

Abstract
A disulfide-bridged cyclic RGD peptide, named iRGD (internalizing RGD, c(CRGDK/RGPD/EC)), is known to facilitate tumor targeting as well as tissue penetration. After the RGD motif-induced targeting on αv integrins expressed near tumor tissue, iRGD encounters proteolytic cleavage to expose the CendR motif that promotes penetration into cancer cells via the interaction with neuropilin-1. Based on these proteolytic cleavage and internalization mechanism, we designed an iRGD-based monolithic imaging probe that integrates multiple functions (cancer-specific targeting, internalization and fluorescence activation) within a small peptide framework. To provide the capability of activatable fluorescence signaling, we conjugated a fluorescent dye to the N-terminal of iRGD, which was linked to the internalizing sequence (CendR motif), and a quencher to the opposite C-terminal. It turned out that fluorescence activation of the dye/quencher-conjugated monolithic peptide probe requires dual (reductive and proteolytic) cleavages on both disulfide and amide bond of iRGD peptide. Furthermore, the cleavage of the iRGD peptide leading to fluorescence recovery was indeed operative depending on the tumor-related angiogenic receptors (αvβ3 integrin and neuropilin-1) in vitro as well as in vivo. Compared to an ‘always fluorescent’ iRGD control probe without quencher conjugation, the dye/quencher-conjugated activatable monolithic peptide probe visualized tumor regions more precisely with lower background noise after intravenous injection, owing to the multifunctional responses specific to tumor microenvironment. All these results, along with minimal in vitro and in vivo toxicity profiles, suggest potential of the iRGD-based activatable monolithic peptide probe as a promising imaging agent for precise tumor diagnosis.
Keywords: Internalizing RGD (iRGD), activatable probe, peptide monolith, tumor imaging
Graphical abstract
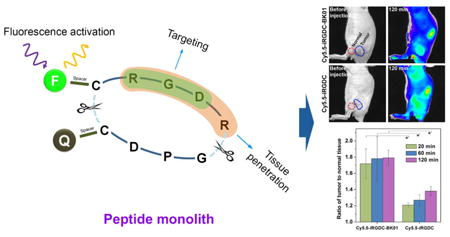
1. Introduction
Selective molecular imaging of cancer cells at the malignant tumor sites allows for accurate diagnosis of cancer. To this end, many synaphic targeting agents including antibodies, peptides, nucleic acids, and small molecules, have been discovered to provide specific interaction with targets overexpressed in tumor microenvironment [1-6]. Chemical conjugation of contrast agents to such targeting ligands has widely been employed to develop molecular imaging probes for cancer theranostics [7, 8]. In general, however, the circulation of the untargeted imaging probes remaining in the body produces inevitable background signals, leading to a low target-to-background imaging contrast.
To minimize such a background noise coming from off-target tissues, fluorescent probes that are initially inactive but activatable only in the target tissue in response to the specific microenvironment, have been devised [8, 9]. Typically, these probes are comprised of fluorescence resonance energy transfer (FRET) or self-quenching systems whose fluorescence can be turned on by cleavage or conformational change of the specific moiety in the probe. For tumor imaging, unusual tumor microenvironments, such as hypoxic condition, low pH, or a variety of overexpressed biomarkers, are employed to trigger the fluorescence activation [10-15]. Tumor-specific signals with no activation in off-target tissues can minimize the background noise, to yield a high contrast for tumor imaging.
Among tumor targeting agents, peptides afford advantages such as low toxicity, low immunogenicity, fast renal clearance, and high specificity. Because of the versatility of peptides, tumor targeting peptides, such as RGD and NGR peptides, have served cancer diagnostics and therapeutics [2, 16-18]. Furthermore, the peptide-based small theranostic system has relatively simple, compact, biocompatible, and unidirectional structures, demonstrating remarkable theranostic performances and low side effects over the nanoparticle-based system [19]. Recently, Ruoslahti group discovered a tumor-homing peptide by phase display, named iRGD (internalizing RGD, CRGDK/RGPD/EC), which shows more efficient tumor penetration than conventional RGD peptides [20]. Tumor homing of the disulfide-bridged cyclic iRGD peptide follows the consecutive steps: the RGD sequence binds to αv integrins on tumor endothelium, then iRGD undergoes a proteolytic cleavage to expose the CendR motif (R/KXXR/K) that triggers neuropilin-1 (NRP-1)-mediated internalization into cells [20, 21]. These distinct attributes of iRGD have been utilized to improve the tumor targeting and internalization of imaging agents and drugs [22-29].
In this study, we introduce monolithic functionalization of iRGD with the capability of fluorescence activation as a new design concept for a peptide-based smart probe that integrates multiple functions of cancer targeting, internalization and diagnostic signal activation within a small molecular framework. In the probe design, the tumoral proteolytic cleavage of iRGD is employed to modulate the quenching/dequenching states of fluorescence. Scheme 1 shows the design of an initially quenched iRGD probe that is cyclized via a disulfide linkage and conjugated with a fluorescent dye on the N-terminus of iRGD, a part of the internalizing sequence (CendR motif), and a quencher on the opposite C-terminus. The iRGD-derived multifunctional nature differentiates this monolithic peptide probe from other dye/quencher-combined activatable systems that necessitate further modifications for cancer targeting and internalization [11-13, 30-32]. Here, we report the synthesis and fluorescence turn-on characteristics of the resulting iRGD probe, as well as the in vitro dependence of signaling on tumor-related angiogenic receptors (αvβ3 and NRP-1) in cells and tumor spheroids. We also demonstrate that upon intravenous injection of the iRGD monolith into a tumor-bearing mouse model, the iRGD-derived multiple functions (cancer targeting, internalization and proteolytic reaction) interplay cooperatively to enhance the diagnostic imaging contrast by tumor-specific fluorescence activation.
Scheme 1.
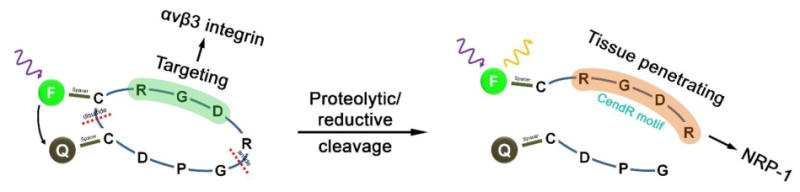
Schematic of fluorescence recovery process of dye/quencher-conjugated iRGD peptide. After targeting on αvβ3 integrin and cleavage near tumor environment, the CendR motif with turned-on fluorescence is internalized into cancer cells via NRP-1.
2. Materials and Methods
2.1. General method
Unless otherwise noted, all solvents and reagents were obtained from commercial suppliers and used without further purification. The peptide fragments analysis and purification were carried out on an Agilent Technologies 1200 HPLC system (Agilent, USA) equipped with UV-VIS and fluorescence detector using XBridge BEH C18 Column (10 μm, 150 mm × 4.6 mm, Waters Corp., USA) and XBridge BEH130 Prep C18 (10 μm, 250 mm × 10 mm, Waters Corp., USA). Mass spectra were acquired on a MALDI-TOF, Voyager-DETM STR Biospectrometry Workstation (Applied Biosystems Inc., USA) and a high-resolution electrospray ionization mass spectrometry (HR-ESI-MS, Thermo Finnigan, LTQ-Orbitrap). Fluroescence spectra were acquired by a fluorescence spectrophotometer (Hitachi F-7000, Japan). Cell images were collected using an Axioimager M1 microscope (Zeiss, Germany) and tumor spheroids were observed with FluoView FV10i confocal laser scanning microscope (Olympus, Japan). UV absorption for WST-1 assay was measured by SpectraMax M5 (Molecular Devices, USA). All in vivo data and images were taken on an IVIS Spectrum imaging system (Caliper, USA). The histological images were obtained from optical microscope (BX 51, Olympus, Japan).
2.2. Solid-phase Peptide Synthesis of iRGD Peptides and Control Peptides
Synthesis of Fmoc-Ahx-c(CRGDRGPDC)-Ahx-K-NH2 (iRGDK, 1)
iRGDK peptide (Fmoc-Ahx-c(CRGDRGPDC)-Ahx-K-NH2, iRGDK) was synthesized manually on Fmoc-Rink amide core–shell type resin (0.2 g, 0.30 mmol/g) [33] using Fmoc/tBu solid-phase procedure. For coupling reaction, the resin was treated with pre-activated amino acid solution, which was prepared with Fmoc-amino acid (0.18 mmol, 3 equiv.), HBTU (0.18 mmol, 3 equiv.), HOBt (0.18 mmol, 3 equiv.) and DIPEA (0.36 mmol, 6 equiv.) in NMP (4 mL). All amino acid couplings were performed for 1 h at 25 °C. After the coupling reaction, the resin was washed with NMP (5 mL × 3), CH2Cl2 (5 mL × 3), and MeOH (5 mL × 3). The Fmoc group was removed using 20% piperidine/NMP (4 mL, 5 min + 10 min). Completion of each coupling step was monitored by the Kaiser test. After the last amino acid coupling, on-bead cyclization (S-S) was performed by treating the resin containing linear peptide with the S-Acm protecting groups with Tl(tfa)3 (0.12 mmol, 2 equiv.) in NMP (4 mL) for 2 h. The final peptide was cleaved with 4 mL of TFA:thioanisole:TIS:H2O (85:5:5:5, v/v) solution for 1 h and was recovered by ether precipitation as a crude white solid powder. For the peptide analysis, a flow rate of 1.0 mL/min and a 20 min-gradient of 10–80% of solvent B followed by a 5 min-constant flow of 100% solvent B (solvent A, 0.1% TFA in water; solvent B, 0.1% TFA in acetonitrile) was used with XBridge BEH C18 Column (10 μm, 150 mm × 4.6 mm). For the peptide purification, a flow rate of 4.0 mL/min and a 20 min-gradient of 10–80% of solvent B followed by a 5 min-constant flow of 100% solvent B (solvent A, 0.1% TFA in water; solvent B, 0.1% TFA in acetonitrile) was used with XBridge BEH130 Prep C18 (10 μm, 250 mm × 10 mm). Absorbance was measured at 230 nm. The lyophilized peptide was obtained by freeze drying of HPLC fraction containing the product (23 mg). The yield was calculated based on the initial loading level of resins (yield: 25%). iRGDK (1) was analyzed by MALDI-TOF (calculated exact mass = 1551.7201 for C68H103N20O18S2 (iRGDK) [M+H]+, found 1551.2295).
Synthesis of H-Ahx-c(CRGDRGPDC)-Ahx-C-NH2 (iRGDC, 2)
Another iRGD peptide (H-Ahx-c(CRGDRGPDC)-Ahx-C-NH2, iRGDC) was synthesized by the same procedure as used for iRGDK (1). After the last Fmoc deprotection, the peptide was cleaved from the resin by 4 mL of TFA:thioanisole:1,2-ethanedithiol:anisole (90:5:3:2) solution for 1 h and was recovered by ether precipitation. The crude peptide was treated with the methanolic 2,2-dipyridyl disulfide solution (10 mL, 1 mg/mL) for 1 h. Disulfide formation was monitored by HPLC. The final peptide was obtained by deprotection of t-butyl on the C-terminal cysteine with TFA:TFMSA:thioanisole:EDT (80:8:8:4, v/v). HPLC conditions for analysis and purification of the peptide were the same as used for iRGDK (1). After HPLC purification, the lyophilized peptide was obtained by freeze drying of HPLC fraction containing the product (12 mg). The yield was calculated based on the initial loading level of resins (yield: 15%). iRGDC (2) was analyzed by HRMS (calculated exact mass = 1304.5662 for C50H86N19O16S3 (iRGDC) [M+H]+ and [M+2H]2+, found 1304.5789 and 652.7883).
Synthesis of Fmoc-Ahx-c(CGGGGGGGC)-Ahx-K-NH2 (CG7CK, 3)
Glycine cyclic peptide was synthesized by the same procedure as used for iRGDK (1). The final lyophilized peptide was obtained in 17% yield (12 mg). CG7CK (3) was analyzed by MALDI-TOF (calculated exact mass = 1197.5185 for C53H77N14O14S2 (CG7CK) [M+H]+, found 1197.6894).
2.3. Dye and Quencher Conjugation on iRGD Peptides
Synthesis of AF488-iRGDK-dab (4)
Dabcyl-OSu (dab-OSu, 1.06 mg, 2.90 μmol) and DIPEA (0.67 μL, 3.86 μmol) were added to iRGDK (3 mg, 1.93 μmol) in DMF (0.5 mL). The mixture was stirred at room temperature for 2 h. Reaction progress was monitored by HPLC. After ether precipitation, the Fmoc group was deprotected by 20% piperidine/DMF (0.5 mL) for 10 min. The crude peptide was washed with diethylether (2 mL × 3). Then, Alexa Fluor 488 NHS ester (AF488-NHS, Invitrogen, USA, 1 mg, 1.55 μmol) and DIPEA (0.67 μL, 3.86 μmol) were added to the crude peptide dissolved in DMF (0.5 mL) at room temperature. The reaction was completed in 1 h, monitored by HPLC. HPLC flow conditions for analysis and purification of the peptide were the same as used for iRGDK (1). UV was measured at 230 nm, and fluorescence detection used excitation at 495 nm and emission at 519 nm. After HPLC purification, the final lyophilized AF488-iRGDK-dab was obtained by freeze drying of HPLC fraction containing the product (∼ 1.3 mg, yield: 32%). AF488-iRGDK-dab (4) was analyzed by MALDI-TOF (calculated exact mass = 2096.7512 for C89H118N25O27S4 (AF488-iRGDK-dab) [M+H]+, found 2096.8908).
Synthesis of Cy5.5-iRGDC-BK01 (5)
QFlamma Black-I maleimide (BK01-maleimide, Bioacts, Korea, 1.55 mg, 1.84 μmol) and DIPEA (0.53 μL, 3.07 μmol) were added to iRGDC (2 mg, 1.53 μmol) in DMF (0.5 mL). The mixture was stirred at room temperature for 1 h. Reaction completion was monitored by HPLC. After ether precipitation, Cy5.5 NHS ester (Cy5.5-NHS, GE Healthcare Life Sciences, USA, 2.08 mg, 1.84 μmol) and DIPEA (0.53 μL, 3.07 μmol) were added to the crude peptide dissolved in DMF (0.5 m). The mixture was stirred at room temperature for 1 h, monitored by HPLC. HPLC flow conditions for analysis and purification of the peptide were the same as used for iRGDK (1). UV was measured at 230 nm, and fluorescence detection used excitation at 675 nm and emission at 690 nm. After HPLC purification, the final lyophilized Cy5.5-iRGDC-BK01 was obtained by freeze drying of HPLC fraction containing the product (∼ 1.2 mg, yield: 26%). Cy5.5-iRGDC-BK01 (5) was analyzed by MALDI-TOF (calculated exact mass = 3045.9224 for C130H165N28O40S9 (Cy5.5-iRGDC-BK01) [M+H]+, found 3045.9224).
Synthesis of AF488-CG7CK-dab (6)
AF488-CG7CK-dab was synthesized from CG7CK (2 mg, 1.67 μmol) by the same procedure as used for AF488-iRGDK-dab (4). After purification process by HPLC, AF488-CG7CK-dab was obtained in 24% yield (∼ 0.7 mg). The final peptide was analyzed by HRMS (calculated exact mass = 1740.5340 for C74H90N19O23S4 (AF488-CG7CK-dab) [M-H]-, found 1740.8339).
Synthesis of Cy5.5-iRGDC (7)
iRGDC (2 mg, 1.53 μmol) dissolved in DMF (0.5 mL) was treated with Cy5.5-NHS (1.55 mg, 1.84 μmol) and DIPEA (0.53 μL, 3.07 μmol). The mixture was stirred at room temperature for 1 h. Reaction completion was monitored by HPLC. After ether precipitation, the peptide was purified by HPLC. HPLC conditions for analysis and purification of the peptide were the same as used for Cy5.5-iRGDC-BK01 (5). The final peptide was obtained by freeze drying of HPLC fraction containing the product (∼ 1.3 mg, yield: 39%). Cy5.5-iRGDC (7) was analyzed by MALDI-TOF (calculated exact mass = 2202.7232 for C91H127N21O29S7 (Cy5.5-iRGDC) [M+H]+, found 2202.9662).
2.4. In Vitro Cell Internalization of iRGD peptide
A431, PC-3, and U-87 MG cells (3 × 104 cells/well) were seeded onto 8-well chamber slides (Nunc Lab Tek) and grown overnight. The cells were incubated with 5 nM of AF488-iRGDK-dab (4) and AF488-CG7CK-dab (6) for 1 h at 37 °C. The cells were washed with PBS (1×, pH 7.4) and fixed in 4% (v/v) paraformaldehyde for 10 min at 4 °C. After PBS (1×, pH 7.4) washing, the cells were finally mounted with ProLong Gold antifade reagent with DAPI (Life Technologies). Fluorescence images were collected using an Axioimager M1 microscope (Zeiss).
2.5. WST-1 Cell Proliferation Assay
To determine cell viability under AF488-iRGDK-dab (4) and AF488-CG7CK-dab (6), cell proliferation was measured by WST-1 assay according to the manufacturer's manual (Clontech). In brief, A431, PC-3, and U-87 MG cells (1 × 104 cells/well) were seeded onto 96-well plates and incubated for 24 h. The culture medium was replaced with 200 μL of serum-free medium without or with AF488-iRGDK-dab (4) and AF488-CG7CK-dab (6), followed by incubation for 24 h at 37 °C. After the cells were washed twice with serum-free culture medium, each well was treated with WST-1 for 2 h and then the absorbance was measured at 450 nm using SpectraMax M5 (Molecular Devices, USA).
2.6. Tumor Spheroids Internalization of iRGD peptide
For spheroid generation, U-87 MG cells (2 × 106 cells/well) were seeded into Ultra Low Attachment (ULA) 24-well flat-bottomed plates (Costar®). After the plate was incubated for 4 days at 37 °C (5% CO2, 95% humidity), cells were maintained a sphere form. For AF488-iRGDK-dab (4) and AF488-CG7CK-dab (6) treatment, the 4-day-old U-87 MG spheroids were transferred to a new ULA plate with medium, and then were incubated for 1 h in the presence of 1 μM of AF488-iRGDK-dab (4) or AF488-CG7CK-dab (6). After 1 h, U-87 MG spheroids were collected in V-bottomed 15-ml Falcon tubes and allowed to sediment. Supernatant was removed by gentle pipetting and pellets were washed once with PBS (1×, pH 7.4). Then, 1 mL of 4% paraformaldehyde (PFA) was added for overnight at 4 °C. After washing with PBS (1×, pH 7.4), 250 μL of Hoechst 33342 solution was gently added to the fixed U-87 MG spheroids for 40 min to stain the nucleic acid. After spheroids were washed once with PBS, U-87 MG spheroids were embedded in 1% Agarose Gel on a glass bottom dish (MatTek Corporation). U-87 MG spheroids were observed with FLUOVIEW FV10i confocal microscope (Olympus).
2.7. In Vivo/Ex Vivo Fluorescence Imaging and Histological Analysis
The animal study was performed according to the procedures approved by the animal care and use committee of Korea Institute of Science and Technology. For tumor xenograft and in vivo imaging, BALB/c mice (6-week-old male; Orient Bio Inc., Korea) were anaesthetized with intraperitoneal injection of the solution (300 μL) of tiletamine hydrochloride (1 mg/mL), zolazepam hydrochloride (1 mg/mL), and xylazine hydrochloride (0.2 mg/mL) in saline. Tumor xenografts were established by subcutaneous inoculation of U-87 MG and PC-3 cells (1 × 107 cells suspended in the culture medium of 80 μL) into the thigh of mice. After 2 weeks, Cy5.5-iRGDC-BK01 and Cy5.5-iRGDC (10 nmol, 200 μL) were intravenously injected into U-87 MG and PC-3 tumor-bearing mice. In vivo imaging was carried out at certain time points with anaesthetized mice by IVIS Spectrum (Caliper, USA) equipped with excitation (675/30 nm) and emission (720/20 nm) filters. The tumor and major organs were resected at 3 h after intravenous injection and directly imaged by IVIS Spectrum. For inhibition test, anti-αvβ3 antibody or anti-NRP-1 antibody (0.1 mg/mL, 100 μL) was intravenously injected into the U-87 MG tumor-bearing mouse 15 min before the peptide injection.
For histological analysis, the major organs were excised from U-87 MG tumor-bearing mice in 3 days after intravenous injection of PBS (control) or Cy5.5-iRGDC-BK01. The excised organs were fixed with 4% formaldehyde solution, embedded in paraffin, and sliced into ∼5 μm thickness. The sections were stained with haematoxylin and eosin (H&E), and visualized by optical microscope (BX 51, Olympus, Japan).
3. Results and Discussion
3.1. Synthesis of dye/quencher-conjugated activatable iRGD probes
As sketched in Scheme 2, iRGD peptides were synthesized using the Fmoc/tBu strategy of solid-phase peptide synthesis (SPPS) on Rink amide linker coupled core–shell type resin.[33] After amino acid coupling, on-bead cyclization was performed using thallium(III) trifluoroacetate to make disulfide bridge in iRGD peptide.[34] As the proteolytic cleavage event generates the tissue-penetrating moiety (CendR motif) on the N-terminal fragment of iRGD,[20, 21] we conjugated a fluorescent dye on the N-terminus with a quencher attached to the opposite C-terminus to construct activatable fluorescence turn-on system. For quencher conjugation, a lysine (iRGDK, 1) or a cysteine (iRGDC, 2) was added to the C-terminus of iRGD peptides, depending on the type and feature of quenchers. To ensure access to intrinsic iRGD-binding site and to enhance the further coupling efficiency, a spacer residue, 6-aminohexanoic acid, was incorporated on both termini of the peptides. AF488-iRGDK-dab (4) was synthesized in three steps from the iRGDK peptide (1): Dabcyl-NHS as a quencher was coupled with the ε-amine of lysine on the C-terminus. After Fmoc deprotection on the N-terminus of iRGDK with a 20% piperidine solution, AF488-NHS as a fluorescent dye was conjugated on the deprotected amine. For in vivo studies, a near-infrared (NIR) cyanine dye (Cy5.5-NHS) and a wide absorbable quencher (BK01-maleimide, λabs: 400∼705 nm) were conjugated on both termini of iRGDC (2) via an orthogonal coupling method using amine-NHS ester and thiol-maleimide reactions (Cy5.5-iRGDC-BK01, 5). As a control peptide without targeting and internalizing moieties, AF488-CG7CK-dab (6) was synthesized from CG7CK (3) in the same synthetic method as AF488-iRGDK-dab. All the synthesized iRGD peptide derivatives (1–6) were fully characterized by HPLC, MALDI-TOF mass, and HRMS (Supplementary data).
Scheme 2.
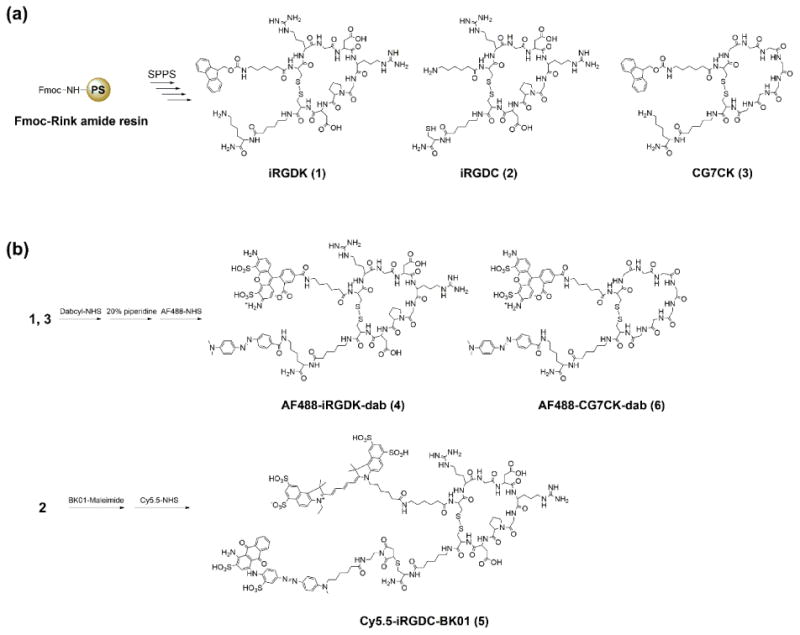
Synthetic routes for (a) functional cyclic peptides and (b) dye/quencher conjugated peptide probes.
3.2. Fluorescence recovery of the quenched iRGD probe
To evaluate the proteolytic cleavage-induced fluorescence activation, the dye/quencher-conjugated iRGD probe (AF488-iRGDK-dab, 4) was incubated in the presence of DTT and trypsin. The intact fluorescence-quenched AF488-iRGDK-dab showed very weak fluorescence. As the concentration of DTT increased under excessive trypsin condition and vice versa, however, its fluorescence gradually increased with a linear correlation (Fig. 1a,b). It turned out that the fluorescence-quenched state is preserved in the absence of DTT or trypsin, indicating that the fluorescence activation necessarily requires both cleavages on disulfide and amide of the iRGD probe. The quenching efficiency of the intact AF488-iRGDK-dab in PBS buffer (pH 7.4) was estimated as 96% that was determined based on the fluorescence intensity ratio between the intact and the fully dequenched solutions at the same concentration (Fig. 1c).[35] It was observed that in the absence of both DTT and trypsin, the quenched fluorescence of intact AF488-iRGDK-dab was stably maintained at various pH (pH 4.4–12) and in human serum for 2 h (quenching efficiency: 94–95%, Fig. 1c), validating that physiological environments other than an abnormally raised proteolytic condition have a minimal influence on the fluorescence recovery. Taken together, it is suggested that the fluorescence activation of the dye/quencher-conjugated iRGD probes are exclusively responsive to the coexistence of bioreductants and proteases, being useful for monitoring the tumoral microenvironment.
Fig. 1.
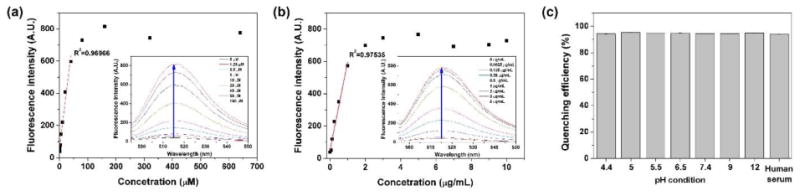
Fluorescence responses of AF488-iRGDK-dab (10 μM) to (a) various concentrations of DTT (0–640 μM) under the excessive trypsin condition (10 μg/mL) in PBS (pH 7.4), and (b) various concentrations of trypsin (0–10 μg/mL) under the excessive DTT condition (640 μM) in PBS (pH 7.4). The fluorescence spectra were obtained after mixing at 37 °C for 1 h (ex/em = 488/515 nm). (c) Quenching stability of AF488-iRGDK-dab (10 μM) at various pH (4.4–12) and human serum solution. Quenching efficiencies were estimated based on the fluorescence intensity of AF488-iRGDK-dab after mixing at 37 °C for 2 h (ex/em = 488/515 nm).
3.3. In vitro fluorescence activation in cancer cells (2D) and tumor spheroids (3D)
To confirm the roles of receptors (αvβ3 integrin and NRP-1) for the cancer cell binding and internalization of iRGD (Scheme 1) [20], three types of cancer cells (A-431, PC-3, and U-87 MG) were treated with fluorescence-quenched peptides (AF488-iRGDK-dab, 4 and AF488-CG7CK-dab, 6) and observed with a fluorescence microscope (Supplementary Fig. S1). After 1 h incubation, the fluorescence of AF488-iRGDK-dab was still quenched in A-431 cells that are devoid of both αvβ3 integrin and NRP-1. [36, 37] In contrast, clear fluorescence signals of the AF488 dye attached to AF488-iRGDK-dab were observed in PC-3 and U-87 MG cells expressing both the receptors.[38-40] The iRGD-free control peptide without targeting and internalizing functions, AF488-CG7CK-dab, showed no fluorescence dequenching response in all the cancer cells studied. These results elucidate that the receptor-mediated multistep mechanism involved in the function of iRGD is indeed operative depending on both αvβ3 integrin and NRP-1 in cancer cells. The iRGD probe showed minimal cytotoxicity (Supplementary Fig. S2), further validating its utility for biomedical uses.
For in-depth evaluation of the iRGD functions in tumor microenvironment, we prepared an U-87 MG tumor spheroid model that is known to closely mimic tumor tissues with many features of tumor microenvironment [41, 42]. In particular, tumor spheroids establish active protease secretion by cells into extracellular matrix [43, 44], which is suitable to assess the cleavage and the penetration of the iRGD probe. After treating the spheroids with AF488-iRGDK-dab and AF488-CG7CK-dab for 1 h, vertical sections of the tumor spheroids were collected from top to bottom by confocal fluorescence microscopy. As shown in Fig. 2, the quenched fluorescence of AF488-iRGDK-dab was dramatically recovered in the tumor spheroid, whereas that of AF488-CG7CK-dab was not activated under the same condition. Furthermore, the dequenched fluorescence of AF488-iRGDK-dab was widespread within the entire tumor spheroid (Supplementary Fig. S3, S4), indicating prominent tumor penetration and accumulation of the fluorescence-labeled iRGD fragment generated by the proteolytic cleavage event.
Fig. 2.
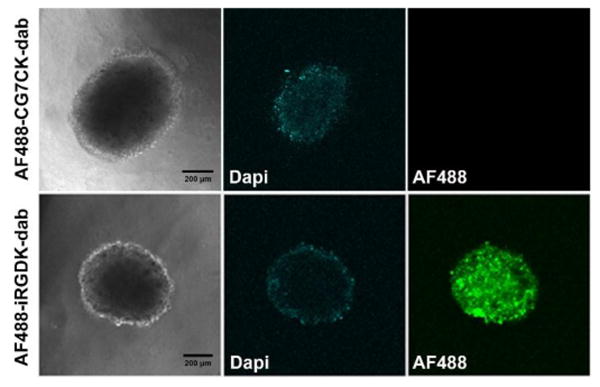
Confocal images of U-87 MG spheroids treated with AF488-iRGDK-dab and AF488-CG7CK-dab (1 μM) for 1 h. All images were extracted at the middle of the spheroids. Green, Alexa Fluor 488; blue, nucleus-staining DAPI. Scale bar: 200 μm.
3.4. In vivo imaging of tumors in mice with the iRGD peptide
With successful in vitro fluorescence recovery in response to the tumoral angiogenic receptors, we applied the dye/quencher-conjugated monolithic iRGD probe to the in vivo tumor diagnosis via systemic administration. For in vivo imaging, Cy5.5-iRGDC-BK01 (5) was used as an activatable probe with deep tissue-penetrating NIR fluorescence. For comparison, an iRGD peptide without quencher conjugation (Cy5.5-iRGDC, 6) was used as an ‘always fluorescent’ control. ‘Fluorescence activatable’ or ‘always fluorescent’ iRGD peptides were intravenously injected into the mice bearing a U-87 MG tumor and comparatively imaged at selected time points (excitation and emission at 675 and 720 nm, respectively). It was observed that both the intravenously injected iRGD peptides exhibit good blood circulation throughout the whole body and fast renal clearance, along with significant fluorescence signals from the tumor (in vivo fluorescence images in Supplementary data). As shown in Fig. 3, the ‘always fluorescent’ iRGD peptide (Cy5.5-iRGDC) displayed strong signals in both tumoral as well as normal tissues, which diminished the diagnostic contrast at the tumor. In the case of ‘activatable’ iRGD probe (Cy5.5-iRGDC-BK01), however, the initial weak signal that is mostly localized at the tumor was gradually intensified with time, resulting in more evident tumor visualization. To quantify the imaging contrast, tumor-to-normal tissue ratios (T/N) were determined with signal intensities from the regions indicated blue and red circles in Fig. 3. At initial time points after intravenous injection (20, 60, 120 min), Cy5.5-iRGDC-BK01 indeed provided notably higher T/N ratios than those of Cy5.5-iRGDC with statistical significance (p < 0.05) (Fig. 4a). Such a better imaging contrast is attributable to the tumor-specific signaling ability of Cy5.5-iRGDC-BK01 with minimal nonspecific fluorescence activation in background normal tissues. In addition to the enhanced contrast, ‘activatable’ Cy5.5-iRGDC-BK01 provided more sustaining tumor signals than ‘always fluorescent’ iRGD peptide (Supplementary Fig. S5), revealing an advantageous feature of the activatable iRGD probe for tumor visualization. All these comparative results confirm that the exclusive fluorescence activation by the tumoral proteolytic cleavage event is operative for enhancing the imaging precision in tumor diagnosis. Furthermore, the ‘activatable’ iRGD probe (Cy5.5-iRGDC-BK01) showed minimal in vivo toxicity with no conspicuous abnormities in major organs including heart, lung, liver, spleen, and kidney (Supplementary Fig. S6), validating its potential as a biomedical imaging probe.
Fig. 3.
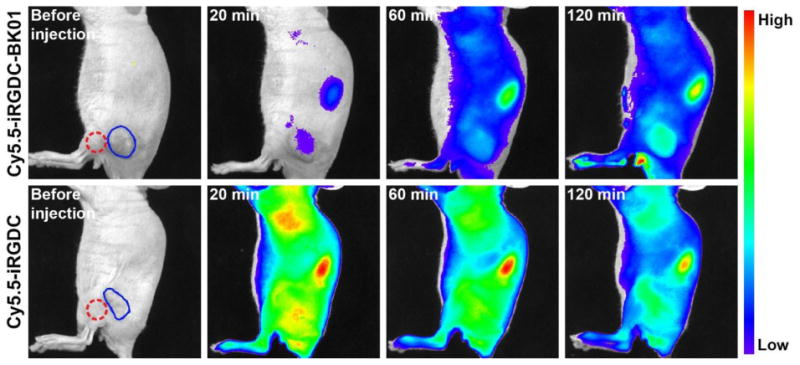
In vivo fluorescence images of U-87 MG tumor-bearing mice before and after intravenous injection of Cy5.5-iRGDC-BK01 and Cy5.5-iRGDC (50 μM). Blue and red circles indicate the tumor and the normal thigh regions nearby the tumor, respectively. The scales of the fluorescence intensity were adjusted to make maximum-to-minimum ratios of average tumor intensity the same.
Fig. 4.
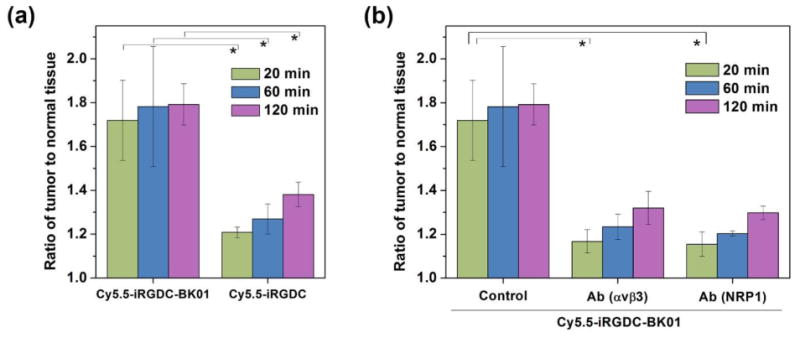
In vivo fluorescence intensity ratios of iRGD probes between tumor and normal tissues. (a) Comparison between probes with and without quencher conjugation (Cy5.5-iRGDC-BK01 and Cy5.5-iRGDC). (b) Receptor inhibition test by anti-αvβ3 antibody or anti-NRP-1 antibody to block binding or internalization function of Cy5.5-iRGDC-BK01. All tumor-to-normal fluorescence intensity ratios were calculated from the ROIs of tumor and normal tissues (blue and red circles in Fig. 3, respectively) at selected time points (20, 60, and 120 min). Error bars are presented as standard deviation (n = 3), and the statistical analysis was evaluated according to one-way ANOVA (*p < 0.05).
We further examined the dependence of in vivo tumor imaging performance on the angiogenic receptors (αvβ3 integrin and NRP-1) by inhibition test. For competitive inhibition, U-87 MG tumor-bearing mice were pre-administered with anti-αvβ3 or anti-NRP-1 antibody before the Cy5.5-iRGDC-BK01 injection. By such pretreatment, cancer targeting or cell internalization of the iRGD peptide is expected to be blocked, which would minimize the probe localization at tumor microenvironment and thus attenuate proteolytic fluorescence activation. Indeed, the fluorescence contrasts (T/N ratios) of the Cy5.5-iRGDC-BK01 were considerably reduced in the anti-αvβ3 or anti-NRP-1 antibody-pretreated mice (Fig. 4b). These results clearly evidence that the intrinsic attributes of iRGD (cancer targeting and internalization) cooperatively assist tumoral proteolytic reaction for tumor-specific signaling, to enhance the diagnostic precision.
4. Conclusion
We have synthesized a fluorescence-activatable multifunctional monolithic probe based on the cyclic iRGD peptide for precise tumor imaging. It has been demonstrated that both reductive and proteolytic cleavages of the probe (on disulfide and peptide linkages, respectively) are necessary for the fluorescence activation of the dye/quencher-conjugated monolithic iRGD probe. It turned out that such cleavage-induced fluorescence recovery is strongly dependent on the angiogenic receptors (αvβ3 and NRP-1) under both in vitro and in vivo conditions. Unlike the ‘always fluorescent’ iRGD control, the dye/quencher-conjugated iRGD peptide showed the background-minimized tumor-specific signal and thus greatly improved the fluorescence contrast between tumor and normal tissues. Besides the outstanding imaging performance, the activatable iRGD-based peptide monolith revealed minimal in vitro and in vivo toxicity profiles, holding potential for biomedical imaging uses.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (2014M3C1A3054141, 2012K1A1A2A01055811), the Korea Health Industry Development Institute (HI15C1540), and the Intramural Research Program of KIST.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 2.Shadidi M, Sioud M. Selective targeting of cancer cells using synthetic peptides. Drug Resist Updat. 2003;6:363–371. doi: 10.1016/j.drup.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Heath VL, Bicknell R. Anticancer strategies involving the vasculature. Nat Rev Clin Oncol. 2009;6:395–404. doi: 10.1038/nrclinonc.2009.52. [DOI] [PubMed] [Google Scholar]
- 4.Koopmans KP, Neels ON, Kema IP, Elsinga PH, Links TP, de Vries EGE, Jager PL. Molecular imaging in neuroendocrine tumors: Molecular uptake mechanisms and clinical results. Crit Rev Oncol Hematol. 2009;71:199–213. doi: 10.1016/j.critrevonc.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Mayer G. The Chemical Biology of Aptamers. Angew Chem Int Ed. 2009;48:2672–2689. doi: 10.1002/anie.200804643. [DOI] [PubMed] [Google Scholar]
- 6.Chari RVJ, Miller ML, Widdison WC. Antibody–Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew Chem Int Ed. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 7.Rao J, Dragulescu-Andrasi A, Yao H. Fluorescence imaging in vivo: recent advances. Curr Opin Biotechnol. 2007;18:17–25. doi: 10.1016/j.copbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi H, Ogawa M, Alford R, Choyke PL, Urano Y. New Strategies for Fluorescent Probe Design in Medical Diagnostic Imaging. Chem Rev. 2009;110:2620–2640. doi: 10.1021/cr900263j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi H, Choyke PL. Target-Cancer-Cell-Specific Activatable Fluorescence Imaging Probes: Rational Design and in Vivo Applications. Acc Chem Res. 2010;44:83–90. doi: 10.1021/ar1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urano Y, Asanuma D, Hama Y, Koyama Y, Barrett T, Kamiya M, Nagano T, Watanabe T, Hasegawa A, Choyke PL, Kobayashi H. Selective molecular imaging of viable cancer cells with pH-activatable fluorescence probes. Nat Med. 2009;15:104–109. doi: 10.1038/nm.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinstain R, Savariar EN, Felsen CN, Tsien RY. In Vivo Targeting of HydrogenPeroxide by Activatable Cell-Penetrating Peptides. J Am Chem Soc. 2014;136:874–877. doi: 10.1021/ja411547j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santra S, Kaittanis C, Santiesteban OJ, Perez JM. Cell-Specific, Activatable, andTheranostic Prodrug for Dual-Targeted Cancer Imaging and Therapy. J Am Chem Soc. 2011;133:16680–16688. doi: 10.1021/ja207463b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MH, Han JH, Kwon PS, Bhuniya S, Kim JY, Sessler JL, Kang C, Kim JS. Hepatocyte-Targeting Single Galactose-Appended Naphthalimide: A Tool for Intracellular Thiol Imaging in Vivo. J Am Chem Soc. 2011;134:1316–1322. doi: 10.1021/ja210065g. [DOI] [PubMed] [Google Scholar]
- 14.Ueki N, Lee S, Sampson NS, Hayman MJ. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat Commun. 2013;4 doi: 10.1038/ncomms3735. [DOI] [PubMed] [Google Scholar]
- 15.Zhuang YD, Chiang PY, Wang CW, Tan KT. Environment-Sensitive Fluorescent Turn-On Probes Targeting Hydrophobic Ligand-Binding Domains for Selective Protein Detection. Angew Chem Int Ed. 2013;52:8124–8128. doi: 10.1002/anie.201302884. [DOI] [PubMed] [Google Scholar]
- 16.Arap W, Pasqualini R, Ruoslahti E. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Xie J, Chen X. Peptides and Peptide Hormones for Molecular Imaging and Disease Diagnosis. Chem Rev. 2010;110:3087–3111. doi: 10.1021/cr900361p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosca EV, Koskimaki JE, Rivera CG, Pandey NB, Tamiz AP, Popel AS. Anti-Angiogenic Peptides for Cancer Therapeutics. Current Pharm Biotechnol. 2011;12:1101–1116. doi: 10.2174/138920111796117300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar R, Shin WS, Sunwoo K, Kim WY, Koo S, Bhuniya S, Kim JS. Small conjugate-based theranostic agents: an encouraging approach for cancer therapy. Chem Soc Rev. 2015;44:6670–6683. doi: 10.1039/c5cs00224a. [DOI] [PubMed] [Google Scholar]
- 20.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci USA. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye Y, Zhu L, Ma Y, Niu G, Chen X. Synthesis and evaluation of new iRGD peptide analogs for tumor optical imaging. Bioorg Med Chem Lett. 2011;21:1146–1150. doi: 10.1016/j.bmcl.2010.12.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su S, Wang H, Liu X, Wu Y, Nie G. iRGD-coupled responsive fluorescent nanogel for targeted drug delivery. Biomaterials. 2013;34:3523–3533. doi: 10.1016/j.biomaterials.2013.01.083. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Ji M, Wong MK, Joo KI, Wang P. Enhanced Therapeutic Efficacy of iRGD-Conjugated Crosslinked Multilayer Liposomes for Drug Delivery. Biomed Res Int. 2013;2013:11. doi: 10.1155/2013/378380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu G, Gao X, Hu Q, Kang T, Liu Z, Jiang M, Miao D, Song Q, Yao L, Tu Y, Pang Z, Chen H, Jiang X, Chen J. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials. 2013;34:5138–5148. doi: 10.1016/j.biomaterials.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 27.Shen J, Meng Q, Sui H, Yin Q, Zhang Z, Yu H, Li Y. iRGD Conjugated TPGS Mediates Codelivery of Paclitaxel and Survivin shRNA for the Reversal of Lung Cancer Resistance. Mol Pharmaceutics. 2013;11:2579–2591. doi: 10.1021/mp400576f. [DOI] [PubMed] [Google Scholar]
- 28.Nie X, Zhang J, Xu Q, Liu X, Li Y, Wu Y, Chen C. Targeting peptide iRGD-conjugated amphiphilic chitosan-co-PLA/DPPE drug delivery system for enhanced tumor therapy. J Mater Chem B. 2014;2:3232–3242. doi: 10.1039/c3tb21744b. [DOI] [PubMed] [Google Scholar]
- 29.Peng ZH, Kopeček J. Enhancing Accumulation and Penetration of HPMA Copolymer-Doxorubicin Conjugates in 2D and 3D Prostate Cancer Cells via iRGD Conjugation with an MMP-2 Cleavable Spacer. J Am Chem Soc. 2015;137:6726–6729. doi: 10.1021/jacs.5b00922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komatsu T, Johnsson K, Okuno H, Bito H, Inoue T, Nagano T, Urano Y. Real-Time Measurements of Protein Dynamics Using Fluorescence Activation-Coupled Protein Labeling Method. J Am Chem Soc. 2011;133:6745–6751. doi: 10.1021/ja200225m. [DOI] [PubMed] [Google Scholar]
- 31.Arai S, Yoon SI, Murata A, Takabayashi M, Wu X, Lu Y, Takeoka S, Ozaki M. Fluorescent “Turn-on” system utilizing a quencher-conjugated peptide for specific protein labeling of living cells. Biochem Biophys Res Commun. 2011;404:211–216. doi: 10.1016/j.bbrc.2010.11.095. [DOI] [PubMed] [Google Scholar]
- 32.Myochin T, Hanaoka K, Iwaki S, Ueno T, Komatsu T, Terai T, Nagano T, Urano Y. Development of a Series of Near-Infrared Dark Quenchers Based on Si-rhodamines and Their Application to Fluorescent Probes. J Am Chem Soc. 2015;137:4759–4765. doi: 10.1021/jacs.5b00246. [DOI] [PubMed] [Google Scholar]
- 33.Cho HJ, Lee TK, Kim JW, Lee SM, Lee YS. Controllable Core-Shell-Type Resin for Solid-Phase Peptide Synthesis. J Org Chem. 2012;77:9156–9162. doi: 10.1021/jo301716d. [DOI] [PubMed] [Google Scholar]
- 34.Fujii N, Otaka A, Funakoshi S, Bessho K, Watanabe T, Akaji K, Yajima H. Studies on Peptides. CLI. Syntheses of Cystine-Peptides by Oxidation of S-Protected Cysteine-Peptides with Thallium(III) Trifluoroacetate. Chem Pharm Bull. 1987;35:2339–2347. doi: 10.1248/cpb.35.2339. [DOI] [PubMed] [Google Scholar]
- 35.Marras SAE, Kramer FR, Tyagi S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002;30:e122. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haubner R, Weber WA, Beer AJ, Vabuliene E, Reim D, Sarbia M, Becker KF, Goebel M, Hein R, Wester HJ, Kessler H, Schwaiger M. Noninvasive visualization of the activated alpha v beta 3 integrin in cancer patients by positron emission tomography and F-18 Galacto-RGD. PLoS Med. 2005;2:244–252. doi: 10.1371/journal.pmed.0020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slimani H, Guenin E, Briane D, Coudert R, Charnaux N, Starzec A, Vassy R, Lecouvey M, Perret YG, Cao A. Lipopeptide-based liposomes for DNA delivery into cells expressing neuropilin-1. J Drug Targeting. 2006;14:694–706. doi: 10.1080/10611860600947607. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, Chen X. Quantitative PET Imaging of Tumor Integrin αvβ3 Expression with 18F-FRGD2. J Nucl Med. 2006;47:113–121. [PMC free article] [PubMed] [Google Scholar]
- 39.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 Is Expressed by Endothelial and Tumor Cells as an Isoform-Specific Receptor for Vascular Endothelial Growth Factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 40.Hu B, Guo P, Bar-Joseph I, Imanishi Y, Jarzynka MJ, Bogler O, Mikkelsen T, Hirose T, Nishikawa R, Cheng SY. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF//SF autocrine pathway. Oncogene. 2007;26:5577–5586. doi: 10.1038/sj.onc.1210348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutherland R. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240:177–184. doi: 10.1126/science.2451290. [DOI] [PubMed] [Google Scholar]
- 42.Phung YT, Barbone D, Broaddus VC, Ho M. Rapid Generation of In Vitro Multicellular Spheroids for the Study of Monoclonal Antibody Therapy. J Cancer. 2011;2:507–514. doi: 10.7150/jca.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dautzenberg IJC, van den Wollenberg DJM, van den Hengel SK, Limpens RWA, Barcena M, Koster AJ, Hoeben RC. Mammalian orthoreovirus T3D infects U-118 MG cell spheroids independent of junction adhesion molecule-A. Gene Ther. 2014;21:609–617. doi: 10.1038/gt.2014.34. [DOI] [PubMed] [Google Scholar]
- 44.Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Multicellular tumor spheroids: An underestimated tool is catching up again. J Biotechnol. 2010;148:3–15. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.