

There is a renewed interest in immunotherapy for soft tissue and bone sarcoma. This review details evidence for an immune response in sarcomas and the different therapeutic modalities in immunotherapy for sarcomas, focusing on the current state of immunotherapy clinical trials in sarcomas, immune checkpoint inhibitors, vaccine trials, and adoptive cell therapy.
Keywords: Sarcoma, Immunotherapy, Vaccines, Soft tissue sarcoma, Adoptive T‐cell therapy, Immune checkpoint inhibitors, Bone sarcoma
Abstract
Soft tissue and bone sarcomas are a rare and heterogeneous form of cancer. With standard of care treatment options including surgery, radiation, and chemotherapy, the long‐term survival is still low for high‐risk soft tissue sarcoma patients. New treatment strategies are needed. Immunotherapy offers a new potential treatment paradigm with great promise. Immunotherapy of soft tissue sarcomas dates back to Dr. Coley's first use of toxins in the late 1800s. A variety of strategies of immunotherapy have been tried in soft tissue and bone sarcomas, including various vaccines and cytokines, with limited success. Results of these early clinical trials with vaccines and cytokines were disappointing, but there are reasons to be optimistic. Recent advances, particularly with the use of adoptive T‐cell therapy and immune checkpoint inhibitors, have led to a resurgence of this field for all cancer patients. Clinical trials utilizing adoptive T‐cell therapy and immune checkpoint inhibitors in soft tissue and bone sarcomas are under way. This paper reviews the current state of evidence for the use of immunotherapy, as well as current immunotherapy strategies (vaccines, adopative T‐cell therapy, and immune checkpoint blockade), in soft tissue and bone sarcomas. By understanding the tumor microenviroment of sarcomas and how it relates to their immunoresponsiveness, better immunotherapy clinical trials can be designed, hopefully with improved outcomes for soft tissue and bone sarcoma patients.
Implications for Practice.
Immunotherapy is a promising treatment paradigm that is gaining acceptance for the management of several cancers, including melanoma, renal cell carcinoma, prostate cancer, and lung cancer. There is a long history of immunotherapy in the treatment of soft tissue and bone sarcomas, although with little success. It is important to understand past failures to develop future immunotherapy treatment strategies with an improved possibility of success. This article reviews the history of and current state of immunotherapy research in the treatment of soft tissue and bone sarcomas, with particular regard to vaccine trials, adoptive T‐cell therapy, and immune checkpoint blockade.
Introduction
Sarcomas are a rare set of cancers in adults, representing approximately 1% of all adult malignancies [1]. In adults in the U.S., approximately 13,000 cases of soft tissue sarcomas (STS) [1] and 3,000 cases of bone sarcomas are reported each year [2]. The primary management for localized STS is complete surgical resection with adjuvant or neoadjuvant radiation therapy in selected cases [1]. Neoadjuvant or adjuvant chemotherapy for STS is often utilized with limited data for efficacy [2]. Excluding non‐gastrointestinal stromal tumor (GIST), the prognosis of STS has not changed significantly over the past 20 years, despite vigorous investigation [3], [4]. Standard therapy remains structured around chemotherapy (doxorubicin, ifosfamide, dacarbazine, gemcitabine/docetaxel); however, patients incur substantial toxicities, and these strategies rarely result in cure. With these standard chemotherapies, the median overall survival for patients with metastatic soft tissue sarcoma is approximately 1.5 to 2 years. Recently, several agents received approved indications by the U.S. Food and Drug Administration (FDA) for the treatment of metastatic STS, including pazopanib [5], [6], trabectedin [7], and eribulin [8], [9], [10]. These agents have marginally improved progression‐free survival and overall survival but do not lead to durable responses or cure [4].
Despite the advances in the above chemotherapeutics, novel therapies are needed. One emerging strategy is the field of immuno‐oncology, in which the goal is to manipulate the immune system to generate a response against the tumor. The immuno‐oncology field has seen a revival in solid tumor oncology, with FDA approvals in prostate cancer, melanoma, renal cell carcinoma, and non‐small cell lung cancer (NSCLC), among others. These exciting outcomes in immune‐oncology have led to renewed consideration of this approach for sarcomas. Recently, immunotherapy strategies have improved the treatment and prognosis of metastatic prostate cancer [11], metastatic malignant melanoma [12], [13], [14], [15], [16], [17], metastatic NSCLC [18], [19], [20], [21], metastatic renal cell carcinomas [22], [23], [24], [25], and Hodgkin's lymphoma [26]. The most successful of these strategies involve immune checkpoint inhibitors. These accomplishments have led to renewed consideration of immunotherapy for soft tissue and bone sarcoma. This review details evidence for an immune response in sarcomas and the different therapeutic modalities in immunotherapy for sarcomas, focusing on the current state of immunotherapy clinical trials in sarcomas, immune checkpoint inhibitors, vaccine trials, and adoptive cell therapy.
Materials and Methods
An in‐depth literature search was conducted in Ovid Medline and PubMed using the search terms sarcomas, soft tissue sarcoma, bone sarcoma, immunotherapy, vaccines, immune checkpoint blockade, anti‐CTLA‐4 antibody, tremelimumab, ipilimumab, anti‐PD‐1 antibody, nivolumab, pembrolizumab, and anti‐PD‐L1 antibody. A ClinicalTrials.gov search for all clinical trials involving immunotherapy and sarcomas was conducted.
Mechanisms of Action
The immune system plays a critical role in the surveillance, prevention, and development of cancer. Evading immune system destruction has been established as a hallmark of cancer [27]. The concept of tumor surveillance was first described by Burnet [28]. The theory was further revised to encompass the current “immunoediting of cancer,” including the three phases of elimination, equilibrium, and escape [29]. First, in the elimination phase, the immune system can recognize and destroy potential malignant tumor cells. Second, equilibrium is the process by which the immune system selects and ultimately sculpts tumor cells with an ever‐increasing ability to survive immune system attack. Third, during the escape phase, the immunologically sculpted tumor cells expand uncontrollably in the immunocompetent host [30]. A corollary of this theory is that tumors escape immune destruction by developing tolerance to and altering the tumor microenvironment [31].
Evaluation of immunotherapy is still in its early stages. Most immunotherapy trials in soft tissue and bone sarcomas to date have been negative, although there have been suggestions of positive responses. It is imperative to be able to manipulate the immune system in such a way as to induce an antitumoral response. Current sarcoma immunotherapies have failed in this regard. The immunological milieu of the sarcoma microenvironment plays an important role, but its evaluation, albeit critically important, is in the early stages of predicting response of sarcomas to immunotherapy. Components of this immunological milieu include cytokines, tumor infiltrating lymphocytes (TILs) and associated macrophages, expression of immune checkpoint inhibitors such as cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4), programmed cell death‐1 (PD‐1) and programmed death‐ligand 1 (PD‐L1), and major histocompatibility complex (MHC) antigen expression. All of these components may be important for prognosis and responses of tumors to immunologically targeted therapies and are potential therapeutics or therapeutic targets [32]. The human adaptive immune response requires two activation signals; for example, activation of CD8+ cytotoxic T lymphocytes requires signaling via the T‐cell receptor (TCR) and a costimulatory molecule.
Initial immunotherapy strategies sought to stimulate the immune system through the use of signaling molecules such as interleukin‐2 (IL‐2), which can activate cytotoxic T cells, or interferon‐alpha [33], [34]. These approaches were not successful. In addition to co‐stimulatory molecules, multiple co‐inhibitory molecules exist, such as CTLA‐4 or the interaction of PD‐1 with PD‐L1 or PD‐L2. Current immunotherapy trials are targeting these interactions with monoclonal antibodies, essentially “taking the brakes off” the immune system. However, if there is no underlying immune response, simply taking the brakes off will be insufficient. In tumors that lack a sufficient immune response, the immune system will have to be reprogrammed to lead to an antitumor immune response through the use of sarcoma‐directed vaccines or adaptive T‐cell strategies.
Sarcoma Clinical Trials in Immunotherapy
Immune Checkpoint Blockade.
An encouraging approach to immunotherapy is checkpoint blockade, namely the removal of the “brakes” of the immune system [35]. Activation of T cells requires two signals: one through the T‐cell receptor and a second costimulatory signal that ultimately proceeds through B7‐1 with CD28. When T cells are activated, CTLA‐4 is upregulated and competes for binding of B7‐1 with CD28. Because CTLA‐4 has a greater affinity for B7‐1, it acts as a negative regulator of T‐cell activation. The two currently available monoclonal anti‐CTLA‐4 antibodies are tremelimumab and ipilimumab. Ipilimumab has shown clinical activity and is now approved for the treatment of metastatic melanoma [36]. A completed phase I trial of ipilimumab in children and adolescents with treatment‐resistant cancer, NCT00556881 (https://clinicaltrials.gov/ct2/show/NCT01445379), included sarcomas, but no results are reported yet. A second phase II study with ipilimumab in patients with synovial sarcoma, NCT00140855 (https://www.clinicaltrials.gov/ct2/show/NCT00140855), was stopped early due to poor accrual and no objective responses [37]. According to the Response Evaluation Criteria in Solid Tumors (RECIST), there were no documented radiographic responses, and the time to progression ranged from 0.47 to 2.1 months [37]. Final results from two additional studies of immune (CTLA‐4) checkpoint blockade in sarcomas, a phase I trial of ipilimumab and dasatinib for recurrent or metastatic GIST and a phase I trial of ipilimumab for pediatric solid tumors including sarcoma, are awaited. Initial results of the ipilimumab and dasatinib study reported at the Connective Tissue Oncology Society meeting in 2015 showed no response per RECIST or Immune‐Related Response Criteria (ir‐RC), although stable disease was seen in 9 of 16 patients [38]. These results are disappointing.
Research suggests that anthracyclines enhance tumor infiltration of interferon‐gamma‐producing CD8+ T cells and that cyclophosphamide depletes CD4+ CD25+ T regulatory cells [39], [40]. Combinations of classic cytotoxic chemotherapy with immune checkpoint inhibitors may enhance the efficacy of immunotherapy and should be considered in the future. Furthermore, the site of immune checkpoint blockade may influence the antitumor activity where blocking PD‐1 or PD‐L1 at the sites of T‐cell activity may be more beneficial than blocking CTLA‐4.
PD‐1 and PD‐L1 are another immune checkpoint pathway [41], [42]. PD‐1 is usually expressed on activated T cells. PD‐1 binds to PD‐L1 or PD‐L2, resulting in an inhibitor signal and T‐cell inactivation. The two FDA‐approved anti‐PD‐1 antibodies are nivolumab and pembrolizumab, and the safety of anti‐PD‐1 antibodies and anti‐PD‐L1 antibodies have been clinically proven [43], [44]. Combination therapy with nivolumab and ipilimumab has already yielded novel, successful strategies in melanoma. Nivolumab is now approved for metastatic non‐small cell lung cancer, melanoma, renal cell carcinoma, and Hodgkin's lymphoma [45]. Pembrolizumab is now approved for metastatic melanoma and non‐small cell lung cancer.
PD‐1 and PD‐L1 are expressed on many human tumors in varying degrees, including sarcomas [46], although the impact of the presence of PD‐1 and PD‐L1 expression in the tumor microenvironment is still debated. PD‐1 and PD‐L1 expression have been evaluated in GIST, STS, and uterine sarcomas. In one study, tumor PD‐L1 expression was noted in 12% of all soft STS, and 29% of GIST patients’ PD‐1 expression was noted in 22% of specimens [46]. There was a significant correlation between tumor PD‐L1 and PD‐1 expression and CD8+ tumor infiltrating lymphocytes but no correlation between PD‐L1 or PD‐1 expression and overall survival [46]. In a second study, PD‐L1 expression was seen in 65% of STS tumor specimens and lymphocyte PD‐1 expression in 58% of STS specimens [47], although the sample numbers were small. Both positive PD‐1 and PD‐L1 expression were seen in 75% of epithelioid sarcomas, 80% of angiosarcomas, 82% of undifferentiated sarcomas, 50% synovial sarcomas, and 30% of leiomyosarcomas [47]. In addition, expression of PD‐L1, infiltration by PD‐1‐positive lymphocytes, and the PD‐1/PD‐L1 pattern were all independent predictors of worse overall survival and worse event‐free survival in a multivariate analysis [47]. PD‐L1 expression is also seen in osteosarcoma [48]. Notably, these studies used different assays to detect PD‐L1 and PD‐1, namely the DAKO 5H‐1 antibody in the former study and the Santa Cruz antibody in the later study. In uterine sarcomas, a sample of 42 patients showed 100% positivity for PD‐L1 using the Abcam antibody (Abcam, Cambridge, UK, http://www.abcam.com/) [49].
These four reports indicate varying expression levels of PD‐1 and PD‐L1 in sarcoma patients, with the suggestion of decreased overall survival in patients with higher PD‐1 and PD‐L1 expression, suggesting that blocking PD‐1 and PD‐L1 could be therapeutically beneficial. Nonetheless, PD‐1 and PD‐L1 expression has not been directly correlated with chances of response [50], and PD‐1 and PD‐L1 expression within treated tumors should be carefully evaluated to determine whether expression predicts response. Moreover, the heterogeneity of expression of PD axis markers raises the important issue of what control populations would be suitable for judging the clinical value of such interventions.
There are several clinical trials using anti‐PD‐1 and/or anti‐PD‐L1 antibodies in sarcomas. First, SARC028 was a phase II trial of the anti‐PD‐1 antibody pembrolizumab in patients with unresectable, recurrent, or metastatic soft tissue or bone sarcomas [51]. The primary endpoint was an objective response rate per RECIST 1.1, and secondary outcomes included progression‐free survival, overall survival, and response per ir‐RC. Results presented at the American Society of Clinical Oncology 2016 annual meeting showed a partial response rate of 0% (0 of 10) in leiomyosarcomas, 11% (1 of 9) in synovial sarcomas, 22% (2 of 9) in liposarcomas, and 44% (4 of 9) in undifferentiated pleomorphic sarcomas [51]. The study met its secondary endpoint with a progression‐free survival of 55% at 12 weeks [51]. The correlative biomarker analysis is still pending. It will be essential to understand the prognostic or predictive value of TILs, MHC, PD‐1, and PD‐L1 expression in sarcomas to determine for which patients immunotherapy may be appropriate. A second trial of anti‐PD‐1 therapy in ten patients with uterine leiomyosarcomas failed to show any response [52], although a single patient with uterine leiomyosarcoma treated with pembrolizumab had complete tumor remission for over 2 years [53]. It will be important to understand the role of single agent or combination CTLA‐4, PD‐1, or PD‐L1 blockade in sarcoma treatment. Finally, other costimulatory molecules or inhibitory molecules in early development, such as BTLA, LAG‐3, TIM3, VISTA, OX40, and CD73, may be future targets in sarcoma treatment to further rev up or release the brakes of the immune system. Table 1 lists immune checkpoint inhibitor trials in sarcomas.
Table 1. Current and ongoing anti‐PD‐1 and anti‐PD‐L1 inhibitor trials in patients with soft tissue and bone sarcomas, as of April 30, 2017.
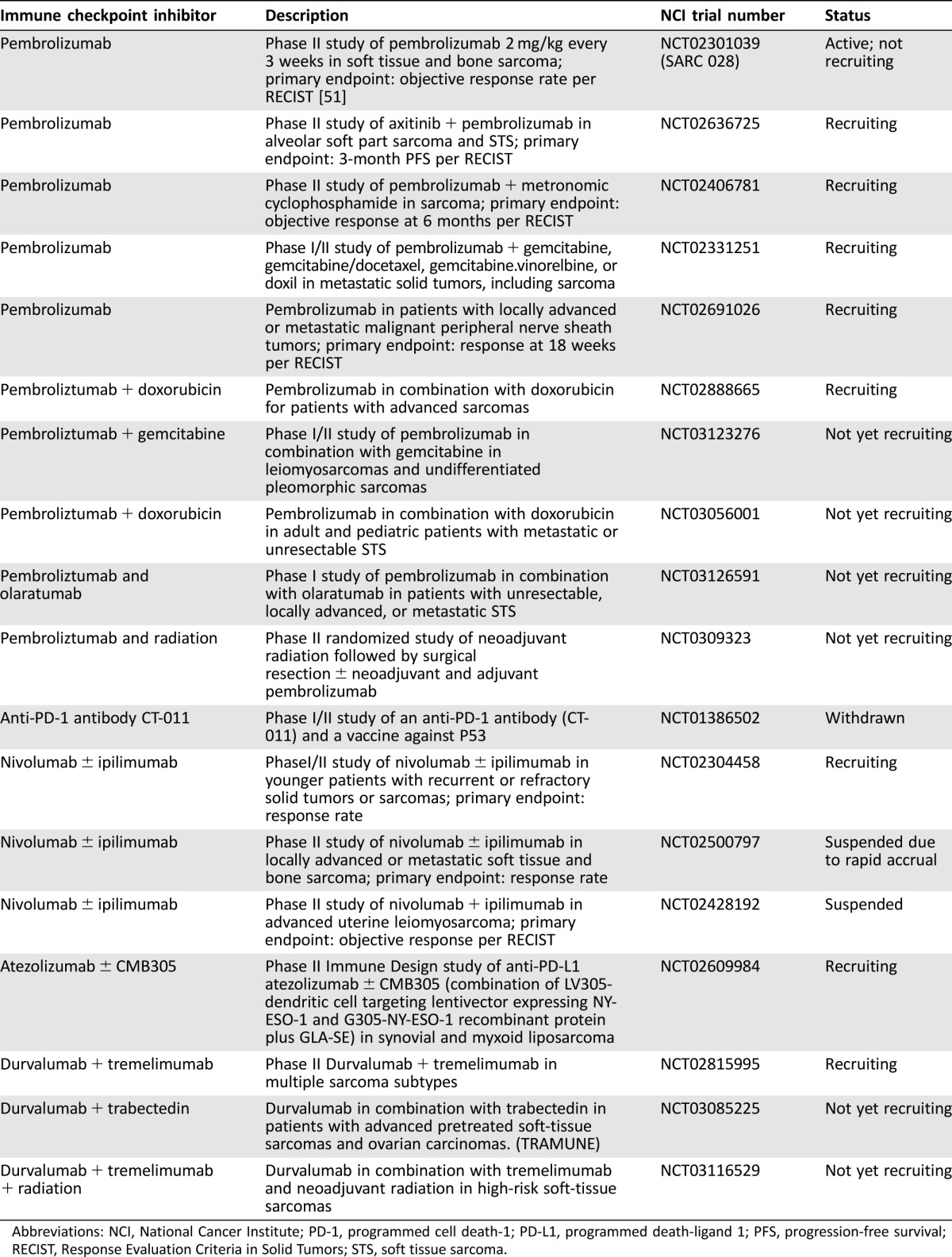
Abbreviations: NCI, National Cancer Institute; PD‐1, programmed cell death‐1; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival; RECIST, Response Evaluation Criteria in Solid Tumors; STS, soft tissue sarcoma.
Vaccines.
Vaccines were one of the first immunotherapy strategies used to treat cancer. Marcove et al. introduced the first osteosarcoma vaccine in 1970 [54]. Theoretically, vaccines are elegantly designed to target only the cancer and not normal tissues, resulting in little harm to the patient. Vaccines may be able to help induce an antitumor immunologic response in immunologically silent tumors. In practice, however, most vaccine studies have yielded modest results despite a large number of clinical trials. Currently, there are only two FDA‐approved vaccines used for metastatic prostate cancer and unresectable melanoma [11], [13].
The central theme in cancer vaccine development is the identification of tumor‐specific or tumor‐associated peptide fragments with recognition by MHC molecules to eventually trigger the immune system. Vaccines have utilized neoantigens derived from whole tumor cells, tumor cell lysates, and cancer‐related peptides [55], [56]. Somatic mutations can give rise to neoantigens, as can the breakpoints of cancer‐specific fusion proteins [57]. Many sarcoma subtypes contain unique genetic abnormalities and/or chromosomal translocations (supplemental online Table 1), which could serve as possible targets for vaccine development. Additionally, many sarcomas express tumor‐specific or differentiation antigens that are not expressed on most normal tissues. These antigens, such as MAGE‐1, disialogangliosides (GD2 and GD3), and NY‐ESO‐1, have previously been described in other cancers, for example, in melanoma and testicular cancer [58], [59], and have been observed in sarcomas as well (Table 2). Thus, there are a multitude of potential neoantigen targets in sarcomas. However, without a second signal provided by a variety of adjuvants, including granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), IL‐2, interferon, heat shock proteins, second peptides, tumor DNA or mRNA, and radiation, there will only be a minimal immune response [60], [61]. Benefits of immunotherapy with adjuvants alone are only suggested but not definitively proven with interferon‐α2b and muramyl tripeptide in osteosarcoma [62]. Toll‐like receptor agonists are new adjuvants that can profoundly enhance T‐cell‐based immunotherapy [63], [64]. The efficacy of vaccines may be improved by combination with cytokines, toll‐like receptors, or other adjuvants (to rev up the immune system), chemotherapy or radiation (to increase the release of neoantigens), or checkpoint inhibitors, such as anti‐CTLA, anti‐PD‐1, or anti‐PD‐L1 antibody (to release the immune system).
Table 2. Neoantigens associated with soft tissue and bone sarcomas.
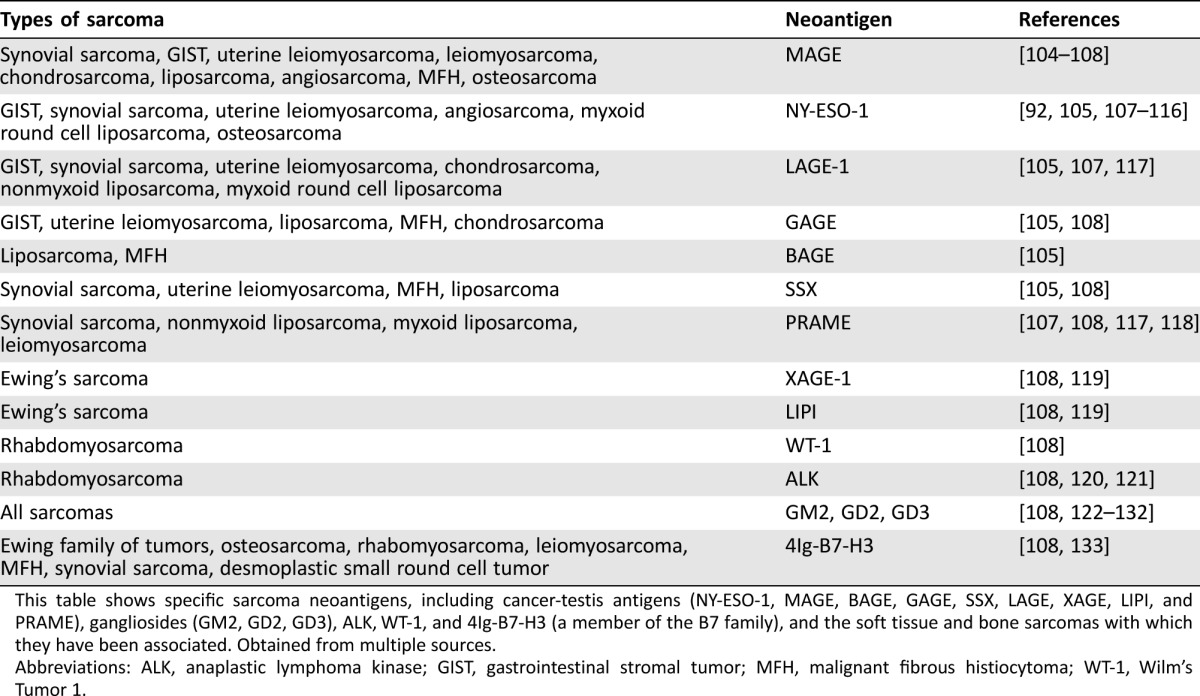
This table shows specific sarcoma neoantigens, including cancer‐testis antigens (NY‐ESO‐1, MAGE, BAGE, GAGE, SSX, LAGE, XAGE, LIPI, and PRAME), gangliosides (GM2, GD2, GD3), ALK, WT‐1, and 4Ig‐B7‐H3 (a member of the B7 family), and the soft tissue and bone sarcomas with which they have been associated. Obtained from multiple sources.
Abbreviations: ALK, anaplastic lymphoma kinase; GIST, gastrointestinal stromal tumor; MFH, malignant fibrous histiocytoma; WT‐1, Wilm's Tumor 1.
Vaccines using peptides derived from the breakpoints of sarcoma‐specific fusion proteins are promising. In one study, six patients with synovial sarcomas were treated with a SYT‐SSX peptide vaccine; induction of peptide‐specific cytotoxic T lymphocytes (CTLs) occurred in four of six patients, and one patient had stable disease for 2 months per RECIST [65]. The addition of interferon‐α resulted in peptide‐specific CTLs in 9 patients and stable disease in 7 of 21 patients for 6 to 57 months, but no clinical responses [66]. Another study of a personalized peptide vaccine for bone and soft tissue sarcomas showed stable disease in 6 patients for 5.7 to 33 months [67]. The patient with stable disease for 33 months had synovial sarcoma. A study of 25 patients with sarcomas treated with irradiated tumor cells and interferon‐γ or GM‐CSF showed a difference in survival of 8.2 months versus 16.6 months, comparing those patients who did not have a positive immune response to those who had a positive response, although there were no clinical responses [68], [69]. These studies support the development of a tumor‐specific immune response resulting in a sustained clinical benefit in some patients despite no radiographic improvement. Alternative vaccine approaches use dendritic cell vaccines combined with radiation in the neoadjuvant setting prior to surgical resection. A phase I trial showed the feasibility of this approach, with 52.9% of patients developing a tumor‐specific immune response [70]. This approach requires further study in sarcomas. Other promising approaches in sarcomas include NY‐ESO‐1‐targeted vaccines in combination with an anti‐PD‐L1 antibody, and these studies are ongoing. Table 3 summarizes the current, completed, and ongoing vaccine trials in sarcomas.
Table 3. Completed, current, and ongoing vaccine or adoptive cellular therapy trials in patients with soft tissue and bone sarcoma, as of August 2016.

Abbreviations: CAR, chimeric antigen receptor; CTL, cytotoxic T lymphocyte; EBRT, external beam radiotherapy; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HER2, human epidermal growth receptor 2; IL, interleukin; INF, interferon; KLH, keyhole limpet hemocyanin; MDSC, myeloid‐derived suppressor cell; N/A, not applicable; NCI, National Cancer Institute; NK, natural killer; NSCLC, non‐small cell lung cancer; OS, overall survival; PNET, primitive neuroectodermal tumor; pts, patients; VEGF, vascular endothelial growth factor; XRT, radiation.
One possible reason for the limited benefit of past vaccines in sarcomas is the lack of MHC class I expression in some bone and soft tissue sarcomas. MHC molecules present antigen in cells of the adaptive immune system, including cytotoxic CD8+ T cells. Without MHC class I expression on the surface of tumor cells, there can be no primary signal to activate the immune system. In 2006, Tsukahara et al. showed that loss or downregulation of class I MHC/human lymphocyte antigen (HLA) was seen in 62% of soft tissue sarcomas and 52% of osteosarcomas [71]. The loss of HLA I expression was recently shown in 39% of undifferentiated pleomorphic sarcomas, 80% of fibrosarcomas, and 88% of dermatofibrosarcomas [72]. For patients with osteosarcomas and high levels of expression of class I MHC/HLA molecules, compared with patients with no expression, overall survival and event‐free survival significantly improved [73]. Regarding Ewing's sarcoma, loss of HLA expression was shown in 79% of Ewing's tumors [74]. Furthermore, downregulated versus high levels of HLA class I expression and presence of CD8+ T‐cell infiltration were shown to be independent prognostic markers for overall survival in Ewing's sarcoma [75], [76]. Thus, both bone and soft tissue sarcomas show loss of MHC class I expression, which correlated with the prognosis, presumably due to the ability of MHC class I negative tumor cells to better evade the immune system in bone tumors.
Another basis for the disappointing early studies with ipilimumab, nivolumab, and pembrolizumab in sarcomas could relate to the fact that translocation‐associated sarcomas have a low mutation rate, resulting in fewer neoantigens. As many as 25% of sarcomas have a specific translocation or gene fusion (supplemental online Table 1); the remaining sarcomas are genetically complex, such as leiomyosarcoma, osteosarcoma and undifferentiated pleomorphic sarcomas [77]. These sarcomas may have more neoantigens that could lead to tumor‐specific immune responses than translocation‐associated sarcomas. Certainly, immune checkpoint blockade has been the most effective in tumors with high mutational load, such as melanoma, renal cell carcinoma, and non‐small cell lung cancer [77]. Future STS trials should evaluate MHC expression to correlate it with treatment responses as well as the genetic complexity of the tumors. Arguably, vaccine strategies must profoundly change before they can be used as adequate therapeutics [56].
Adoptive Cell Transfer.
A new strategy for inducing tumor immune responses or reprogramming the immune system involves the ex vivo expansion of lymphocytes, termed adoptive cell transfer. These lymphocytes, whether T cells or natural killer (NK) cells, are the main effectors of the adaptive immune response. T lymphocytes infused back into the patient can be nonspecific or altered by cytokines to become active cytotoxic cells, such as lymphokine‐activated killer or cytokine‐induced killer cells. Tumor‐infiltrating lymphocytes can be used, or cytotoxic T lymphocytes selected for recognizing tumor‐associated antigens or genetically engineered to recognize specific tumor antigens [78].
Tumor‐infiltrating lymphocytes are particularly attractive targets because, historically, TILs have been a potential marker for the immune responsiveness of a tumor, indicating that some TILs may represent an immune response against a tumor. TIL have been described in a variety of tumors, including melanoma, renal cell carcinoma, breast cancer, prostate adenocarcinoma, head and neck cancer, ovarian cancer, bladder cancer, esophageal cancer, lung cancer, and colorectal cancer [79]. Evidence has been gathered to support TIL influence on outcomes in some of these tumors [80], such as colon cancer [81], non‐small cell lung cancer [82], and melanoma [83], with the type, density, location, and functional orientation of TILs entering into consideration. High‐density TH1 cluster‐differentiated eight (CD8+) T cells correlated with improved disease‐free survival and overall survival [83]. TILs have been reported in sarcomas since 1990, when Balch et al. found TIL in 36% of patients with sarcomas [79]. Additionally, TILs have been evaluated in GIST, STS, Ewing's sarcomas, osteosarcoma, and uterine sarcomas. One report showed high levels (defined as >5%) of CD3+ T cells in 44% of patients, of CD4+ T cells in 8% of patients, and of CD8+ T cells in 22% of patients. Most of these patients had GIST with occasional TILs seen in other STS [46]. In a sample of 91 patients with GIST, TILs composed of both CD3+ T cells and NK cells independently correlated with an improved progression‐free survival in both univariate and multivariate analysis, accounting for known GIST risk factors such as size, location, and mitotic rate [84]. For non‐GIST soft tissue sarcomas, the presence of TILs and impact on prognosis varied. TILs have been only occasionally reported in specific subtypes, such as angiosarcoma, liposarcoma, synovial sarcoma, and high‐grade sarcoma not otherwise specified [46], [85], [86].
Considering the numerous STS subtypes and the small sample sizes, these reports may not be representative of the general population. In terms of effect on survival, Katenkamp reported no impact of TIL on survival in 160 patients with STS [87]. However, in a sample of 249 patients with STS, Sorbye et al. showed on a univariate analysis that increased numbers of CD4+ T cells and CD20+ B cells TIL correlated with improved disease‐specific survival and that there was also a trend for improved survival with CD8+ T cells [88]. Concerning bone sarcomas in Ewing's sarcoma, CD8+ TIL correlated significantly with improved overall survival [75], and higher numbers of T regulatory cells (CD4+ CD25hi FoxP3) at diagnosis correlated with metastatic disease [89]. In osteosarcoma, the presence of moderate to marked lymphocyte infiltration significantly correlated with relapse‐free survival [90]. Although the presence of TILs and their impact on survival have been noted in several sarcoma subtypes, whether this is a sufficient predictor of response to immunologically targeted therapies remains unknown. Given the heterogeneous nature of TILs, further processing steps to produce cytotoxic T lymphocytes selected or genetically engineered to recognize tumor‐associated or tumor‐specific antigens may be required to improve the efficacy of adoptive cell transfer—in other words, to better reprogram the immune system.
The feasibility and safety of producing large numbers of autologous antitumor‐specific cytotoxic T lymphocytes have been demonstrated for several solid tumors, including one patient with soft tissue sarcoma [91]. In a phase I/II study, six patients with refractory metastatic synovial sarcoma were treated with genetically modified autologous T cells recognizing NY‐ESO‐1 and IL‐2 after lympho‐depletion with cyclophosphamide and fludarabine. There were four partial responses lasting 5, 8, 10, and 18 months [92]. A follow‐up report from the same study showed 11 of 18 responses (61%) in patients with synovial sarcoma per RECIST lasting from 3 to 47 months, with one patient experiencing a complete response for 20 months [93]. The 5‐year overall survival for synovial sarcoma patients was 38%, an encouraging result for patients with sarcomas refractory to standard chemotherapy. Given the variety of tumor‐specific or differentiation antigens in sarcomas, this approach may apply to a wide range of sarcoma patients. Several active clinical trials using T cells specific to NY‐ESO‐1, MAGE, GD2, and human epidermal growth receptor 2 (HER2) in sarcomas are ongoing. Overall, adoptive cell transfer is a promising new therapy for patients with metastatic cancer, with success in metastatic melanoma that might apply to a wide range of solid tumors, including sarcomas [94]. However, tumor progression can occur despite high levels of antitumor‐specific T cells [95], so the production of a robust T‐cell response may not suffice to induce tumor regression in less immune‐sensitive tumors, and a combination immunotherapy approach may be more appropriate.
Future Directions
The field of immunotherapy is rapidly expanding [96], [97]. Some of the most promising strategies are deregulating the immune system by immune checkpoint blockade and reprogramming the immune system via adoptive cell transfer with re‐infusion of ex vivo expanded TIL or genetically engineered T lymphocytes [98]. These techniques may even be synergistic. A further extension of this technique is chimeric antigen receptor (CAR) T cells, with a genetically modified TCR to a specific tumor‐associated antigen. The generation and safety of CAR T cells has been shown in phase I trials, with a CAR T cell targeted against CD19 in patients with acute lymphoblastic leukemia [99], and remarkable responses have been observed [100]. Many sarcomas express GD2, NY‐ESO‐1, and MAGE, suggesting opportunities to study CAR T cells specific against these antigens in patients with sarcomas. In that regard, specific cataloging of sarcoma‐related neoantigens would be of particular interest as a basis for considering CAR‐related strategies. Additional immunotherapeutic strategies of interest in sarcoma are inhibitors of CD47 and indoleamine‐2,3‐dioxygenase (IDO). CD47 is a “don't‐eat‐me” signal that helps tumors escape phagocytosis by macrophages. Anti‐CD47 therapy is effective in leiomyosarcoma cell lines [101]. IDO is an intracellular enzyme, which leads to effector T‐cell energy through downregulation of tryptophan [102]. High IDO expression is seen in osteosarcoma and other sarcomas, potentially correlating with survival, and thus is an attractive immunotherapeutic strategy [103].
Primary resistance to immunotherapy may be due to an immunologically quiet tumor, without a preexisting antitumor response. Additionally, loss of MHC class I expression prevents the primary activation signal of the immune system. These mechanisms can lead to a lack of response to immune checkpoint inhibitors. Resistance may also develop after initial response to immunotherapy. Loss of MHC expression is again a possible mechanism. Loss of PTEN was shown to be another possible mechanism in uterine leiomyosarcoma resistance to immunotherapy after an initial response [53].
Conclusion
Immunotherapy recently has shown successes in bladder cancer, prostate cancer, melanoma, renal cell cancer, Hodgkin's lymphoma, and, most surprisingly, non‐small cell lung cancer. Extensive preclinical evidence suggests a possible role for the immune responsiveness of sarcomas; however, future clinical trials will need to evaluate the sarcoma tumor microenvironment and immunological milieu, including cytokines, TIL, MHC, and PD‐1 and PD‐L1 expression, to better understand the markers that predict the immune responsiveness of patients with sarcomas to design more effective immunotherapeutic sarcoma trials. Although initial sarcoma immunotherapeutic trials were disappointing, some early successes with dendritic cell vaccines or adoptive cell transfer treatment of sarcomas suggest a potential pathway for future clinical trials. Additionally, while single‐agent immune checkpoint blockade of CTLA‐4 has been ineffective, combinations with blockade of PD‐1 and PD‐L1 offer intriguing avenues of investigation in sarcomas, as does the use of CAR T cells. Overall, immunotherapy is a promising strategy, but current strategies will need to be refined for use in sarcoma patients.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
We thank Gabriela Steier for her support in the editing of this review. We thank Dr. Melissa Burgress for providing information on SARC028. This study was supported by a grant from NCI and NIH (P30 CA134274). M.J.N. is currently affiliated with the Center for Sarcoma and Bone Oncology, Dana‐Farber Cancer Institute, Boston, MA.
Author Contributions
Conception/design: Michael J. Nathenson
Collection and/or assembly of data: Michael J. Nathenson
Data analysis and interpretation: Michael J. Nathenson
Manuscript writing: Anthony Conley, Michael J. Nathenson, Edward Sausville
Final approval of manuscript: Anthony Conley, Michael J. Nathenson, Edward Sausville
Disclosures
The authors indicated no financial relationships.
References
- 1. Siegel R, Ma J, Zou Z et al. Cancer statistics, 2014. CA Cancer J Clin 2014;64:9–29. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute. Seer Cancer Statistics Review, 1975–2012. Available at http://seer.cancer.gov/csr/1975_2012/. Accessed April 2015.
- 3. Jacobs AJ, Michels R, Stein J et al. Improvement in overall survival from extremity soft tissue sarcoma over twenty years. Sarcoma 2015;2015:279601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weitz J, Antonescu CR, Brennan MF. Localized extremity soft tissue sarcoma: Improved knowledge with unchanged survival over time. J Clin Oncol 2003;21:2719–2725. [DOI] [PubMed] [Google Scholar]
- 5. Kawai A, Araki N, Hiraga H et al. A randomized, double‐blind, placebo‐controlled, phase III study of pazopanib in patients with soft tissue sarcoma: Results from the Japanese subgroup. Jpn J Clin Oncol 2016;46:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van der Graaf WT, Blay JY, Chawla SP et al. Pazopanib for metastatic soft‐tissue sarcoma (PALETTE): A randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2012;379:1879–1886. [DOI] [PubMed] [Google Scholar]
- 7. Demetri GD, von Mehren M, Jones RL et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: Results of a phase III randomized multicenter clinical trial. J Clin Oncol 2016;34:786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai A, Araki N, Naito Y et al. Phase 2 study of eribulin in patients with previously treated advanced or metastatic soft tissue sarcoma. Jpn J Clin Oncol 2017;47:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schöffski P, Chawla S, Maki RG et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open‐label, multicentre, phase 3 trial. Lancet 2016;387:1629–1637. [DOI] [PubMed] [Google Scholar]
- 10. Schöffski P, Ray‐Coquard IL, Cioffi A et al. Activity of eribulin mesylate in patients with soft‐tissue sarcoma: A phase 2 study in four independent histological subtypes. Lancet Oncol 2011;12:1045–1052. [DOI] [PubMed] [Google Scholar]
- 11. Kantoff PW, Higano CS, Shore ND et al. Sipuleucel‐T immunotherapy for castration‐resistant prostate cancer. N Engl J Med 2010;363:411–422. [DOI] [PubMed] [Google Scholar]
- 12. Eggermont AM, Suciu S, Santinami M et al. Adjuvant therapy with pegylated interferon alfa‐2b versus observation alone in resected stage III melanoma: Final results of EORTC 18991, a randomised phase III trial. Lancet 2008;372:117–126. [DOI] [PubMed] [Google Scholar]
- 13. Andtbacka RH, Kaufman HL, Collichio F et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015;33:2780–2788. [DOI] [PubMed] [Google Scholar]
- 14. Robert C, Long GV, Brady B et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–330. [DOI] [PubMed] [Google Scholar]
- 15. Robert C, Ribas A, Wolchok JD et al. Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: A randomised dose‐comparison cohort of a phase 1 trial. Lancet 2014;384:1109–1117. [DOI] [PubMed] [Google Scholar]
- 16. Robert C, Thomas L, Bondarenko I et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–2526. [DOI] [PubMed] [Google Scholar]
- 17. Errico A. Melanoma: Checkmate 067: Frontline nivolumab improves PFS alone or in combination with ipilimumab. Nat Rev Clin Oncol 2015;12:435. [DOI] [PubMed] [Google Scholar]
- 18. Borghaei H, Paz‐Ares L, Horn L et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med 2015;373:1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gettinger SN, Horn L, Gandhi L et al. Overall survival and long‐term safety of nivolumab (anti‐programmed death 1 antibody, BMS‐936558, ONO‐4538) in patients with previously treated advanced non‐small‐cell lung cancer. J Clin Oncol 2015;33:2004–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brahmer J, Reckamp KL, Baas P et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med 2015;373:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kazandjian D, Suzman DL, Blumenthal G et al. FDA approval summary: Nivolumab for the treatment of metastatic non‐small cell lung cancer with progression on or after platinum‐based chemotherapy. The Oncologist 2016;21:634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenberg SA, Yang JC, Topalian SL et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high‐dose bolus interleukin 2. JAMA 1994;271:907–913. [PubMed] [Google Scholar]
- 23. McDermott DF, Drake CG, Sznol M et al. Survival, durable response, and long‐term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol 2015;33:2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Motzer RJ, Escudier B, McDermott DF et al. Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med 2015;373:1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Motzer RJ, Rini BI, McDermott DF et al. Nivolumab for metastatic renal cell carcinoma: Results of a randomized phase II trial. J Clin Oncol 2015;33:1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen R, Zinzani PL, Fanale MA et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 28. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res 1970;13:1–27. [DOI] [PubMed] [Google Scholar]
- 29. Dunn GP, Bruce AT, Ikeda H et al. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol 2002;3:991–998. [DOI] [PubMed] [Google Scholar]
- 30. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329–360. [DOI] [PubMed] [Google Scholar]
- 31. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest 2007;117:1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhardwaj N. Harnessing the immune system to treat cancer. J Clin Invest 2007;117:1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dagher R, Long LM, Read EJ et al. Pilot trial of tumor‐specific peptide vaccination and continuous infusion interleukin‐2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: An inter‐institute NIH study. Med Pediatr Onocl 2002;38:158–164. [DOI] [PubMed] [Google Scholar]
- 34. Bielack SS, Smeland S, Whelan JS et al. Methotrexate, doxorubicin, and cisplatin (MAP) plus maintenance pegylated interferon alfa‐2b versus MAP alone in patients with resectable high‐grade osteosarcoma and good histologic response to preoperative map: First results of the EURAMOS‐1 good response randomized controlled trial. J Clin Oncol 2015;33:2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pardoll DM. Immunology beats cancer: A blueprint for successful translation. Nat Immunol 2012;13:1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hodi FS, O'Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maki RG, Jungbluth AA, Gnjatic S et al. A pilot study of anti‐CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma 2013;2013:168145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. D'Angelo SP, Shoushtari AN, Keohan ML et al. Combined KIT and CTLA‐4 blockade in patients with refractory GIST and other advanced sarcomas: A phase IB study of dasatinib plus ipilimumab. Clin Cancer Res 2017;23:2972–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mattarollo SR, Loi S, Duret H et al. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 2011;71:4809–4820. [DOI] [PubMed] [Google Scholar]
- 40. Ghiringhelli F, Menard C, Puig PE et al. Metronomic cyclophosphamide regimen selectively depletes C4+C25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 2007;56:641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen DS, Irving BA, Hodi FS. Molecular pathways: Next‐generation immunotherapy: Inhibiting programmed death‐ligand 1 and programmed death‐1. Clin Cancer Res 2012;18:6580–6587. [DOI] [PubMed] [Google Scholar]
- 42. Topalian SL, Drake CG, Pardoll DM. Targeting the PD‐1/B7‐H1(PD‐L1) pathway to activate anti‐tumor immunity. Curr Opin Immunol 2012;24:207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brahmer JR, Tykodi SS, Chow LQ et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kasamon YL, de Claro RA, Wang Y et al. FDA approval summary: Nivolumab for the treatment of relapsed or progressive classical Hodgkin lymphoma. The Oncologist 2017;22:585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. D'Angelo SP, Shoushtari AN, Agaram NP et al. Prevalence of tumor‐infiltrating lymphocytes and PD‐L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol 2015;46:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim JR, Moon YJ, Kwon KS et al. Tumor infiltrating PD1‐positive lymphocytes and the expression of PD‐L1 predict poor prognosis of soft tissue sarcomas. PloS One 2013;8:e82870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shen JK, Cote GM, Choy E et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol Res 2014;2:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vanderstraeten A, Luyten C, Verbist G et al. Mapping the immunosuppressive environment in uterine tumors: Implications for immunotherapy. Cancer Immunol Immunother 2014;63:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lizée G, Overwijk WW, Radvanyi L et al. Harnessing the power of the immune system to target cancer. Ann Rev Med 2013;64:71–90. [DOI] [PubMed] [Google Scholar]
- 51. Tawbi H, Reinke DK, Reed DR et al. Pembrolizumab (p) in patients with advanced soft tissue (sts) and bone sarcomas (bs): Upated efficacy results of multicenter phase II study SARC028 and correlates of response. Poster #2570708 presented at: 21st Annual Meeting of the Connective Tissue Oncology Society; November 11, 2016; Lisbon, Portugal.
- 52. Ben‐Ami E, Barysauskas CM, Solomon S et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017;123:3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. George S, Miao D, Demetri GD et al. Loss of PTEN is associated with resistance to anti‐PD‐1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 2017;46:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marcove RC, Southam CM, Levin A et al. A clinical trial of autogenous vaccine in osteogenic sarcoma in patients under the age of twenty‐five. Surg Forum 1971;22:434–435. [PubMed] [Google Scholar]
- 55. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: Moving beyond current vaccines. Nat Med 2004;10:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maki RG. Future directions for immunotherapeutic intervention against sarcomas. Curr Opin Oncol 2006;18:363–368. [DOI] [PubMed] [Google Scholar]
- 57. Worley BS, van den Broeke LT, Goletz TJ et al. Antigenicity of fusion proteins from sarcoma‐associated chromosomal translocations. Cancer Res 2001;61:6868–6875. [PubMed] [Google Scholar]
- 58. Scanlan MJ, Gure AO, Jungbluth AA et al. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immun Rev 2002;188:22–32. [DOI] [PubMed] [Google Scholar]
- 59. Simpson AJ, Caballero OL, Jungbluth A et al. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 2005;5:615–625. [DOI] [PubMed] [Google Scholar]
- 60. Goldberg JM. Immunotherapy of sarcomas. Curr Opin Oncol 2013;25:390–397. [DOI] [PubMed] [Google Scholar]
- 61. Burgess M and Tawbi H. Immunotherapeutic approaches to sarcoma. Curr Treat Options Oncol 2015;16:26. [DOI] [PubMed] [Google Scholar]
- 62. Meyers PA, Schwartz CL, Krailo MD et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival: A report from the Children's Oncology Group. J Clin Oncol 2008;26:633–638. [DOI] [PubMed] [Google Scholar]
- 63. Nelson MH, Bowers JS, Bailey SR et al. Toll‐like receptor agonist therapy can profoundly augment the antitumor activity of adoptively transferred CD8(+) t cells without host preconditioning. J Immunother Cancer 2016;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paulos CM, Kaiser A, Wrzesinski C et al. Toll‐like receptors in tumor immunotherapy. Clin Cancer Res 2007;13:5280–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kawaguchi S, Wada T, Ida K et al. Phase I vaccination trial of SYT‐SSX junction peptide in patients with disseminated synovial sarcoma. J Transl Med 2005;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kawaguchi S, Tsukahara T, Ida K et al. SYT‐SSX breakpoint peptide vaccines in patients with synovial sarcoma: A study from the Japanese Musculoskeletal Oncology Group. Cancer Sci 2012;103:1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takahashi R, Ishibashi Y, Hiraoka K et al. Phase II study of personalized peptide vaccination for refractory bone and soft tissue sarcoma patients. Cancer Sci 2013;104:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dillman R, Barth N, Selvan S et al. Phase I/II trial of autologous tumor cell line‐derived vaccines for recurrent or metastatic sarcomas. Cancer Biother Radiopharm 2004;19:581–588. [DOI] [PubMed] [Google Scholar]
- 69. Dillman RO, Wiemann M, Nayak SK et al. Interferon‐gamma or granulocyte‐macrophage colony‐stimulating factor administered as adjuvants with a vaccine of irradiated autologous tumor cells from short‐term cell line cultures: A randomized phase 2 trial of the cancer biotherapy research group. J Immunother 2003;26:367–373. [DOI] [PubMed] [Google Scholar]
- 70. Finkelstein SE, Iclozan C, Bui MM et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo‐adjuvant treatment of high‐risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys 2012;82:924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tsukahara T, Kawaguchi S, Torigoe T et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti‐pan HLA class I monoclonal antibody, EMR8‐5. Cancer Sci 2006;97:1374–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Meissner M, König V, Hrgovic I et al. Human leucocyte antigen class I and class II antigen expression in malignant fibrous histiocytoma, fibrosarcoma and dermatofibrosarcoma protuberans is significantly downregulated. J Eur Acad Dematol Venereol 2010;24:1326–1332. [DOI] [PubMed] [Google Scholar]
- 73. Nada OH, Ahmed NS, Abou Gabal HH. Prognostic significance of HLA EMR8‐5 immunohistochemically analyzed expression in osteosarcoma. Diag Pathol 2014;9:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Berghuis D, de Hooge AS, Santos SJ et al. Reduced human leukocyte antigen expression in advanced‐stage Ewing sarcoma: Implications for immune recognition. J Pathol 2009;218:222–231. [DOI] [PubMed] [Google Scholar]
- 75. Berghuis D, Santos SJ, Baelde HJ et al. Pro‐inflammatory chemokine‐chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T‐lymphocyte infiltration and affect tumour progression. J Pathol 2011;223:347–357. [DOI] [PubMed] [Google Scholar]
- 76. Yabe H, Tsukahara T, Kawaguchi S et al. Prognostic significance of HLA class I expression in Ewing's sarcoma family of tumors. J Surg Oncol 2011;103:380–385. [DOI] [PubMed] [Google Scholar]
- 77. Wheeler DA, Wang L. From human genome to cancer genome: The first decade. Genome Res 2013;23:1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pedrazzoli P, Secondino S, Perfetti V et al. Immunotherapeutic intervention against sarcomas. J Cancer 2011;2:350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Balch CM, Riley LB, Bae YJ et al. Patterns of human tumor‐infiltrating lymphocytes in 120 human cancers. Arch Surg 1990;125:200–205. [DOI] [PubMed] [Google Scholar]
- 80. Pagès F, Galon J, Dieu‐Nosjean MC et al. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010;29:1093–1102. [DOI] [PubMed] [Google Scholar]
- 81. Galon J, Costes A, Sanchez‐Cabo F et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313:1960–1964. [DOI] [PubMed] [Google Scholar]
- 82. Kilic A, Landreneau RJ, Luketich JD et al. Density of tumor‐infiltrating lymphocytes correlates with disease recurrence and survival in patients with large non‐small‐cell lung cancer tumors. J Surg Res 2011;167:207–210. [DOI] [PubMed] [Google Scholar]
- 83. Bogunovic D, O'Neill DW, Belitskaya‐Levy I et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci USA 2009;106:20429–20434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rusakiewicz S, Semeraro M, Sarabi M et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res 2013;73:3499–3510. [DOI] [PubMed] [Google Scholar]
- 85. Kuhnen C, Mentzel T, Sciot R et al. Dedifferentiated liposarcoma with extensive lymphoid component. Pathol Res Pract 2005;201:347–353. [DOI] [PubMed] [Google Scholar]
- 86. Kraus MD, Guillou L, Fletcher CD. Well‐differentiated inflammatory liposarcoma: An uncommon and easily overlooked variant of a common sarcoma. Am J Surg Pathol 1997;21:518–527. [DOI] [PubMed] [Google Scholar]
- 87. Katenkamp D, Hunerbein R. The prognostic significance of inflammatory cells in malignant human soft tissue tumors: Malignancy grading [in German]. Zentralbl Pathol 1992;138:21–25. [PubMed] [Google Scholar]
- 88. Sorbye SW, Kilvaer T, Valkov A et al. Prognostic impact of lymphocytes in soft tissue sarcomas. PloS One 2011;6:e14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Brinkrolf P, Landmeier S, Altvater B et al. A high proportion of bone marrow T cells with regulatory phenotype (CD4+CD25hiFoxP3+) in Ewing sarcoma patients is associated with metastatic disease. Intl J Cancer 2009;125:879–886. [DOI] [PubMed] [Google Scholar]
- 90. Goorin AM, Perez‐Atayde A, Gebhardt M et al. Weekly high‐dose methotrexate and doxorubicin for osteosarcoma: The Dana‐Farber Cancer Institute/The Children's Hospital: Study III. J Clin Oncol 1987;5:1178–1184. [DOI] [PubMed] [Google Scholar]
- 91. Montagna D, Turin I, Schiavo R et al. Feasibility and safety of adoptive immunotherapy with ex vivo‐generated autologous, cytotoxic T lymphocytes in patients with solid tumor. Cytotherapy 2012;14:80–90. [DOI] [PubMed] [Google Scholar]
- 92. Robbins PF, Morgan RA, Feldman SA et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY‐ESO‐1. J Clin Oncol 2011;29:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Robbins PF, Kassim SH, Tran TL et al. A pilot trial using lymphocytes genetically engineered with an NY‐ESO‐1‐reactive T‐cell receptor: Long‐term follow‐up and correlates with response. Clin Cancer Res 2015;21:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rosenberg SA, Restifo NP, Yang JC et al. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008;8:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rosenberg SA, Sherry RM, Morton KE et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen‐specific CD8+ T cells in patients with melanoma. J Immunol 2005;175:6169–6176. [DOI] [PubMed] [Google Scholar]
- 96. Reichert JM. Antibodies to watch in 2016. MAbs 2016;8:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Reichert JM. Antibodies to watch in 2015. MAbs 2015;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer: What clinicians need to know. Nat Rev Clin Oncol 2011;8:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lee DW, Kochenderfer JN, Stetler‐Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose‐escalation trial. Lancet 2015;385:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Brentjens RJ, Davila ML, Riviere I et al. CD19‐targeted t cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci Transl Med 2013;5:177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Edris B, Weiskopf K, Volkmer AK et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci USA 2012;109:6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Moon YW, Hajjar J, Hwu P et al. Targeting the indoleamine 2,3‐dioxygenase pathway in cancer. J Immunother Cancer 2015;3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Urakawa H, Nishida Y, Nakashima H et al. Prognostic value of indoleamine 2,3‐dioxygenase expression in high grade osteosarcoma. Clin Exp Metastasis 2009;26:1005–1012. [DOI] [PubMed] [Google Scholar]
- 104. Antonescu CR, Busam KJ, Iversen K et al. MAGE antigen expression in monophasic and biphasic synovial sarcoma. Hum Pathol 2002;33:225–229. [DOI] [PubMed] [Google Scholar]
- 105. Ayyoub M, Taub RN, Keohan ML et al. The frequent expression of cancer/testis antigens provides opportunities for immunotherapeutic targeting of sarcoma. Cancer Immun 2004;4:7. [PubMed] [Google Scholar]
- 106. Sudo T, Kuramoto T, Komiya S et al. Expression of MAGE genes in osteosarcoma. J Orthop Res 1997;15:128–132. [DOI] [PubMed] [Google Scholar]
- 107. D'Angelo SP, Tap WD, Schwartz GK et al. Sarcoma immunotherapy: Past approaches and future directions. Sarcoma 2014;2014:391967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gouw LG, Jones KB, Sharma S et al. Sarcoma immunotherapy. Cancers (Basel) 2011;3:4139–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Maki RG. Soft tissue sarcoma as a model disease to examine cancer immunotherapy. Curr Opin Oncol 2001;13:270–274. [DOI] [PubMed] [Google Scholar]
- 110. Jungbluth AA, Antonescu CR, Busam KJ et al. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY‐ESO‐1 but not MAGE‐A1 or CT7. Intl J Cancer 2001;94:252–256. [DOI] [PubMed] [Google Scholar]
- 111. Jungbluth AA, Chen YT, Stockert E et al. Immunohistochemical analysis of NY‐ESO‐1 antigen expression in normal and malignant human tissues. Intl J Cancer 2001;92:856–860. [DOI] [PubMed] [Google Scholar]
- 112. Pollack SM, Jungbluth AA, Hoch BL et al. NY‐ESO‐1 is a ubiquitous immunotherapeutic target antigen for patients with myxoid/round cell liposarcoma. Cancer 2012;118:4564–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Maki RG. Immunity against soft‐tissue sarcomas. Curr Oncol Rep 2003;5:282–287. [DOI] [PubMed] [Google Scholar]
- 114. Francescutti V and Skitzki JJ. Sarcomas and the immune system: Implications for therapeutic strategies. Surg Oncol Clin N Am 2012;21:341–355. [DOI] [PubMed] [Google Scholar]
- 115. Endo M, de Graaff MA, Ingram DR et al. NY‐ESO‐1 (CTAG1B) expression in mesenchymal tumors. Mod Pathol 2015;28:587–595. [DOI] [PubMed] [Google Scholar]
- 116. Lai JP, Rosenberg AZ, Miettinen MM et al. NY‐ESO‐1 expression in sarcomas: A diagnostic marker and immunotherapy target. Oncoimmunology 2012;1:1409–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Skubitz KM, Pambuccian S, Manivel JC et al. Identification of heterogeneity among soft tissue sarcomas by gene expression profiles from different tumors. J Transl Med 2008;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Pollack SM, Loggers ET, Rodler ET et al. Immune‐based therapies for sarcoma. Sarcoma 2011;2011:438940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Foell JL, Hesse M, Volkmer I et al. Membrane‐associated phospholipase A1 beta (LIPI) is an Ewing tumour‐associated cancer/testis antigen. Pediatr Blood Cancer 2008;51:228–234. [DOI] [PubMed] [Google Scholar]
- 120. Pillay K, Govender D, Chetty R. ALK protein expression in rhabdomyosarcomas. Histopathology 2002;41:461–467. [DOI] [PubMed] [Google Scholar]
- 121. Corao DA, Biegel JA, Coffin CM et al. ALK expression in rhabdomyosarcomas: Correlation with histologic subtype and fusion status. Pediatr Dev Pathol 2009;12:275–283. [DOI] [PubMed] [Google Scholar]
- 122. Hamilton WB, Helling F, Lloyd KO et al. Ganglioside expression on human malignant melanoma assessed by quantitative immune thin‐layer chromatography. Int J Cancer 1993;53:566–573. [DOI] [PubMed] [Google Scholar]
- 123. Zhang S, Cordon‐Cardo C, Zhang HS et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer 1997;73:42–49. [DOI] [PubMed] [Google Scholar]
- 124. Zhang S, Zhang HS, Cordon‐Cardo C et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: Protein antigens. Clin Cancer Res 1998;4:2669–2676. [PubMed] [Google Scholar]
- 125. Zhang S, Zhang HS, Cordon‐Cardo C et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: II. Blood group‐related antigens. Int J Cancer 1997;73:50–56. [DOI] [PubMed] [Google Scholar]
- 126. Chang HR, Cordon‐Cardo C, Houghton AN et al. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer 1992;70:633–638. [DOI] [PubMed] [Google Scholar]
- 127. Perez CA, Ravindranath MH, Soh D et al. Serum anti‐ganglioside IgM antibodies in soft tissue sarcoma: Clinical prognostic implications. Cancer J 2002;8:384–394. [DOI] [PubMed] [Google Scholar]
- 128. Cheung NK, Saarinen UM, Neely JE et al. Monoclonal antibodies to a glycolipid antigen on human neuroblastoma cells. Cancer Res 1985;45:2642–2649. [PubMed] [Google Scholar]
- 129. Wu ZL, Schwartz E, Seeger R et al. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res 1986;46:440–443. [PubMed] [Google Scholar]
- 130. Schulz G, Cheresh DA, Varki NM et al. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res 1984;44:5914–5920. [PubMed] [Google Scholar]
- 131. Kailayangiri S, Altvater B, Meltzer J et al. The ganglioside antigen G(D2) is surface‐expressed in Ewing sarcoma and allows for MHC‐independent immune targeting. Br J Cancer 2012;106:1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ziebarth AJ, Felder MA, Harter J et al. Uterine leiomyosarcoma diffusely express disialoganglioside GD2 and bind the therapeutic immunocytokine 14.18‐IL2: Implications for immunotherapy. Cancer Immunol Immunother 2012;61:1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Modak S, Kramer K, Gultekin SH et al. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res 2001;61:4048–4054. [PubMed] [Google Scholar]
- 134. Mahvi DM, Shi FS, Yang NS et al. Immunization by particle‐mediated transfer of the granulocyte‐macrophage colony‐stimulating factor gene into autologous tumor cells in melanoma or sarcoma patients: Report of a phase I/Ib study. Hum Gene Ther 2002;13:1711–1721. [DOI] [PubMed] [Google Scholar]
- 135. Goldberg J, Grier HE, Fisher DE et al. GM‐CSF secreting autologous tumor vaccines for the microphthalmia transcription factor associated tumors: Abstract presented at: American Association for Cancer Research Special Conference in Cancer Research Tumor Immunology: New Perspectives; December 2–5, 2008; Miami, FL.
- 136. Kamstock D, Elmslie R, Thamm D et al. Evaluation of a xenogeneic VEGF vaccine in dogs with soft tissue sarcoma. Cancer Immunol Immunother 2007;56:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Matsuzaki A, Suminoe A, Hattori H et al. Immunotherapy with autologous dendritic cells and tumor‐specific synthetic peptides for synovial sarcoma. J Pediatr Hematol Oncol 2002;24:220–223. [DOI] [PubMed] [Google Scholar]
- 138. Suminoe A, Matsuzaki A, Hattori H et al. Immunotherapy with autologous dendritic cells and tumor antigens for children with refractory malignant solid tumors. Pediatr Transplant 2009;13:746–753. [DOI] [PubMed] [Google Scholar]
- 139. Geiger JD, Hutchinson RJ, Hohenkirk LF et al. Vaccination of pediatric solid tumor patients with tumor lysate‐pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res 2001;61:8513–8519. [PubMed] [Google Scholar]
- 140. Geiger J, Hutchinson R, Hohenkirk L et al. Treatment of solid tumours in children with tumour‐lysate‐pulsed dendritic cells. Lancet 2000;356:1163–1165. [DOI] [PubMed] [Google Scholar]
- 141. Mackall CL, Rhee EH, Read EJ et al. A pilot study of consolidative immunotherapy in patients with high‐risk pediatric sarcomas. Clin Cancer Res 2008;14:4850–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Himoudi N, Wallace R, Parsley KL et al. Lack of T‐cell responses following autologous tumour lysate pulsed dendritic cell vaccination, in patients with relapsed osteosarcoma. Clin Transl Oncol 2012;14:271–279. [DOI] [PubMed] [Google Scholar]
- 143. Goletz TJ, Mackall CL, Berzofsky JA, et al. Molecular alterations in pediatric sarcomas: Potential targets for immunotherapy. Sarcoma 1998;2:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Panagopoulos I, Gorunova L, Bjerkehagen B, et al. Chromosome aberrations and hey1‐ncoa2 fusion gene in a mesenchymal chondrosarcoma. Oncology reports 2014;32:40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Panagopoulos I, Hoglund M, Mertens F, et al. Fusion of the ews and chop genes in myxoid liposarcoma. Oncogene 1996;12:489–494. [PubMed] [Google Scholar]
- 146. Scappaticci FA and Marina N. New molecular targets and biological therapies in sarcomas. Cancer treatment reviews 2001;27:317–326. [DOI] [PubMed] [Google Scholar]
- 147. Tomescu O and Barr FG. Chromosomal translocations in sarcomas: Prospects for therapy. Trends in molecular medicine 2001;7:554–559. [DOI] [PubMed] [Google Scholar]
- 148. Bennicelli JL and Barr FG. Chromosomal translocations and sarcomas. Current opinion in oncology 2002;14:412–419. [DOI] [PubMed] [Google Scholar]
- 149. Sato Y, Nabeta Y, Tsukahara T, et al. Detection and induction of ctls specific for syt‐ssx‐derived peptides in hla‐a24+ patients with synovial sarcoma. The Journal of Immunology 2002;169:1611–1618. [DOI] [PubMed] [Google Scholar]
- 150. Skubitz KM and Skubitz AP. Characterization of sarcomas by means of gene expression. The Journal of laboratory and clinical medicine 2004;144:78–91. [DOI] [PubMed] [Google Scholar]
- 151. Todd R and Lunec J. Molecular pathology and potential therapeutic targets in soft‐tissue sarcoma. Expert review of anticancer therapy 2008;8:939–948. [DOI] [PubMed] [Google Scholar]
- 152. Linehan DC, Bowne WB, Lewis JJ. Immunotherapeutic approaches to sarcoma. Seminars in surgical oncology 1999;17:72–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.