

Abstract
Background
Bifidobacterium longum is a common member of the human gut microbiota and is frequently present at high numbers in the gut microbiota of humans throughout life, thus indicative of a close symbiotic host-microbe relationship. Different mechanisms may be responsible for the high competitiveness of this taxon in its human host to allow stable establishment in the complex and dynamic intestinal microbiota environment. The objective of this study was to assess the genetic and metabolic diversity in a set of 20 B. longum strains, most of which had previously been isolated from infants, by performing whole genome sequencing and comparative analysis, and to analyse their carbohydrate utilization abilities using a gene-trait matching approach.
Results
We analysed their pan-genome and their phylogenetic relatedness. All strains clustered in the B. longum ssp. longum phylogenetic subgroup, except for one individual strain which was found to cluster in the B. longum ssp. suis phylogenetic group. The examined strains exhibit genomic diversity, while they also varied in their sugar utilization profiles. This allowed us to perform a gene-trait matching exercise enabling the identification of five gene clusters involved in the utilization of xylo-oligosaccharides, arabinan, arabinoxylan, galactan and fucosyllactose, the latter of which is an abundant human milk oligosaccharide (HMO).
Conclusions
The results showed high diversity in terms of genes and predicted glycosyl-hydrolases, as well as the ability to metabolize a large range of sugars. Moreover, we corroborate the capability of B. longum ssp. longum to metabolise HMOs. Ultimately, their intraspecific genomic diversity and the ability to consume a wide assortment of carbohydrates, ranging from plant-derived carbohydrates to HMOs, may provide an explanation for the competitive advantage and persistence of B. longum in the human gut microbiome.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4388-9) contains supplementary material, which is available to authorized users.
Keywords: Bifidobacterium longum, Pan-genome, Carbohydrates, Comparative genomes, Gene-trait-matching, Microbiota
Background
The human gut microbiota harbours a complex and dynamic microbial community, which plays various functional roles to support human physiology and health [1]. This so-called “hidden organ” elicits important metabolic, structural and protective activities, and influences behaviour and brain function through the proposed “gut-brain-axis” [2, 3]. Bifidobacteria are among the first colonisers of the gut and represent common intestinal inhabitants throughout life, being purported as beneficial for the host [4–6]. Certain Bifidobacterium species are more frequently detected as gut commensals during early life and childhood, when the microbiota undergoes a succession to an adult-like composition after weaning [5, 7]. Bifidobacteria are Gram-positive, high G + C content, rod shaped, non-motile and non-spore-forming bacteria, belonging to the Actinobacteria phylum [8]; currently the genus Bifidobacterium includes 51 species and 10 subspecies, of which eight have been only recently identified (http://www.bacterio.net/bifidobacterium.html). Functional genome analysis has been vital to unravel the genetic strategies adopted by bifidobacteria to colonize the intestinal tract, a feature that appears to be facilitated by their adaptation to a glycan-rich environment [9]. More than 13% of the so-called clusters of orthologous gene (COG) families in the Bifidobacterium pan-genome are associated with carbohydrate metabolism [9]. Consequently, this saccharolytic genotype allows bifidobacteria to metabolize a variety of carbohydrates (host- and diet-derived substrates), which are not digested by the host’s digestive enzymes [10], to provide a competitive advantage in the complex gut environment.
Among the bifidobacteria colonizing the human gut, Bifidobacterium longum deserves a special mention due to its prevalence (and sometimes abundance) in both infants and adults [11, 12]. Despite the shift in microbiota composition following the transition from an exclusively milk-based diet to solid foods, it has been demonstrated that specific B. longum strains may persist in the intestinal microbiota over time [13, 14]; in addition, strains can be newly acquired by the adult host [15], being associated with health benefits for the host [16]. The B. longum taxon currently recognizes four subspecies: longum, infantis, suis and suillum [17, 18]. Isolates of the latter two subspecies are of animal origin [17, 19], while the former two are considered to be characteristic of the human gut microbiota. Genomic and functional studies have indicated that B. longum ssp. infantis is more specialized in the metabolism of milk glycans and for this reason is more adapted to the gut of a breast-feeding infant [20], being involved in the metabolism of HMOs [21]. In spite of the close genetic relatedness, B. longum ssp. longum strains are distinctly different from their B. longum ssp. infantis relatives when assessing carbohydrate utilization, with the former possessing various enzymes that are active on plant-derived oligosaccharides (while not encoding the same versatile enzymatic ability to degrade HMOs) [22, 23]. These metabolic capabilities exemplify the specific strategies followed by these two subspecies for the adaptation to a particular niche.
The correct assembly of the intestinal microbiota plays a key role in health and well-being of the host. The mechanisms of bifidobacterial establishment and persistence in the gut microbiota are far from being fully understood. However, in the particular case of B. longum, their diversity and capabilities to metabolize a large range of carbohydrates, considered to be the result of gene duplication and horizontal gene transfer [9], seem to make them particularly powerful microbial competitors in the complex human gut environment [24]. For these reasons, investigating metabolic abilities related to sugar fermentation and corresponding genetic functions is important to fully understand and appreciate how and which microbes can persist in the gut. In this context, the objective of this study was to explore the intraspecific genomic diversity within a pool of B. longum strains by performing whole genome sequencing and comparative analysis, and to analyse their carbohydrate utilization abilities using a gene-trait matching (GTM) approach.
Results
General genome features
In order to determine the genetic content, diversity and general characteristics of B. longum, especially strains previously isolated from babies (Table 1), we performed genome sequencing of 20 selected isolates in order to perform a comparative analysis against a number of publicly available B. longum genomes (for salient properties of these genomes see Additional file 1: Table S1). The sequencing and assembly efforts resulted in a set of unoriented contigs ranging in number from 20 to 72 per strain. In order to facilitate a coherent comparative analysis, we carried out a uniform open reading frame (ORF) prediction for all our 20 sequenced genomes in addition to the fully-sequenced B. longum genomes retrieved from public databases (https://www.ncbi.nlm.nih.gov/genome/genomes/183). As outlined in Table 1, the number of predicted ORFs ranged from 2189 for B. longum APC1503 to 1761 for B. longum APC1477, with an average value of 1961 ± 107 identified ORFs per genome. The average percentage G + C content was calculated to be 59.99 ± 0.20%, and ranged from 59.6% for B. longum APC1465 to 60.4% for B. longum DPC6316. The genome sizes follow a normal distribution with an average value of 2.39 Mbp, where B. longum APC1503 has the largest genome (2.56 Mbp) and B. longum APC1478 the smallest (2.22 Mbp), which is presumed to be equal or at least very close to the actual genome size as Illumina-based genome sequencing does not allow complete closure of the bacterial chromosome [25].
Table 1.
Bifidobacterium longum genomes sequenced and analysed in this study
| Genomes | Source of Isolation | Contigs | No. of ORFs | Genome Size (bp) | GC content (%) | No. of unique genes | Plasmids (Rep+) | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject | Age | Delivery | Feeding | ||||||||
| B. longum APC 1461 | A | 27 w | CS | BM | 37 | 1987 | 2,418,994 | 60.0 | 60 | No | [49] |
| B. longum APC 1462 | B | 1 w | NB | BM | 27 | 1992 | 2,417,784 | 60.2 | 4 | Yes | [49] |
| B. longum APC 1464 | B | 1 w | NB | BM | 31 | 1918 | 2,346,522 | 60.0 | 3 | Yes | [49] |
| B. longum APC 1465 | C | 4 w | NB | BM | 57 | 2032 | 2,452,211 | 59.6 | 6 | Yes | [49] |
| B. longum APC 1466 | C | 27 w | NB | BM | 51 | 2026 | 2,419,982 | 59.8 | 0 | Yes | [49] |
| B. longum APC 1468 | C | 27 w | NB | BM | 45 | 1994 | 2,395,158 | 60.1 | 12 | Yes | [49] |
| B. longum APC 1472 | E | 1 w | CS | BM | 50 | 1927 | 2,364,041 | 60.1 | 3 | Yes | [49] |
| B. longum APC 1473 | F | 27 w | CS | BM | 39 | 1874 | 2,317,071 | 59.8 | 6 | No | [49] |
| B. longum APC 1476 | G | 27 w | NB | BM | 48 | 2151 | 2,532,540 | 60.0 | 1 | Yes | [49] |
| B. longum APC 1477 | H | 1 w | NB | BM | 24 | 1761 | 2,228,807 | 59.8 | 0 | No | [49] |
| B. longum APC 1478 | H | 4 w | NB | BM | 21 | 1766 | 2,223,352 | 59.8 | 1 | No | [49] |
| B. longum APC 1480 | J | 4 w | NB | BM | 27 | 2084 | 2,477,750 | 59.9 | 6 | No | [49] |
| B. longum APC 1482 | D | 1 w | NB | FM | 72 | 1899 | 2,337,438 | 60.1 | 17 | No | [49] |
| B. longum APC 1503 | D | 1 w | NB | FM | 39 | 2189 | 2,562,703 | 59.7 | 23 | No | [49] |
| B. longum APC 1504 | I | 1 w | NB | BM | 51 | 1883 | 2,310,288 | 60.2 | 14 | No | [49] |
| B. longum DPC 6316 | K | 25 y | ND | ND | 32 | 1992 | 2,393,969 | 60.4 | 23 | Yes | [50] |
| B. longum DPC 6317 | L | 3 d | ND | ND | 20 | 1987 | 2,448,630 | 60.2 | 8 | No | [50] |
| B. longum DPC 6320 | M | 64 y | ND | ND | 25 | 1852 | 2,330,371 | 59.9 | 9 | No | [50] |
| B. longum DPC 6321 | N | 3 d | ND | ND | 28 | 1941 | 2,382,360 | 59.9 | 24 | Yes | [50] |
| B. longum DPC 6323 | O | 4 d | ND | ND | 52 | 1970 | 2,396,960 | 60.2 | 7 | Yes | [50] |
w weeks, y years, d days, CS C-Section, NB natural birth, BM breast-milk, FM formula milk, ND not determined or unknown
Comparative analyses
Employing a BLASTP-mediated comparative genomic approach coupled with hierarchical clustering analysis using the MCL algorithm (see Materials and Methods), allowed us to define a pool of 1200 gene families that are common among the 20 B. longum APC/DPC genomes and fully sequenced genomes of B. longum (Additional file 1: Table S1; Fig. 1a, b), thereby representing the predicted core genome. Core genes are present at least once in every examined genome, and in this case account for ~33% of the total gene families obtained in our analysis (Fig. 1c). The remaining 67% (which in total constitute 2433 gene families) represent genes that make up the variable or dispensable genome, which consists of genes that are present in some but not all genomes (Fig. 1b, c). In line with what had been established previously [23] regarding accessory gene functions (variable genome), we observed gene families involved in the process of colonization of, and adaptation to the host environment (e.g. surface proteins, sortase-dependent pili, exopolysaccharide production, R/M systems), as well as hypothetical proteins and mobile genetic elements. Despite the fact that fragmented genome assemblies do not allow a precise estimate of the occurrence, number and chromosomal position of mobile DNA elements in genomes, a search for genes encoding plasmid replication functions (PF01051 and PF01446) revealed that at least 10 of the 20 sequenced genomes appear to contain plasmid(s) (Table 1).
Fig. 1.
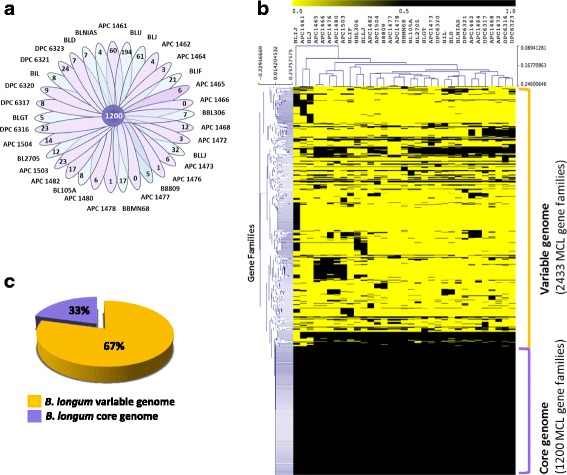
Comparative analysis of B. longum genomes. a Venn diagram displaying core gene families obtained by MCL clustering, and unique genes of B. longum APC/DPC strains and B. longum complete genomes. b Hierarchical clustering heatmap representing the variability of B. longum in terms of presence/absence of gene families. c Pie chart indicating the percentage of variable and core with respect to the total of gene families, resulting from the MCL clustering algorithm. APC 1461: B. longum APC 1461; APC 1462: B. longum APC 1462; APC 1464: B. longum APC 1464; APC 1465: B. longum APC 1465; APC 1466: B. longum APC 1466; APC 1468: B. longum APC 1468; APC 1472: B. longum APC 1472; APC 1473: B. longum APC 1473; APC 1476: B. longum APC 1476; APC 1477: B. longum APC 1477; APC 1478: B. longum APC 1478; APC 1480: B. longum APC 1480; APC 1482: B. longum APC 1482; APC 1503: B. longum APC 1503; APC 1504: B. longum APC 1504; BLIJ: B. longum ssp. infantis ATCC15697; BLJ: B. longum ssp. longum JDM301; BLIF: B. longum ssp. longum 157F; BBL306: B. longum ssp. longum CCUG30698; BLLJ: B. longum ssp. longum JCM1217; B8809: B. longum ssp. longum NCIMB8809; BBMN68: B. longum ssp. longum BBMN68; BL105A: B. longum ssp. longum 105A; BL2705: B. longum ssp. longum NCC2705; BLGT: B. longum ssp. longum GT15; BIL: B. longum ssp. longum F8; BLD: B. longum ssp. longum DJO10A; BLNIAS: B. longum ssp. longum KACC9156; DPC 6316: B. longum DPC 6316; DPC 6317: B. longum DPC 6317; DPC 6320: B. longum DPC 6320; DPC 6321: B. longum DPC 6321; DPC 6323: B. longum DPC 6323
Furthermore, this analysis also revealed the presence of 608 unique or strain-specific genes (i.e. genes that are present in just one of the analysed genomes), of which 227 are present in the B. longum APC/DPC genomes. These genes represent the presumed truly unique genes (TUGs) of each B. longum strain, and range between 60 in B. longum APC1461 to 0 in B. longum APC1466 and B. longum APC 1477 (Fig. 1a). The large percentage of genes observed within the dispensable genome is a reflection of a high level of diversity among the B. longum APC/DPC strains, and also with respect to publicly available genomes. Interestingly, strains isolated from the same baby generally cluster together based on hierarchical clustering analysis (Fig. 1b). For example, two pairs of B. longum APC/DPC strains (APC1465/APC1466 from subject C and APC1477/APC1478 from subject H) which had been isolated from samples collected at different dates, grouped together based on their gene content. In contrast, B. longum strains APC1462/APC1464, isolated from the same baby from samples collected in the same week exhibit clear genomic differences (Fig. 1b). Based on genome alignment, presence of SNPs and gene content it is likely that strain pair APC1477/APC1478, both isolated from the same subject H within a relatively short time frame (1 and 4 weeks), represent clonal derivatives of the same strain (we detected just 2 different genes, 24 SNPs and an average nucleotide identity of 99.9%) (Additional files 2, 3 and 4). Strains APC1465/APC1466 were isolated from subject C at an age of 4 and 27 weeks, respectively, and despite being closely related, they do contain a considerable number of genomic differences (we detected 78 different genes and an average nucleotide identity of 99.7%) (Additional files 2, 3 and 4). Interestingly, strain pair APC1462/APC1464, isolated from subject B within the same week, elicits the most striking differences (we detected 242 different genes and a lower average nucleotide identity of 99.1%). It is worth mentioning that among the genetic differences between these three strain pairs, we found functions predicted to represent mobile genetic elements (e.g. integrases, phage proteins as well as a considerable amount of hypothetical proteins) (Additional file 4). This suggests that horizontally transferred DNA plays an important role in causing genome diversity in this species.
Finally, B. longum APC1461 is distinct from all other sequenced B. longum APC/DPC strains, being the most diverse among them in terms of gene content.
Pan-genome of B. longum species computation
In order to calculate the total gene repertoire encountered in the newly sequenced B. longum APC/DPC genomes and the degree of overlap and diversity with respect to other B. longum genomes sequenced and publicly available, a pan-genome computation was performed in two steps employing the PGAP pipeline [26]. Firstly, we calculated the pan-genome of 37 genomes, including 17 publicly available B. longum complete sequenced genomes (Additional file 1: Table S1) and the 20 newly sequenced B. longum APC/DPC genomes. This analysis generated a total of 5970 gene families for this species (Fig. 2a). The pan-genome curve was determined as the total of gene families as a function of the number of included genomes, and was shown to display an asymptotic trend with a growth rate of an average of 169 families per genome in the first 17 iterations, decreasing to an average of 55 in the final seven additions. From this calculation, after the inclusion of the 31st genome a minor increase in the pan genome size is observed. Consistent with the above, the analysis of the core-genome function shows an asymptotic trend and stabilizes after the 22nd genome iteration reaching a value of 1163 gene families at the last iteration (Fig. 2b). The trends observed in both pan-genome and core-genome functions indicate that the pan-genome calculation for the included 37 genomes is not yet fully closed, but approaching closure. For this reason, we conclude that the number of genomes analysed here provides a comprehensive and near complete overview of the gene repertoire of the B. longum species. Secondly, we calculated the pan-genome repertoire of just our 20 newly sequenced APC/DPC genomes. The total number of identified gene families in this case was 4309 with a core-genome size of 1295, showing an asymptotic trend consistent with what was observed for the 37 B. longum strains (Fig. 2). A comparison between the two-calculated pan-genomes revealed that 72% of the identified gene families are present in both, while 28% of the gene families (1698) identified in B. longum species are absent in the 20 newly sequenced APC/DPC genomes. As expected, analysis of the 1698 gene families that did not have representatives in the newly sequenced strains showed that 60% of these are predicted to encode hypothetical proteins, transposases, putative prophage-related proteins and generic transporters. This confirms that the high genetic diversity observed within the B. longum APC/DPC strains is partly attributable to what are presumed to be horizontally transferred genes.
Fig. 2.
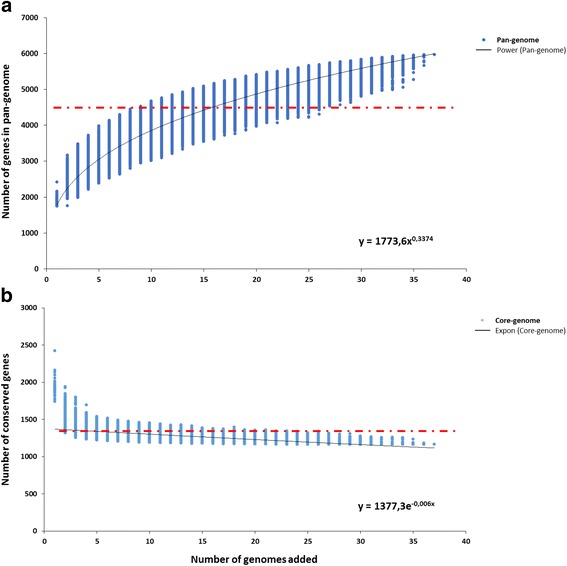
Pan-genome and core-genome of B. longum. a The pan-genome plot is represented by the accumulated number of new genes against the number of genomes added. b The core-genome plot is represented by the accumulated number of genes attributed to the core-genomes against the number of added genomes. The deduced mathematical function is reported. The red line represents the pan-genome and core-genome attributed to the 20 B. longum APC/DPC strains
Phylogenetic analysis
For the purpose of analysing the phylogenetic relationships between the B. longum APC/DPC strains and other B. longum strains, a phylogenetic supertree was created based on sequence similarities among the gene families that constitute the core genome. The selection of families was performed based on comparative analyses and their presence in single copy between the complete B. longum genomes, other B. longum draft genomes available in the NCBI public database at the time of the study (Additional file 1: Table S1) and Bifidobacterium breve UCC2003 (which was selected as the outlier). The resulting phylogenetic tree (Fig. 3) reveals the presence of two major branches among the analysed members of the B. longum species. The first branch constitutes members of the subspecies infantis taxonomic group, which contains 11 representatives (Fig. 3, Group A). The second branch is split into two different clades (Fig. 3, Groups B & C). As expected, we observed that most of the genomes annotated as B. longum ssp. longum fall into the same subclade (Fig. 3, Group C), in which most of the B. longum APC/DPC genomes were positioned and uniformly spread, with the genome of B. longum APC1461 being the only exception. As expected, each of the four pairs of strains isolated from same babies, B. longum APC1462/APC1464, B. longum APC1477/APC1478, B. longum APC1465/APC1466 and B. longum APC1503/APC1482, also cluster together. Consistent with previous findings [22], the animal–derived isolates (B. longum ssp. suis LMG21814, DSM20211 and AGR2137) also cluster together (Fig. 3, Group B). Interestingly, B. longum APC1461 was shown to belong to this group. The genomes of B. longum ssp. longum JDM301, B. longum ssp. longum CMCCP0001 and B. longum ssp. longum BXY01 cluster together as part of a distinct sub-clade of Group B (Fig. 3, Group B), as had previously been observed [22].
Fig. 3.
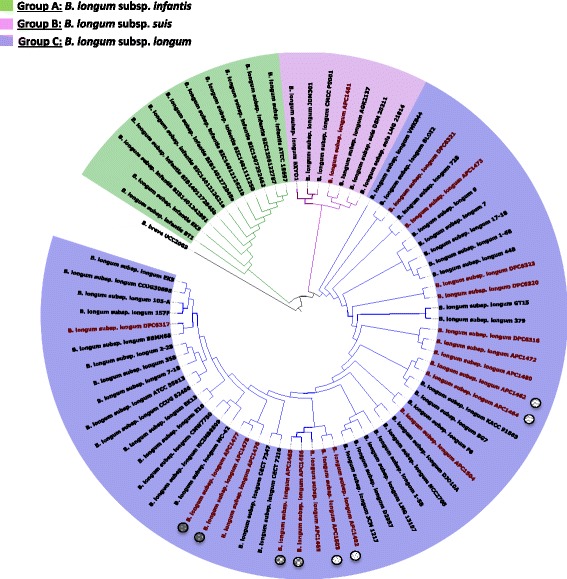
Phylogenetic analysis of B. longum. Phylogenetic supertree showing the relationship between 73 complete and incomplete B. longum strains and B. breve UCC 2003 as an outlier. B. longum APC/DPC strains are coloured in red. Grey circles with different fillings represent isolates from the same infant
Glycosyl-hydrolase prediction in B. longum species
It is known that more than 13% of the genes present in bifidobacterial genomes are predicted to be involved in sugar metabolism, allowing bifidobacteria to metabolize a wide range of carbohydrates [9]. In order to assess the carbohydrate fermentation capabilities of the B. longum APC/DPC strains, an in silico prediction of glycosyl hydrolases (GHs) encoded in their genomes was performed using the Cazy database (Fig. 4a; Additional file 1: Table S2). This analysis revealed that the B. longum pan-genome contains genes encoding GHs that belong to 22 different GH families (Fig. 4b). Distributed across various strains, GHs were found that were putatively assigned to ferment plant-derived carbohydrates and classified as members of GH31 (α-D-xyloside xylohydrolase), GH32 (β-fructofuranosidase), GH43 (Ara-f)(3)-Hypβ-L-arabinobiosidase, arabinan-endo-1,5-α-L-arabinosidase, xylan-1,4-β-xylosidase, L-arabinofuranosidase), GH51 (L-arabinofuranosidase), GH121 ((Ara-f)(3)-Hypβ-L-arabinobiosidase), GH53 (arabinogalactan-endo-β-1,4-galactanase) or GH127 (L-arabinofuranosidase). Our analysis also revealed the presence of enzymes with predicted α-galactosidase (GH36, GH27), β-galactosidase (GH2, GH42), β-glucosidase (GH1, GH3) and, though at lower abundance, α-glucosidase (GH13), α-mannosidase (GH38), α-amylase (GH13) and cyclomaltodextrinase (GH13) activities (Fig. 4a; Additional file 1: Table S2). The GHs from the in silico profile allowed us to identify certain GHs that appear to be characteristic of our B. longum APC/DPC collection. The most distinctive profile we found was that of strain B. longum APC1461 (Fig. 4a). Given the phylogenomic analysis, this strain seems to belong to a separate phylogenetic group (and may represent a separate subspecies) with genomic characteristics that appear to be a mix between those of ssp. longum and ssp. infantis members. This also is reflected by the in silico GH profile which predicts a reduced number of glycolytic activities involved in hydrolysis of plant-derived saccharidic substrates, while lacking a complete HMO utilization cluster.
Fig. 4.
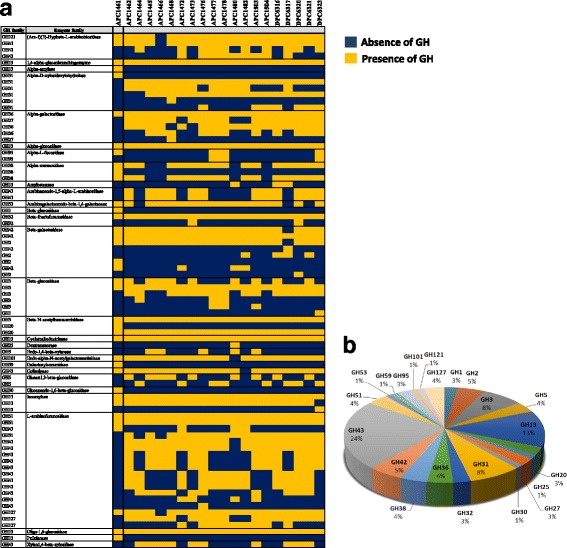
The predicted glycosyl hydrolases in B. longum. a Heatmap displaying the in silico prediction of the GH family members identified in the B. longum genomes. b Pie chart indicating the percentage of each GH family identified only in B. longum APC/DPC genomes
In vitro bifidobacterial growth on different carbohydrate sources
In order to assess and validate the in silico analyses, in vitro growth assays of the B. longum APC/DPC strains were performed involving 31 carbohydrates as sole carbon sources (Additional file 1: Table S3; Fig. 5a; Additional file 5). In this manner, we observed that all strains were able to grow in the presence of lactose, which was therefore used as a positive control in each batch culture. All strains exhibited good growth in glucose, arabinose, arabinogalactan and GOS. In addition, just two strains, B. longum APC1461 and DPC6321, were capable of (limited) growth with mannose as the sole carbon source; similarly, just three strains, B. longum APC1464, APC1477 and DPC6317, were able to achieve (limited) growth when amylopectin was used as the sole carbon source. None of the 20 strains tested were able to metabolize N-acetyl galactosamine, N-acetyl mannosamine, sialic acid, pullulan, starch, inulin, xylan, mucin, lacto-N-neotetraose (LNnT) or fucose. The fermentation capabilities for the remainder of the tested carbohydrates were shown to be variable among the 20 strains (Fig. 5a). The APC1461 strain showed the most variable sugar utilization pattern, being the only strain that was not capable of growth on xylose. In contrast with other strains tested in this study, B. longum DPC6320, DPC6321, APC1480 and APC1503 did not exhibit any appreciable growth on galactose as the sole carbon source. Interestingly, B. longum APC1461 and APC1503 were not able to utilize fructooligosaccharides (FOS) and sucrose, whereas 10 B. longum strains were shown to only attain intermediate growth, while the remaining eight strains displayed good growth in the presence of these two carbohydrates (Fig. 5a). A similar trend was found in the case of utilization of two different sources of galactan: pectin galactan and potato galactan. Seventeen out of the 20 strains were shown to be able to degrade both sugars, while strains B. longum APC1464, DPC6317 and DPC6321 did not show those metabolic capabilities. Notably, xylo-oligosaccharides (XOS) supported growth of half of the bifidobacterial strains tested, whereas arabinan supported growth of 12 strains.
Fig. 5.
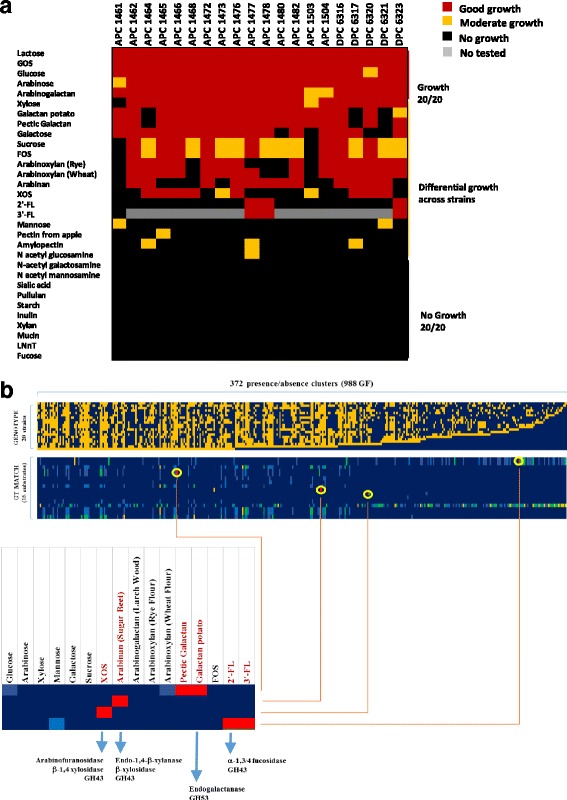
Evaluation of carbohydrate utilization by B. longum strains. a Heatmap showing the growth performance of B. longum APC/DPC strains on different carbon sources at 12 h. b Heatmap displaying the in silico gene-trait matching exercise performed based on the association between the presence/absence of GH families predicted and growth/no growth phenotype of the B. longum APC/DPC strains
We tested two types of arabinoxylan (isolated from rye or from wheat) (Additional file 1: Table S3) and growth was observed in the presence of both carbohydrates for 14 strains, with the exception of B. longum APC1477, which was not able to use rye arabinoxylan though it was able to metabolize wheat arabinoxylan. Interestingly, good growth was observed for 2′-fucosyllactose (2’-FL) or 3’-FL as a unique carbon source for three out of 20 tested B. longum strains (Fig. 5a).
In silico gene-trait-matching
An in silico evaluation of the role of specific genes associated with sugar metabolism found in our B. longum genomes was performed using a gene-trait matching (GTM) analysis based on the association between the presence or absence of gene families, and growth/no growth phenotype of the 20 B. longum APC/DPC strains. The analysis was performed on those carbohydrates with a differential utilization profile by the B. longum strains. The analyses allowed the identification of five clusters of gene families that are associated with the utilization of XOS, arabinan, arabinoxylan, galactan and fucosyllactose (Fig. 5b; Table 2; Additional file 5). Strain-specific growth on XOS corresponds with Cluster A, which consists of genes predicted to encode a 1,4-beta-xylosidase, a transporter system and a LacI-type transcriptional regulator (Table 2). Growth on arabinan was shown to correspond to Cluster B, which includes genes putatively encoding three endo-1,4-beta-xylanases and one beta-xylosidase, which is absent in all strains that are not able to grow on arabinan. The genotype/phenotype association also allowed us to identify a cluster of genes assignable to arabinoxylan utilization in 12 out of 20 strains. This cluster (Cluster C) is predicted to encode an exo-alpha-L-arabinofuranosidase II, three beta-xylosidases and one alpha-arabinofuranosidase I. Despite a partial match (<85% of phenotype/genotype concordance), 14 strains were able to metabolize arabinoxylan of which ten contain Cluster C. Moreover, three strains which were unable to grow on arabinoxylan were negative for this cluster. In contrast we observed that Cluster B may also be involved in arabinoxylan utilization, since an in silico association between the presence/absence of both clusters and the (in)ability to metabolize arabinoxylan was evident. Nine out of the 20 strains were able to grow in the presence of arabinan and arabinoxylan, and in five of these cases, both gene clusters (B and C) were present. However, when strains were shown to be unable to grow in the presence of arabinan, yet capable of growth on arabinoxylan (B. longum APC1466, DPC6323, APC1465, APC1473, APC1504) Cluster C was always present. Additionally, when strains were shown to metabolize arabinan yet did not grow on arabinoxylan (B. longum APC1478, APC1480), Cluster C was absent. Nevertheless, in the three cases of no growth observed on arabinan and arabinoxylan growth, we could only match one strain with the absence of Cluster B and Cluster C. Based on the above results, we hypothesize that both clusters are involved in the utilization of arabinoxylan, resulting in the observed ambiguous assignment. Furthermore, the observed strain-specific growth profile distribution on galactan matches with Cluster D, which contains a predicted endogalactanase. Furthermore, the lack-of-growth phenotype associated with galactan is associated with a truncation in the endogalactanase gene (resulting in a 107 amino acid-specifying pseudo-gene instead of the full gene which encodes a protein of 897 amino acids) which is believed to prevent its proper functioning. Finally, just three strains were able to utilize 2’-FL and 3’-FL as unique carbon sources and this specific growth profile allowed us to identify a gene cluster (Cluster E) encoding a predicted alpha-1,3/4-fucosidase surrounded by a LacI-like transcriptional regulator, ABC transporter system, a predicted fucose isomerase and another putative GH family 95 (Fig. 6; Table 2).
Table 2.
Gene-trait matching with functions resulting from hierarchical clustering analysis
| Carbohydrate | Gene cluster | Functions |
|---|---|---|
| XYLO-OLIGOSACCHARIDES (XOS) | A | Hypothetical protein |
| Putative outer membrane protein | ||
| Galactoside O-acetyltransferase | ||
| Alpha-L-arabinofuranosidase | ||
| Beta-1,4-xylosidase | ||
| ABC transporter permease | ||
| ABC transporter permease | ||
| Lactose ABC transporter substrate-binding protein | ||
| LacI family transcriptional regulator | ||
| NADH-dependent butanol dehydrogenase 1 | ||
| ARABINAN | B | Endo-1,4-beta-xylanase |
| Endo-1,4-beta-xylanase | ||
| Beta-xylosidase | ||
| Endo-1,4-beta-xylanase | ||
| ARABINOXYLAN | C | ABC transporter, permease protein, probably fructooligosaccharide porter |
| ABC transporter, permease protein, probably fructooligosaccharide porter | ||
| ABC transporter, extracellular SBP, probably fructooligosaccharide porter | ||
| Exo-alpha-L-arabinofuranosidase II | ||
| Lipase | ||
| LacI family transcriptional regulator | ||
| Beta-xylosidase | ||
| Beta-xylosidase | ||
| Beta-xylosidase o endo-arabinase | ||
| Alpha-arabinofuranosidase I | ||
| GALACTAN | D | Solute-binding protein of ABC transporter system for sugars galactan metabolism |
| ABC transporter permease | ||
| ABC transporter permease | ||
| Beta-galactosidase galactan metabolism | ||
| Transcriptional regulator LacI family galactan metabolism | ||
| Glycosyl hydrolases family 53 Endogalactanase galactan metabolism | ||
| 2,5-diketo-D-gluconic acid reductase | ||
| FUCOSYLLACTOSE (FL) | E | LacI family transcriptional regulator |
| putative ABC transporter permease | ||
| ABC transporter permease | ||
| ABC transporter substrate binding component | ||
| Mandelate racemase/muconate lactonizing protein | ||
| Short chain dehydrogenase | ||
| Hypothetical protein | ||
| Dihydrodipicolinate synthase | ||
| Predicted fucose isomerase | ||
| Alpha-1,3/4-fucosidase | ||
| Hypothetical protein (Glycosyl hydrolases family 95) |
Glycosyl hydrolases involved in carbohydrate utilization of each cluster are italicized
Fig. 6.
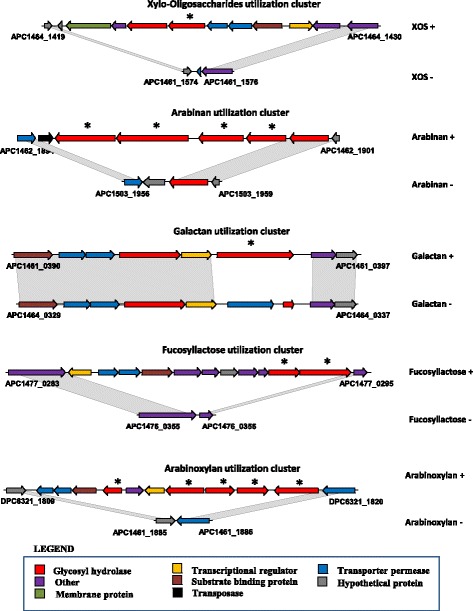
Carbohydrate utilization clusters predicted by GTM. Locus map showing gene clusters putatively involved in the utilization of various sugars by certain B. longum strains. Glycosyl hydrolases involved in carbohydrate utilization of each cluster are also highlighted (*)
Discussion
Bifidobacterium longum is one of the most abundant members of the human intestinal microbiota and is widely used as a probiotic [11, 16]. B. longum ssp. longum is commonly present at high numbers in the gut microbiota of humans throughout life, thus indicative of a close symbiotic host-microbe relationship [5, 11]. Different mechanisms may be responsible for the highly competitive nature of this taxon in its human host to allow stable establishment in the complex and dynamic intestinal microbiota environment. Genomic tools and the increased number of publicly available bifidobacterial sequences have enabled the analysis of genomic diversity and functionality in different species [9]. In this study, we carried out an analysis of the intra-species genomic diversity of 20 B. longum strains and we determined their capabilities to metabolize different carbohydrates.
We sequenced the genomes of 20 strains and performed a comparative genomic analysis using other publicly available B. longum genome sequences. Despite the fact that our genome sequencing, being based on Illumina short reads, did not permit full closure of the bacterial chromosome, we could observe that the average G + C % content and obtained genome size are in line with other B. longum strains sequenced [27, 28]. In contrast to observations for B. breve or B. bifidum pan-genomes [29, 30], the total predicted ORFs per genome was shown to exhibit a wide range (from 1761 to 2189), indicating that B. longum shows a higher level of genetic diversity than other bifidobacterial taxa, which has also been observed for the pangenome of B. adolescentis [31]. MCL comparative genome analysis also revealed high genomic diversity among the B. longum APC/DPC strains. The most diverse strain in terms of gene content was B. longum APC1461, which harbours the highest numbers of truly unique genes (TUGs).
The B. longum genomes analysed in this study allowed us to describe the pan-genome of B. longum species, which was defined as not fully closed but approaching a closed pan-genome, being consistent with that reported by O’Callaghan and coworkers [22]. The description of the pan-genome of other species such as B. breve, B. bifidum, and B. animalis ssp. lactis showed an essentially closed trend [29, 30, 32]. In contrast, the B. adolescentis pan-genome has been defined as an open pan-genome [31]. This indicates that B. longum shows higher genetic diversity than other bifidobacterial taxa, with the exception of B. adolescentis, which has also been observed to possess a wide range of predicted ORFs per genome.
The phylogenetic tree based on sequence similarity of core genes placed all but one (i.e. APC1461) of the B. longum APC/DPC strains in Group C of the phylogenetic super-tree, represented by members of the subspecies longum taxonomic group. The resulting consensus tree is in concordance with a previous one published by O’Callaghan and coworkers [22]. Group B includes representatives of the subspecies suis taxonomic group, which comprises strains isolated from pigs and from a calf [17, 19]. A new subspecies, B. longum ssp. sullium, was recently proposed by differentiating the subspecies suis taxonomic group on their ability/inability to produce urease, where the newly proposed subspecies is urease-negative [18]. However, the inclusion of this novel subspecies in the phylogenetic analysis was not possible as there is currently no representative sequenced genome available. B. longum APC1461 was found to carry a urease gene cluster (EC 3.5.1.5), indicating that this strain resembles subspecies suis rather than subspecies sullium. B. longum APC1461 is the first isolate (faecal isolate of a baby) of this subspecies to be included as a member in this clade which up until now only contains isolates of animal origin. The rest of B. longum APC/DPC strains were positioned clearly, yet spread uniformly across the B. longum clade, showing a good representation of those strains in this subspecies taxonomic group and being consistent with the genetic diversity observed.
The pan-genome analysis enabled an in silico prediction of 22 GH families involved in carbohydrate metabolism. Members of GH43 and GH13 families represented the largest proportion of all GHs predicted in APC/DPC genomes, accounting for 24% and 13%, respectively, and being consistent with previous observations for the Bifidobacterium genus [33]. These enzyme families are involved in the degradation of a wide range of carbohydrates, including plant-derived polysaccharides, such as amylopectin, maltodextrin, melibiose, (arabino)xylan, amylose or trehalose [34]. Other GH families such as GH31, GH51, GH53 or GH127, involved in plant-derived glycan metabolism, were commonly found in APC/DPC genomes. This enzymatic machinery is proposed to degrade the complex plant polysaccharides into mono- and oligosaccharides, which can then be taken up by ABC-type transporters (for further internal hydrolysis and metabolism). The B. longum APC/DPC genomes are enriched in genes that are predicted to be involved in the metabolism of plant-derived carbohydrates that are expected to be present in an adult diet [22]. However, the APC/DPC genomes also encode genes involved in the metabolism of host-derived glycans. Members of the families GH95, with fucosidase activity; GH20, hexosaminidase activity; or GH38, mannosidase activity, all exhibit a strain-specific presence in APC/DPC genomes. An endo-alpha-N-acetylgalactosaminidase (GH101), which hydrolyses the O-glycosidic bond in mucin-type O-glycan, was previously found in B. longum JCM1217 among others [35, 36], and is also present in all B. longum APC/DPC genomes with the only exception being strain APC1480. However, there was a noted absence of genes encoding sialidases or lacto-N-biosidases, which are involved in the utilization of other host-derived glycans, including particular HMOs [34].
The identification of genes or gene clusters which correlate with growth or non-growth on a particular carbohydrate was facilitated by the use of a gene-trait matching (GTM) approach, in concordance with other GTM analyses in different microorganisms [37]. We observed a strain-specific growth profile by B. longum APC/DPC strains on galactan, related to the presence of an endo-beta-1,4-galactanase GH53) (GalA), which has previously been reported to be involved in galactan metabolism by B. longum [38, 39]. Certain bifidobacterial strains/species are able to degrade xylo-oligosaccharides (XOS) and arabinoxylans (AX), complex molecules which contain arabinose and xylose substituents [40]. Both in vivo and in vitro studies have indicated the bifidogenic effect and other health benefits elicited by these potential prebiotics [40, 41]. The GTM approach applied here allowed us to identify a gene cluster (B) for arabinan utilization with a predicted endo-1,4-β-xylanase (GH5) as a key hydrolytic enzyme. Moreover, a predicted 1,4-β-xylosidase (GH43) in Cluster A is predicted to be crucial for XOS utilization, and a similar situation has previously been identified for B. adolescentis [42]. B. bifidum and B. longum ssp. infantis are known to be able to metabolize many different HMOs, and in particular the most abundant HMOs found in breast milk, fucosylated HMOs (2′-FL and 3′-FL) [43–45]. B. longum ssp. longum strains typically do not grow on 2′-FL as sole carbon source [46, 47], though a very recent study has described a B. longum ssp. longum strain capable of growing on 2′-FL or 3′-FL as the unique carbon source [48]. Interestingly, we found that three out of 20 B. longum APC/DPC strains exhibit vigorous growth on these sugars. This finding was supported by the prediction of a gene cluster (E) by GTM encoding an α-fucosidase, a key enzyme in HMO utilization [46] and a substrate binding component of an ABC transporter, previously found to be essential for FL intracellular utilization and being a key stable colonization factor of gut microbes in infants [47]. The positive strain found by Garrido and coworkers for 2′-FL and 3′-FL utilization showed a similar cluster of genes by transcriptomics [48], verifying our observations and the validity of our GTM approach as gene cluster predictor.
Conclusions
In this works we studied 20 B. longum strains which had been isolated (mostly from infants) to explore their intra-species genomic diversity and their metabolic and genetic variation for carbohydrate consumption. According to our comparative genomic analysis, the results depict a snapshot of their substantial genetic diversity in terms of genes and predicted GHs, as well as their ability to metabolize a large range of sugars. The genomic/ phenotype of B. longum ssp. longum strains is consistent with an adult diet as exemplified by an extensive enzymatic machinery and ability of strains to degrade plant-derived carbohydrates. Furthermore, we found strains able to grow on particular HMOs. Indeed, GTM analysis predicted the key gene cluster of FL utilization, corroborating the capability of B. longum ssp. longum species to metabolise HMOs, although further biochemical analyses are required to confirm these findings. Ultimately, these observations may provide an explanation for the persistence of B. longum species in the gut throughout life.
Methods
Bacterial strains and growth conditions
The 20 bifidobacterial strains used in this study (Table 1) had previously been isolated from infant faeces [49, 50] except in two cases where the strains originate from adult faeces, and deposited in the APC Culture Collection (APC Microbiome Institute, Ireland). Bifidobacteria were routinely grown on de Man-Rogosa-Sharpe (MRS) medium (Difco Laboratories, Detroit, MI) supplemented with 0.05% w/v cysteine-HCl (Sigma, St. Louis, MO) (MRSC) and incubated at 37 °C under anaerobic conditions (10% H2, 10% CO2, and 80% N2) in a chamber Mac 500 (Don Whitley Scientific, West Yorkshire, UK). For solid medium, 2% (w/v) agar (Oxoid, Basingstoke, UK) was added. Prior to each DNA extraction, bacteria were sub-cultured twice and overnight cultures were used.
Genomic DNA extraction, sequencing and data assembly
DNA was isolated by the use of a DNeasy Blood & Tissue Kit (Qiagen, Sussex, UK) following the manufacturer’s instructions. Briefly, the overnight culture was centrifuged, washed with PBS (Sigma, St. Louis, MO) and incubated for 1 h at 37 °C in an enzymatic lysis buffer with lysozyme (50 mg/ml) (Sigma, St. Louis, MO). Then, the manufacturer’s protocol was followed with the extra addition of RNAse (Sigma, St. Louis, MO). Sequencing and assembly was performed by Eurofins Genetic Services Ltd. (Ebersberg, Germany). The genomic DNA was sequenced on an Illumina MiSeq platform using chemistry v3 with paired-end sequencing. The draft genomes were de novo assembled following a pipeline that incorporates Velvet software (v1.2.10) [51] and a multi-kmer approach [52].
Contig analysis and general feature prediction
A preliminary comparative genomic analysis (methods explained in the next section) was conducted to align the 20 B. longum APC/DPC genomes with all complete genomes of B. longum available in the NCBI public database at the time of the study (Additional file 1: Table S1), in order to select the most appropriate genome of reference for ordering contigs prior to annotation. The alignment of the 20 B. longum draft genome sequences was performed against a number of closely related, fully sequenced and publicly available B. longum genomes (Additional file 1: Table S1). The B. longum JDM301, B. longum BBMN68, B. longum NCIMB 8809, B. longum DJ010A and B. longum GT15 genomes were thus selected as reference genomes so as to determine presumed contig order and orientation of each draft genome. MAUVE (v2.3.1) was used to reorder contigs based on the reference genome along with Artemis software (v.14) for visualization and manual editing of the beginning of each genome at the dnaA gene. Prediction of putative open reading frames (ORFs) was carried out using Prodigal predictor v2.50 software (https://github.com/hyattpd/Prodigal). Identified ORFs were then automatically annotated on the basis of BLASTP analysis [53]. Functional assignment was performed against the non-redundant protein database provided by the National Centre for Biotechnology (https://www.ncbi.nlm.nih.gov). Artemis was also used for inspecting and editing, where necessary, the ORF finding outputs and the associated BLASTP results. Moreover, annotations were further refined and verified using the protein family (Pfam) (http://pfam.xfam.org) database. Glycosyl hydrolases were predicted and annotated based on similarity to the Carbohydrate-Active enZYmes (CAZy) (http://www.cazy.org/) database, Enzyme Commission number (EC) database (http://enzyme.expasy.org) using annot8r pipeline (http://www.nematodes.org/bioinformatics/annot8r) and Pfam alignments.
Comparative genomics
An all-versus-all BLASTP alignment [53] (50% identity; e-value 1e-4 cut-off) was performed on the extracted protein sequences from each strain. The BLAST output were used as an input for the clustering into protein families sharing the same function using the Markov Cluster Algorithm (MCL) with an inflation index of 2.5, as previously described [29]. The obtained gene families were classified as belonging to either the core or to the variable genome based on their presence in either all strains or in a subset of the investigated strains, respectively. In the orthologue extraction an additional filter for paralogues was applied by selecting only those families that were shown to contain a single protein member for each genome.
Pangenome analyses
In order to predict the possible dynamic changes of genome size at the genus level, the sizes of pan-genome, core genome and unique gene, were computed. For all the genomes used in this study, a pangenome calculation was done using PGAP [26], as previously described [29]. The ORF content of each genome was organized in functional gene clusters using the gene family (GF) method implemented in the PGAP pipeline. A pan-genome profile and a core genome profile were built using all possible BLAST combinations for each genome being sequentially added.
Phylogenetic analyses
The supertree computation was performed from the alignment of a set of orthologous genes obtained from a BLAST-based comparative approach as indicated above, with the APC/DPC strains plus all B. longum genomes available in the NCBI public database at the time of writing. Each set of orthologous proteins was aligned using MUSCLE [54] and phylogenetic trees were constructed using the maximum-likelihood in PhyML [55]. The resulting consensus tree was computed using the Consense module from Phylip package v3.69 using the majority rule method (http://evolution.genetics.washington.edu/phylip.html) and phylogenetic data were submitted to TreeBASE database (http://treebase.org/treebase-web/home.html).
Bifidobacterial growth on different carbohydrate sources and GTM associations
For evaluation of growth on the different carbohydrates sources, strains were cultured on modified MRS (mMRS) medium manually prepared to obtain the following composition: tryptone (10.0 g/L), yeast extract (5 g/L), beef extract (10.0 g/L), K2HPO4 (3.0 g/L), KH2PO4 (3.0 g/L), tri-ammonium citrate (2.0 g/L), pyruvic acid (0.2 g/L), cysteine-HCl (0.3 g/L), Tween-80 (1 mL), MgSO4·7H2O (0.575 g/L), MnSO4·4H2O (0.12 g/L), FeSO4·7H2O (0.034 g/L). Prior to autoclaving, mMRS medium was adjusted to pH 6.8.
Thirty-one carbohydrates were tested in this study (Additional file 1: Table S3). Solutions of most of them were prepared by dissolution at 5% (w/v) in distilled water and sterilized by filtration (0.45 μm). They were then added to mMRS medium at final concentration of 0.5% (v/v). Since certain carbohydrates do not easily dissolve in water (pullulan, starch, amylopectin, inulin, arabinoxylan (wheat and rye), pectin galactan, mucin, galactan potato, pectin apple, arabinan, arabinogalactan, xylan and amylopectin), they were added directly to mMRS medium at a final concentration of 0.5% (w/v) and autoclaved at 121 °C for 15 min.
For growth assays, Bifidobacterium strains were cultured at 37 °C under anaerobic conditions in 10 ml of MRSC. Afterwards they were sub-cultured twice, first at 2% (v/v) into 10 mL of fresh medium during 8 h, and then at 1% (v/v) into 10 mL of MRSC fresh medium overnight. The next day, strains were inoculated at 1% (v/v) into 10 mL of mMRS medium, previously supplemented with the soluble carbohydrates at a final concentration of 0.5% (v/v) or directly to mMRS with non-soluble carbohydrate autoclaved. Prior to inoculation, both media were supplemented with cysteine-HCl at a final concentration of 0.05%.
Growth of the bacterial strains was evaluated by optical density (OD600 nm) using UV-1280 spectrophotometer (Schimadzu Corporation, Kyoto, Japan). Measurements were taken manually over a 24-h period at different time points: 0, 6, 9, 12 and 24 h. mMRS supplemented with 0.05% w/v cysteine-HCl and without the addition of a carbohydrate source served as a negative control. Based on the OD600 nm values at 12 h, bacterial growth was categorized as good growth (+) with an OD600 nm higher than 0.5 and moderate growth (+−) with an OD600 nm between 0.4 and 0.5. A cut-off value of 0.4 OD600 nm was used to discriminate strains which were not able to grow in the given carbohydrates.
Following completion of the fermentation assessment of B. longum strains, an in silico genotype/phenotype gene-trait matching (GTM) exercise was performed, correlating the presence/absence of genes obtained from comparative analysis with an observed phenotype.
The analysis was performed on a subset of gene families obtained after exclusion of the ones present in all the strains (core-genome). A further filter was included which excluded from the analysis that fraction of genes not involved in carbon source utilization (e.g. transposases, R/M systems, CRISPR systems and prophages).
After obtaining the final number of families, a further reduction of the dataset in clusters of unique combination of occurrence was performed which allowed us to obtain the “genotype” binary matrix (values 0 for absence and 1 for presence of a gene family). This obtained matrix contained 372 clusters on the rows and 20 strains on the columns. A second binary matrix was also similarly generated containing the fermentation profile and constituting the “phenotype”. In this case, a value of 0 was assigned for OD600 < 0.3 and value of 1 for OD600 > 0.4 (taken at 12 h of growth curve). The “phenotype” binary matrix contained 16 carbohydrates on the rows and 20 strains on the columns, organized in the same order as in the genotype.
Following alignment of these two matrices, the matching percentage between each row of the two matrices and the result was represented as a heatmap. The positive matches obtained from the analysis (> 95% of match between “genotype” and “phenotype”) as well as the relative genomic surrounding regions were further inspected and compared with additional information retrieved from alternative available databases such as EC (Enzyme Classification) database [56] and PFAM (http://pfam.sanger.ac.uk) alignments, in order to further support the obtained results [29].
Additional files
Bifidobacterium longum genomes publicly available used for different analysis along the study. Table S2. EC annotation numbers of the in silico glycosyl hydrolases predicted. Table S3. Carbohydrates used for in vitro assays. (DOC 152 kb)
Excel file containing the SNPs identified between Bifidobacterium longum strain pairs APC1465 - APC1466, APC1477 - APC1478, and APC1462 - APC1464. (XLSX 1103 kb)
Mauve representation containing the genome alignment of Bifidobacterium longum strain pairs APC1465 - APC1466, APC1477 - APC1478 and APC1462 - APC1464. (PPTX 229 kb)
Excel file containing the list of MCL cluster which differ between Bifidobacterium longum strain pairs APC1465 - APC1466, APC1477 - APC1478 and APC1462 - APC1464. (XLSX 31 kb)
Presence and absence of carbohydrate clusters predicted and growth on those carbohydrates of all the strains. (XLS 62 kb)
Acknowledgements
The authors acknowledge Orafti, Glycom, Longlife and Hayashibara for the gift of carbohydrates.
Funding
This work was supported by The APC Microbiome Institute (under Science Foundation Ireland [SFI] grant number: SFI/12/RC/2273) and by Department of Agriculture Food and Marine (DAFM) funded INFANTMET (Infant Nutrition for Programming the Gut Microbiota in Neonates) Project (Ref No. 10FDairy). The funding bodies played no role in the design of the study and collection, analysis and interpretation of the data, or in writing of the manuscript.
Availability of data and materials
The sequences described in this study have been deposited at GenBank database under the following accession numbers: PJDN00000000, PJDO00000000, PJDP00000000, PJDQ00000000, PJDR00000000, PJDS00000000, PJDT00000000, PJDU00000000, PJDV00000000, PJDW00000000, PJDX00000000, PJDY00000000, PJDZ00000000, PJEA00000000, PJEB00000000, PJEC00000000, PJED00000000, PJEE00000000, PJEF00000000, PJEG00000000.
Abbreviations
- 2’-FL
2′-fucosyllactose
- AX
Arabinoxylans
- BM
Breast-milk
- COG
Cluster of Orthologous Gene
- CS
C-Section
- D
Days
- FM
Formula milk
- FOS
Fructooligosaccharides
- G + C
Guanine + cytosine
- GHs
Glycosyl hydrolases
- GTM
Gene-trait matching
- HMOs
Human milk oligosaccharides
- LNnT
Lacto-N-neotetraose
- MCL
Markov Cluster Algorithm
- NA
Not applicable
- NB
Natural birth
- ORF
Open reading frame
- W
Weeks
- XOS
Xylo-oligosaccharides
- Y
Years
Authors’ contributions
RPR, DVS, CS designed research and revised the manuscript giving critically improvement. SA, FB conducted research, analysed data and wrote the paper. MOCM, CAR provided essential reagents and materials and, revised the manuscript giving critically improvement. CAR sought ethical approval for the studies from which the strains were derived. CS has primary responsibility for final content. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Ethical approval was obtained from the Clinical Research Ethics Committee of the Cork Teaching Hospitals and written informed consent was obtained from adult volunteers and from the parents of all babies, for previous publications with the bacterial strains from which the genomes originated, and referenced in this manuscript.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4388-9) contains supplementary material, which is available to authorized users.
Contributor Information
Silvia Arboleya, Email: Silvia.Arboleya@ipla.csic.es.
Francesca Bottacini, Email: francesca.bottacini@ucc.ie.
Mary O’Connell-Motherway, Email: M.OConnellMotherway@ucc.ie.
C. Anthony Ryan, Email: tony.ryan@ucc.ie.
R. Paul Ross, Email: p.ross@ucc.ie.
Douwe van Sinderen, Email: d.vansinderen@ucc.ie.
Catherine Stanton, Phone: +353 25 42606, Email: Catherine.Stanton@teagasc.ie.
References
- 1.Clemente Jose C, Ursell Luke K, Parfrey Laura W, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Hara AM, Shanahan F. The gut Flora as a forgotten organ. EMBO Rep. 2006;7(7):688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandhu KV, Sherwin E, Schellekens H, Stanton C, Dinan TG, Cryan JF. Feeding the microbiota-gut-brain axis: diet, microbiome, and neuropsychiatry. Transl Res. 2017;179:223–44. [DOI] [PubMed]
- 4.Turroni F, Marchesi JR, Foroni E, Gueimonde M, Shanahan F, Margolles A, van Sinderen D, Ventura M. Microbiomic analysis of the bifidobacterial population in the human distal gut. ISME J. 2009;3(6):745–751. doi: 10.1038/ismej.2009.19. [DOI] [PubMed] [Google Scholar]
- 5.Arboleya S, Watkins C, Stanton C, Ross RP. Gut Bifidobacteria populations in human health and aging. Front Microbiol. 2016;7:1204. doi: 10.3389/fmicb.2016.01204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 8.Leahy SC, Higgins DG, Fitzgerald GF, van Sinderen D. Getting better with bifidobacteria. J Appl Microbiol. 2005;98(6):1303–1315. doi: 10.1111/j.1365-2672.2005.02600.x. [DOI] [PubMed] [Google Scholar]
- 9.Milani C, Lugli GA, Duranti S, Turroni F, Bottacini F, Mangifesta M, Sanchez B, Viappiani A, Mancabelli L, Taminiau B, et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl Environ Microbiol. 2014;80(20):6290–6302. doi: 10.1128/AEM.02308-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pokusaeva K, Fitzgerald GF, van Sinderen D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011;6(3):285–306. doi: 10.1007/s12263-010-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, Cagnasso P, Bizzarri B, de’Angelis GL, Shanahan F, et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl Environ Microbiol. 2009;75(6):1534–1545. doi: 10.1128/AEM.02216-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, Kerr C, Hourihane J, Murray D, Fuligni F, et al. Diversity of Bifidobacteria within the infant gut microbiota. PLoS One. 2012;7(5):e36957. doi: 10.1371/journal.pone.0036957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shkoporov AN, Khokhlova EV, Kulagina EV, Smeianov VV, Kafarskaia LI, Efimov BA. Application of several molecular techniques to study numerically predominant Bifidobacterium spp. and Bacteroidales order strains in the feces of healthy children. Biosci Biotechnol Biochem. 2008;72(3):742–748. doi: 10.1271/bbb.70628. [DOI] [PubMed] [Google Scholar]
- 14.Shkoporov AN, Efimov BA, Khokhlova EV, Chaplin AV, Kafarskaya LI, Durkin AS, McCorrison J, Torralba M, Gillis M, Sutton G, et al. Draft genome sequences of two pairs of human intestinal Bifidobacterium longum subsp. longum strains, 44B and 1-6B and 35B and 2-2B, consecutively isolated from two children after a 5-year time period. Genome Announc. 2013;1(3):e00234-13. [DOI] [PMC free article] [PubMed]
- 15.Maldonado-Gomez MX, Martinez I, Bottacini F, O'Callaghan A, Ventura M, van Sinderen D, Hillmann B, Vangay P, Knights D, Hutkins RW, et al. Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell Host Microbe. 2016;20(4):515–526. doi: 10.1016/j.chom.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Arboleya S, Stanton C, Ryan CA, Dempsey E, Ross PR. Bosom buddies: the symbiotic relationship between infants and Bifidobacterium longum ssp. longum and ssp. infantis. Genetic and Probiotic features. Annu Rev Food Sci Technol. 2016;7:1–21. doi: 10.1146/annurev-food-041715-033151. [DOI] [PubMed] [Google Scholar]
- 17.Mattarelli P, Bonaparte C, Pot B, Biavati B. Proposal to reclassify the three biotypes of Bifidobacterium longum as three subspecies: Bifidobacterium longum subsp. longum subsp. nov., Bifidobacterium longum subsp. infantis comb. nov. and Bifidobacterium longum subsp. suis comb. nov. Int J Syst Evol Microbiol. 2008;58(Pt 4):767–772. doi: 10.1099/ijs.0.65319-0. [DOI] [PubMed] [Google Scholar]
- 18.Yanokura E, Oki K, Makino H, Modesto M, Pot B, Mattarelli P, Biavati B, Watanabe K. Subspeciation of Bifidobacterium longum by multilocus approaches and amplified fragment length polymorphism: description of B. Longum subsp. suillum subsp. nov., isolated from the faeces of piglets. Syst Appl Microbiol. 2015;38(5):305–314. doi: 10.1016/j.syapm.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Kelly WJ, Cookson AL, Altermann E, Lambie SC, Perry R, Teh KH, Otter DE, Shapiro N, Woyke T, Leahy SC. Genomic analysis of three Bifidobacterium species isolated from the calf gastrointestinal tract. Sci Rep. 2016;6:30768. doi: 10.1038/srep30768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sela DA, Mills DA. Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol. 2010;18(7):298–307. doi: 10.1016/j.tim.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, Lapidus A, Rokhsar DS, Lebrilla CB, German JB, et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A. 2008;105(48):18964–18969. doi: 10.1073/pnas.0809584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Callaghan A, Bottacini F, O'Connell Motherway M, van Sinderen D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genomics. 2015;16:832. doi: 10.1186/s12864-015-1968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Ren F, Zhao L, Jiang L, Hao Y, Jin J, Zhang M, Guo H, Lei X, Sun E, et al. Starch and starch hydrolysates are favorable carbon sources for Bifidobacteria in the human gut. BMC Microbiol. 2015;15:54. doi: 10.1186/s12866-015-0362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fushinobu S. Unique sugar metabolic pathways of Bifidobacteria. Biosci Biotechnol Biochem. 2010;74(12):2374–2384. doi: 10.1271/bbb.100494. [DOI] [PubMed] [Google Scholar]
- 25.Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, Ahn TH, Karpinets T, Lund O, Kora G, Wassenaar T, et al. Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics. 2015;15(2):141–161. doi: 10.1007/s10142-015-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. PGAP: pan-genomes analysis pipeline. Bioinformatics (Oxford, England) 2012;28(3):416–418. doi: 10.1093/bioinformatics/btr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaplin AV, Efimov BA, Smeianov VV, Kafarskaia LI, Pikina AP, Shkoporov AN. Intraspecies genomic diversity and long-term persistence of Bifidobacterium longum. PLoS One. 2015;10(8):e0135658. doi: 10.1371/journal.pone.0135658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bottacini F, Medini D, Pavesi A, Turroni F, Foroni E, Riley D, Giubellini V, Tettelin H, van Sinderen D, Ventura M. Comparative genomics of the genus Bifidobacterium. Microbiology (Reading, England) 2010;156(Pt 11):3243–3254. doi: 10.1099/mic.0.039545-0. [DOI] [PubMed] [Google Scholar]
- 29.Bottacini F, O'Connell Motherway M, Kuczynski J, O'Connell KJ, Serafini F, Duranti S, Milani C, Turroni F, Lugli GA, Zomer A, et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genomics. 2014;15:170. doi: 10.1186/1471-2164-15-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duranti S, Milani C, Lugli GA, Turroni F, Mancabelli L, Sanchez B, Ferrario C, Viappiani A, Mangifesta M, Mancino W, et al. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ Microbiol. 2015;17(7):2515–2531. doi: 10.1111/1462-2920.12743. [DOI] [PubMed] [Google Scholar]
- 31.Duranti S, Milani C, Lugli GA, Mancabelli L, Turroni F, Ferrario C, Mangifesta M, Viappiani A, Sanchez B, Margolles A, et al. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci Rep. 2016;6:23971. doi: 10.1038/srep23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milani C, Duranti S, Lugli GA, Bottacini F, Strati F, Arioli S, Foroni E, Turroni F, van Sinderen D, Ventura M. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl Environ Microbiol. 2013;79(14):4304–4315. doi: 10.1128/AEM.00984-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Callaghan A, van Sinderen D. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol. 2016;7:925. doi: 10.3389/fmicb.2016.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milani C, Lugli GA, Duranti S, Turroni F, Mancabelli L, Ferrario C, Mangifesta M, Hevia A, Viappiani A, Scholz M, et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci Rep. 2015;5:15782. doi: 10.1038/srep15782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujita K, Oura F, Nagamine N, Katayama T, Hiratake J, Sakata K, Kumagai H, Yamamoto K. Identification and molecular cloning of a novel glycoside hydrolase family of core 1 type O-glycan-specific endo-alpha-N-acetylgalactosaminidase from Bifidobacterium longum. J Biol Chem. 2005;280(45):37415–37422. doi: 10.1074/jbc.M506874200. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki R, Katayama T, Kitaoka M, Kumagai H, Wakagi T, Shoun H, Ashida H, Yamamoto K, Fushinobu S. Crystallographic and mutational analyses of substrate recognition of endo-alpha-N-acetylgalactosaminidase from Bifidobacterium longum. J Biochem. 2009;146(3):389–398. doi: 10.1093/jb/mvp086. [DOI] [PubMed] [Google Scholar]
- 37.Smokvina T, Wels M, Polka J, Chervaux C, Brisse S, Boekhorst J, van Hylckama Vlieg JE, Siezen RJ. Lactobacillus paracasei comparative genomics: towards species pan-genome definition and exploitation of diversity. PLoS One. 2013;8(7):e68731. doi: 10.1371/journal.pone.0068731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, Zwahlen M-C, Desiere F, Bork P, Delley M, et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99(22):14422–14427. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinz SW, Pastink MI, van den Broek LA, Vincken JP, Voragen AG. Bifidobacterium longum endogalactanase liberates galactotriose from type I galactans. Appl Environ Microbiol. 2005;71(9):5501–5510. doi: 10.1128/AEM.71.9.5501-5510.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivière A, Moens F, Selak M, Maes D, Weckx S, De Vuyst L. The ability of Bifidobacteria to degrade Arabinoxylan oligosaccharide constituents and derived oligosaccharides is strain dependent. Appl Environ Microbiol. 2014;80(1):204–217. doi: 10.1128/AEM.02853-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aachary AA, Prapulla SG. Xylooligosaccharides (XOS) as an emerging Prebiotic: microbial synthesis, utilization, structural characterization, bioactive properties, and applications. Compr Rev Food Sci Food Saf. 2011;10(1):2–16. doi: 10.1111/j.1541-4337.2010.00135.x. [DOI] [Google Scholar]
- 42.Amaretti A, Bernardi T, Leonardi A, Raimondi S, Zanoni S, Rossi M. Fermentation of xylo-oligosaccharides by Bifidobacterium adolescentis DSMZ 18350: kinetics, metabolism, and beta-xylosidase activities. Appl Microbiol Biotechnol. 2013;97(7):3109–3117. doi: 10.1007/s00253-012-4509-y. [DOI] [PubMed] [Google Scholar]
- 43.Sela DA, Garrido D, Lerno L, Wu S, Tan K, Eom H-J, Joachimiak A, Lebrilla CB, Mills DA. Bifidobacterium longum subsp. infantis ATCC 15697 α-Fucosidases are active on fucosylated human milk oligosaccharides. Appl Environ Microbiol. 2012;78(3):795–803. doi: 10.1128/AEM.06762-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagae M, Tsuchiya A, Katayama T, Yamamoto K, Wakatsuki S, Kato R. Structural basis of the catalytic reaction mechanism of novel 1,2-alpha-L-fucosidase from Bifidobacterium bifidum. J Biol Chem. 2007;282(25):18497–18509. doi: 10.1074/jbc.M702246200. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz-Moyano S, Totten SM, Garrido DA, Smilowitz JT, German JB, Lebrilla CB, Mills DA. Variation in consumption of human milk oligosaccharides by infant gut-associated strains of Bifidobacterium breve. Appl Environ Microbiol. 2013;79(19):6040–6049. doi: 10.1128/AEM.01843-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bunesova V, Lacroix C, Schwab C. Fucosyllactose and L-fucose utilization of infant Bifidobacterium longum and Bifidobacterium kashiwanohense. BMC Microbiol. 2016;16(1):248. doi: 10.1186/s12866-016-0867-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuki T, Yahagi K, Mori H, Matsumoto H, Hara T, Tajima S, Ogawa E, Kodama H, Yamamoto K, Yamada T, et al. A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nat Commun. 2016;7:11939. doi: 10.1038/ncomms11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garrido D, Ruiz-Moyano S, Kirmiz N, Davis JC, Totten SM, Lemay DG, Ugalde JA, German JB, Lebrilla CB, Mills DA. A novel gene cluster allows preferential utilization of fucosylated milk oligosaccharides in Bifidobacterium longum subsp. longum SC596. Sci Rep. 2016;6:35045. doi: 10.1038/srep35045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barrett E, Deshpandey AK, Ryan CA, Dempsey EM, Murphy B, O'Sullivan L, Watkins C, Ross RP, O'Toole PW, Fitzgerald GF, et al. The neonatal gut harbours distinct bifidobacterial strains. Arch Dis Child Fetal Neonatal Ed. 2015;100(5):F405–F410. doi: 10.1136/archdischild-2014-306110. [DOI] [PubMed] [Google Scholar]
- 50.Barrett E, Ross RP, Fitzgerald GF, Stanton C. Rapid screening method for analyzing the conjugated linoleic acid production capabilities of bacterial cultures. Appl Environ Microbiol. 2007;73(7):2333–2337. doi: 10.1128/AEM.01855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Schroder J, Schmidt B. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics (Oxford, England) 2013;29(3):308–315. doi: 10.1093/bioinformatics/bts690. [DOI] [PubMed] [Google Scholar]
- 53.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 54.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 56.Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28(1):304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bifidobacterium longum genomes publicly available used for different analysis along the study. Table S2. EC annotation numbers of the in silico glycosyl hydrolases predicted. Table S3. Carbohydrates used for in vitro assays. (DOC 152 kb)
Excel file containing the SNPs identified between Bifidobacterium longum strain pairs APC1465 - APC1466, APC1477 - APC1478, and APC1462 - APC1464. (XLSX 1103 kb)
Mauve representation containing the genome alignment of Bifidobacterium longum strain pairs APC1465 - APC1466, APC1477 - APC1478 and APC1462 - APC1464. (PPTX 229 kb)
Excel file containing the list of MCL cluster which differ between Bifidobacterium longum strain pairs APC1465 - APC1466, APC1477 - APC1478 and APC1462 - APC1464. (XLSX 31 kb)
Presence and absence of carbohydrate clusters predicted and growth on those carbohydrates of all the strains. (XLS 62 kb)
Data Availability Statement
The sequences described in this study have been deposited at GenBank database under the following accession numbers: PJDN00000000, PJDO00000000, PJDP00000000, PJDQ00000000, PJDR00000000, PJDS00000000, PJDT00000000, PJDU00000000, PJDV00000000, PJDW00000000, PJDX00000000, PJDY00000000, PJDZ00000000, PJEA00000000, PJEB00000000, PJEC00000000, PJED00000000, PJEE00000000, PJEF00000000, PJEG00000000.