

Abstract
Background
Runs of homozygosity (ROH) are continuous homozygous segments of the DNA sequence. They have been applied to quantify individual autozygosity and used as a potential inbreeding measure in livestock species. The aim of the present study was (i) to investigate genome-wide autozygosity to identify and characterize ROH patterns in Gyr dairy cattle genome; (ii) identify ROH islands for gene content and enrichment in segments shared by more than 50% of the samples, and (iii) compare estimates of molecular inbreeding calculated from ROH (FROH), genomic relationship matrix approach (FGRM) and based on the observed versus expected number of homozygous genotypes (FHOM), and from pedigree-based coefficient (FPED).
Results
ROH were identified in all animals, with an average number of 55.12 ± 10.37 segments and a mean length of 3.17 Mb. Short segments (ROH1–2 Mb) were abundant through the genomes, which accounted for 60% of all segments identified, even though the proportion of the genome covered by them was relatively small. The findings obtained in this study suggest that on average 7.01% (175.28 Mb) of the genome of this population is autozygous. Overlapping ROH were evident across the genomes and 14 regions were identified with ROH frequencies exceeding 50% of the whole population. Genes associated with lactation (TRAPPC9), milk yield and composition (IRS2 and ANG), and heat adaptation (HSF1, HSPB1, and HSPE1), were identified. Inbreeding coefficients were estimated through the application of FROH, FGRM, FHOM, and FPED approaches. FPED estimates ranged from 0.00 to 0.327 and FROH from 0.001 to 0.201. Low to moderate correlations were observed between FPED-FROH and FGRM-FROH, with values ranging from −0.11 to 0.51. Low to high correlations were observed between FROH-FHOM and moderate between FPED-FHOM and FGRM-FHOM. Correlations between FROH from different lengths and FPED gradually increased with ROH length.
Conclusions
Genes inside ROH islands suggest a strong selection for dairy traits and enrichment for Gyr cattle environmental adaptation. Furthermore, low FPED-FROH correlations for small segments indicate that FPED estimates are not the most suitable method to capture ancient inbreeding. The existence of a moderate correlation between larger ROH indicates that FROH can be used as an alternative to inbreeding estimates in the absence of pedigree records.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4365-3) contains supplementary material, which is available to authorized users.
Keywords: Bos indicus, Dairy traits, Inbreeding coefficients, ROH islands
Background
Autozygosity occurs when chromosomal segments arising from a common ancestor are identical by descent (IBD) and inherited from both parents on to the offspring genome [1]. This pattern of inheritance gives rise to continuous IBD homozygous segments characterized as runs of homozygosity (ROH) [2], which can be a consequence of several population phenomena [3]. The development of high-density SNP arrays to scan the genome for ROH has been proposed as a useful method to distinguish non-autozygotic segments that are identical by state (IBS) from those autozygotic and IBD [4].
As the occurrence of ROH tend to be revealed in the genome, its identification and characterization can provide an insight into how population structure and demography have evolved over time [5–7]. ROH can disclose the genetic relationships among individuals, estimating with a high accuracy the autozygosity at the individual and population levels [8–11] and can elucidate about selection pressure events [10, 12, 13]. As the expected length of the autozygous segment follows an exponential distribution with mean equal to 1/2g morgans, where g is equal to the number of generations since the common ancestor, the number of generations from the selection events can be inferred from the length and frequency of ROH [4].
The autozygosity based on ROH can help to improve the understanding of genetic selection process of quantitative traits as the selection is one of the main forces that tend to print homozygous stretches on the genome [14]. According to Zhang et al. [13], ROH patterns are not randomly distributed across the genomes, and ROH islands are seen to be distributed and shared among individuals, which is likely the result of selection events. Therefore, ROH can be used to explore signatures of selection [12, 14], since genomic regions sharing ROH potentially contain alleles associated with genetic improvement in livestock [6] and are of interest for breeding programs [14]. ROH can also be an accurate estimator of inbreeding considering that high levels of inbreeding increase the frequency of homozygous alleles [10].
Studies have considered pedigree-based estimates of inbreeding (FPED) since Wright [15], although the availability of whole-genome marker panels has widespread the use of genomic information in animal breeding [16]. Pedigree-based relatedness is calculated from statistical expectations of the probable proportion of genomic identity by descent, while genotype-based estimates show the current relatedness among individuals [17]. Molecular approaches based on inbreeding coefficient estimates derived from ROH (FROH) and based on genomic relationship matrix (FGRM) [18] are meaningfulness to avoid drawbacks of using pedigrees to analyze inbreeding. FROH are worth to estimate genome-wide autozygosity as it captures the influence of relatedness among founders. FROH also takes into account the stochastic nature of recombination and mutations loads [19], and the potential bias resulting from selection [20] as well.
The first Gyr (Bos primigenius indicus) animals in Brazil had arrived in 1912, and most of the bulls were imported between 1914 and 1921, being then incorporated in crosses [21]. Those imported animals were first consumed for beef cattle purpose, and some breeders started to use them for milk production. Gyr animals have been intensely applied as the basis for crosses with taurine dairy breeds due to its rusticity and greater tolerance to the tropical environment [22]. The mating between imported animals invariably led to a steep increase in inbreeding rate in the population, resulting in genetic gains and fixation of favorable alleles. Over time, the deleterious effects associated with boosted homozygosity arising from inbreeding are predisposed to reduce the genetic gains, implicating in a clear loss of genetic variability (reviewed by Peripolli et al. [23]). Hence, the intense use of founders’ animals to create the first Gyr dairy lines presumably triggered the autozygosity. This outcome is due in part to the inexistence of a breeding program at the time [24], the limited number of animals imported from India and the small number of proven sires mated to disseminate the breed [25]. Therefore, maintaining genetic variability in the Gyr cattle in Brazil is a demanding issue, since Brazil is recognized as a Gyr genetic supplier to some tropical countries that have deficiencies in milk production. Genome-wide autozygosity is an upcoming research area with a growing interest in characterizing and comprehending the mechanisms involved in it, so as to preserve a long-term viability and sustainability of Gyr breeding programs.
The aim of this work was to assess genome-wide autozygosity in Gyr cattle to identify and characterize ROH patterns, as well as to investigate ROH islands for dairy gene content in segments shared by more than 50% of the population. Further, we aimed to compare estimates of molecular inbreeding calculated from FROH, FGRM and based on the observed versus expected number of homozygous genotypes (FHOM) with those obtained from FPED.
Results
Genomic distribution of runs of homozygosity
ROH were identified in all 2908 individuals, totaling 161,362 homozygous segments among overall samples. On an individual animal basis, the average number of ROH per animal was 55.12 ± 10.37, with values ranging from 17 to 121. The mean ROH length was 3.17 Mb and the longest segment was 108.97 Mb in length (33,050 SNPs) found on BTA8. The number of ROH per chromosome was greater for BTA5 (10,670 segments) and tended to decrease with chromosome length. The major fraction of chromosome residing in ROH was found on BTA25 (11.98% of chromosomal length in a ROH) (Fig. 1).
Fig. 1.
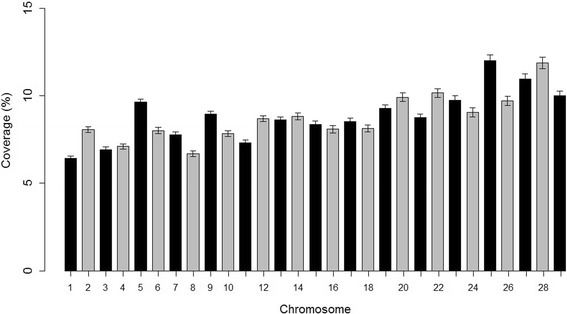
Average percentage of chromosome coverage by runs of homozygosity of minimum length of 1 Mb. The error bars indicate the standard error
Descriptive statistic of ROH number and length by classes is given in Table 1. The total length of ROH for Gyr is composed mostly of a high number of shorter segments (ROH1–2 Mb). These segments accounted for approximately 60% of all ROH detected, which contributed, however, for less than 25% of the cumulative ROH length. While shorter ROH were abundant throughout the genome, the proportion of the genome covered by them was relatively small. In contrast, larger ROH (ROH>16 Mb) were at least twenty-five fold less abundant than shorter ROH (ROH1–2 Mb) and still covered a higher proportion of the genome than small and medium ROH.
Table 1.
Descriptive statistics of runs of homozygosity number (n ROH) and length (in Mb) by ROH length class (ROH1 − 2 MB, ROH2 − 4 MB, ROH4 − 8 MB, ROH8–16 MB, and ROH>16 Mb)
| Class | n ROH | Percent | Mean length | Standard deviation | Genome coverage (%) |
|---|---|---|---|---|---|
| ROH1–2 Mb | 95,892 | 59.42 | 1.34 | 0.27 | 1.77 |
| ROH2–4 Mb | 35,395 | 21.93 | 2.77 | 0.55 | 1.34 |
| ROH4–8 Mb | 17,843 | 11.05 | 5.54 | 1.12 | 1.36 |
| ROH8–16 Mb | 8518 | 5.27 | 10.98 | 2.17 | 1.46 |
| ROH>16 Mb | 3714 | 2.30 | 25.23 | 10.06 | 2.33 |
The animal displaying the highest autozygosity exhibited a ROH genome coverage encompassing 730.21 Mb of the total autosomal genome extension covered by markers (29.20% of the cattle genome), with 71 ROH ≥ ROH1–2 Mb, and a mean ROH length of 10.28 Mb. The least inbred animal exhibited a ROH genome coverage encompassing 48.81 Mb (1.95% of the cattle genome), with 32 ROH ≥ ROH1–2 Mb, and a mean ROH length of 1.52 Mb. Differences among animals regarding the number of ROH and the length of the genome covered by them were observed (Fig. 2). The sum of all ROH per animal allowed the estimation of the percentage of the genome that is autozygous and an average value of 7.01% (175.28 Mb) was observed.
Fig. 2.
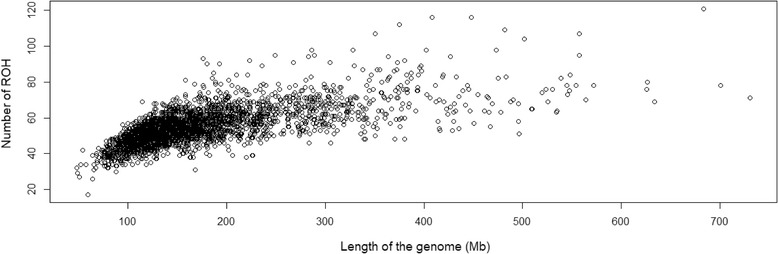
Number of ROH per individual and the length of the genome covered by ROH
Gene characterization in ROH islands
Overlapping ROH were evident across the genome, and their genomic distribution was non-uniform both in length and position across chromosomes. A total of 14 regions with outlying ROH frequencies on BTA2, BTA6, BTA10, BTA12, and BTA14 were identified (Additional file 1). Among the described ROH islands, the strongest pattern was observed on BTA2 (78,394,916:87,587,063 bp), with an overlapping ROH region present in 92% of the samples (Fig. 3). The majority of SNPs within ROH regions showed higher linkage disequilibrium (LD) levels than the estimates obtained for the entire chromosome (Additional file 2).
Fig. 3.
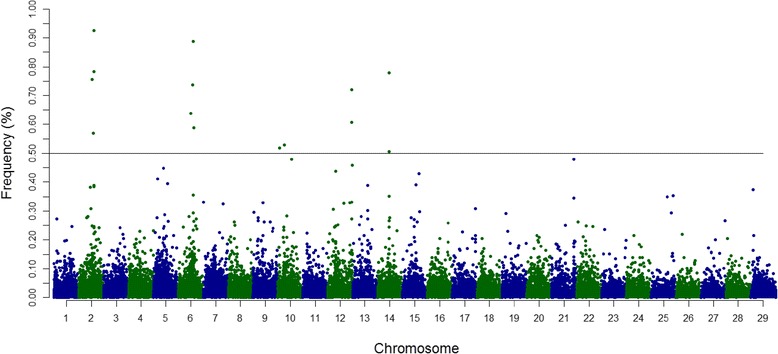
Manhattan plot of the distribution of runs of homozygosity (ROH) islands in the Gyr cattle genome. The X-axis represents the distribution of ROH across the genome, and the Y-axis shows the frequency (%) of overlapping ROH shared among samples
A relevant number of genes (n = 282) inside these ROH islands were observed (Additional file 1), in which several of them play important role in the mammary gland biology and have a prominent importance in milk, dairy traits, and heat adaptation. Gene ontology (GO) and pathway analysis (KEEG) were performed by DAVID tool [26, 27] to obtain a broad functional insight into the set of genes. An enrichment of genes involved in several GO terms (14 molecular functions, 23 biological processes, and seven cellular components) and KEGG pathways was observed (Additional file 3).
Pedigree and genomic inbreeding
Descriptive statistics for FPED and FROH coefficients are presented in Table 2. Among FROH estimates, it can be observed an increase in variation with ROH length, being evidenced by the coefficient of variation (CV).
Table 2.
Descriptive statistics of the pedigree-based inbreeding coefficient (FPED) and genomic inbreeding coefficients based on runs of homozygosity (FROH) for different lenghts (FROH1–2 Mb, FROH2–4 Mb, FROH4–8 Mb, FROH8–16 Mb, and FROH > 16 Mb) for genotyped animals (n)
| Inbreeding coefficient | Mean | Median | Minimum | Maximum | Coefficient of Variation (%) | n |
|---|---|---|---|---|---|---|
| FPED | 0.019 | 0.004 | 0.000 | 0.327 | 3.38 | 2758 |
| FROH1–2 Mb | 0.017 | 0.017 | 0.006 | 0.037 | 20.70 | 2758 |
| FROH2–4 Mb | 0.013 | 0.013 | 0.001 | 0.039 | 35.30 | 2757 |
| FROH4–8 Mb | 0.013 | 0.012 | 0.001 | 0.063 | 55.63 | 2740 |
| FROH8–16 Mb | 0.014 | 0.012 | 0.003 | 0.082 | 73.04 | 2422 |
| FROH > 16 Mb | 0.023 | 0.016 | 0.006 | 0.201 | 97.75 | 1533 |
Low to moderate correlations were observed between FPED-FROH and it increased with ROH length (Fig. 4). Additionally, FPED slightly correlated with FGRM (0.23). The correlations between FGRM-FROH were higher than those between FPED-FROH for all ROH classes described. FHOM highly correlated with FROH over than 4 Mb, FPED, and FGRM.
Fig. 4.
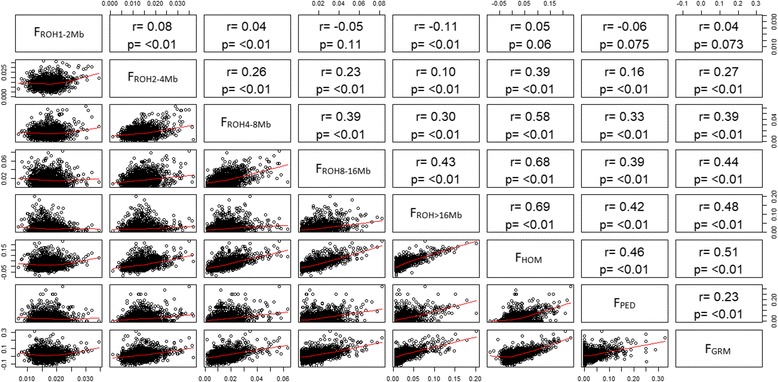
Scatterplots (lower panel) and correlations (upper panel) of genomic inbreeding coefficients FROH (FROH 1–2 Mb, FROH 2–4 Mb, FROH 4–8 Mb, FROH 8–16 Mb, and FROH > 16 Mb) and FGRM, and pedigree-based inbreeding coefficients (FPED)
The inbreeding evolution (FPED and FROH) for animals born between 1980 and 2012 is shown in Fig. 5 and the genotyping sampling of animals per inbreeding coefficient in Table 2. The FPED evolution showed a tendency to slightly increase over time (Fig. 5a), while FROH tended to decrease for segments higher than 4 Mb (Fig. 5d-f).
Fig. 5.
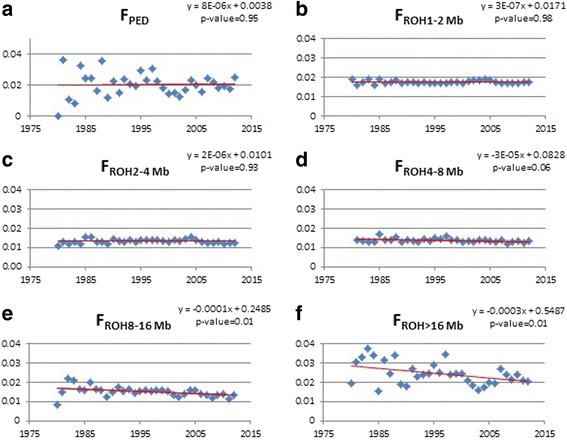
Inbreeding evolution over the past 30 years for pedigree-based inbreeding (FPED) and FROH (FROH1–2 Mb, FROH2–4 Mb, FROH4–8 Mb, FROH8–16 Mb, and FROH > 16 Mb) coefficients. Linear regression (red) in function of the year (x-axis) and inbreeding (y-axis). Each blue dot represents the average FPED and FROH observed per year
Discussion
Genomic runs of homozygosity patterns
The greatest number of ROH per chromosome was described on BTA5, however, results in taurine breeds [6, 28, 29] have evidenced the highest number of ROH on BTA1. The longest ROH was found on BTA8 with 108.97 Mb in length and similar results on BTA8 were reported by Kim et al. [10] in a contemporary Holstein cow (87.13 Mb) and Mastrangelo et al. [28] in Cinisara cattle breed (112.65 Mb).
The number of generations of inbreeding can be inferred from the extent of ROH since their extension is expected to correlate to ancient and recent inbreeding due to recombination events [1]. Therefore, due to recent inbreeding, ROH are expected to be longer since recombination did not have enough time to break up these IBD segments, while short ROH tend to reflect ancient inbreeding because the segments have been broken down by repeated meiosis [30]. The presence of segments larger than 10 Mb is traceable to inbreeding from recent common ancestors that occurred only five generations ago [4], and 78% of the animals comprised in this study presented at least one homozygous segment extending over 10 Mb. The reflection of a recent parental relatedness for animals with segments longer than 10 Mb was confirmed when analyzing the pedigree back in only two generations, in which animals were seen to be inbred by their grand and great-grandparents.
The highest autozygosity value per animal was similar to those reported in the literature for dairy breeds. Purfield et al. [6] observed that dairy breeds were the most autozygous animals among several studied breeds, and had on average 700.3 Mb of their genome classified as ROH. Mastrangelo et al. [28] observed a close value for the Reggiana dairy breed (725.2 Mb) and also did Szmatoła et al. [31] for Holstein cattle with 25% of their genome located in ROH. It is noteworthy to highlight that Marras et al. [14] described that dairy breeds had a higher sum of all ROH than did beef breeds. The higher autozygosity observed in dairy breeds can be explained by the intense artificial selection and the repeatedly use of superior and proven sires for reproduction by artificial insemination [10]. In the Gyr cattle, it can be attributed to the rapid growth and dissemination of the breed over the last years, in which a small number of proven sires with high breeding value were frequently used [32].
Animals with the same length of the genome covered by ROH displayed a variable number of segments, which is likely a consequence of the distinct distances from the common ancestor, as also described by Mészáros et al. [33]. Overall, the autozygotic proportion of the genome found in this population was considered low given the Gyr dissemination historical process. A similar value was achieved by Marras et al. [14] (7% for Marchigiana beef breed). Gyr cattle presented a lower genome average autozygosity compared to previous studies reported by Ferenčaković et al. [8] (9% for Austrian dual purpose Simmental, Brown Swiss, and Tyrol Grey bulls) and Kim et al. [10] (10% for Holstein cattle), and a higher autozygosity than results obtained by Zavarez et al. [11] (4.58% for Nellore cattle).
Runs of homozygosity islands and gene functional annotation
The overlapping ROH regions observed across the genome suggest that these regions are likely a sign of ROH islands shared among animals [9]. ROH islands can be defined as genomic regions with reduced genetic diversity and, consequently, high homozygosity around the selected locus that might harbor targets of positive selection and are under strong selective pressure [34]. The strongest ROH island pattern on BTA2 (78,394,916:87,587,063 bp) present in 92% of the samples showed an enrichment of genes involved with the immunity system. Similarly, Marras et al. [14] reported a ROH in 90% of the samples in Piedmontese cattle, although it was located at the beginning of BTA2, closest to the myostatin (MSTN) locus. Karimi [12] identified the most common pattern in indicine breeds on BTA21, with a value exceeding 93% of individuals.
The high LD levels found in the majority of SNPs within the ROH islands are not surprisingly since selection in cattle has possibly acted to maintain conserved ROH regions originated from IBD segments. These segments are likely to have experienced fewer recombination events and they are expected to display high levels of LD. Besides, a study on human populations has shown a correlation between extensive LD, locally low recombination rates and high incidence of ROH [2].
Several genomic regions with significant SNPs (−log10(p) > 4) based on the integrated Haplotype Score (iHS) were identified for the Gyr cattle by Utsunomiya et al. [35], using a subset of Gyr animals comprised in this study. Of the significant SNPs, seven of them were located within ROH islands described here on BTA2, BTA6, and BTA10 (Additional file 4).
When analyzing genomic positions of the identified ROH islands, the results pointed out by Szmatoła et al. [31] directly overlaps with some of the islands found in our study, with similar regions identified on BTA2 for Holstein, Polish Red and Limousin breeds, on BTA6 for Polish Red, Limousin and Simmental breeds, and on BTA14 for the Simmental cattle (Additional file 4). The Simmental breed also showed a ROH island on BTA6 located closely to the one described in this study for the Gyr cattle (70,117,799:81,603,050 bp). Karimi [12] and Sölkner et al. [36] study on Brahman, Gyr and Nellore cattle also identified ROH islands in some chromosomes as those described in this study. Although the islands on BTA10 and BTA12 were not found to be located at the same genomic region as in our study, the described island on BTA10 was found closest to ours. ROH islands identified on BTA6 were also described in Italian Holstein cattle [37], dairy and beef breeds [14], and in Tyrol Grey cattle [33], but none of them overlapped with those previously described for the Gyr cattle in this study. It is worth to highlight that BTA6 is well documented to harbor genes that affect milk production traits [38–41], thus, a high autozygosity in chromosomal regions may be an indicator of signatures of selection for dairy traits. Further, ROH islands were found overlapping in cattle breeds selected for different purposes, suggesting that selection pressure can also be undergoing on traits other than those specific to dairy or beef traits.
The GO analyses showed several enriched terms for the ROH gene list. A total of 10 genes were identified related to cell differentiation biological process (GO:0030154), in which we highlight the TRAPPC9 (trafficking protein particle complex 9) gene on BTA14. Interestingly, this gene was found to have significant effects on mastitis-related traits in Chinese Holstein herds [42]. Polymorphisms in TRAPPC9 gene has been associated with milk production traits in Holstein cattle [43]. Jiang et al. [44] observed a higher TRAPPC9 mRNA expression level in the mammary gland of lactating cows than in the other tissues, such as heart, liver, lung, kidney, ovary, uterus, and muscle.
Seven genes identified in ROH islands were related to positive regulation of cell migration (GO:0030335) biological process. Of these, the IRS2 (insulin receptor substrate 2), ATP8A1 (ATPase phospholipid transporting 8A1), GABRG1 (gamma-aminobutyric acid type A receptor gamma1 subunit), and GABRAG2 (gamma-aminobutyric acid type A receptor gamma2 subunit) genes have been previously associated with dairy traits. The IRS2 gene on BTA12 encodes the insulin receptor substrate 2, a cytoplasmic signaling molecule that mediates effects of insulin, insulin-like growth factor 1 and other cytokines (provided by RefSeq, Jul 2008). Insulin infusion has been shown to increase milk and protein yields, and reduce milk fat content and yield in lactating goats. It also decreased net uptake of C10:0, C14:0, C16:0, trans-C16:1 and >C18:0 fatty acids, and increased mammary blood flow by 42% [45]. The ATP8A1, GABRG1, and GABRAG2 genes on BTA6 laid within the region with highest iHS score as reported by Hayes et al. [46] in Norwegian Red cattle, a breed which has been intensely selected for milk production.
The nuclear stress granule (GO:0097165) cellular component was substantially enriched (p ≤ 0.05), which contains the HSF1 (heat shock transcription factor 1) gene on BTA14. This gene encodes a heat-shock transcription factor, and its transcription is rapidly induced after heat stress (provided by RefSeq, Jul 2008). Heat shock transcription factors and heat shock proteins (HSP) play a crucial role in environmental stress adaptation and thermal balance since it allows cells to adapt to gradual environmental changes, being an immunoregulatory agent upon controlling the balance between survival and an effective immune system in order to adjust to stress [47]. Kumar et al. [48] observed a higher abundance of HSP family genes during summer and winter compared to the mid-spring season in Bos indicus cattle and Murrah buffaloes, and the magnitude of increase was higher during summer as compared to winter. Among their findings, a significantly (p ≤ 0.001) higher HSF1 mRNA expression during the summer as compared to the mid-spring season was also observed. These findings are consistent with the zebu cattle adaptation traits, in which we highlight its greater ability to tolerate poor feed and inconsistent climate. Li et al. [49] identified polymorphisms in HSF1 gene associated with thermal tolerance in Holstein cattle. In addition to the HSF1 gene, other heat shock genes were found within a ROH island on BTA2, such as HSPD1 (heat shock protein family D (Hsp60) member 1) and HSPE1 (heat shock protein family E (Hsp10) member 1).
We also encountered a number of genes within ROH islands that have been reported to have a prominent importance in milk-related traits on BTA2 (STAT1 and INSIG2 genes) [50, 51], BTA10 (ANG gene) [52] and BTA14 (EEF1D, CRH, DGAT1, and CYP11B1 genes) [53–60]. In addition, mammary gland development-related genes were also described on BTA6 (IGFBP7 gene) [61, 62] and BTA14 (EEF1D gene) [44, 63].
A total of seven KEGG pathways were identified as being enriched (p ≤ 0.05) and the GABAergic synapse (bta04727) was the most significant (p < 0.001) KEGG pathway found (Additional file 3). Gamma-aminobutyric acid (GABA) is an inhibitory neurotransmitter in the mammalian central nervous system and the GABAergic synapse pathway has been associated with animal feed intake and weight gain [64]. Among the others KEGG pathways identified, the ones related to environmental information processing were highlighted, such as neuroactive ligand-receptor interaction (bta04080), PI3K-Akt signaling pathway (bta04151), and AMPK signaling pathway (bta04152) with 11, 10, and 5 genes identified within ROH islands, respectively. PI3K-Akt signaling pathway regulates key cellular functions such as transcription, translation, growth, proliferation, and survival. This pathway has been associated with prolactin signaling, mammary development, and involution in Holstein-Friesian and Jersey breeds [65]. AMPK signaling pathway acts as a sensor of cellular energy status leading to a concomitant inhibition of energy-consuming biosynthetic pathways and activation of ATP-producing catabolic pathways.
Instead of being randomly distributed across the genomes, ROH patterns were seen clustering in specific genomic regions among individuals. These regions were screened for genes under selection and several ROH islands harboring dairy-related genes have been identified, suggesting a directional selection for milk and mastitis-related traits, mammary gland development, and environmental adaptation traits. Surprisingly, BTA14 has shown an enrichment of genes affecting traits of interest for dairy breeders. BTA2 and BTA6 also have shown ROH islands previously described in the literature, and these chromosomes along with BTA10 also revealed signatures of selection previously identified for the Gyr breed [33]. These findings suggest that these chromosomes are likely to contain traces of selection since ROH patterns are not expected to be randomly distributed over the genomes [13]. Also, they evidenced that ROH can reveal signatures of selection since ROH islands described in here corroborated with footprints of recent positive selection previously described for the Gyr cattle [35].
Inbreeding coefficients
The higher the CV was, the greater the differences between the mean and median were for each FROH length (Table 2). Thus, given the dissimilarity among the CV, it is assumed that the mean should not be used as the best measurement of central tendency, indicating that the median should be applied instead for FPED and FROH coefficients. The average FPED and FROH were low for the Gyr cattle, and the FPED estimate was lower than those reported by Reis Filho et al. [24] and Santana Junior et al. [32] for Brazilian Gyr cattle, with values of 2.82 and 1.92%, respectively.
The age of inbreeding can be defined as the distance with the common ancestor and there is an approximate correlation with the length of the ROH [4, 66]. Under the assumption that 1 cM equals to 1 Mb [4], calculated FROH are expected to correspond to the reference ancestral population dating 50 (FROH1–2 Mb), 20 (FROH2–4 Mb), 12.5 (FROH4–8 Mb), 6 (FROH8–16 Mb), and 3 (FROH > 16 Mb) generations ago. Zavarez et al. [11] observed that incomplete pedigree fails to capture remote inbreeding and estimates based on FPED are only comparable with FROH calculated over large ROH. Thus, given the average pedigree depth of three generations, FPED estimate should be comparable with FROH > 16 Mb. The variation between these two estimates can be attributed to the fact that FPED assumes that the entire genome does not undergo selection [20] and recombination events, therefore, it does not take into account potential bias from these events [67]. In addition, it should be underlined that pedigree relatedness is estimated from statistical expectations of the probable IBD genomic proportion, whereas genotype-based estimates show the actual relatedness among individuals [17] and can provide greater accuracy on relatedness.
The increasing correlation between FPED-FROH with ROH length may be explained by considering that ROH reflect both past and recent relatedness and that FPED estimates are based on pedigree records which may not extend back many generations [9, 14]. When longer ROH reflecting recent relatedness are considered to calculate FROH, the FPED-FROH correlation tends to be higher [14, 68]. Several authors have described a high FPED-FROH correlation when a deeper number of described generations are available in the pedigree [6, 8, 9, 14, 29], suggesting that the correlation between these parameters increases with pedigree deep. Ferenčaković et al. [8, 9] observed FPED-FROH correlations values ranging from 0.61 to 0.67 and 0.50 to 0.72, respectively, for pedigrees with more than five generations. Purfield et al. [6] used a complete generation equivalents higher than six and obtained FPED-FROH correlations of 0.73 for ROH > 10 Mb and 0.71 for ROH > 1 Mb, both with the reduced panel. Marras et al. [14] observed high FPED-FROH correlations using pedigree with four, seven and ten generations, with values ranging from 0.56 to 0.74. Gurgul et al. [29] also reported the highest FPED-FROH correlation for animals with seven complete generations of pedigree data, with an average value of 0.45. In the present study, a small number of generations were available to estimate FPED, which may have introduced biased FPED values as the pedigree was not able to cover ancient relatedness.
The slight correlation between FPED-FGRM concurs with the results obtained by Pryce et al. [69]. VanRaden et al. [70] reported higher correlations for Holstein (0.59), Jersey (0.68), and Brown Swiss (0.61) animals. Hayes and Goddard [71] also obtained higher correlations for Australian Angus bulls (0.69). Lower correlations between these estimates were reported by Marras et al. [14], Gurgul et al. [29], and Zhang et al. [72]. In the dairy industry, genomic inbreeding coefficients of genotyped animals are commonly calculated from FGRM [73]. Two out of three reasons hypothesized by Pryce et al. [69] might explain the poor correlation found out in our study: (i) FGRM is strongly dependent on allele frequencies, and population with divergent allele frequencies can lead to misleading IBD results; and (ii) pedigree completeness.
It is well addressed in the literature that incomplete pedigree information reduces estimates of inbreeding and leads to underestimated values [74, 75], as well as missing or incorrect pedigree information. Hence, accurate estimates of FPED depend on a well-structured pedigree dataset. When analyzing the Gyr pedigree structure, it was observed that 72.96% of the animals available in the pedigree dataset had both known sire and dam information, and 3.52% had only known sire and 1.16% known dam information. On the basis of the results, FPED estimate might have been underestimated as well as its correlations with other inbreeding measurements due in part to the poor pedigree depth and pedigree incompleteness.
Several studies also have found a low to moderate FGRM-FROH correlation for dairy breeds [14, 28]. In Holstein cattle, moderate to high correlations were described by Bjelland et al. [73] (0.81). Pryce et al. [69] observed a correlation of 0.62 in Holstein and Jersey populations, and Zavarez et al. [11] correlations ranging from 0.41 to 0.74 in Nellore cattle based on ROH of different minimum lengths. Further, the moderate to high correlations between FROH and the two other estimates of genomic inbreeding (FGRM and FHOM) suggest that the proportion of the genome in ROH can be an accurate estimator of the IBD genomic proportion.
The inbreeding evolution illustration (Fig. 5) stress out a significant (p < 0.01) decline in FROH > 8–16 Mb and FROH > 16 Mb, and it is worth to highlight that these coefficients reflect an inbreeding up to six and just three generations ago, respectively. The FROH > 8–16 Mb and FROH > 16 Mb coefficients reduction since the 80’s happen together with the creation of the Brazilian Dairy Gyr Breeding Program (PNMGL) and the implementation of the Gyr progeny testing, both in 1985. Probably, these facts suggest that different proven sires from divergent lines started to be incorporated into the population, and previously closed herds started to make use of these genetically evaluated sires in their breeding programs. Mating between herds increased after 2002, a fact that may have strongly contributed to reducing the average inbreeding by increasing the genetic exchange [32]. Additionally, Santana Junior et al. [32] reported that the degree of nonrandom mating was close to zero at the end of the last decade, indicating that better mating decisions were taken by the breeders to avoid mating between relatives, changing the mating policy and decreasing the genomic inbreeding level in these populations over time.
These findings reinforce the importance of effective breeding programs for maintaining genetic diversity and suitable inbreeding levels, contributing to a better understanding of the population structure and providing the basis to overcome challenges. Given the Gyr breed growth background, in which a small number of founder animals was imported to Brazil to disseminate the breed, information regarding genetic diversity within the Gyr cattle is therefore essential for genetic improvement and conservation programs.
Conclusions
Despite the reduced genetic basis and the limited number of animals imported to form the first Gyr dairy lines, the autozygotic proportion of the genome was considerably low in this population. Hence, maintaining a low autozygosity is crucial in cattle breeding populations, avoiding inbreeding depression [76] and reduced response in breeding programs [77]. Several common ROH islands have been found in the Gyr genome, suggesting that ROH might be used to identify genomic regions under selection signatures [78, 79]. Common islands on BTA2 and BTA14 are supposed to be a sign of strong selection for dairy and environmental adaptation traits as several genes associated with them were identified. Low correlations between FPED-FROH may be partly due to the relatively shallow depth of the pedigree, indicating that FPED is not the most suitable method to capture ancient inbreeding. The existence of moderate to high correlations between FROH and other genomic inbreeding measures suggests that the levels of autozygosity derived from ROH can be used as an accurate estimator of individual inbreeding levels [6, 8, 29, 73]. In addition, when analyzing the inbreeding evolution for the past 30 years, it can be seen a clear decay in FROH for segments higher than 4 Mb, reinforcing the importance of effective breeding programs and mating management. Our findings contribute to the understanding of the inbreeding effects when assessing genome-wide autozygosity, and how selection can shape the distribution of ROH islands in the cattle genome. Hence, this approach may contribute to comprehend the evolutionary process of the Gyr breed, i.e. selection and domestication process [80, 81], and provide the basis to overcome future challenges.
Methods
Animals and genotyping
The animals used in this study comprise the progeny test program from the National Program for Improvement of Dairy Gir (PNMGL), headed by Embrapa Dairy Cattle (Juiz de Fora, Minas Gerais, Brazil) in cooperation with the Brazilian Association of Dairy Gyr Breeders (ABCGIL) and the Brazilian Association of Zebu Breeders (ABCZ). The objective of the program is to promote the genetic improvement of the Gyr dairy cattle, through the identification and selection of genetically superior bulls for fat, protein and total solids in milk, as well as traits associated with animal conformation and management.
A total of 19 dams and 563 sires born between 1964 and 2013 were genotyped with the BovineHD BeadChip (Illumina Inc., San Diego, CA, USA), containing 777,962 markers; 1664 dams with the BovineSNP50 BeadChip (Illumina Inc., San Diego, CA, USA), that contains 54,609 SNP; and 662 dams with the GGP-LD Indicus BeadChip (GeneSeek® Genomic Profiler Indicus 30 K), that contains 27,533 markers.
Imputation was implemented using the FIMPUTE 2.2 software [82], and lower density panels were imputed to the HD level. Imputation accuracy was 0.99, in accordance with the results presented by Boisin et al. [83] using the same population (0.98). SNPs unsigned to any chromosome and mapped to sexual chromosomes were removed from the dataset. The animals genotyped with the BovineHD BeadChip (Illumina Inc., San Diego, CA, USA) were used as reference population for imputation. The missing genotypes were imputed in the reference population and all the markers were retained. Prior imputation, samples were edited for call rate (< 90%). After editing the reference and imputed genotypes, a total of 2908 animals and 735,236 SNPs were retained for the analyses.
Runs of homozygosity
ROH were identified in every individual using PLINK v1.90 [84]. The PLINK software uses a sliding window of a specified length or number of homozygous SNPs to scan along each individual’s genotype at each SNP marker position to detect homozygous segments [4]. The parameters and thresholds applied to define a ROH were (i) a sliding window of 50 SNPs across the genome; (ii) the proportion of homozygous overlapping windows was 0.05; (iii) the minimum number of consecutive SNPs included in a ROH was 100; (iv) the minimum length of a ROH was set to 1 Mb; (v) the maximum gap between consecutive homozygous SNPs was 500 kb; (vi) a density of one SNP per 50 kb; and (vii) a maximum of five SNPs with missing genotypes and up to one heterozygous genotype were allowed in a ROH. The ROH were defined by a minimum of 1 Mb in length to avoid short and common ROH that occur throughout the genome due to LD [6]. ROH were classified into five length classes: 1–2, 2–4, 4–8, 8–16, and >16 Mb, identified as ROH1–2 Mb, ROH2–4 Mb, ROH4–8 Mb, ROH8–16 Mb, and ROH>16 Mb, respectively.
Pedigree and genomic inbreeding coefficients
Four types of inbreeding coefficients (FPED, FROH, FGRM, and FHOM) were taken into account. Pedigree-based inbreeding coefficients (FPED) were estimated for all animals using pedigree records from a dataset containing 101,351 animals born between 1946 and 2015. The pedigree data was provided by Embrapa Dairy Cattle (Juiz de Fora, Minas Gerais, Brazil). The average pedigree depth was approximately three generations ranging from 0 to 7.85. The FPED was estimated through the software INBUPGF90 [85]. Genomic inbreeding coefficients based on ROH (FROH) were estimated for each animal according to McQuillan et al. [86]:
where LROHj is the length of ROHj, and Ltotal is the total size of the autosomes covered by markers. Ltotal was taken to be 2,510,605,962 bp, based on the consensus map. For each animal FROH (FROH1–2 Mb, FROH2–4 Mb, FROH4–8 Mb, FROH8–16 Mb, and FROH > 16 Mb) was calculated based on ROH distribution of five minimum different lengths (ROHj): 1–2, 2–4, 4–8, 8–16, and >16 Mb, respectively. A second measure of genomic inbreeding was calculated from a Genomic relationship matrix (G) and was denoted as FGRM. The G matrix was calculated according to the method described by VanRaden et al. [70] using the following formula:
where Z is a genotype matrix that contains the 0-2p values for homozygotes, 1–2p for heterozygotes, and 2-2p for opposite homozygotes, where Pi is the reference allele frequency at locus ith. The diagonal elements of the matrix G represent the relationship of the animal with itself, thus, it was used to assess the genomic inbreeding coefficient. Inbreeding based on the observed versus expected number of homozygous genotypes (FHOM) was calculated in PLINK v1.90 [84] by computing observed and expected autosomal homozygous genotypes counts for each sample, as follows:
Spearmann’s correlation coefficients between the inbreeding measures were estimated.
Gene prospection in shared ROH regions
The homozygous segments shared by more than 50% of the samples were chosen as an indication of possible ROH islands throughout the genome. The --homozyg-group function implemented in PLINK v1.90 [84] was used to assess ROH islands shared among individuals. The Map Viewer of the bovine genome UMD3.1.1 was used for identification of genes in ROH regions, available at “National Center for Biotechnology Information” (NCBI Map Viewer - https://www.ncbi.nlm.nih.gov/mapview/). Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.8 tool [26, 27] was used to identify significant (p ≤ 0.05) Gene Ontology (GO) terms and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways using the list of genes from ROH islands and the Bos taurus annotation file as background.
Additional files
Gene content inside runs of homozygosity overlapping regions (ROH Islands). (DOCX 27 kb)
Mean linkage disequilibrium (LD) estimated considering a physical distance lower than 100 kb between markers for each Bos taurus autosome and within each runs of homozygosity island. (DOCX 17 kb)
Gene Ontology (GO) terms and KEGG pathways enriched (p < 0.05) based on runs of homozygosity islands. (DOCX 30 kb)
Runs of homozygosity islands and signatures of selection located within or closely to those islands observed in the present study. (DOCX 19 kb)
Acknowledgements
E.P received a scholarship from Coordination for the Improvement of Higher Education Personnel (CAPES) in cooperation with the Brazilian Agricultural Research Corporation (EMBRAPA). N.B.S. was supported by postdoctoral fellowship from CAPES. F.B, D.P.M and M.V.G.B.S held productivity research fellowships from The Brazilian National Council for Scientific and Technological Development (CNPQ).
Funding
Marcos V. G. B. Silva was supported by the Embrapa (Brazil) SEG 02.09.07.008.00.00 “Genomic Selection in Dairy Cattle in Brazil,” CNPq PVE 407246/2013–4 “Genomic Selection in Dairy Gyr and Girolando Breeds,” and FAPEMIG CVZ PPM 00606/16 “Identification of Signatures of Selection using Next Generation Sequencing Data” appropriated projects.
Availability of data and materials
The genomic information used in this study belongs to Brazilian Agricultural Research Corporation (Embrapa), so we do not have authorization to share the data.
Abbreviations
- ABCGIL
Brazilian Association of Dairy Gyr Breeders
- ABCZ
Brazilian Association of Zebu Breeders
- CV
Coefficient of variation
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- FGRM
Genomic relationship matrix-based estimates of inbreeding
- FHOM
Inbreeding estimate based on observed and expected autosomal homozygous genotypes
- FPED
Pedigree-based estimates of inbreeding
- FROH
ROH-based estimates of inbreeding
- G
Genomic relationship matrix
- GO
Gene Ontology
- IBD
Identical by descent
- IBS
Identical by state
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LD
Linkage disequilibrium
- PNMGL
National Program for Improvement of Dairy Gir
- ROH
Runs of homozygosity
Authors’ contributions
EP, DPM, MAM, JCCP, MVGBS, and FB conceived and designed the experiment. EP, RVV, and FB carried out the data analyses. EP, NBS, ALFL, RI, DPM, RVV, MAM, JCCP, MVGBS, and FB interpreted the results and drafted the manuscript. NBS, DPM, ALFL, RI, MAM, JCCP, RVV, and MVGBS helped to draft and revise the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The DNA was extracted from semen bought from an artificial insemination center and therefore no specific ethical approval is needed (Brazil law number 11794, from October 8th, 2008, Chapter 1, Art. 3, paragraph III). All the samples were obtained with the consent of the artificial insemination center to use for research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4365-3) contains supplementary material, which is available to authorized users.
Contributor Information
Elisa Peripolli, Email: elisa_peripolli@hotmail.com.
Nedenia Bonvino Stafuzza, Email: nedeniabs@gmail.com.
Danísio Prado Munari, Email: danisio@fcav.unesp.br.
André Luís Ferreira Lima, Email: andre_lfl@yahoo.com.br.
Renato Irgang, Email: rirgang@hotmail.com.
Marco Antonio Machado, Email: marco.machado@embrapa.br.
João Cláudio do Carmo Panetto, Email: joao.panetto@embrapa.br.
Ricardo Vieira Ventura, Email: rventura@uoguelph.ca.
Fernando Baldi, Email: fernandobaldiuy@gmail.com.
Marcos Vinícius Gualberto Barbosa da Silva, Email: marcos.vb.silva@embrapa.br.
References
- 1.Broman KW, Weber JL. Long homozygous chromosomal segments in reference families from the centre d’Etude du polymorphisme humain. Am J Hum Genet. 1999;65:1493–1500. doi: 10.1086/302661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 3.Falconer DS, Mackay TFC. Introduction to quantitative genetics. Essex: Pearson; 1996. [Google Scholar]
- 4.Howrigan DP, Simonson MA, Keller MC. Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics. 2011;12:460. doi: 10.1186/1471-2164-12-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosse M, Megens HJ, Madsen O, Paudel Y, Frantz LAF, Schook LB, et al. Regions of Homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genet. 2012;8:e1003100. doi: 10.1371/journal.pgen.1003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Purfield DC, Berry DP, McParland S, Bradley DG. Runs of homozygosity and population history in cattle. BMC Genet. 2012;13:70. doi: 10.1186/1471-2156-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrero-Medrano JM, Megens H-J, Groenen MAM, Ramis G, Bosse M, Pérez-Enciso M, et al. Conservation genomic analysis of domestic and wild pig populations from the Iberian peninsula. BMC Genet. 2013;14:106. doi: 10.1186/1471-2156-14-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferenčaković M, Hamzic E, Gredler B, Curik I, Sölkner J. Runs of Homozygosity reveal genome-wide Autozygosity in the Austrian Fleckvieh cattle. Agric Conspec Sci. 2011;76:325–329. [Google Scholar]
- 9.Ferenčaković M, Hamzić E, Gredler B, Solberg TR, Klemetsdal G, Curik I, et al. Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations. J Anim Breed Genet. 2013;130:286–293. doi: 10.1111/jbg.12012. [DOI] [PubMed] [Google Scholar]
- 10.Kim ES, Cole JB, Huson H, Wiggans GR, Van Tassel CP, Crooker BA, et al. Effect of artificial selection on runs of homozygosity in U.S. Holstein cattle. PLoS One. 2013;8:e80813. doi: 10.1371/journal.pone.0080813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zavarez LB, Utsunomiya YT, Carmo AS, Neves HHR, Carvalheiro R, Ferencakovic M, et al. Assessment of autozygosity in Nellore cows (Bos Indicus) through high-density SNP genotypes. Front Genet. 2015;6:1–8. doi: 10.3389/fgene.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karimi S. Runs of Homozygosity Patterns in Taurine and Indicine Cattle Breeds (Master thesis). Vienna: BOKU - University of Natural Resources and Life Sciences; 2013. p. 53.
- 13.Zhang Q, Guldbrandtsen B, Bosse M, Lund MS, Sahana G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genomics. 2015;16:542. doi: 10.1186/s12864-015-1715-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marras G, Gaspa G, Sorbolini S, Dimauro C, Ajmone-Marsan P, Valentini A, et al. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim Genet. 2014;46:110–121. doi: 10.1111/age.12259. [DOI] [PubMed] [Google Scholar]
- 15.Wright S. Coefficients of inbreeding and relationship. Am Nat. 1922;56:330–338. doi: 10.1086/279872. [DOI] [Google Scholar]
- 16.Meuwissen THE, Hayes BJ, Goddard ME. Prediction of Total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visscher PM, Medland SE, Ferreira MAR, Morley KI, Zhu G, Cornes BK, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2006;2:0316–0325. doi: 10.1371/journal.pgen.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 19.Keller MC, Visscher PM, Goddard ME. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics. 2011;189:237–249. doi: 10.1534/genetics.111.130922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curik I, Sölkner J, Stipic N. Effects of models with finite loci, selection, dominance, epistasis and linkage on inbreeding coefficients based on pedigree and genotypic information. J Anim Breed Genet. 2002;119:101–115. doi: 10.1046/j.1439-0388.2002.00329.x. [DOI] [Google Scholar]
- 21.Santiago AA. O Zebu na Índia, no Brasil e no mundo. Instituto Campineiro de ensino agrícola: Campinas; 1986. [Google Scholar]
- 22.Queiroz SA, Lôbo RB. Genetic relationship, inbreeding and generation interval in registered Gir cattle in Brazil. J Anim Breed Genet. 1993;110:228–233. doi: 10.1111/j.1439-0388.1993.tb00734.x. [DOI] [PubMed] [Google Scholar]
- 23.Peripolli E, Munari DP, Silva MVGB, Lima ALF, Irgang R, Baldi F. Runs of homozygosity: current knowledge and applications in livestock. Anim Genet. 2016;48:255–271. doi: 10.1111/age.12526. [DOI] [PubMed] [Google Scholar]
- 24.Filho Reis JC, da Verneque Silva R, RDA T, Lopes PS, FSS R, FLB T. Inbreeding on productive and reproductive traits of dairy Gyr cattle. Rev Bras Zootec. 2015;44:174–179. doi: 10.1590/S1806-92902015000500002. [DOI] [Google Scholar]
- 25.Wang Y. Runs of Homozygosity Patterns in Taurine and Indicine Cattle Breeds (Master thesis). Vienna: BOKU-University of Natural Resources and Life Sciences; 2015. p. 37.
- 26.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 28.Mastrangelo S, Tolone M, Di Gerlando R, Fontanesi L, Sardina MT, Portolano B. Genomic inbreeding estimation in small populations: evaluation of runs of homozygosity in three local dairy cattle breeds. Animal. 2016;10:746–754. doi: 10.1017/S1751731115002943. [DOI] [PubMed] [Google Scholar]
- 29.Gurgul A, Szmatoła T, Topolski P, Jasielczuk I, Żukowski K, Bugno-Poniewierska M. The use of runs of homozygosity for estimation of recent inbreeding in Holstein cattle. J Appl Genet. 2016;57:527–530. doi: 10.1007/s13353-016-0337-6. [DOI] [PubMed] [Google Scholar]
- 30.Kirin M, McQuillan R, Franklin CS, Campbell H, Mckeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity. PLoS One. 2010;5:e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szmatoła T, Gurgul A, Ropka-molik K, Jasielczuk I, Tomasz Z, Bugno-poniewierska M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest Sci. 2016;188:72–80. doi: 10.1016/j.livsci.2016.04.006. [DOI] [Google Scholar]
- 32.Santana Junior ML, Pereira RJ, Bignardi AB, El Faro L, Tonhati H, Albuquerque LG. History, structure, and genetic diversity of Brazilian Gir cattle. Livest Sci. 2014;163:26–33. doi: 10.1016/j.livsci.2014.02.007. [DOI] [Google Scholar]
- 33.Mészáros G, Boison AS, Pérez O’Brien AM, Ferenčaković M, Curik I, da Silva MV, et al. Genomic analysis for managing small and endangered populations : a case study in Tyrol Grey cattle. Front Genet. 2015;6:1–12. doi: 10.3389/fgene.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012;91:275–292. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Utsunomiya YT, Pérez O’Brien AM, Sonstegard TS, Van Tassell CP, do Carmo AS, Mészáros G, et al. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8:e64280. doi: 10.1371/journal.pone.0064280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sölkner J, Karimi Z, Pérez O’Brien AM, Mészáros G, Eaglen S, Boison SA, et al. Extremely non-uniform : patterns of runs of Homozygosity in bovine populations. Vancouver: 10th World Congr. Genet. Appl. to Livest. Prod; 2014. [Google Scholar]
- 37.Gaspa G, Marras G, Sorbolini S, Marsan PA, Williams JL, Valentini A, et al. Genome-wide Homozygosity in Italian Holstein cattle using HD SNP panel. Vancouver: 10th World Congr. Genet. Appl. to Livest. Prod; 2014. [Google Scholar]
- 38.Cohen M, Reichenstein M, Everts-Van Der Wind A, Heon-Lee J, Shani M, Lewin HA, et al. Cloning and characterization of FAM13A1 - a gene near a milk protein QTL on BTA6: evidence for population-wide linkage disequilibrium in Israeli Holsteins. Genomics. 2004;84:374–383. doi: 10.1016/j.ygeno.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Cohen-Zinder M, Seroussi E, Larkin DM, Loor JJ, Everts-Van Der Wind A, Lee JH, et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005;15:936–944. doi: 10.1101/gr.3806705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schnabel RD, Kim J-J, Ashwell MS, Sonstegard TS, Van Tassell CP, Connor EE, et al. Fine-mapping milk production quantitative trait loci on BTA6: analysis of the bovine osteopontin gene. Proc Natl Acad Sci U S A. 2005;102:6896–6901. doi: 10.1073/pnas.0502398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schopen GCB, Visker MHPW, Koks PD, Mullaart E, van Arendonk JAM, Bovenhuis H. Whole-genome association study for milk protein composition in dairy cattle. J Dairy Sci. 2011;94:3148–3158. doi: 10.3168/jds.2010-4030. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Ma P, Liu J, Zhang Q, Zhang Y, Ding X, et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015;16:111. doi: 10.1186/s12863-015-0263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kai W, GuanBin L, TeMin H, ZhenFang W, BaoLi S, DeWu L. Associations of polymorphisms in TRAPPC9, DGAT1, ATP1A1and GHR genes with milk production traits in Holstein dairy cow in southern China. J S C Agric Univ. 2016;37:2016. [Google Scholar]
- 44.Jiang L, Liu X, Yang J, Wang H, Jiang J, Liu L, et al. Targeted resequencing of GWAS loci reveals novel genetic variants for milk production traits. BMC Genomics. 2014;15:1105. doi: 10.1186/1471-2164-15-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bequette BJ, Kyle CE, Crompton LA, Buchan V, Hanigan MD. Insulin regulates milk production and mammary gland and hind-leg amino acid fluxes and blood flow in lactating goats. J Dairy Sci Sci. 2001;84:241–255. doi: 10.3168/jds.S0022-0302(01)74474-8. [DOI] [PubMed] [Google Scholar]
- 46.Hayes BJ, Lien S, Nilsen H, Olsen HG, Berg P, Maceachern S, et al. The origin of selection signatures on bovine chromosome 6. Anim Genet. 2008;39:105–111. doi: 10.1111/j.1365-2052.2007.01683.x. [DOI] [PubMed] [Google Scholar]
- 47.Morange M. HSFs in Development. In: Starke K, Gaestel M, editors. Molecular Chaperones in Health and Disease. Handbook of Experimental Pharmacology, vol 172. Berlin: Springer; 2006. 10.1007/3-540-29717-0_7.
- 48.Kumar A, Ashraf S, Goud TS, Grewal A, Singh SV, Yadav BR, et al. Expression profiling of major heat shock protein genes during different seasons in cattle (Bos Indicus) and buffalo (Bubalus Bubalis) under tropical climatic condition. J Therm Biol. 2015;51:55–64. doi: 10.1016/j.jtherbio.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Li QL, Ju ZH, Huang JM, Li JB, Li RL, Hou MH, et al. Two novel SNPs in HSF1 gene are associated with thermal tolerance traits in Chinese Holstein cattle. DNA Cell Biol. 2011;30:247–254. doi: 10.1089/dna.2010.1133. [DOI] [PubMed] [Google Scholar]
- 50.Cobanoglu O, Zaitoun I, Chang YM, Shook GE, Khatib H. Effects of the signal transducer and activator of transcription 1 (STAT1) gene on milk production traits in Holstein dairy cattle. J Dairy Sci. 2006;89:4433–4437. doi: 10.3168/jds.S0022-0302(06)72491-2. [DOI] [PubMed] [Google Scholar]
- 51.Rincon G, Islas-Trejo A, Castillo AR, Bauman DE, German BJ, Medrano JF. Polymorphisms in genes in the SREBP1 signalling pathway and SCD are associated with milk fatty acid composition in Holstein cattle. J Dairy Res. 2012;79:66–75. doi: 10.1017/S002202991100080X. [DOI] [PubMed] [Google Scholar]
- 52.Li C, Cai W, Zhou C, Yin H, Zhang Z, Loor JJ, et al. RNA-Seq reveals 10 novel promising candidate genes affecting milk protein concentration in the Chinese Holstein population. Sci Rep. 2016;6:26813. doi: 10.1038/srep26813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang L, Liu J, Sun D, Ma P, Ding X, Yu Y, et al. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS One. 2010;5:e13661. doi: 10.1371/journal.pone.0013661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jansen S, Aigner B, Pausch H, Wysocki M, Eck S, Benet-Pagès A, et al. Assessment of the genomic variation in a cattle population by re-sequencing of key animals at low to medium coverage. BMC Genomics. 2013;14:446. doi: 10.1186/1471-2164-14-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Argov-Argaman N, Mida K, Cohen B-C, Visker M, Hettinga K. Milk fat content and DGAT1 genotype determine lipid composition of the milk fat globule membrane. PLoS One. 2013;8:e68707. doi: 10.1371/journal.pone.0068707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grisart B, Farnir F, Karim L, Cambisano N, Kim J-J, Kvasz A, et al. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc Natl Acad Sci U S A. 2004;101:2398–2403. doi: 10.1073/pnas.0308518100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thaller G, Kramer W, Winter A, Kaupe B, Erhardt G, Fries R. Effects of DGAT1 variants on milk production traits in Jersey cattle. J Anim Sci. 2003;81:1911–1918. doi: 10.2527/2003.8181911x. [DOI] [PubMed] [Google Scholar]
- 58.Schennink A, Stoop WM, Visker MHPW, Heck JML, Bovenhuis H, Van Der Poel JJ, et al. DGAT1 underlies large genetic variation in milk-fat composition of dairy cows. Anim Genet. 2007;38:467–473. doi: 10.1111/j.1365-2052.2007.01635.x. [DOI] [PubMed] [Google Scholar]
- 59.Kaupe B, Brandt H, Prinzenberg EM, Erhardt G. Joint analysis of the influence of CYP11B1 and DGAT1 genetic variation on milk production, somatic cell score, conformation, reproduction, and productive lifespan in German Holstein cattle. J Anim Sci. 2007;85:11–21. doi: 10.2527/jas.2005-753. [DOI] [PubMed] [Google Scholar]
- 60.Maryam J, Babar ME, Nadeem A, Yaqub T, Hashmi AS. Identification of functional consequence of a novel selection signature in CYP11b1 gene for milk fat content in Bubalus Bubalis. Meta Gene. 2015;6:85–90. doi: 10.1016/j.mgene.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartella V, De Marco P, Malaguarnera R, Belfiore A, Maggiolini M. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signal. 2012;24:1515–1521. doi: 10.1016/j.cellsig.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 62.Kleinberg DL, Barcellos-Hoff MH. The pivotal role of insulin-like growth factor I in normal mammary development. Endocrinol Metab Clin N Am. 2011;40:461–471. doi: 10.1016/j.ecl.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 63.Xie Y, Yang S, Cui X, Jiang L, Zhang S, Zhang Q, et al. Identification and expression pattern of two novel alternative splicing variants of EEF1D gene of dairy cattle. Gene. 2014;534:189–196. doi: 10.1016/j.gene.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 64.Fan H, Wu Y, Zhou X, Xia J, Zhang W, Song Y, et al. Pathway-based genome-wide association studies for two meat production traits in Simmental cattle. Sci Rep. 2015;5:18389. doi: 10.1038/srep18389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raven L-A, Cocks BG, Goddard ME, Pryce JE, Hayes BJ. Genetic variants in mammary development, prolactin signalling and involution pathways explain considerable variation in bovine milk production and milk composition. Genet Sel Evol. 2014;46:29. doi: 10.1186/1297-9686-46-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curik I, Ferenčaković M, Sölkner J. Inbreeding and runs of homozygosity: a possible solution to an old problem. Livest Sci. 2014;166:26–34. doi: 10.1016/j.livsci.2014.05.034. [DOI] [Google Scholar]
- 67.Carothers AD, Rudan I, Kolcic I, Polasek O, Hayward C, Wright AF, et al. Estimating human inbreeding coefficients: comparison of genealogical and marker heterozygosity approaches. Ann Hum Genet. 2006;70:666–676. doi: 10.1111/j.1469-1809.2006.00263.x. [DOI] [PubMed] [Google Scholar]
- 68.Saura M, Fernández A, Varona L, Fernández AI, de Cara MÁR, Barragán C, et al. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data. Genet Sel Evol. 2015;47:1. doi: 10.1186/s12711-014-0081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pryce JE, Haile-Mariam M, Goddard ME, Hayes BJ. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genet Sel Evol. 2014;46:71. doi: 10.1186/s12711-014-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.VanRaden PM, Olson KM, Wiggans GR, Cole JB, Tooker ME. Genomic inbreeding and relationships among Holsteins, jerseys, and Brown Swiss. J Dairy Sci. 2011;94:5673–5682. doi: 10.3168/jds.2011-4500. [DOI] [PubMed] [Google Scholar]
- 71.Hayes BJ, Goddard ME. Technical note: prediction of breeding values using marker-derived relationship matrices. J Anim Sci. 2008;86:2089–2092. doi: 10.2527/jas.2007-0733. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Q, Calus MPL, Guldbrandtsen B, Lund MS, Sahana G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015;16:88. doi: 10.1186/s12863-015-0227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bjelland DW, Weigel KA, Vukasinovic N, Nkrumah JD. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J Dairy Sci. 2013;96:4697–4706. doi: 10.3168/jds.2012-6435. [DOI] [PubMed] [Google Scholar]
- 74.Lutaaya E, Misztal I, Bertrand JK, Mabry W. Inbreeding in populations with incomplete pedigrees. J Anim Breed Genet. 1999;116:475–480. doi: 10.1046/j.1439-0388.1999.00210.x. [DOI] [Google Scholar]
- 75.Cassell BG, Adamec V, Pearson RE. Effect of incomplete pedigrees on estimates of inbreeding and inbreeding depression for days to first service and summit milk yield in Holsteins and jerseys. J Dairy Sci. 2003;86:2967–2976. doi: 10.3168/jds.S0022-0302(03)73894-6. [DOI] [PubMed] [Google Scholar]
- 76.Miglior F, Szkotnicki B, Burnside EB. Analysis of levels of inbreeding and inbreeding depression in Jersey cattle. J Dairy Sci. 1992;75:1112–1118. doi: 10.3168/jds.S0022-0302(92)77856-4. [DOI] [PubMed] [Google Scholar]
- 77.Weigel KA. Controlling inbreeding in modern breeding programs. J Dairy Sci. 2001;84:E177–E184. doi: 10.3168/jds.S0022-0302(01)70213-5. [DOI] [Google Scholar]
- 78.Metzger J, Karwath M, Tonda R, Beltran S, Águeda L, Gut M, et al. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genomics. 2015;16:764. doi: 10.1186/s12864-015-1977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Purfield DC, McParland S, Wall E, Berry DP. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS One. 2017;12:e0176780. doi: 10.1371/journal.pone.0176780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Machugh DE, Shriver MD, Loftus RT, Cunningham P, Bradley DG. Domestication and Phylogeography of Taurine and zebu cattle. Genetics. 1997;146:1071–1086. doi: 10.1093/genetics/146.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan EKF, Nagaraj SH, Reverter A. The evolution of tropical adaptation: comparing taurine and zebu cattle. Anim Genet. 2010;41:467–477. doi: 10.1111/j.1365-2052.2010.02053.x. [DOI] [PubMed] [Google Scholar]
- 82.Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478. doi: 10.1186/1471-2164-15-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boison SA, Santos DJA, Utsunomiya AHT, Carvalheiro R, Neves HHR, O’Brien AMP, et al. Strategies for single nucleotide polymorphism (SNP) genotyping to enhance genotype imputation in Gyr (Bos Indicus) dairy cattle: comparison of commercially available SNP chips. J Dairy Sci. 2015;98:4969–4989. doi: 10.3168/jds.2014-9213. [DOI] [PubMed] [Google Scholar]
- 84.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aguilar I, Misztal I. Technical note: recursive algorithm for inbreeding coefficients assuming nonzero inbreeding of unknown parents. J Dairy Sci. 2008;91:1669–1672. doi: 10.3168/jds.2007-0575. [DOI] [PubMed] [Google Scholar]
- 86.McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, et al. Runs of Homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene content inside runs of homozygosity overlapping regions (ROH Islands). (DOCX 27 kb)
Mean linkage disequilibrium (LD) estimated considering a physical distance lower than 100 kb between markers for each Bos taurus autosome and within each runs of homozygosity island. (DOCX 17 kb)
Gene Ontology (GO) terms and KEGG pathways enriched (p < 0.05) based on runs of homozygosity islands. (DOCX 30 kb)
Runs of homozygosity islands and signatures of selection located within or closely to those islands observed in the present study. (DOCX 19 kb)
Data Availability Statement
The genomic information used in this study belongs to Brazilian Agricultural Research Corporation (Embrapa), so we do not have authorization to share the data.