target interactions and provide recommendations for future drug-target interaction curation efforts.
This meets expectations given the enormous resource commitment to targeting kinases in oncology following the break-through drug imatinib [63]. For instance, Davis et al. performed an extensive and comprehensive analysis of kinase inhibitor selectivity, including both approved and investigational stage drugs [59,64]. Experimental binding results for select approved cancer drugs from their analysis are included in our aggregated resource due to our data collection from the bioactivity database BindingDB. Among this set of highly tested kinase inhibitors, we see variation in the number of interacting targets for each drug (Supp. Figure 4). As we threshold the experimental binding evidence to stronger binding affinities (10,000nM, 1000nM, 100nM), we see that some drugs have a small number of targets meeting strong binding affinity criteria, such as afatinib, imatinib, and lapatinib, while other drugs have a seemingly high number of targets, such as bosutinib, crizotinib, dasatinib, and sunitinib. Due to the high number of experimentally tested targets for this subset of drugs, we can perform deeper data quality analysis and in particular investigate the contribution of different experimental binding activity types.
Imatinib and Vandetanib Use Cases
We highlight two use cases for the drugs imatinib and vandetanib. Both of these drugs are protein kinase inhibitors and have extensive binding activity information available across a large number of targets. Using the Cancer Targetome evidence framework, we assess the experimental evidence supporting target binding for imatinib and vandetanib at the strict threshold of 100nM. In Figure 3A we show all targets for imatinib with experimental binding evidence under 100nM. While there are a total of fourteen targets with assay evidence under 100nM, tyrosine-protein kinase ABL1 (ABL1), the canonical target of imatinib [1,63,65] notably has low nanomolar assay evidence across all four binding assay types (KD, Ki, IC50, and EC50). For KD, Ki, and IC50 assay evidence, ABL1 has multiple low nanomolar assay values, which lends more confidence to ABL1 being a biological target of the drug imatinib. Furthermore, for each of the four binding assay types, ABL1 has either the lowest or second-lowest assay value for target interactions with imatinib (Figure 3A). The case of imatinib serves as an example where evidence of the canonical “primary” target can be seen in experimental binding data. In the cases where a target other than ABL1 occupies the best or close to the best assay value (epithelial discoidin domain-containing receptor 1 (DDR1), platelet-derived growth factor alpha (PDGFRA), and platelet-derived growth factor beta (PDGFRB)), there is binding assay support from only one or two of the binding assay types rather than all four binding types, as in the case of ABL1.
Figure 3. A. Imatinib Target Interactions Under 100nM. Colored by Target, bin width=1nM.
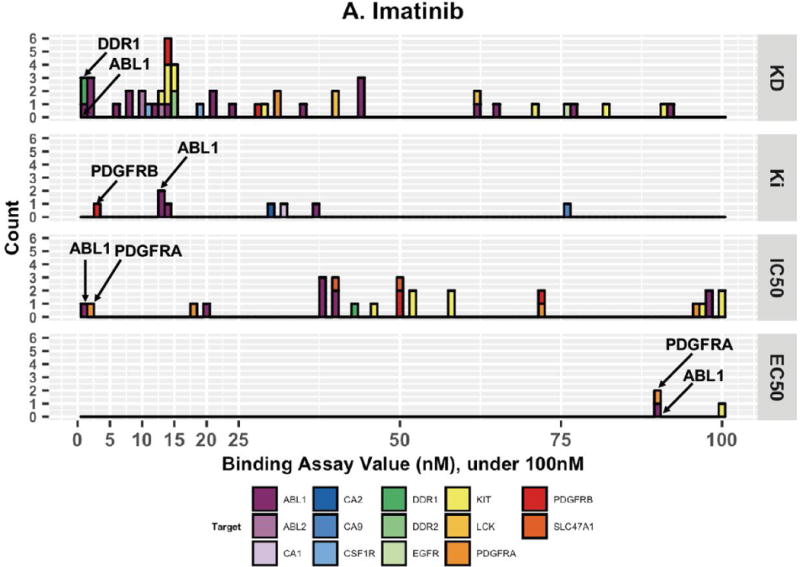
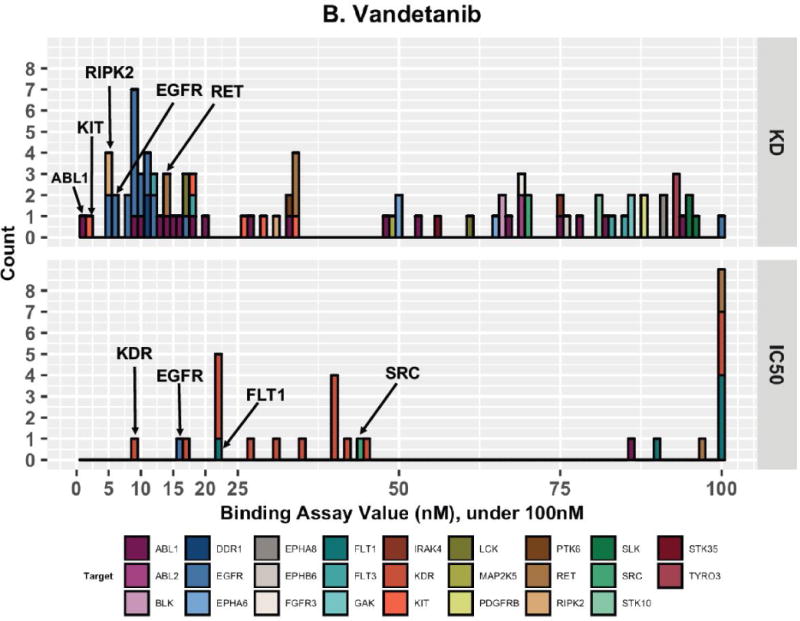
Imatinib has experimental binding evidence for fourteen different targets under 100nM. ABL1 stands out as it has many low nanomolar assay results and it occupies the best or second best assay value for each binding assay type. B. Vandetanib Target Interactions Under 100nM. Colored by Target, bin width=1nM. Vandetanib has experimental binding evidence for interacting with twenty-six different targets under 100nM. KD and IC50 assay evidence provide strong support for different targets for vandetanib (EGFR and KDR, respectively). Vandetanib does not have any targets supported by EC50 assay evidence and does not have any targets supported by <100,000nM Ki assay evidence.
In Figure 3B we show all targets for vandetanib with experimental binding evidence under 100nM. In total, there are twenty-six unique targets meeting these criteria but we see a striking discordance in the type of binding assay support available for these targets. Experimental KD values indicate that tyrosine-protein kinase ABL1 (ABL1), mast/stem cell growth factor receptor Kit (KIT), receptor-interacting serine/threonine-protein kinase 2 (RIPK2), epidermal growth factor receptor (EGFR), and proto-oncogene tyrosine-protein kinase receptor Ret (RET) have very low nanomolar experimental evidence. Experimental IC50 values indicate that vascular endothelial growth factor receptor 2 (KDR or VEGFR2), EGFR, vascular endothelial growth factor receptor 1 (FLT1), and proto-oncogene tyrosine-protein kinase Src (SRC) all have evidence for interaction at very low nanomolar evidence. According to KD assay evidence, EGFR is strongly supported as a target (multiple low nanomolar assay values), while according to IC50 assay evidence, KDR is strongly supported as a target. For vandetanib, there were no EC50 binding assay values available and no Ki binding assay values under 100,000nM available.
Interestingly, vandetanib is considered to be a dual KDR and EGFR inhibitor, or in some cases a multiple kinase inhibitor for EGFR, KDR, and RET [63,66,67]. A literature search reveals that while originally designed to inhibit KDR, vandetanib exhibited additional activity with EGFR in preliminary lead candidate stages [67]. These results prompted further testing which established vandetanib as inhibiting EGFR in mouse cells, human cancer cells, and in seven human cell lines lacking the target KDR [68]. This example highlights the rich contextual information for drug-target interactions that is currently not captured in drug-target interaction or bioactivity resources.
Next Steps for Drug-Target Interaction Evidence Curation
A unified Cancer Targetome framework provides researchers with access to cancer drug-target relationships from the public domain that are accompanied by transparent literature and experimental binding evidence lineage. The proposed evidence framework allows researchers to prioritize drug-target relationships according to the evidence criteria that are best suited to their research aims. Transparent and well-evidenced drug-target interactions will enable higher confidence and more informed decision making in the prioritization of drugs and targets in precision oncology efforts.
However, examining the factors needed for the creation of the Cancer Targetome reveals critical unmet needs. In particular, the vandetanib use case highlights the need for binding assay metadata. While we were able to retrieve and assess experimental binding affinities between vandetanib and many biological targets, we must also consider the information that is not captured in this process. Namely, we are currently not able to capture metadata such as the cell line used in experimental binding assays, tumor or non-tumor status of the cell line, and whether the cell line is derived from patient cells. The availability of this metadata would allow for further tiering of drug-target binding evidence to aid target prioritization. For instance, the category we have proposed for experimental binding evidence (Level III) could be further subdivided into tiers indicating whether the interaction has been tested in non-cancer cells, cancer cells, or cells that are patient-derived. Further tiering could be used to capture metadata indicating whether other targets were knocked down or remained functional during the experimental binding assay for the target of interest. This metadata is invaluable to prioritization of drug-target binding information in precision oncology, where it is critical to know whether experimental evidence was obtained using cancer or non-cancer cell lines.
Mapping Drug-Target Interactions to Pathways
Given the dysregulation that can occur in multiple pathways in cancer, there has been increasing attention and effort dedicated to targeting cellular pathways, particularly through the use of combination drug therapies [22,69]. We conducted a simple pathway analysis to assess the targeted pathway coverage of approved cancer drugs. Briefly, we mapped all targets participating in drug-target relationships to Reactome pathways using increasingly strict supporting evidence requirements. Reactome is a comprehensive open source pathway resource widely used by the research community [70]. Cellular pathways in Reactome are organized in a hierarchical manner, allowing for smooth pathway navigation and improved integration with external data resources. We designate those biological pathways containing one or more drug targets as “light” or potentially targetable by approved antineoplastic drugs. Conversely, biological pathways containing no drug targets are “dark” or currently out of scope for approved antineoplastic drugs. While a considerable portion of pathways (approximately 60%) are light to antineoplastic drugs when we consider any type of supporting evidence for drug-target interactions, this should be considered the most liberal estimate of potentially targetable pathways (Table 2). A more reasonable estimate is obtained when we require drug-target relationships to be supported by experimental binding evidence with a reported assay value of less than 100nM. This estimate indicates that there is strong evidence for approved antineoplastic drugs targeting approximately 39% of Reactome pathways. Depending upon distribution of key molecular aberrations for a given patient among the light and dark pathways, the evidence based curation as presented and envisioned herein will refine selection of therapeutics and in some cases could dramatically limit therapeutic options. We highlight the NOTCH Signaling pathway in Box 1, which contains several dark child pathways. Dark pathways that are currently out of scope of FDA-approved cancer drugs present areas for future cancer therapeutics development.
Table 2. Light Pathways of the Cancer Targetome, by Supporting Evidence Level.
All unique targets involved in drug-target relationships (supported by indicated evidence levels) were mapped to Reactome pathways. The number of unique targets within the set of drug-target relationships is shown in the second column. A pathway is considered light if it contains at least one drug target.
| Evidence Level Required | Number of Unique Targets | Number of Light Pathways | Total Number of Pathways | Percent Light Pathways |
|---|---|---|---|---|
| Levels I, II, III | 658 | 1214 | 2008 | 60.46% |
|
|
||||
| Levels II, III* | 651 | 1213 | 2008 | 60.40 % |
|
|
||||
| Level III | 558 | 1139 | 2008 | 56.72 % |
|
|
||||
| Level III Exact | 511 | 1091 | 2008 | 54.33 % |
|
|
||||
| Level III Exact, Threshold <100nM | 246 | 790 | 2008 | 39.34 % |
|
|
||||
Box 1. Pathway Example: NOTCH Signaling.
Dark pathways are of particular interest for future drug discovery and development efforts as they are currently outside the scope of approved cancer agents. We highlight the NOTCH signaling pathway, which is light at the topmost hierarchical level but contains several dark child pathways (Figure I). Dysregulated NOTCH signaling has been implicated in breast, prostate, lung, head and neck, and central nervous system cancers as well as T-cell leukemia and has thus been identified as a therapeutic target of interest [71–74]. Three of the five child pathways of NOTCH signaling are currently dark to cancer drugs (Signaling by NOTCH2, Signaling by NOTCH3, and Signaling by NOTCH4). We highlight the light child pathway Pre-NOTCH Expression and Processing in the figure inset, which shows that there are two drugs potentially interacting with two targets in this pathway. Arsenic trioxide putatively interacts with transcription factor AP-1 (JUN, UniProt P05412) and G1/S-specific cyclin-D1 (CCND1, UniProt P24385), while vinblastine sulfate putatively interacts with JUN. However, all three of these drug-target relationships have Level II evidence only, as there is no accompanying experimental binding evidence. Therefore, if we assess light pathway coverage while requiring at least experimental binding evidence for drug-target interactions, this nested pathway goes dark. We use this example to illustrate that the classification of a particular pathway as light or dark to approved cancer drugs is directly impacted by the strength of supporting evidence for the drug-target interactions involving the pathway of interest.
Figure I. Signaling by NOTCH Pathway.
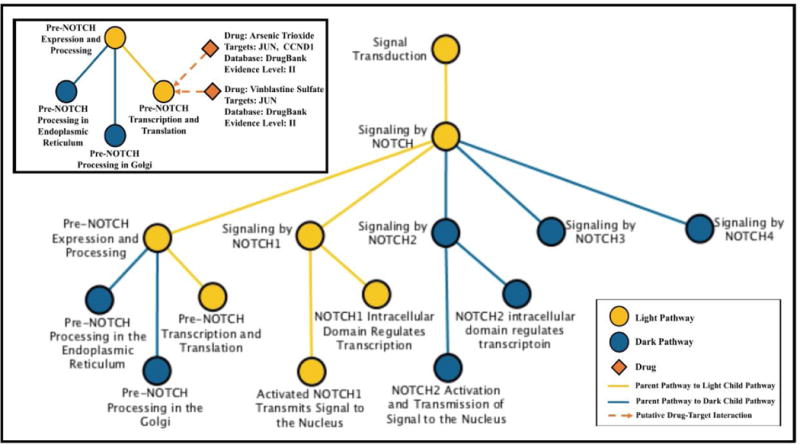
Signaling by NOTCH is a light or potentially targetable pathway when considering drug-target relationships supported by any level of evidence. Main Figure. Of the five child pathways in Signaling by NOTCH, two are light (gold) and three are dark (blue) to current approved antineoplastic drugs. Inset. The light child pathway Pre-NOTCH Expression and Processing contains two targets, JUN and CCND1 that are putatively targeted by antineoplastic drugs. This pathway is light when including drug-target interactions of Level II evidence, but goes dark when Level III evidence is required.
Concluding Remarks
We foresee the possibility that these analyses will allow weighting the level, extent and type of evidence to guide prioritization of drugs moving to the clinic, for better synchronization of preclinical promise and patient benefit. Recently, attention has been drawn to the need for evidence quantification of patient-specific alterations in tumors in order to guide decisions about actionable therapies [5]. A similar characterization of evidence is also needed for drug-target and drug-pathway interactions if we hope to unite drug-target information with patient-specific information and develop targeted therapies (Box 2 and Outstanding Questions). In particular, evidence characterization frameworks accommodate the inherent uncertainty in the targetome space due to multiple types of supporting evidence.
Box 2. Mapping Evidence Levels to Precision Oncology Applications.
The appropriate level of evidence to require when including drug-target interactions in precision oncology applications will be heavily context-dependent. In Figure 1, we detailed several examples of precision oncology applications that have a key dependency on drug-target interactions. For applications that are exploratory or hypothesis-generating in nature, such as computational and predictive modeling (Figure 1 Panel B), the use of drug-target interactions supported by Level I or Level II will often be appropriate. Such applications would benefit from casting a wider net of drug-target interactions so that all options can be explored. Similarly, exploratory work geared towards drug repurposing (Figure 1 Panel C), such as the inclusion of FDA-approved drugs on a screening panel for an indication other than the drug’s primary one, may also benefit from liberal evidence requirements that allow for investigation of all possibly relevant drug-target interactions. While additional Level III experimental binding evidence would lend support to these interactions being potentially relevant for human physiology, this will always be necessary at the discovery stage. Applications involving the planned use of a drug in a patient, however, will require (at a minimum) rigorous Level III experimental binding assay evidence. These applications could include off-label use of a drug, design of combination therapies (Figure 1 Panel D), or inclusion of an already-approved drug in a clinical trial for an alternate indication (Figure 1 Panel A). In these examples, choice of therapy may be driven by a patient’s particular molecular aberrations if there is substantial evidence that those aberrations can be targeted by an existing pharmaceutical therapy. The requirements for evidence supporting such drug-target interactions must be very rigorous - meaning very low nanomolar binding evidence for a drug-target interaction, ideally across binding assay types and from multiple, independent sources. As mentioned previously, experimental metadata (such as cell line information) will also be necessary for rigorous evaluation and prioritization of drug-target interactions. For clinical applications of drugs in patients, we emphasize that drug-target interaction evidence (even rigorously supported evidence) is intended to supplement but never to replace an oncologist’s or tumor board’s expertise and recommendations. We envision the use of this information as one line of evidence among the many that are evaluated by medical experts when deciding the best course of action for a patient.
Outstanding Questions.
How will we prioritize drug-target annotations in a way that reflects weight of supporting experimental evidence, i.e. both qualitative and quantitative evidence?
What is the best way to handle discordant binding assay values for the same drug-target interaction? Additional assay metadata such as cell line, cancer status, and target knockdowns can provide critical context that would affect prioritization of drug-target annotations within precision oncology pipelines. In the absence of this data, how do we best incorporate this uncertainty?
How well does the public domain drug-target interaction space approximate the fully tested drug-target interaction space? We must consider that publicly available data is incomplete as much of the tested drug-target interaction space is proprietary.
How will our knowledge base change as we add incoming information on promising new therapies, for instance from targeted immune checkpoint inhibitors? While our data collection included immune checkpoint inhibitors such as ipilimumab and nivolumab, there was limited target information available for these drugs from the public resources used here. As immune checkpoint inhibitor therapy is showing considerable promise, we would expect more publicly available data for these drugs is on the horizon and will soon be accessible to drug-target interaction databases.
How do we develop similar evidence frameworks for drug-target annotations in other therapeutic areas? Given that precision oncology is at the forefront of targeted therapy development, we would expect this domain to be one of the best characterized areas. Will the heterogeneity in annotation and data availability prevent the ultimate promise of drug repurposing across therapeutic domains?
Given the recent attention and dedication of resources to investigate understudied areas of the druggable genome by the NIH Illuminating the Druggable Genome Consortium, we believe this work will be of current interest to the larger precision medicine community. This has implications for other therapeutics areas of interest with respect to guided investigation into understudied and underdeveloped therapeutic drugs, targets, and pathways.
Supplementary Material
Trends.
Precision oncology, which aims to rationally select treatments for patients based on their genetic information, has a key dependency on drug to target annotation that is often overlooked.
While “patient-specific” treatment broadly encompasses all aspects of a patient’s health, such as additional diagnoses, other prescribed medications, or even adverse effects experienced in response to therapy, our scope for this article is focused narrowly on use of the term “patient-specific” to mean those biological targets specific to a patient’s cancerous cells that may be modulated to have a therapeutic effect.
Drug-target annotation is often heavily biased towards primary targets with limited or difficult to find information on secondary targets.
Resources for drug-target interactions differ in coverage, consistency, and evidence curation which makes it challenging for researchers to obtain credible and reproducible drug to target annotation.
Acknowledgments
This work was supported by the National Library of Medicine Informatics Training Grant T15LM007088 (A.B.), the National Cancer Institute 1R01CA192405 (G.C., M.K.M, S.K.M), the National Human Genome Research Institute 2U41HG003751 (G.W.), and National Center for Advancing Translational Sciences 5UL1TR000128 (G.W., S.K.M.) We thank Dr. Beth Wilmot, Eisa Mahyari, Xiaoming Ouyang, and Kristen Stevens for their discussions and input on the manuscript and Arvin Paranjpe and the OHSU Technology Transfer and Business Development for thoughtful comments.
Glossary
- biomarker
a biological indicator used here to refer to presence of a particular genetic mutation
- Cancer Targetome
a unified concept for all target annotations of FDA-approved cancer drugs aggregated across the public domain and encompassing multiple types of supporting evidence
- combinatorial drug treatment
drug regimen composed of more than one drug
- druggable genome
subset of human genome that encodes proteins targeted by pharmaceutical drugs
- drug-target interaction
a physical binding relationship between a drug molecule and a target entity
- polypharmacology
disposition of drugs to bind to more than one biological target
- precision oncology
treatment rationale that aims to match patients with therapies based on their genetic information for improved outcome
- target
biological entity of interest whose activity inside cells is modulated by drug
- targeted therapy
drug that interacts with a particular biological entity often by design
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no conflicts of interest to declare.
References
- 1.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Prasad V, et al. Precision oncology: origins, optimism, and potential. Lancet Oncol. 2016;17:e81–e86. doi: 10.1016/S1470-2045(15)00620-8. [DOI] [PubMed] [Google Scholar]
- 3.What Precisely Is Precision Oncology—and Will It Work? - The ASCO Post. [Online]. Available: http://www.ascopost.com/issues/january-25-2017/what-precisely-is-precision-oncology-and-will-it-work/. [Accessed: 26-May-2017]
- 4.Saad ED, et al. Precision medicine needs randomized clinical trials. Nat Rev Clin Oncol. 2017;14:317–323. doi: 10.1038/nrclinonc.2017.8. [DOI] [PubMed] [Google Scholar]
- 5.Andre F, et al. Prioritizing targets for precision cancer medicine. Ann Oncol. 2014;25:2295–2303. doi: 10.1093/annonc/mdu478. [DOI] [PubMed] [Google Scholar]
- 6.Watson IR, et al. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013;14:703–718. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hudson (Chairperson) TJ, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLendon R, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen DT, et al. Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2017;45:D995–D1002. doi: 10.1093/nar/gkw1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biankin AV, et al. Patient-centric trials for therapeutic development in precision oncology. Nature. 2015;526:361–370. doi: 10.1038/nature15819. [DOI] [PubMed] [Google Scholar]
- 11.Kim ES, et al. The BATTLE Trial: Personalizing Therapy for Lung Cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The I-SPY 1 TRIAL Investigators et al. Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657) Breast Cancer Res Treat. 2012;132:1049–1062. doi: 10.1007/s10549-011-1895-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Redig AJ, Jänne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol. 2015;33:975–977. doi: 10.1200/JCO.2014.59.8433. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Chavez A, et al. Molecular Profiling and Targeted Therapy for Advanced Thoracic Malignancies: A Biomarker-Derived, Multiarm, Multihistology Phase II Basket Trial. J Clin Oncol. 2015;33:1000–1007. doi: 10.1200/JCO.2014.58.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie L, et al. Drug Discovery Using Chemical Systems Biology: Identification of the Protein-Ligand Binding Network To Explain the Side Effects of CETP Inhibitors. PLoS Comput Biol. 2009;5:e1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pauwels E, et al. Predicting drug side-effect profiles: a chemical fragment-based approach. BMC Bioinformatics. 2011;12:169. doi: 10.1186/1471-2105-12-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johns DG, et al. On-and Off-Target Pharmacology of Torcetrapib. Drugs. 2012;72:491–507. doi: 10.2165/11599310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Berger SI, Iyengar R. Role of systems pharmacology in understanding drug adverse events. Wiley Interdiscip Rev Syst Biol Med. 2011;3:129–135. doi: 10.1002/wsbm.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao S, et al. Systems Pharmacology of Adverse Event Mitigation by Drug Combinations. Sci Transl Med. 2013;5:206ra140–206ra140. doi: 10.1126/scitranslmed.3006548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dudley JT, et al. Exploiting drug-disease relationships for computational drug repositioning. Brief Bioinform. 2011;12:303–311. doi: 10.1093/bib/bbr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corsello SM, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23:405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 23.Drews J. Genomic sciences and the medicine of tomorrow. Nat Biotechnol. 1996;14:1516–1518. doi: 10.1038/nbt1196-1516. [DOI] [PubMed] [Google Scholar]
- 24.Drews J, Ryser S. Classic drug targets. Nat Biotechnol. 1997;15:1350–1350. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- 25.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 26.Imming P, et al. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 27.Overington JP, et al. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 28.Rask-Andersen M, et al. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 29.Rask-Andersen M, et al. The Druggable Genome: Evaluation of Drug Targets in Clinical Trials Suggests Major Shifts in Molecular Class and Indication. Annu Rev Pharmacol Toxicol. 2014;54:9–26. doi: 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- 30.Santos R, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paolini GV, et al. Global mapping of pharmacological space. Nat Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 32.Wishart DS. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, et al. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002;30:412–415. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanehisa M, et al. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pawson AJ, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffith M, et al. DGIdb: mining the druggable genome. Nat Methods. 2013;10:1209–1210. doi: 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ursu O, et al. DrugCentral: online drug compendium. Nucleic Acids Res. 2017;45:D932–D939. doi: 10.1093/nar/gkw993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hecker N, et al. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012;40:D1113–D1117. doi: 10.1093/nar/gkr912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhn M, et al. STITCH 4: integration of protein-chemical interactions with user data. Nucleic Acids Res. 2014;42:D401–D407. doi: 10.1093/nar/gkt1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koscielny G, et al. Open Targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 2017;45:D985–D994. doi: 10.1093/nar/gkw1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Bajorath J. Compound promiscuity: what can we learn from current data? Drug Discov Today. 2013;18:644–650. doi: 10.1016/j.drudis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Bento AP, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilson MK, et al. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–D1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2011;40:D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014;42:D1075–D1082. doi: 10.1093/nar/gkt978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellis LJ, et al. Collation and data-mining of literature bioactivity data for drug discovery. Biochem Soc Trans. 2011;39:1365–1370. doi: 10.1042/BST0391365. [DOI] [PubMed] [Google Scholar]
- 49.Southan C, et al. Quantitative assessment of the expanding complementarity between public and commercial databases of bioactive compounds. J Cheminformatics. 2009;1:10. doi: 10.1186/1758-2946-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Southan C, et al. Comparing the Chemical Structure and Protein Content of ChEMBL, DrugBank, Human Metabolome Database and the Therapeutic Target Database. Mol Inform. 2013;32:881–897. doi: 10.1002/minf.201300103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiikkainen P, Franke L. Analysis of Commercial and Public Bioactivity Databases. J Chem Inf Model. 2012;52:319–326. doi: 10.1021/ci2003126. [DOI] [PubMed] [Google Scholar]
- 52.Li Q, et al. PubChem as a public resource for drug discovery. Drug Discov Today. 2010;15:1052–1057. doi: 10.1016/j.drudis.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, et al. Evidence-Based and Quantitative Prioritization of Tool Compounds in Phenotypic Drug Discovery. Cell Chem Biol. 2016;23:862–874. doi: 10.1016/j.chembiol.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 54.Koutsoukas A, et al. In Silico Target Predictions: Defining a Benchmarking Data Set and Comparison of Performance of the Multiclass Naïve Bayes and Parzen-Rosenblatt Window. J Chem Inf Model. 2013;53:1957–1966. doi: 10.1021/ci300435j. [DOI] [PubMed] [Google Scholar]
- 55.Mervin LH, et al. Target prediction utilising negative bioactivity data covering large chemical space. J Cheminformatics. 2015;7 doi: 10.1186/s13321-015-0098-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim H, et al. Large-Scale Off-Target Identification Using Fast and Accurate Dual Regularized One-Class Collaborative Filtering and Its Application to Drug Repurposing. PLoS Comput Biol. 2016;12:e1005135. doi: 10.1371/journal.pcbi.1005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Y, Bajorath J. Many structurally related drugs bind different targets whereas distinct drugs display significant target overlap. RSC Adv. 2012;2:3481. [Google Scholar]
- 58.Afzal AM, et al. A multi-label approach to target prediction taking ligand promiscuity into account. J Cheminformatics. 2015;7 doi: 10.1186/s13321-015-0071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis MI, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 60.Finan C, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang H, et al. Therapeutic target database update 2016: enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 2016;44:D1069–D1074. doi: 10.1093/nar/gkv1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mestres J, et al. Data completeness—the Achilles heel of drug-target networks. Nat Biotechnol. 2008;26:983–984. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- 63.Wu P, et al. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 64.Karaman MW, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 65.Druker BJ, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 66.Knowles PP, et al. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J Biol Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 67.Wedge SR, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–4655. [PubMed] [Google Scholar]
- 68.Ciardiello F, et al. Antitumor effects of ZD6474, a small molecule vascular endothelial growth factor receptor tyrosine kinase inhibitor, with additional activity against epidermal growth factor receptor tyrosine kinase. Clin Cancer Res Off J Am Assoc Cancer Res. 2003;9:1546–1556. [PubMed] [Google Scholar]
- 69.Jimeno A, Hidalgo M. Multitargeted therapy: Can promiscuity be praised in an era of political correctness? Crit Rev Oncol Hematol. 2006;59:150–158. doi: 10.1016/j.critrevonc.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 70.Fabregat A, et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44:D481–D487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013;139:95–110. doi: 10.1016/j.pharmthera.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuan X, et al. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20–27. doi: 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 73.Sioutos N, et al. NCI Thesaurus: A semantic model integrating cancer-related clinical and molecular information. J Biomed Inform. 2007;40:30–43. doi: 10.1016/j.jbi.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 74.Guerrero-Preston R, et al. Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics. 2014;9:1031–1046. doi: 10.4161/epi.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.