

Abstract
Cerebral blood flow (CBF) regulation is essential for normal brain function. The mammalian brain has evolved a unique mechanism for CBF control known as neurovascular coupling. This mechanism ensures a rapid increase in the rate of CBF and oxygen delivery to activated brain structures. The neurovascular unit composed of astrocytes, mural vascular smooth muscle cells and pericytes, and endothelia, regulates neurovascular coupling. This article examines the cellular and molecular mechanisms within the neurovascular unit contributing to CBF control, and neurovascular dysfunction in neurodegenerative disorders such as Alzheimer's disease.
Introduction
Brain receives one fifth of the cardiac output, and consumes one fifth of the body's oxygen (O2) and glucose1. Both, O2 and glucose are delivered to neurons by cerebral blood flow (CBF) and transport across the blood-brain barrier (BBB)1–3. Thus, brain functions depend on healthy blood vessels and cardiovascular system. If CBF stops, brain functions stop in seconds, and irreversible damage to neurons occurs in minutes4.
The CBF is maintained by a coordinated action of interconnected blood vessels, which in human brain form a 400-mile long vascular network1. Within this network, cerebral arteries, arterioles and capillaries supply brain with O2, energy metabolites and nutrients. The cerebral venous return removes carbon dioxide (CO2) and metabolic waste products from brain into systemic circulation for clearance by lungs, kidney and liver, respectively.
The mammalian brain has evolved a unique mechanism for regional CBF control known as neurovascular coupling or functional hyperemia5. This mechanism ensures a rapid increase in the rate of CBF to activated brain structures2,5.Under physiological conditions, the capacity of increased CBF and O2 delivery exceeds metabolic demand and oxygen consumption by activated brain sites providing a large gradient for oxygen diffusion to brain cells furthest from capillaries2,6,7. Different cell types within the neurovascular unit (NVU) including astrocytes, mural cells, such as vascular smooth muscle cells (VSMCs) and pericytes, and endothelial cells, contribute to neurovascular coupling1–3,5. The NVU cellular composition differs along the vascular tree (Figure 1). However, within the NVU, mural cells of the vessel wall are the ones with contractile properties that allow direct control of the vessel diameter and blood flow.
Figure 1. A schematic representation of the neurovascular unit showing cellular elements regulating cerebral blood flow along the vascular tree.
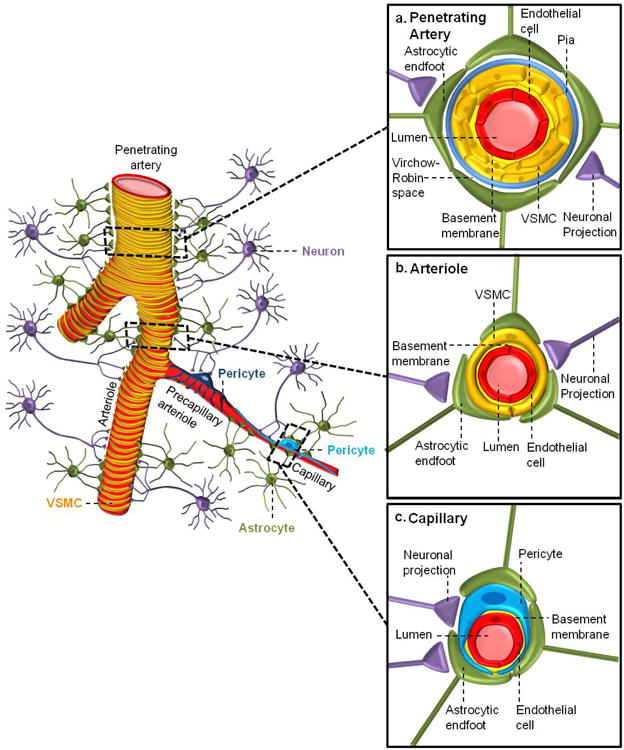
Different cell types of the neurovascular unit (NVU) including neurons, astrocytes, mural cells – vascular smooth muscle cells (VSMCs) and pericytes, and endothelium, regulate cerebral blood flow at different levels of the vascular tree. The cellular composition of the NVU differs along the vascular tree, but the principal cellular components all remain represented, as illustrated here. a) At the level of penetrating arteries, the NVU is composed of endothelial cells making up the inner layer of the vessel wall, covered by a thin extracellular basement membrane and ringed by one to three layers of VSMCs, and ensheathed by pia. The Virchow-Robin space containing the cerebrospinal fluid is between pia and the glia limitans formed by astrocytic endfeet. Both VSMCs and astrocytes are innervated by local neurons. b) Arterioles differ in that there is only one layer of VSMCs, and astrocyte coverage and innervation of the vessel wall and endothelial inner layer display continuity with penetrating arteries, and brain capillaries, above and below the arteriole level, respectively. In addition to VSMCs, precapillary arterioles may also contain transitional pericytes, a cell type between pericyte and VSMCs. c) At the capillary level, the NVU is composed of endothelial cells that share a common basement membrane with pericytes. Pericytes stretch their processes along and around capillaries and make direct interdigitated or “peg-socket”-like contacts with endothelial cells. Pericytes and endothelial cells are covered by astrocyte endfeet. Both astrocytes and pericytes are innervated by local neurons similar as shown for astrocytes and VSMCs in the upper segments of the vascular tree.
Because of the tight interactions between CBF and neuronal activity, the CBF provides important insights into how the brain works. This forms the basis for functional imaging to evaluate the brain's functional networks8,9 and responses to different tasks10–12. Disrupted functional connectivity and neurovascular uncoupling resulting from a mismatch between CBF, O2 delivery and neuronal activity, are found in the early stages of many neurological disorders including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS) and stroke1,3,5. Therefore, understanding the cellular and molecular mechanisms underlying physiological and/or pathophysiological CBF responses is critical for understanding brain functions in health and disease.
In this Review, we discuss CBF regulation by the NVU, and neurovascular dysfunction in neurodegenerative disorders with a focus on AD, because neurovascular deficits are well documented in this disorder compared to other neurodegenerative diseases. First, we examine the role of astrocytes, mural cells, and endothelia in neurovascular coupling, and their interactions with neurons, in the arterial, arteriolar and capillary segments of brain microcirculation. Next, we examine cell-signaling pathways mediating dilation and constriction of cerebral blood vessels resulting in CBF increases and reductions, respectively. Finally, the article considers the link between neurovascular dysfunction and neurodegeneration in AD.
Arterial and arteriolar component
The vast network of arteries, arterioles, and capillaries deliver O2 and nutrients to the brain in a highly regulated manner. The arteries that feed the brain split off the pial arteries that run along the surface of the brain, diving down into the parenchyma, narrowing and branching into arterioles and capillaries. As the size and level of branching change, so too does the cellular composition of the NVU (Figure 1). In this section, we will focus on CBF regulation by arteries and arterioles, role of VSMCs in regulating arteriolar and arterial diameter, and signal transduction pathways within the NVU regulating VSMC tone.
Control of arterial and arteriolar blood flow by VSMCs
Where arteries begin their dive into the brain parenchyma, the NVU is composed of endothelial cells making up the blood vessels surrounded by one to three layers of VSMCs, astrocyte endfeet forming glia limitans that separates cerebrospinal fluid (CSF)-containing Virchow-Robin spaces from brain interstitial fluid (ISF), and neuronal afferent projections13–15. In the brain parenchyma, the arteries narrow into arterioles with a single layer of VSMCs and closer apposition of astrocyte endfeet (Figure 1). Arteries and arterioles ensure blood delivery to the brain at a fairly uniform pressure, such that the pulsatile effect of heartbeat is less pronounced in the downstream cerebral microvessels.
During functional hyperemia, arterioles dilate and this dilation propagates at a high-speed in a retrograde direction to upstream arteries, including branches of pial arteries12,14,16–19. Studies in rats have shown that dilation may extend more than 1 mm away from the center of the responding brain region12. In addition to regulating CBF delivery to the activated brain sites, studies in rats20 and mice21 have demonstrated that arterioles deliver the majority of O2 during the resting state, which is considered to provide a safe margin of O2 supply to cerebral tissue22. Recent studies in pericyte-deficient mice indicated, however, that capillaries and capillary pericytes also play a role in O2 delivery to the brain23. More work is needed to determine the exact contributions of arteriolar versus capillary O2 delivery under physiological and pathophysiological conditions, particularly in regards to the timing of arteriolar and capillary dilation in response to neuronal stimulus, which remains a topic of debate12,3,19,23–27, as discussed below.
The key signaling pathways regulating arteriolar and arterial relaxation and contraction are illustrated in Figure 2, and described in detail below.
Figure 2. Arteriolar regulation of cerebral blood flow.
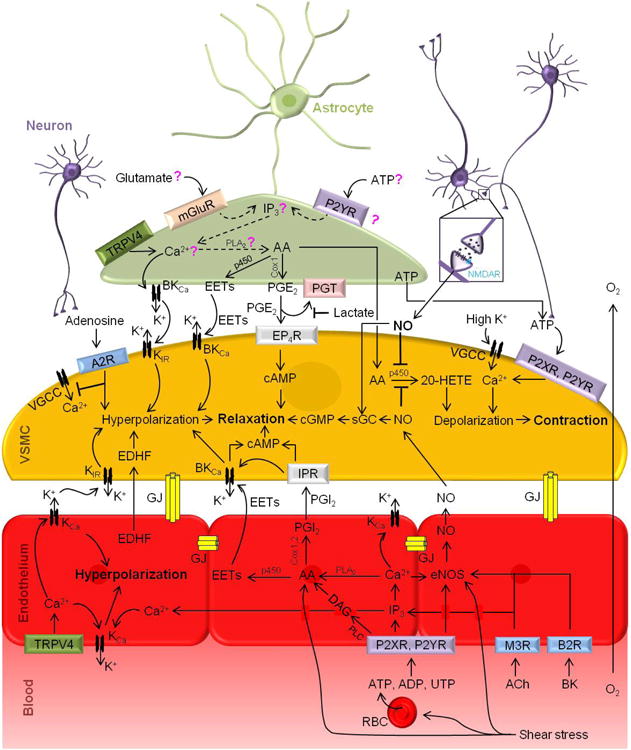
Neurons. Nitric oxide (NO) is a major moderator of functional hyperemia. NO produced by neurons acts directly on VSMCs, leading to VSMC hyperpolarization and relaxation. Adenosine triphosphate (ATP) and adenosine released by neuronal activity can also act directly on VSMCs through purinergic P2XR and P2YR receptors resulting in constriction, or adenosine 2A receptors (A2R) resulting in relaxation, respectively. Neuronal-mediated large increases in extracellular [K+] activate VSMC voltage-gated calcium channels (VGCCs), resulting in VSMCs intracellular [Ca2+] increases, leading to depolarization and contraction.
Astrocytes. The role of astrocytes in neurovascular coupling to arterioles is controversial. Glutamate or ATP released from neurons are postulated to act on metabotropic glutamate receptors (mGluR) or P2YR on astrocytes, respectively, to initiate 1,4,5-trisphosphate (IP3)-dependent intracellular [Ca2+] increase, which has been shown by some studies to contribute to neurovascular coupling, but disputed by others. According to some studies, intracellular [Ca2+] rise launches signaling cascades in the astrocytes and release of vasoactive ions and molecules from astrocyte endfeet to VSMCs, mainly K+ ions from large conductance calcium- activated potassium channels (BKCa), arachidonic acid (AA), through phospholipase A2 (PLA2) pathway, and AA metabolic products epoxyeicosatetraenoic acids (EETs), and prostaglandin E2 (PGE2), via cytochrome P450 (p450) and cyclooxygenase 1 (Cox1), respectively. Extracellular Ca2+ intake from transient receptor potential vanilloid channel 4 (TRPV4) provides another means of increasing intracellular [Ca2+]. Dashed lines and question marks indicate pathways for which there is limited or conflicting data in the literature.
VSMCs. EETs and moderate increases in extracellular [K+] both act on VSMC potassium channels including BKCa and inward rectifier potassium channels (KIR), resulting in hyperpolarization and relaxation of the VSMCs (left). However, large increases in extracellular [K+] activate VGCCs, resulting in intracellular [Ca2+] increases leading to VSMCs depolarization and contraction (right). PGE2, which acts through prostaglandin EP4 receptors (EP4R) on VSMCs, generates cyclic adenosine monophosphate (cAMP) from intracellular ATP, also producing hyperpolarization and relaxation. PGE2 levels can be modulated by extracellular lactate levels, which can block reuptake of PGE2 by prostaglandin transporters (PGT). Lactate levels depend on the oxygen content of the tissue. Conversely, AA taken in by VSMCs can be metabolized to 20-hydroxyeicosatetraenoic acid (20-HETE), a potent VSMC depolarizer, resulting in VSMC contraction. NO released by neurons or endothelial cells can block VSMC 20-HETE production, modulating VSMC contraction and favoring relaxation through facilitation of conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP) via soluble guanylate cyclase (sGC). Adenosine released by neurons acts on A2R and blocks VSMC VGCC activation leading to VSMCs hyperpolarization and relaxation. In contrast, neuronal release of ATP to VSMCs P2XR and P2YR can increase intracellular [Ca2+], resulting in depolarization and contraction.
Endothelium. Vasoactive compounds, including ATP, adenosine diphosphate (ADP), uridine triphosphate (UTP), acetylcholine (ACh), and bradykinin (BK), in the blood stream can bind their respective receptors (P2XR, P2YR, muscarinic M3R, and B2R) to initiate signaling pathways in endothelial cells similar to the pathways in astrocytes, with the addition of the diacyl-glycerol (DAG) AA pathway mediated by phospholipase C (PLC), generating vasoactive molecules that are released to VSMCs. Intracellular [Ca2+] increases initiated by receptor-mediated signaling of endothelium can produce endothelial AA, EETs, and prostacyclin (PGI2), which act on VSMCs similarly to PGE2, to generate VSMC hyperpolarization and relaxation. The intracellular [Ca2+] rise can also activate endothelial nitric oxide synthase (eNOS) leading to NO production, and endothelial KCa channels, releasing K+ that can act on VSMCs as well as hyperpolarizing the endothelium. Endothelial-derived hyperpolarizing factor (EDHF) can also trigger VSMC hyperpolarization. Additionally, shear stress on the endothelial vessel walls and red blood cells (RBCs) due to blood flow triggers ATP release from RBCs and signaling pathways, including activation of endothelial eNOS, and direct production of AA and its metabolites. Endothelial and endothelial-VSMCs gap junctions (GJs) facilitate retrograde endothelial signal propagation and signaling to VSMCs.
Astrocyte-mediated regulation of VSMCs tone
Astrocytes contribute to the regulation of neural circuit formation28, synapse formation29, neuronal calcium oscillation30, plasticity and memory31, and inflammation in neurodegenerative diseases32. However, whether astrocytes and astrocytic calcium, Ca2+, play a role in the regulation of arteriolar tone, remains a controversial topic as examined below.
Earlier reports were not conclusive as to whether an increase in the intracellular Ca2+ in astrocytes leads to arteriolar constriction or dilation. Photolysis experiments with caged Ca2+ in astrocytes have shown that an increase in the intracellular Ca2+, [Ca2+]i, in the astrocyte endfeet precedes arteriolar constriction in rat and mouse brain slices, which is mediated via the phospholipase A2 (PLA2)-arachidonic acid (AA) pathway in astrocytes followed by free diffusion of AA to VSMCs and conversion into 20-hydroxyeicosatetraenoic acid (20-HETE) via cytochrome p450 (p450) causing VSMCs deplolarization and contraction33 (Figure 2). Using similar rat brain slices another study, however, found exactly the opposite -- that an increase in [Ca2+]i in astrocytes leads to arteriolar dilation, not constriction34. This study has shown that dilation and VSMC relaxation is mediated by metabotropic glutamate receptors (mGluR) and cyclooxygenase (Cox) product prostaglandin E2 (PGE2)34 (Figure 2). The discrepancy between the two studies has been reconciled in part by acknowledging different experimental conditions, such as that slices in the earlier study were preincubated with NG-nitro-l-arginine methyl ester (l-NAME) to block nitric oxide (NO) formation and preconstrict blood vessels34, which has been shown to abolish arteriolar constriction by mGluR agonists leading to a moderate vasodilation33.
The first in vivo study imaging the activity of astrocytes labeled with the Ca2+ indicator rhod-2 in the somatosensory cortex of adult mice found that photolysis of caged Ca2+ in astrocytic endfeet ensheathing small pial and penetrating arteries leads to vasodilation, which is blocked by Cox1 inhibitors35. This study confirmed the role of PLA2-AA pathway in astrocytes and showed that astrocytes in vivo can metabolize AA to epoxyeicosatetraenoic acids (EETs) or PGE2 via cyp450 and Cox1, respectively35. Subsequently, EETs released from astrocytes activate large-conductance calcium-activated potassium (BKCa) channels in VSMCs, whereas PGE2 acts through cyclic adenosine monophosphate (cAMP), both leading to hyperpolarization and VSMCs relaxation36.
A recent study using genetically encoded Ca2+ sensor in astrocytes also demonstrated that physiological activation of neuron terminals triggers [Ca2+]i increases in astrocyte processes increasing local red blood cell (RBC) flow in brain capillaries37. Although suggestive of a major role of capillaries in functional hyperemia37, this study did not directly show whether astrocytic Ca2+ regulates neurovascular coupling to capillary pericytes and/or VSMCs in arterioles, or to both. Another recent in vivo study found that astrocytic Ca2+ does not regulate arteriolar responses26.
Several studies disputed the role of astrocytes and astrocytic [Ca2+]i in regulation of arteriolar responses. For example, stimulus-induced arteriolar dilation persists in inositol 1,4,5-triphosphate (IP3) type-2 receptor (R2) knock-out (KO) mice, in which the primary mechanism of astrocytic [Ca2+]i increase - the release of Ca2+ from intracellular stores following activation of an IP3-dependent pathway38 (Figure 2) - is lacking39. Additionally, in vivo astrocytic Ca2+ increase in response to sensory stimulus was delayed relative to arteriolar dilation, suggesting that astrocytes do not play a role in arteriolar responses via a Ca2+-dependent mechanism39. Others have demonstrated that G-protein-coupled receptors (GPCRs)-IP3R-dependent Ca2+ signal in astrocytes does not mediate arteriolar neurovascular coupling in mice implying that mGluR and purinergic receptors in astrocytes are not involved in regulation of arteriolar dilation40. Additionally, mGluR3, the only mGluR expressed in adult mouse astrocytes, did not generate Ca2+ elevations in response to agonists, suggesting glutamatergic signaling per se is insufficient to trigger astrocytic Ca2+ signaling41. These recent studies39–41 questioned whether mGluR33 and purinergic receptors42 in astrocytes can sense glutamate and adenosine triphosphate (ATP), respectively, contributing to regulation of the arteriolar tone. More recent findings confirmed that astrocytes do not mediate neurovascular coupling to arterioles, and that dilation of arterioles depends on N-methyl-D-aspartate receptor (NMDA) activation and Ca2+-dependent generation of NO by interneurons26.
The non-selective transient receptor potential vanilloid channel 4 (TRPV4) have been shown to stimulate Ca2+-induced Ca2+ release in astrocytic end feet, which amplifies neurovascular coupling43,44 by activating the BKCa channels releasing K+ into the extracellular space36. The moderate increase in extracellular [K+] activates inward rectifier potassium (KIR) channels in VSMCs resulting in hyperpolarization and relaxation, as indicated by ex vivo and in vivo studies45. Interestingly, pathologically high levels of the extracellular [K+], as seen during cortical spreading depolarization, activate voltage-gated calcium channels (VGCCs) in VSMCs depolarizing the cells, which results in VSMCs contraction46.
The metabolic state of the tissue influences both arteriolar dilation and constriction47. Under normoxic pO2 conditions (normal 20% O2), lactate levels in brain slices are elevated compared to hyperoxic pO2 conditions (high 95% O2), which inhibits PGE2 reuptake by prostaglandin transporter in astrocytes leading to elevated PGE2 levels in the extracellular space causing VSMCs relaxation47. In high pO2, however, the AA is converted in VSMCs to 20-HETE leading to arteriolar constriction47 (Figure 2). Although this O2 modulatory effect has been shown using ex vivo preparations of brain slices and retina explants47,48, it has not been confirmed in blood flow studies in hyperoxic animals48, leaving open to question whether pO2 has a role in blood flow regulation in vivo. In contrast, the effects of lactate on modifying vessel dilation in slices47 have been translated in vivo as demonstrated in miniature pig retina49 and guinea pig cochlear organ50, both showing vasodilation after systemic or local lactate administration.
The molecular pathways in astrocytes that generate signals to VSMCs as suggested by some initial studies, but not reproduced by more recent studies, are illustrated in Figure 2. Whether astrocytes are involved in neurovascular coupling to arterioles and arteries remains controversial topic that merits further investigation. Some questions to be addressed are: Can astrocytes contribute to neurovascular coupling by modifying neuronal synaptic activity? Do neurons differentially signal to astrocytes on arterioles and capillaries? Do different types of astrocytes communicate with VSMCs and pericytes?
Direct neuron-mediated regulation of VSMCs tone
Early studies have shown that activation of neuronal NMDA receptors in rabbits leads to concentration-dependent NO-mediated dilatation of pial arterioles51. Subsequent studies have demonstrated that mircroperfusion of NMDA in the striatum of newborn sheep52 or functional stimulation of the somatosensory cortex of rats53 both lead to CBF increases through NO production, establishing neuronal NO as a major regulator of the CBF5. Activation of purinergic P2XR54 and P2YR55 ATP-gated channels in VSMCs has been shown to lead to an increase in [Ca2+]i causing VSMCs contraction. It has been suggested that ATP from both neurons56 and astrocytes57 can activate VCMCs purinergic receptors. Additionally, adenosine released from neurons58 is a potent endothelium-independent vasodilator that acts as a direct VSMCs relaxant2,5,23.
Neurotransmitters including vasoactive intestinal polypeptide, dopamine, substance P, serotonin, gamma-aminobutyric acid, noradrenaline, neuropeptide Y, somatostatin and acetylcholine have all been reported to mediate vascular changes5,12. Different types of inter-neurons have been suggested to control local CBF responses possibly through astrocytes or pericytes10,13. In addition to the role of cholinergic afferents modulating CBF via acetylcholine release13, studies in mice59 and neonatal rats60 have demonstrated that noradrenaline release from locus coeruleus afferents generates vasoconstriction. Recent studies using a combination of optogenetic stimulation and mutiphoton imaging in vivo in mice, have shown both excitatory and inhibitory neurons can signal arteriolar dilation, but only inhibitory neurons releasing neuropeptide Y were able to cause arteriolar constriction61. Currently, more information is needed to determine the action and the exact function of each of the neurotransmitter systems, their afferents and effects on the cells that make up the NVU.
Endothelium-mediated regulation of VSMCs tone
Endothelial cells modulate vascular tone by retrograde propagation of vasodilation16,17. Endothelial hyperpolarization travels distances > 1 mm with limited attenuation, which leads to self-dilation via myoendothelial coupling to VSMCs through myoendothelial gap junctions17 or via a putative endothelial-derived hyperpolarizing factor62. Additionally, endothelial release of NO leads to VSMCs relaxation and arteriolar dilation12. Shear stress on red blood cells (RBC) passing through the vessel can release ATP, which interacts with P2XR and P2YR on endothelial cells resulting in generation of AA, EETs and cAMP17,63. cAMP generates prostacyclin (PGI2), which acts on VSMCs much like PGE2 causing relaxation12.
Mechanical shear stress can also cause direct generation of AA and/or NO via endothelial nitric oxide synthase (eNOS) in endothelial cells to initiate the pathways described above2,17. Endothelial receptor targets besides acetylcholine and ATP include bradykinin, adenosine diphosphate, uridine triphosphate and adenosine12. Receptor binding activates phospholipase C (PLC) or PLA2, which through diacyl-glycerol (DAG) produces EETs and PGI2 that both contribute to VSMCs relaxation via cAMP12 (Figure 2).
While these and many additional studies not included here have allowed us to piece together many aspects of endothelial-mediated arteriole regulatory mechanisms, much of this work comes from in vitro or ex vivo studies, and studies from peripheral tissues. More comprehensive in vivo studies in brain are needed to confirm these mechanisms in the functional regulation of brain arteriole vasculature.
As will be discussed later, the neurovascular coupling, VSMC contractility, and endothelial-dependent and independent mechanisms of arteriolar dilation become dysfunctional in models of neurodegenerative disorders including AD, and in individuals with mild cognitive impairment (MCI) and early stage AD.
Capillary component
Capillaries, the smallest vessels in the brain, branch off of the arterioles and form a rich microvascular network with the largest surface area of 120 cm2 per gram of brain available for transport exchanges of molecules between blood and brain across the endothelium of the BBB1. The BBB is formed by a continuous monolayer of endothelial cells connected by tight junctions, which limits entry of large macromolecules, cells and pathogens into the brain. The BBB allows rapid diffusion of O2 from blood-to-brain and of CO2 from brain-to-blood according to their concentration gradients, and regulates transport of energy metabolites and nutrients into the brain, and clearance of metabolic end products from brain to venous circulation via numerous specialized carrier-mediated and receptor-mediated transport systems in the endothelium15,64.
Brain capillaries are covered by pericytes, which share the basement membrane with endothelial cells15 (Figure 1). Pericytes stretch their processes along and around capillaries, pre-capillary arterioles and post-capillary venules with more longitudinal processes in the middle of capillary bed, more circumferential processes at the arteriole end, and more stellate morphology at the venule end of capillaries65–67. Pericytes and endothelial cells make direct interdigitated or “peg-socket”-like contacts where cytoplasmic protrusions (pegs) of one cell type insert into pockets in the opposing cell membrane (socket) of the other cell type68,69. Although it is not clear which type of junctional proteins mediate in vivo interactions between pericytes and endothelial cells, immunostaining studies suggested involvement of N-cadherin during brain angiogenesis70. Several in vitro culture studies suggested that pericytes express connexin 43 (C×43)71. Expression of C×43 and C×37 in pericytes has been confirmed by single-cell RNA-seq study of different cell types in the mouse cortex and hippocampus72. Whether these connexins mediate pericyte-endothelial, pericyte-pericyte and/or pericyte-astrocyte interactions remains, however, to be determined by future studies.
Pericytes regulate angiogenesis during brain development, formation and maintenance of the BBB, clearance of toxins, and neuroinflammatory and stem cells responses, as recently reviewed73. Studies using multiple models have suggested that pericytes regulate CBF and capillary vascular tone24,66,74–78. Below, we examine CBF control by capillaries, role of pericytes in regulating capillary diameter, and signaling pathways regulating pericyte tone.
Control of capillary blood flow by pericytes
The prevailing view is that pericytes contribute to regulation of capillary diameter and CBF23,24,26,27,65,66,74–78. This view has been challenged, however, by some recent studies25,79. What defines a pericyte and pericyte contractility is discussed in Box 1.
Box 1. Pericyte definition and the question of contractility.
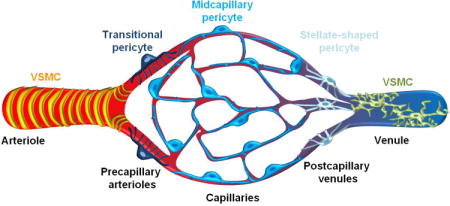
Zimmermann177 included in the definition of pericytes several subtypes that differ by morphology, location within the vascular tree, and function65,73,178. Indeed, a recent morphological study67 indicated that pericytes represent a heterogeneous cell population with different subtypes including “mid-capillary” in the majority of the capillary bed, “transitional” close to vascular smooth muscle cells (VSMCs), and “stellate” on post-capillary venules, as compared to arteriolar ringed VSMCs65,67 (Box 1 figure). Whether different pericyte subtypes have different functions in brain microcirculation such as, for example, regulation of blood-brain barrier (BBB) permeability versus control of blood flow65, remains to be determined.
When it comes to cell contractility, earlier studies have shown that transitional pericytes of pre- and post-capillary microvascular segments express α-smooth muscle actin (SMA)179, a contractile protein typically found in VSMCs25. Using a highly-sensitive immunogold labeling at the ultrastrctural level, SMA expression has been shown in a significant number of mid-capillary pericytes in brain and retina81, which has been corroborated by immunostaining by some studies81,86, but not by others25,79. Besides SMA, other contractile proteins such as myosin and vimentin have been identified in mid-capillary pericytes in brain in situ81. A recent single-cell RNA-seq study demonstrated expression of several contractile proteins in pericytes derived from mouse cortex or hippocampus including skeletal muscle actin, vimentin, desmin, calponin, non-muscle myosin variants, and a low SMA expression72. Importantly, it would be unfortunate to limit one's thinking by assuming that one particular specialized cell type, like VSMCs, must necessarily reflect how the generally occurring phenomenon of contractility in other cell types within the neurovascular unit is achieved, including pericytes.
Although some studies failed to show pericyte contractility in vivo25,79, multiple studies reported that perciytes regulate CBF23,24,26,27,65,66,74–78. Pericytes dilate in response to lactate47,50,78, or contract in response to K+, Ca2+,74 or after application of the thromboxane A2 agonist U46619 in vivo75. They also contract after electrical stimulation or when exposed to neurotransmitters ex vivo in rat retina77 and cerebellar slices24. Human pericytes in culture treated with endothelin-1 also contract76. In vivo studies measuring capillary diameter changes in cortex upon hind limb stimulation23,26 and whisker stimulation24, or in the retina after light stimulation27 also demonstrated active capillary dilation and pericyte ability to regulate capillary tone.
Still, a number of outstanding questions remain to be addressed when it comes to a definition of a pericyte. Some of these questions are: Can a pericyte be molecularly defined by a single cell RNA-seq of a genome-deep quantitative transcriptional profiling compared to other cell types of the neurovascular unit? Do different pericyte subtypes exist in the same organ, and can they be molecularly distinguished from each other in the brain as suggested by morphological studies? Do pericytes have organotypic features, as do endothelial cells, and are pericytes in brain different from pericytes in peripheral organs?
Several studies have shown Ca2+-dependent contraction of pericytes in response to neurotransmitters (e.g., norepinephrine), electrical stimulation or neuronal activity23,24,26,27,74,77,78,80,81. Recent studies demonstrated that astrocytes mediate neurovascular coupling to capillary pericytes but not to arterioles26, and that glial cells in the retina, called Muller cells, also regulate capillary, but not arteriolar diameter in response to light stimulation of neurons27. Pericyte degeneration has been shown to lead to neurovascular uncoupling and reduced oxygen supply to brain, suggesting diminished hemodynamic responses in a pericyte-deficient transgenic model23. Optogenetic experiments indicated initially that pericytes do not contribute to CBF regulation in neural-glial antigen 2-driven NG2-cre channelrhodopsin (ChR2) mice25, which has not been confirmed by another optogenetic study in platelet-derived growth factor receptor beta-driven Pdgfrb-cre ChR2 mice demonstrating that strong 2-photon stimulation leads to pericyte contraction, constriction of capillary lumens and inhibition of RBC flow in vivo (D.A. Hartmann, R.I. Grant and A.Y. Shih, Medical University of South Carolina; personal communication). Whether the differences seen in optogenetic studies are model-dependent and/or light source and duration-dependent, and whether different Cre drivers lead to differential ChR2 expression in different pericyte subpopulations, remains to be examined by future studies.
Recent in vivo studies in mice found that hindlimb stimulation (10s)23,26 or whisker-pad stimulation (15s)24 resulted in capillary dilation ahead of arterioles; this was correlated with pericyte coverage and could be caused by isotonic pericyte relaxation. A shorter stimulation time (2s) produced, however, RBC flow increase without vessel dilation79, and could result from isometric relaxation of pericytes. An earlier study demonstrated that capillaries in the activated rat olfactory glomerulus, but not capillaries adjacent to this glomerulus, actively dilate in response to odor stimulation82. As arterioles do not form a single glomerulus-specific network, their contribution to odor response has not been determined in this model82. Dilations and constrictions of cerebral capillaries have been also observed in rat cortex after forepaw stimulation83 and in response to hypercapnia84. During brain ischemia, pericytes contract and limit CBF by constricting capillaries and trapping blood cells in their lumen, and later die, as shown in a murine model85,86. Pericyte contraction and cell death in response to ischemia has been confirmed ex vivo in brain and retina explants24,77.
While a growing body of evidence supports the role of pericytes in CBF regulation, there is also a growing need to better define pericytes, particularly at the molecular level (Box 1). Future studies should also determine whether all or only some pericyte subpopulations regulate CBF dynamics and/or are more susceptible to ischemic and/or hypoxic insults.
Signaling in astrocytes and pericytes regulating capillary tone
Recent studies have demonstrated that astrocytes mediate neurovascular signaling to capillary pericytes, which involves a rise of [Ca2+]i in astrocytes not by release from stores but by entry through ATP-gated channel - purinergic receptor P2XR, and that Ca2+ generates AA via phospholipase D2 and diacylglycerol kinase rather than PLA226 (Figure 3). The role of a Ca2+-dependent glial cell signaling mechanism in regulating capillary but not arteriole diameter has also been shown in the retina ex vivo and in vivo upon physiological light stimulation27.
Figure 3. Capillary regulation of cerebral blood flow.
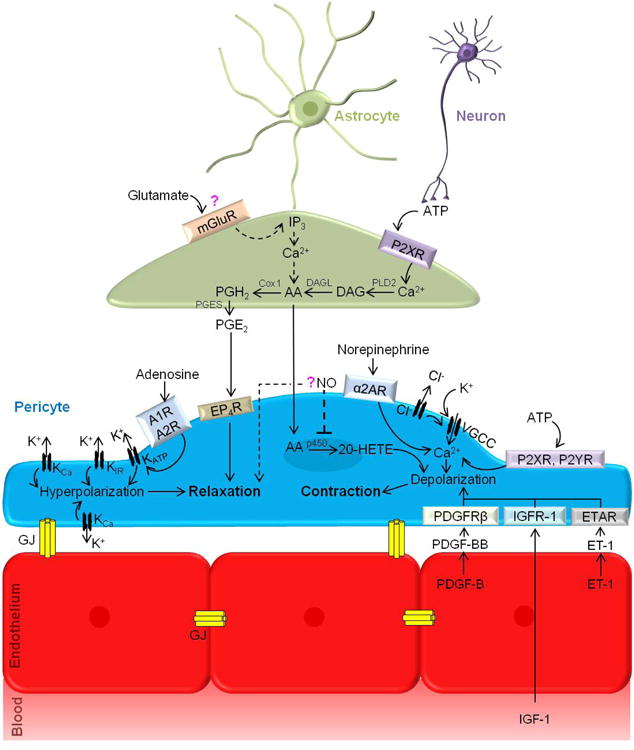
Neuronal ATP can activate P2XRs on astrocytes to produce AA via phospholipase D 2 (PLD2)-mediated production of diacylglycerol (DAG) and subsequent metabolism by DAG lipase (DAGL). Cox1 can metabolize AA to produce PGE2 via prostaglandin H2 (PGH2) and prostaglandin E synthase (PGES), triggering EP4R on pericytes and leading to pericyte relaxation. AA produced in astrocytes can also diffuse into pericytes and form 20-HETE via p450 leading to pericyte depolarization and contraction. Neuronal activity releases glutamate, which has been shown to activate astrocytic mGluRs, triggering an IP3–dependent increase in intracellular [Ca2+] leading to AA production via the PLA2 pathway. However, recent studies contradict this finding (indicated by dashed lines and question mark. Neurotransmitters adenosine, norepinephrine, and ATP have been demonstrated to alter pericyte contractile state. Specifically, adenosine binds A1 and A2 receptors (A1R, A2R) and activates KATP channels leading to hyperpolarization and relaxation. Also, activation of potassium channels including KCa and possibly KIR results in hyperpolarization of pericytes, decreasing Ca2+ entry through voltage-gated channels. Neuronal release of NO, which in pericytes inhibits AA metabolism to 20-HETE, can lead to pericyte relaxation. However, the role of NO in capillary dilation has been questioned (indicated by dashed lines and question mark). Norepinephrine acts through α2-adrenergic receptors (α2AR) leading to increased intracellular [Ca2+], depolarization and contraction. ATP activation of the purinergic receptors P2XR or P2YR on pericytes induces depolarizing currents, and increases intracellular [Ca2+] and pericyte contraction. Additionally, neuronal-mediated large increases in extracellular [K+] activates VGCCs, resulting in pericyte intracellular [Ca2+] increases, depolarization and contraction. Furthermore, several vasoconstrictors, including endothelin-1 (ET-1), platelet-derived growth factor-B (PDGF-BB), both secreted by vascular endothelial cells, or blood-derived insulin-like growth factor (IGF-1), act on their respective receptors (endothelin A receptors (ETAR), Pdgfrβ and IGFR-1) leading to depolarization of pericytes and Ca2+ entry into the cell. Endothelial-endothelial and pericyte-endothelial gap junctions allow fast and direct exchange of small molecules.
Pericytes respond to the AA metabolites PGE2 and 20-HETE to generate relaxation or contraction, respectively, but unlike VSMCs, they do not respond to EETs24,77. Pericytes express both P2X87 and P2Y88 purinergic receptors, which when activated by ATP lead to an increase in Ca2+, depolarization and pericyte contraction. Activation of large- (BKCa) and small- (KCa) conductance potassium channels or ATP-sensitive potassium (KATP) channels results in potassium efflux and hyperpolarization of pericytes, decreasing Ca2+ entry through VGCCs89.
Adenosine that is normally released from neurons and leads to VSMCs relaxation also binds A1 and A2 receptors on pericytes, and activates KATP channels releasing potassium from the cell leading to hyperpolarization and relaxation89. Glutamate-evoked capillary dilation was reduced by blocking NO synthase with L-NG-nitroarginine in brain slices suggesting a role for NO in pericyte relaxation and capillary dilation that was likely mediated by inhibiting the AA conversion to 20-HETE, which blocks depolarization and contraction24. It is unclear if pO2 plays as significant a role in modulating relaxation versus contraction at the capillary level as observed in arterioles24.
Several endothelial-derived vasoactive mediators can also regulate pericyte contraction and relaxation likely via similar mechanisms as in VSMCs89 (Figure 3). A number of questions regarding pericyte signaling in vivo remain, however, to be addressed by future studies including the proposed regulatory neurotransmitter and endothelial signaling pathways. As discussed below, pericytes degenerate in several neurodegenerative disorders including AD and ALS, and are injured during early stages of aging-MCI-AD spectrum. Thus, it is possible that their loss contributes to vascular dysregulation observed in neurological diseases associated with early neurovascular dysfunction as suggested by recent studies in pericyte-deficient mice23.
Alzheimer's vascular dysfunction
In addition to amyloid-β (Aβ), tau pathology and neuron loss, AD is associated with early neurovascular dysfunction, which contributes to disease pathogenesis, as indicated by recent epidemiological, clinical, pathological and experimental studies1,3,5,90–95. Moreover, small vessel disease of the brain has been estimated to contribute wholly or partially to approximately 40% of all dementias worldwide including AD3,96–99.
Multiple risk factors influence AD pathogenesis including genetics, vascular, environment, and lifestyle (Figure 4). According to the two-hit vascular hypothesis of AD1,15, Aβ-independent (hit 1) and Aβ-dependent (hit 2) mechanisms interact and converge on blood vessels leading independently and/or synergistically to neuronal and synaptic dysfunction, neurodegeneration, and cognitive impairment (Figure 4). Besides direct negative effects of CBF reductions and dysregulation, and BBB breakdown and dysfunction on neuronal function and accumulation of Aβ in the brain (hit 1), Aβ-mediated vascular dysfunction (hit 2) could also be an early event in AD pathogenesis5,100. Notably, function of each of the NVU cell types (e.g., VSMCs, pericytes, astrocytes, endothelia) regulating CBF and BBB integrity is affected during different stages of AD by either Aβ-independent or Aβ-dependent mechanisms, and/or both3,5,73,92,100,101, contributing to dementia. Although reduced to two major disease pathways, the two-hit hypothesis considers multiple factors influencing AD.
Figure 4. Neurovascular dysfunction in Alzheimer's disease: two-hit vascular hypothesis.
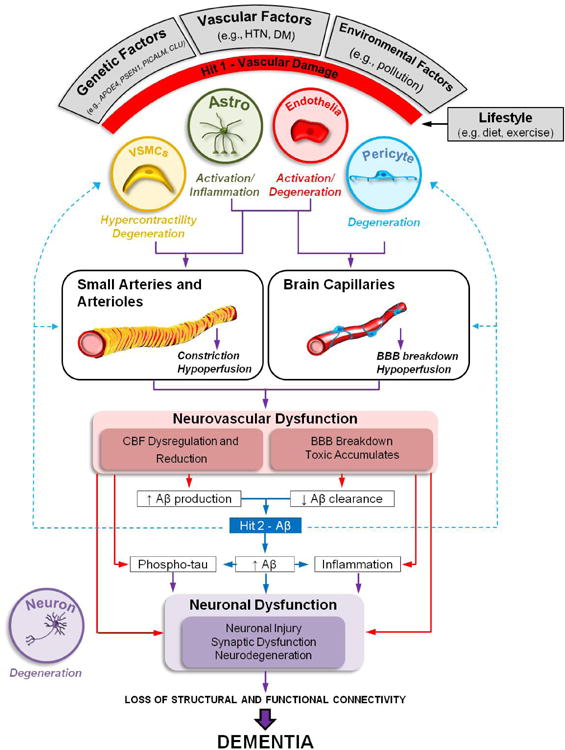
Several genetic risk factors for Alzheimer's disease (AD) (e.g., apolipoprotein E4 gene (APOE4), Presenilin-1 mutations (PSEN1), Phosphatidylinositol Binding Clathrin Assembly Protein (PICALM), Clusterin (CLU), vascular factors (e.g., hypertension (HTN), diabetes mellitus (DM)), and environmental factors (e.g., pollution) lead to neurovascular dysfunction and damage to small arteries, arterioles and brain capillaries via amyloid-β (Aβ)-independent (hit 1, red) and/or Aβ-dependent (hit 2, blue) pathway. Both pathways interact and converge on blood vessels, and can independently or synergistically (purple lines) lead to neuronal injury, synaptic dysfunction and neurodegeneration contributing to dementia. Lifestyle can modify the effects of these hits; for example a moderate exercise and diet have beneficial effects on cardiovascular and cerebrovascular system.
AD affects different cell types of the neurovascular unit. For example, VSMCs hypercontractility and degeneration leads to aberrant responses of small arteries and arterioles, cerebral blood flow (CBF) dysregulation and reductions independently of Aβ or in Aβ-dependent manner. In the Aβ pathway, damage to small arteries and arterioles are often associated with amyloid angiopathy, and rupture of the vessel wall with microhemorrhages. Degeneration of pericytes leads to loss of capillary dilation in response to neuronal stimuli, hypoperfusion and blood-brain barrier (BBB) breakdown with accumulation of blood-derived toxins and fluid in the perivascular spaces. Both, Aβ-independent (e.g., hypoxia, ischemia) and Aβ-dependent mechanisms contribute to changes in capillary circulation. Endothelial damage leads to loss of endothelial-dependent vasodilation, CBF dysregulation and reductions. Activation of astrocytes and microglia mediates inflammatory response and release of vasoactive cytokines and chemokines, further comprising CBF regulation and BBB integrity.
Damage to blood vessels can initiate a cascade of events leading to Aβ accumulation in brain (hit 1), which accelerates the Aβ-dependent pathway of neurodegeneration (hit 2). For example, brain ischemic changes (hit 1) lead to increased Aβ production by stimulating expression of α and γ secretases, enzymes mediating Aβ generation, whereas BBB dysfunction in Aβ clearance receptors lipoprotein receptor and multidrug resistance protein 1 leads to faulty Aβ clearance and retention in brain.
Reduced CBF (hit 1) and elevated Aβ (hit 2) can each independently or synergistically lead to tau phosphorylation (Phospho-tau) and tau pathology in neurons, and worsen neuroinflammation. When combined, they accelerate neuronal damage and injury.
Here, we will review neurovascular deficits during AD pathogenesis, focusing on CBF changes. First, we examine findings in animal models, and next neurovascular dysfunction in humans in relation to cognitive impairment and AD. Many of the molecular and cellular mechanisms underlying arterial, arteriolar and capillary components of CBF regulation that we discussed in the first part of this review have not been studied, however, in animal models of AD, or in humans at risk or diagnosed with AD. Therefore, in the sections below we will also attempt to identify some of the outstanding mechanistic gaps that should be addressed by future studies.
Aβ-independent vascular changes in animal models
Pericyte-deficient transgenic models with undetectable Aβ pathology develop early CBF reductions in gray matter102 and aberrant CBF responses in the presence of initially normal neuronal activity, endothelial-dependent and independent vasodilation, and astrocyte numbers and coverage of the blood vessels23. Pericyte degeneration also leads to early reductions in O2 supply to activated brain23. These vascular changes develop independently of Aβ, and precede neuronal dysfunction and neurodegeneration that develop months later. These findings indicate that pericytes could be an important target in neurological disorders associated with perciyte loss and neurovascular dysfunction. This incudes AD91,103–108, ALS109,110 and stroke24,85,86,111. It is also known that Aβ kills pericytes106,112, which in turn might accelerate neurovascular dysfunction in AD transgenic models overexpressing amyloid precursor protein (APP)105,106 and/or AD patients3,5,100. The relative contributions of Aβ-independent pericyte loss as shown in the ischemic brain24,85,86,111 and Aβ-dependent pericyte degeneration106 remains an important topic for future research of CBF dysregulation in AD.
Hypertension (HTN) is a risk factor for AD. A number of studies have shown that HTN alters functional hyperemia and endothelial function113. Angiotensin II (ANGII)-induced hypertension in rodents attenuates CBF responses to whisker stimulation114 due to diminished endothelium-dependent vasodilation to acetylcholine115. Interestingly, cerebrovascular effects of ANGII are independent on the elevation of blood pressure116–118. Alterations in cerebrovascular responses to endothelium-dependent vasodilators have also been show in rodent models of chronic HTN114,116,119. Relatively little is known, however, about the role of other NVU cell types in the pathogenesis of HTN, and in relation to HTN role in AD pathogenesis. For example, age-dependent loss of pericytes has been shown in ANGII model of HTN120, but the effects on neurovascular coupling and the underlying molecular and cellular mechanisms remain largely unexplored.
Studies in transgenic Notch3R169C mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) expressing the pathogenic Notch3 mutation in VSMCs and pericytes, as in human CADASIL, have shown that both mural cell types accumulate toxic Notch3 aggregates and degenerate121–123. Consistent with loss of mural cells, these mice develop impaired CBF autoregulation124. The exact contributions of VSMCs versus pericytes to CBF dysregulation and possible downstream molecular targets remain, however, presently unknown.
CBF reductions and vascular dysfunction were also found in apolipoprotein E4 (APOE4) transgenic mice with targeted replacement of murine APOE with human APOE4 gene125–127, the major genetic risk factor for AD. It has been also shown that vascular phonotype in APOE4 mice precedes neuronal and synaptic dysfunction125. Whether the same cyclophilin A pathway in pericytes mediating the BBB breakdown in APOE4 mice125 is also involved in CBF dysregulation is not clear at present.
Together, these studies suggest that CBF dysregulation develops early in experimental models of pericyte, VSMCs or endothelial dysfunction, which can lead to neuronal dysfunction and loss independently of Aβ. Little is known, however, about signaling pathways in the NVU that lead to CBF dysregulation in these models. More work is needed to uncover the mechanistic links between dysregulated CBF in the Aβ-independent vascular models and astrocyte-pericyte signaling26,27 and the roles of astrocytic Ca2+ 26,27 and neuronal and endothelial NO5.
Aβ-dependent vascular changes in animal models
Early studies in the isolated rat aorta have shown that Aβ exerts vasoconstrictive properties128. The follow-up in vivo findings in transgenic mice expressing Aβ precursor protein (APP) Swedish mutation indicated that Aβ attenuates acetylcholine-mediated endothelium-dependent vasodilation129. Studies in young Tg2576 mice expressing APP Swedish mutation demonstrated reduced cerebrovascular reactivity to endothelium-dependent vasodilators (e.g., acetylcholine, bradykinin, calcium ionophore A23187) and increased response to vasoconstrictors acting directly on VSMCs (e.g., thromboxane A2 analogue U46619)130, as well as altered neurovascular coupling131. Collectively, these studies suggest that accumulation of low levels soluble Aβ prior to Aβ deposition leads to a global impairment of vascular responses. Aβ mediated generation of ET-1 through the receptor for advanced glycation end products (RAGE) in brain endothelium that binds Aβ has been also shown to led to CBF reductions in Tg2576 mice132,133.
Additional investigations indicated that NADPH-oxidase is a key source of the radicals mediating neurovascular dysfunction caused by Aβ134, which causes endothelial dysfunction by activating transient receptor potential melastatin-2 channels in endothelial cells via poly(ADP)-ribose polymerase pathway135. CD36, another scavenger receptor that binds Aβ, also leads to Aβ-mediated oxidative stress in cerebral blood vessels causing diminished neurovascular coupling136. Interestingly, RAGE regulates CD36 expression136. The exact relationship between CD36 and RAGE in Aβ-induced CBF dysregulation132,133, however, awaits further exploration.
Consistent with these studies, findings in transgenic mice with vasculotropic Dutch and Iowa Aβ mutations crossed with the Swedish APP mutants, i.e., TgSwDI APP mice, and in Tg2576 APP mice have shown that arterial VSMCs exhibit a greatly reduced ability to clear Aβ, which leads to Aβ accumulation in the vessel wall causing cerebral amyloid angiopathy (CAA) and impaired vascular reactivity137. This has been recently confirmed in older APP mice carrying Swedish and Indiana mutations (i.e., hAPPJ20)138.
Similar to AD patients, TgSwDI APP mice and Tg2576 APP mice express low levels of serum response factor (SRF) and myocardin (MYCD), the two transcription factors that control VSMCs differentiation, which leads to elevated expression of several SRF-MYCD-directed VSMCs contractile proteins including SMA, calponin and myosin heavy chain139. This in turn leads a hypercontractile VSMCs phenotype, and diminished endothelium-dependent and endothelium-independent relaxation causing attenuated CBF responses139. Additionally, genes that regulate Ca2+ homeostasis, such as myosin light-chain kinase, calsequestrin 1 and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 are also elevated in AD VSMCs contributing to VSMCs hypercontractile phenotype139. Although, SRF-MYOCD-dependent regulation of VSMCs contractile proteins and Ca2+ homeostasis genes is Aβ-independent139 these changes likely act synergistically with Aβ to accelerate CBF reductions in AD models, and possibly in AD. Finally, Aβ leads to pericyte cell death in human cultured pericytes106,112 and Tg2576 APP mice105,106, which might contribute to CBF dysregulation.
In summary, a body of evidence suggests that Aβ has vasoactive and vasculotoxic effects on cerebral blood vessels affecting different cellular components of the NVU that regulate CBF. Therefore, preventing Aβ accumulation will eliminate Aβ-dependent effects on CBF dysregulation. The question persists, whether removing Aβ will have the same beneficial effects on already damaged vessels. Recent studies have suggested, however, that counteracting the deleterious effects of Aβ after vascular depositions in aged TgSwDI APP mice is not effective in reversing the neurovascular dysfunction owing to VSMCs damage caused by aging and massive Aβ deposition105. Importantly, the BBB dysfunction of Aβ clearance receptors and reduced CBF both promote Aβ accumulation in the brain and vessel wall (hit 1) enhancing Aβ pathogenic effects (Figure 4).
Combined Aβ and vascular models
Here, we briefly illustrate with a few examples interaction of Aβ with some factors negatively affecting cerebral circulation. More detailed discussion on this topic could be found elsewhere97,98.
Regarding HTN, it has been shown that ANGII-induced HTN in mice worsens Aβ-induced neurovascular dysfunction and promotes β-secretase activity increasing amyloidogenic APP processing, which may contribute to the pathogenic interaction between HTN and AD115. ANGII-induced HTN also impairs CBF, cognition and functional connectivity in APP/PS1 mice and decreases functional connectivity in wild type mice140, suggesting another possible mechanism for a link between midlife HTN, decreased cerebral hemodynamics and connectivity.
Cerebral hypoperfusion accelerates CAA in TgSwDI APP mice141. CAA, on the other hand, leads to a more severe cerebrovascular dysfunction than Aβ alone causing intra- and post-ischemic CBF deficits, which exacerbates cerebral infarction, as shown in Tg2576 mice142.
Elevated levels of homocysteine in plasma, hyperhomocysteinemia (HHcy), have been shown to impair action and synthesis of endothelial NO leading to CBF dysregulation143. Induction of HHcy in wild-type mice models vascular dementia by inducing cerebral microhemorrhages and neuroinflammation144. Moreover, HHcy shifts Aβ deposition to the vasculature and exacerbates memory impairment in APP/PS1 mice145.
Cerebrovascular reactivity in Alzheimer's disease
Individuals with early stage, probable AD compared to cognitively normal controls have impaired cerebrovascular reactivity in response to hypercapnia induced by CO2 inhalation and display large CBF fluctuations after repeated sit-stands with no changes in the systemic arterial blood fluctuations146, suggesting that local CBF dysregulation develops early in AD in the presence of intact cardiovascular control of the arterial blood pressure.
Recent studies have shown that individuals carrying APOE4 gene, the major genetic risk factor for AD, compared to non-carriers develop early impaired cerebrovascular reactivity to a memory task and during CO2 inhalation147. Using CO2 inhalation challenge, a larger study in cognitively normal APOE4 carriers compared to APOE4 non-carriers confirmed impaired CBF responses, suggesting that early CBF dysregulation contributes to cognitive impairment in APOE4 carriers148. Using the CO2 inhalation challenge and blood-oxygen level dependent (BOLD)-fMRI, another study found that cerebrovascular deficits in AD could be associated with amyloid deposits, as detected by positron emission tomography (PET) with 11C-Pittsburg compound B149.
Impaired cerebrovascular reactivity indicates damage to the cerebral blood vessels and CBF regulatory mechanisms, which may result in CBF reductions and/or neurovascular uncoupling, as discussed below. The molecular and cellular mechanisms involved remain, however, mostly unknown at present.
Cerebral blood flow reductions in Alzheimer's disease
An earlier large population-based study showed that diminished CBF velocity precedes cognitive decline and hippocampal atrophy150. Additionally, individuals exhibiting greater CBF velocity had larger hippocampal and amygdalar volumes150.
Early studies in individuals with mild cognitive impairment (MCI) with memory loss (e.g., amnestic MCI) have shown CBF reductions151,152 in the posterior cingulate gyrus and precuneus using single positron emission tomography (SPECT), that is also confirmed in individuals with probable AD153. As the posterior cingulate gyrus and precuneus participate early in the pathophysiology of disrupted functional connectivity in AD154, it is possible that initial vascular dysregulation may precede and/or trigger disrupted brain connectivity in these regions. Interestingly, a decrease in regional CBF in the posterior cingulate gyrus and precuneus in early stage AD occurs prior to a loss of gray matter volume153,155 suggesting that CBF reductions precede brain atrophy. Consistent with lower CBF values, and findings demonstrating that glucose transport into the brain depends on the CBF101,156, the same brain regions showing diminished CBF showed diminished brain glucose uptake in early stages of AD, as detected by 18F-fluorodeoxyglucose (FDG)-PET157.
Reductions in regional CBF in the bilateral parietal areas and the precuneus preceded conversion of MCI to AD158,159. Additionally, CBF decline in the frontal, parietal, and temporal cortices preceded the onset of cognitive decline in non-demented APOE4 carriers compared to APOE4 non-carriers160. Consistent with these findings, low glucose uptake by the posterior cingulate, parietal, temporal, and prefrontal cortex was found in young APOE4 carriers compared to APOE4 non-carriers161. More recent in vivo assessment of regional CBF via non-invasive arterial spin labeling (ASL)-MRI, relying on magnetic labeling of arterial blood water, and simultaneous 18F-FDG-PET acquisition confirmed a high correlation between regional brain hypoperfusion and impaired glucose uptake by the brain of AD individuals compared to controls162. Collectively, these findings demonstrate that regional CBF reductions and diminished regional glucose delivery are amongst the earliest functional changes preceding cognitive decline in AD.
A recent large study in healthy controls and MCI and AD patients demonstrated that CBF changes determined by ASL-MRI, and vascular dysregulation are the initial events associated with cognitive decline before changes in classical AD biomarkers Aβ and tau occur94. Another recent study revealed that global CBF was lower in cognitively normal APOE4-carriers compared to APOE4 non-carriers prior to development of amyloid deposits163. Moreover, APOE4 subjects who developed amyloid deposition showed lower global CBF compared to those who were free of amyloid163. These data suggest that APOE4 gene can diminish CBF in humans independently of Aβ, similar as shown in animal studies125, but during AD progression APOE4-induced amyloid deposition results in even greater CBF reductions. A recent contrast-enhanced perfusion MRI study confirmed significantly reduced CBF in parietotemporal regions and the basal ganglia in MCI compared to healthy controls prior to any gray matter brain atrophy164, suggesting that CBF alterations are driven by APOE4 gene164. These recent studies support the role of reduced cerebral perfusion in the development of cognitive impairment prior to reductions in cortical thickness and/or hippocampal atrophy.
Neurovascular uncoupling in Alzheimer's disease
Studies using BOLD-fMRI detected changes in brain activation in AD in the hippocampus in response to visual stimuli and memory encoding tasks165 and face-name association tasks166. BOLD-fMRI studies in cognitively normal individuals with genetic risk for AD (i.e., at least one APOE4 allele) revealed decreased brain activation in key areas engaged during naming and fluency tasks when compared to age-matched controls with no risk factors167. Changes in hemodynamic responses to visual stimuli were also found in patients with CAA compared to healthy elderly controls168. As the diminished BOLD-fMRI responses to different cognitive tasks are regionally specific, collectively BOLD-fMRI findings indicate multiple pathophysiological alterations in neurovascular coupling in early stage AD, and in individuals at genetic risk for AD, demonstrating important role of local CBF dysregulation during AD progression.
Because of the tight relationship between CBF and neuronal activity, the resting state fMRI, also called brain “default-mode network” (DMN), relies on local CBF to provide important insights into how the brain works9,169. DMN typically includes the medial prefrontal cortex, the posterior cingulate and precuneus, inferior parietal lobe, lateral temporal cortex, and hippocampus169–171. A decreased resting-state activity in the posterior cingulate and hippocampus was reported in subjects during early stages of AD when compared to age-matched elderly controls, suggesting disrupted connectivity between these two regions172. In addition, disrupted hippocampal connectivity was also confirmed in the medial prefrontal cortex and cingulate cortex in AD compared to cognitively normal controls173.
A large cross-sectional study found a significant decrease in DMN functional connectivity by fMRI in autosomal dominant AD (ADAD) including presenilin 1 (PSEN1), PSEN2 and APP mutation carriers before clinically evident dementia174. A decrease in functional connectivity with increasing dementia show similarity in ADAD and sporadic AD175. Early disrupted functional connectivity by fMRI in DMN regions including hippocampus, parahippocampus, precuneus, and cingulate, mediotemporal, and orbital cortices has been demonstrated in non-demented APOE4 carriers compared to APOE4 non-carriers prior to amyloid deposits176, suggesting that early neurovascular dysfunction occurs prior to Aβ pathology.
Collectively, these studies provide strong evidence demonstrating that impaired cerebrovascular reactivity, CBF reductions, and CBF dysregulation, and BBB breakdown are early events in the AD pathophysiological cascade. How they relate to each other and disrupted brain connectivity remains to be determined by future studies (Box 2).
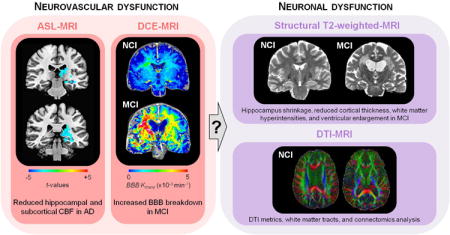
Box 2. Early imaging biomarkers of neurovascular and neuronal dysfunction.
As recently reviewed180, multiple imaging modalities and biomarkers in the living human brain have been used to evaluate neurovascular and neuronal dysfunction during early stages of Alzheimer's disease (AD) pathophysiology. Here, we will briefly examine some of these biomarkers and how they relate to each other. For example, early cerebral blood flow (CBF) studies using transcranial Doppler150 or SPECT151,155 and recent studies using arterial spin labeling (ASL)-MRI94,163 and contrast-enhanced MRI164 have suggested that CBF reductions in the hippocampus, cingulate cortex and precuneus precede cognitive decline, hippocampal and cortical atrophy and/or amyloid deposition during early stages of AD, and are observed in individuals with genetic risk for AD including APOE4 carriers. Similarly, recent studies using dynamic contrast-enhanced (DCE)-MRI have shown increased blood-brain barrier (BBB) permeability in the hippocampus during normal aging with no cognitive impairment (NCI), which worsens with mild cognitive impairment (MCI), but precedes changes in hippocampal volume91. DCE-MRI studies also confirmed BBB breakdown in the hippocampus and cortical and subcortical regions during early stages of AD181,182 and in individuals with vascular cognitive impairment183. Some studies have suggested that neurovascular dysfunction occurs prior to amyloid deposition and tau-mediated neurodegeneration94, and/or changes in Aβ and tau cerebrospinal fluid (CSF) biomarkers91,92. However, longitudinal studies are needed to establish the exact temporal pattern of neurovascular changes relative to brain atrophy, and how they relate to Aβ and tau CSF biomarkers, amyloid and tau lesions, and cognitive decline, particularly in individuals with genetic risk for sporadic AD (i.e., APOE4) and autosomal dominant AD (ADAD) (i.e., PSEN1, PSEN2 and APP mutation carriers). It is also unclear how CBF changes relate to BBB changes and white matter hyperintensities in early stages of NCI and aging-MCI-AD spectrum. It is also almost completely unclear how imaging biomarkers of neurovascular dysfunction relate to disrupted structural connectivity determined by diffusion tensor imaging (DTI)-MRI and DTI-based connectomics analysis184 or functional connectivity by resting state fMRI in default-mode network regions169,171.
Conclusions and perspectives
Recent studies have demonstrated that signaling pathways in astrocytes, VSMCs, pericytes, and endothelia control CBF. Importantly, many of the same pathways are found in different NVU cell types, raising a possibility that targeting more common pathways might result in synergistic cellular responses contributing to CBF control. However, some outstanding questions remain as indicated throughout the review. There is also a large gap in understanding how the basic physiological findings of CBF regulation translate to animal disease models, and humans with healthy brain and neurological disorders associated with neurovascular dysfunction such as AD, as well as between animal disease models and human neurological conditions.
To determine the relative contributions of different cell types and pathways in CBF control, the pathways should be targeted in a cell-specific manner taking advantage of advanced genetic engineering and pharmacological approaches. For example, using cell-specific ablation models and/or disrupting pathways in a cell-specific manner in VSMCs, pericytes, endothelia and astrocytes, coupled with a single-cell RNA sequencing and proteomic analyses, should be able to define more precisely the role of each NVU cell type in neurovascular coupling and maintenance of cerebrovascular integrity. In vivo CBF studies are also needed to better understand the neuronal component of CBF regulation, and the role of different neurotransmitter systems in neurovascular coupling.
Although we know that each cell type regulating CBF is affected in neurodegenerative disorders such as AD, the translation of knowledge from bench to bedside has been slow. Studies in the living human brain have established that aberrant cerebrovascular reactivity, CBF reductions and dysregulated CBF are a prominent feature during early stages across the aging-MCI-AD spectrum. However, the pharmaceutical industry and academia are still not reacting in a way to explore systematically whether treating neurovascular dysfunction will delay onset and/or slow down the neurodegenerative process. The question persists – are we missing an important opportunity by not utilizing the wealth of knowledge generated in the vascular field, and not focusing enough on vascular dysregulation as a major therapeutic target in neurodegenerative disease such as AD?
Acknowledgments
The work of B.V.Z. is supported by the National Institutes of Health grants R01AG023084, R01NS090904, R01NS034467, R01AG039452, R01NS100459, P01AG052350 and Cure for Alzheimer's fund. We thank Melanie Sweeney for careful reading of the manuscript.
References
- 1.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease andother disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attwell D, et al. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 6.Buxton RB. Interpreting oxygenation-based neuroimaging signals: the importance and the challenge of understanding brain oxygen metabolism. Front Neuroenergetics. 2010 doi: 10.3389/fnene.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin AL, Fox PT, Hardies J, Duong TQ, Gao JH. Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. Proc Natl Acad Sci. 2010;107:8446–8451. doi: 10.1073/pnas.0909711107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci U S A. 2003;100:253–258. doi: 10.1073/pnas.0135058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snyder AZ, Raichle ME. A brief history of the resting state: the Washington University perspective. NeuroImage. 2012;62:902–910. doi: 10.1016/j.neuroimage.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–625. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- 11.Lauritzen M, Mathiesen C, Schaefer K, Thomsen KJ. Neuronal inhibition and excitation, and the dichotomic control of brain hemodynamic and oxygen responses. NeuroImage. 2012;62:1040–1050. doi: 10.1016/j.neuroimage.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 12.Hillman EMC. Coupling mechanism and significance of the BOLD signal: a status report. Annu Rev Neurosci. 2014;37:161–181. doi: 10.1146/annurev-neuro-071013-014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cauli B, Hamel E. Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics. 2010;2:9. doi: 10.3389/fnene.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015;163:1064–1078. doi: 10.1016/j.cell.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen BR, Bouchard MB, McCaslin AFH, Burgess SA, Hillman EMC. Highspeed vascular dynamics of the hemodynamic response. NeuroImage. 2011;54:1021–1030. doi: 10.1016/j.neuroimage.2010.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EMC. A Critical Role for the Vascular Endothelium in Functional Neurovascular Coupling in the Brain. J Am Heart Assoc. 2014;3:e000787–e000787. doi: 10.1161/JAHA.114.000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iadecola C, Yang G, Ebner TJ, Chen G. Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J Neurophysiol. 1997;78:651–659. doi: 10.1152/jn.1997.78.2.651. [DOI] [PubMed] [Google Scholar]
- 19.Tian P, et al. Cortical depth-specific microvascular dilation underlies laminar differences in blood oxygenation level-dependent functional MRI signal. Proc Natl Acad Sci U S A. 2010;107:15246–15251. doi: 10.1073/pnas.1006735107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devor A, et al. ‘Overshoot’ of O2 is required to maintain baseline tissue oxygenation at locations distal to blood vessels. J Neurosci Off J Soc Neurosci. 2011;31:13676–13681. doi: 10.1523/JNEUROSCI.1968-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasischke KA, et al. Two-photon NADH imaging exposes boundaries of oxygen diffusion in cortical vascular supply regions. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2011;31:68–81. doi: 10.1038/jcbfm.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakadžić S, et al. Large arteriolar component of oxygen delivery implies a safe margin of oxygen supply to cerebral tissue. Nat Commun. 2014;5:5734. doi: 10.1038/ncomms6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kisler K, et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017 doi: 10.1038/nn.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall CN, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill RA, et al. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron. 2015;87:95–110. doi: 10.1016/j.neuron.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra A, et al. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci. 2016;19:1619–1627. doi: 10.1038/nn.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biesecker KR, et al. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J Neurosci Off J Soc Neurosci. 2016;36:9435–9445. doi: 10.1523/JNEUROSCI.1782-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015;18:942–952. doi: 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai HH, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358–362. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gundersen V, Storm-Mathisen J, Bergersen LH. Neuroglial Transmission. Physiol Rev. 2015;95:695–726. doi: 10.1152/physrev.00024.2014. [DOI] [PubMed] [Google Scholar]
- 31.Lee HS, et al. Astrocytes contribute to gamma oscillations and recognition memory. Proc Natl Acad Sci. 2014;111:E3343–E3352. doi: 10.1073/pnas.1410893111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- 34.Zonta M, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 35.Takano T, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 36.MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otsu Y, et al. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 2015;18:210–218. doi: 10.1038/nn.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agulhon C, et al. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nizar K, et al. In vivo Stimulus-Induced Vasodilation Occurs without IP3 Receptor Activation and May Precede Astrocytic Calcium Increase. J Neurosci. 2013;33:8411–8422. doi: 10.1523/JNEUROSCI.3285-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonder DE, McCarthy KD. Astrocytic Gq-GPCR-Linked IP3R-Dependent Ca2+ Signaling Does Not Mediate Neurovascular Coupling in Mouse Visual Cortex In Vivo. J Neurosci. 2014;34:13139–13150. doi: 10.1523/JNEUROSCI.2591-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piet R, Jahr CE. Glutamatergic and purinergic receptor-mediated calcium transients in Bergmann glial cells. J Neurosci Off J Soc Neurosci. 2007;27:4027–4035. doi: 10.1523/JNEUROSCI.0462-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim KJ, et al. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J Neurosci Off J Soc Neurosci. 2015;35:8245–8257. doi: 10.1523/JNEUROSCI.4486-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longden TA, Nelson MT. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirc N Y N 1994. 2015;22:183–196. doi: 10.1111/micc.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Girouard H, et al. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A. 2010;107:3811–3816. doi: 10.1073/pnas.0914722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordon GRJ, Choi HB, Rungta RL, Ellis-Davies GCR, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mishra A, Hamid A, Newman EA. Oxygen modulation of neurovascular coupling in the retina. Proc Natl Acad Sci U S A. 2011;108:17827–17831. doi: 10.1073/pnas.1110533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brazitikos PD, Pournaras CJ, Munoz JL, Tsacopoulos M. Microinjection of L-lactate in the preretinal vitreous induces segmental vasodilation in the inner retina of miniature pigs. Invest Ophthalmol Vis Sci. 1993;34:1744–1752. [PubMed] [Google Scholar]
- 50.Dai M, Yang Y, Shi X. Lactate dilates cochlear capillaries via type V fibrocyte-vessel coupling signaled by nNOS. AJP Heart Circ Physiol. 2011;301:H1248–H1254. doi: 10.1152/ajpheart.00315.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in brain. Circ Res. 1993;72:476–480. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- 52.Bhardwaj A, et al. P-450 epoxygenase and NO synthase inhibitors reduce cerebral blood flow response to N-methyl-D-aspartate. Am J Physiol Heart Circ Physiol. 2000;279:H1616–1624. doi: 10.1152/ajpheart.2000.279.4.H1616. [DOI] [PubMed] [Google Scholar]
- 53.Buerk DG, Ances BM, Greenberg JH, Detre JA. Temporal dynamics of brain tissue nitric oxide during functional forepaw stimulation in rats. NeuroImage. 2003;18:1–9. doi: 10.1006/nimg.2002.1314. [DOI] [PubMed] [Google Scholar]
- 54K.Kur J, Newman EA. Purinergic control of vascular tone in the retina. J Physiol. 2014;592:491–504. doi: 10.1113/jphysiol.2013.267294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horiuchi T, Dietrich HH, Tsugane S, Dacey RG. Analysis of purine- and pyrimidine-induced vascular responses in the isolated rat cerebral arteriole. Am J Physiol Heart Circ Physiol. 2001;280:H767–776. doi: 10.1152/ajpheart.2001.280.2.H767. [DOI] [PubMed] [Google Scholar]
- 56.Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pascual O, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 58.Lovatt D, et al. Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci. 2012;109:6265–6270. doi: 10.1073/pnas.1120997109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bekar LK, Wei HS, Nedergaard M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32:2135–2145. doi: 10.1038/jcbfm.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kozberg MG, Chen BR, DeLeo SE, Bouchard MB, Hillman EMC. Resolving the transition from negative to positive blood oxygen level-dependent responses in the developing brain. Proc Natl Acad Sci U S A. 2013;110:4380–4385. doi: 10.1073/pnas.1212785110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uhlirova H, et al. Cell type specificity of neurovascular coupling in cerebral cortex. eLife. 2016;5 doi: 10.7554/eLife.14315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wölfle SE, et al. Non-linear relationship between hyperpolarisation and relaxation enables long distance propagation of vasodilatation. J Physiol. 2011;589:2607–2623. doi: 10.1113/jphysiol.2010.202580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ralevic V, Dunn WR. Purinergic transmission in blood vessels. Auton Neurosci Basic Clin. 2015;191:48–66. doi: 10.1016/j.autneu.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 64.Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid β-peptide elimination from the brain. J Neurochem. 2010;115:1077–1089. doi: 10.1111/j.1471-4159.2010.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Attwell D, Mishra A, Hall CN, O'Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016;36:451–455. doi: 10.1177/0271678X15610340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernández-Klett F, Priller J. Diverse functions of pericytes in cerebral blood flow regulation and ischemia. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2015;35:883–887. doi: 10.1038/jcbfm.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hartmann DA, et al. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics. 2015;2:041402. doi: 10.1117/1.NPh.2.4.041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cuevas P, et al. Pericyte endothelial gap junctions in human cerebral capillaries. Anat Embryol (Berl) 1984;170:155–159. doi: 10.1007/BF00319000. [DOI] [PubMed] [Google Scholar]
- 69.Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 70.Gerhardt H, Wolburg H, Redies C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev Dyn Off Publ Am Assoc Anat. 2000;218:472–479. doi: 10.1002/1097-0177(200007)218:3<472::AID-DVDY1008>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 71.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 73.Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19:771–783. doi: 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dai M, Nuttall A, Yang Y, Shi X. Visualization and contractile activity of cochlear pericytes in the capillaries of the spiral ligament. Hear Res. 2009;254:100–107. doi: 10.1016/j.heares.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci U S A. 2010;107:22290–22295. doi: 10.1073/pnas.1011321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neuhaus AA, Couch Y, Sutherland BA, Buchan AM. Novel method to study pericyte contractility and responses to ischaemia in vitro using electrical impedance. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16659495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamanishi S, Katsumura K, Kobayashi T, Puro DG. Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H925–934. doi: 10.1152/ajpheart.01012.2005. [DOI] [PubMed] [Google Scholar]
- 79.Wei HS, et al. Erythrocytes Are Oxygen-Sensing Regulators of the Cerebral Microcirculation. Neuron. 2016;91:851–862. doi: 10.1016/j.neuron.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kawamura H, et al. Effects of angiotensin II on the pericyte-containing microvasculature of the rat retina. J Physiol. 2004;561:671–683. doi: 10.1113/jphysiol.2004.073098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bandopadhyay R, et al. Contractile proteins in pericytes at the blood-brain and blood-retinal barriers. J Neurocytol. 2001;30:35–44. doi: 10.1023/a:1011965307612. [DOI] [PubMed] [Google Scholar]
- 82.Chaigneau E, Oheim M, Audinat E, Charpak S. Two-photon imaging of capillary blood flow in olfactory bulb glomeruli. Proc Natl Acad Sci U S A. 2003;100:13081–13086. doi: 10.1073/pnas.2133652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stefanovic B, et al. Functional reactivity of cerebral capillaries. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2008;28:961–972. doi: 10.1038/sj.jcbfm.9600590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hutchinson EB, Stefanovic B, Koretsky AP, Silva AC. Spatial flow-volume dissociation of the cerebral microcirculatory response to mild hypercapnia. NeuroImage. 2006;32:520–530. doi: 10.1016/j.neuroimage.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 85.Fernández-Klett F, et al. Early loss of pericytes and perivascular stromal cell-induced scar formation after stroke. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2013;33:428–439. doi: 10.1038/jcbfm.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yemisci M, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 87.Kawamura H, et al. ATP: a vasoactive signal in the pericyte-containing microvasculature of the rat retina. J Physiol. 2003;551:787–799. doi: 10.1113/jphysiol.2003.047977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lacar B, Herman P, Hartman NW, Hyder F, Bordey A. S phase entry of neural progenitor cells correlates with increased blood flow in the young subventricular zone. PloS One. 2012;7:e31960. doi: 10.1371/journal.pone.0031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamilton NB, Attwell D, Hall CN. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics. 2010;2 doi: 10.3389/fnene.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toledo JB, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI). Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun. 2013;1:65. doi: 10.1186/2051-5960-1-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Montagne A, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sweeney MD, Sagare AP, Zlokovic BV. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer's disease. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2015;35:1055–1068. doi: 10.1038/jcbfm.2015.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016;15:934–943. doi: 10.1016/S1474-4422(16)30029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. Alzheimer's Disease Neuroimaging Initiative Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934. doi: 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. 2016;1862:887–900. doi: 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wardlaw JM, et al. STandards for ReportIng Vascular changes on nEuroimaging (STRIVE v1). Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–838. doi: 10.1016/S1474-4422(13)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Montine TJ, et al. ADRD 2013 Conference Organizing Committee. Recommendations of the Alzheimer's disease-related dementias conference. Neurology. 2014;83:851–860. doi: 10.1212/WNL.0000000000000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Snyder HM, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2015;11:710–717. doi: 10.1016/j.jalz.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hachinski V. World Stroke Organization. Stroke and Potentially Preventable Dementias Proclamation: Updated World Stroke Day Proclamation. Stroke J Cereb Circ. 2015;46:3039–3040. doi: 10.1161/STROKEAHA.115.011237. [DOI] [PubMed] [Google Scholar]
- 100.Iadecola C. Cerebrovascular effects of amyloid-beta peptides: mechanisms and implications for Alzheimer's dementia. Cell Mol Neurobiol. 2003;23:681–689. doi: 10.1023/A:1025092617651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 102.Bell RD, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sengillo JD, et al. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol Zurich Switz. 2013;23:303–310. doi: 10.1111/bpa.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Halliday MR, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2015 doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Park L, et al. Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke J Cereb Circ. 2014;45:1815–1821. doi: 10.1161/STROKEAHA.114.005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sagare AP, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 107.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 108.Baloyannis SJ, Baloyannis IS. The vascular factor in Alzheimer's disease: a study in Golgi technique and electron microscopy. J Neurol Sci. 2012;322:117–121. doi: 10.1016/j.jns.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 109.Winkler EA, et al. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 2013;125:111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Winkler EA, Sengillo JD, Bell RD, Wang J, Zlokovic BV. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32:1841–1852. doi: 10.1038/jcbfm.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Underly RG, et al. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J Neurosci. 2017;37:129–140. doi: 10.1523/JNEUROSCI.2891-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wilhelmus MMM, et al. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am J Pathol. 2007;171:1989–1999. doi: 10.2353/ajpath.2007.070050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertens Dallas Tex 1979. 2013;62:810–817. doi: 10.1161/HYPERTENSIONAHA.113.01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Capone C, et al. Central Cardiovascular Circuits Contribute to the Neurovascular Dysfunction in Angiotensin II Hypertension. J Neurosci. 2012;32:4878–4886. doi: 10.1523/JNEUROSCI.6262-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Faraco G, et al. Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016;36:241–252. doi: 10.1038/jcbfm.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Capone C, et al. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. AJP Heart Circ Physiol. 2011;300:H397–H407. doi: 10.1152/ajpheart.00679.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kazama K. Angiotensin II Impairs Neurovascular Coupling in Neocortex Through NADPH Oxidase-Derived Radicals. Circ Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- 118.Girouard H. Angiotensin II Attenuates Endothelium-Dependent Responses in the Cerebral Microcirculation Through Nox-2-Derived Radicals. Arterioscler Thromb Vasc Biol. 2006;26:826–832. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- 119.Didion SP, Sigmund CD, Faraci FM, Katusic ZS. Impaired Endothelial Function in Transgenic Mice Expressing Both Human Renin and Human Angiotensinogen Editorial Comment. Stroke. 2000;31:760–765. doi: 10.1161/01.str.31.3.760. [DOI] [PubMed] [Google Scholar]
- 120.Toth P, et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2013;33:1732–1742. doi: 10.1038/jcbfm.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ghosh M, et al. Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol. 2015;78:887–900. doi: 10.1002/ana.24512. [DOI] [PubMed] [Google Scholar]
- 122.Joutel A, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120:433–445. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dziewulska D, Lewandowska E. Pericytes as a new target for pathological processes in CADASIL. Neuropathol Off J Jpn Soc Neuropathol. 2012;32:515–521. doi: 10.1111/j.1440-1789.2011.01290.x. [DOI] [PubMed] [Google Scholar]
- 124.Lacombe P, Oligo C, Domenga V, Tournier-Lasserve E, Joutel A. Impaired cerebral vasoreactivity in a transgenic mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy arteriopathy. Stroke J Cereb Circ. 2005;36:1053–1058. doi: 10.1161/01.STR.0000163080.82766.eb. [DOI] [PubMed] [Google Scholar]
- 125.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wiesmann M, et al. A Dietary Treatment Improves Cerebral Blood Flow and Brain Connectivity in Aging apoE4 Mice. Neural Plast. 2016;2016:6846721. doi: 10.1155/2016/6846721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Alata W, Ye Y, St-Amour I, Vandal M, Calon F. Human apolipoprotein E ɛ4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2015;35:86–94. doi: 10.1038/jcbfm.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 129.Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci Off J Soc Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Iadecola C, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 131.Niwa K, et al. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A. 2000;97:9735–9740. doi: 10.1073/pnas.97.17.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deane R, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 133.Deane R, et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Park L, et al. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci Off J Soc Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Park L, et al. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nat Commun. 2014;5:5318. doi: 10.1038/ncomms6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Park L, et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc Natl Acad Sci U S A. 2011;108:5063–5068. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bell RD, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kimbrough IF, Robel S, Roberson ED, Sontheimer H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer's disease. Brain J Neurol. 2015;138:3716–3733. doi: 10.1093/brain/awv327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chow N, et al. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proc Natl Acad Sci U S A. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wiesmann M, et al. Angiotensin II, hypertension, and angiotensin II receptor antagonism: Roles in the behavioural and brain pathology of a mouse model of Alzheimer's disease. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16667364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Okamoto Y, et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol (Berl) 2012;123:381–394. doi: 10.1007/s00401-011-0925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Milner E, et al. Cerebral amyloid angiopathy increases susceptibility to infarction after focal cerebral ischemia in Tg2576 mice. Stroke. 2014;45:3064–3069. doi: 10.1161/STROKEAHA.114.006078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Toda N, Okamura T. Hyperhomocysteinemia impairs regional blood flow: involvements of endothelial and neuronal nitric oxide. Pflugers Arch. 2016;468:1517–1525. doi: 10.1007/s00424-016-1849-y. [DOI] [PubMed] [Google Scholar]
- 144.Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2013;33:708–715. doi: 10.1038/jcbfm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sudduth TL, Weekman EM, Brothers HM, Braun K, Wilcock DM. β-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res Ther. 2014;6:32. doi: 10.1186/alzrt262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Den Abeelen ASSM, Lagro J, van Beek AHEA, Claassen JAHR. Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer's disease. Curr Alzheimer Res. 2014;11:11–17. doi: 10.2174/1567205010666131119234845. [DOI] [PubMed] [Google Scholar]
- 147.Suri S, et al. Reduced cerebrovascular reactivity in young adults carrying the APOE ε4 allele. Alzheimers Dement J Alzheimers Assoc. 2015;11:648–657.e1. doi: 10.1016/j.jalz.2014.05.1755. [DOI] [PubMed] [Google Scholar]
- 148.Hajjar I, Sorond F, Lipsitz LA. Apolipoprotein E, carbon dioxide vasoreactivity, and cognition in older adults: effect of hypertension. J Am Geriatr Soc. 2015;63:276–281. doi: 10.1111/jgs.13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yezhuvath US, et al. Forebrain-dominant deficit in cerebrovascular reactivity in Alzheimer's disease. Neurobiol Aging. 2012;33:75–82. doi: 10.1016/j.neurobiolaging.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ruitenberg A, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005;57:789–794. doi: 10.1002/ana.20493. [DOI] [PubMed] [Google Scholar]
- 151.Kogure D, et al. Longitudinal evaluation of early Alzheimer's disease using brain perfusion SPECT. J Nucl Med Off Publ Soc Nucl Med. 2000;41:1155–1162. [PubMed] [Google Scholar]
- 152.Minoshima S, et al. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- 153.Hirao K, et al. Functional interactions between entorhinal cortex and posterior cingulate cortex at the very early stage of Alzheimer's disease using brain perfusion single-photon emission computed tomography. Nucl Med Commun. 2006;27:151–156. doi: 10.1097/01.mnm.0000189783.39411.ef. [DOI] [PubMed] [Google Scholar]
- 154.Yuan B, et al. Differential Effects of APOE Genotypes on the Anterior and Posterior Subnetworks of Default Mode Network in Amnestic Mild Cognitive Impairment. J Alzheimers Dis JAD. 2016 doi: 10.3233/JAD-160353. [DOI] [PubMed] [Google Scholar]
- 155.Matsuda H, et al. Longitudinal evaluation of both morphologic and functional changes in the same individuals with Alzheimer's disease. J Nucl Med Off Publ Soc Nucl Med. 2002;43:304–311. [PubMed] [Google Scholar]
- 156.Daulatzai MA. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer's disease. J Neurosci Res. 2016 doi: 10.1002/jnr.23777. [DOI] [PubMed] [Google Scholar]
- 157.Mosconi L, et al. Functional interactions of the entorhinal cortex: an 18F-FDG PET study on normal aging and Alzheimer's disease. J Nucl Med Off Publ Soc Nucl Med. 2004;45:382–392. [PubMed] [Google Scholar]
- 158.Hirao K, et al. The prediction of rapid conversion to Alzheimer's disease in mild cognitive impairment using regional cerebral blood flow SPECT. NeuroImage. 2005;28:1014–1021. doi: 10.1016/j.neuroimage.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 159.Borroni B, et al. Combined 99mTc-ECD SPECT and neuropsychological studies in MCI for the assessment of conversion to AD. Neurobiol Aging. 2006;27:24–31. doi: 10.1016/j.neurobiolaging.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 160.Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010;67:93–98. doi: 10.1001/archneurol.2009.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Reiman EM, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen Y, et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology. 2011;77:1977–1985. doi: 10.1212/WNL.0b013e31823a0ef7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Michels L, et al. Arterial spin labeling imaging reveals widespread and Aβ-independent reductions in cerebral blood flow in elderly apolipoprotein epsilon-4 carriers. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016;36:581–595. doi: 10.1177/0271678X15605847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wirth M, et al. Divergent regional patterns of cerebral hypoperfusion and gray matter atrophy in mild cognitive impairment patients. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16641128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Small SA, Perera GM, DeLaPaz R, Mayeux R, Stern Y. Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer's disease. Ann Neurol. 1999;45:466–472. doi: 10.1002/1531-8249(199904)45:4<466::aid-ana8>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 166.Sperling RA, et al. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:44–50. doi: 10.1136/jnnp.74.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Smith CD, et al. Altered brain activation in cognitively intact individuals at high risk for Alzheimer's disease. Neurology. 1999;53:1391–1396. doi: 10.1212/wnl.53.7.1391. [DOI] [PubMed] [Google Scholar]
- 168.Dumas A, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol. 2012;72:76–81. doi: 10.1002/ana.23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kim SG, Ogawa S. Biophysical and physiological origins of blood oxygenation level-dependent fMRI signals. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2012;32:1188–1206. doi: 10.1038/jcbfm.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dennis EL, Thompson PM. Functional brain connectivity using fMRI in aging and Alzheimer's disease. Neuropsychol Rev. 2014;24:49–62. doi: 10.1007/s11065-014-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Raichle ME, et al. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang L, et al. Changes in hippocampal connectivity in the early stages of Alzheimer's disease: evidence from resting state fMRI. NeuroImage. 2006;31:496–504. doi: 10.1016/j.neuroimage.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 174.Chhatwal JP, et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology. 2013;81:736–744. doi: 10.1212/WNL.0b013e3182a1aafe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Thomas JB, et al. Functional connectivity in autosomal dominant and late-onset Alzheimer disease. JAMA Neurol. 2014;71:1111–1122. doi: 10.1001/jamaneurol.2014.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sheline YI, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J Neurosci Off J Soc Neurosci. 2010;30:17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zimmerman KW. Der feinere Bau der Blutkapillaren. 1923;68:29–36. [Google Scholar]
- 178.Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol Zurich Switz. 2014;24:371–386. doi: 10.1111/bpa.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nehls V, Drenckhahn D. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol. 1991;113:147–154. doi: 10.1083/jcb.113.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Montagne A, et al. Brain imaging of neurovascular dysfunction in Alzheimer's disease. Acta Neuropathol (Berl) 2016;131:687–707. doi: 10.1007/s00401-016-1570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Starr JM, Farrall AJ, Armitage P, McGurn B, Wardlaw J. Blood-brain barrier permeability in Alzheimer's disease: a case-control MRI study. Psychiatry Res. 2009;171:232–241. doi: 10.1016/j.pscychresns.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 182.Van de Haar HJ, et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology. 2016;152244 doi: 10.1148/radiol.2016152244. [DOI] [PubMed] [Google Scholar]
- 183.Taheri S, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke J Cereb Circ. 2011;42:2158–2163. doi: 10.1161/STROKEAHA.110.611731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nir TM, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI). Effectiveness of regional DTI measures in distinguishing Alzheimer's disease, MCI, and normal aging. NeuroImage Clin. 2013;3:180–195. doi: 10.1016/j.nicl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]