

Abstract
Understanding the origin of the catalytic power of enzymes has both conceptual and practical importance. One of the most important finding from computational studies of enzyme catalysis is that a major part of the catalytic power is due to the preorganization of the enzyme active site. Unfortunately, misunderstanding of the non trivial preorganization idea lead some (e.g. Ref 1) to assume that it does not consider the effect of the protein residues. This major confusion reflects a misunderstanding of the statement that the interaction energy of the enzyme group and the transition state is similar to the corresponding interaction between the water molecules and the TS, and that the catalysis is due to the reorganization free energy of the water molecules. Obviously, this finding does not mean that we do not consider the enzyme groups. Another problem is the idea that catalysis is due to substrate preorganization. This more traditional idea is based in some cases on inconsistent interpretation of the action of model compounds, which unfortunately, do not reflect the actual situation in the enzyme active site. The present paper addresses the above problems, clarifying first the enzyme polar preorganization idea and the current misunderstandings. Next we take a specific model compound that was used to promote the substrate preorganization proposal and establish its irrelevance to enzyme catalysis. Overall, we show that the origin of the catalytic power of enzymes cannot be assessed uniquely without computer simulations, since at present this is the only way of relating structure and energetics.
Keywords: Active Site Preorganization, TS Stabilization, Enzyme Catalysis, Empirical Valence Bond, Substrate Preorganization
1. Introduction
1.1 General background
The polar preorganization of enzymes active sites has been proposed2-4 to be the most important factor in enzyme catalysis. However, the corresponding idea is not trivial and its misunderstanding might lead to conceptual problems and to difficulties in quantifying the catalytic power of enzymes. A glaring example is a recent communication,1 where Menger, Nome and coworkers ventured to assume (while quoting Ref. 5) that the preorganization concept never considered the contribution of the enzyme catalytic groups (it was stated that “He (Warshel) claimed that “enzyme catalysis is not due to the interaction between enzyme and substrate (which is what was believed by most people), but rather to a large free-energy penalty for the reorganization of the solvent in the reference reaction.” Thus, he argued that “electrostatic reorganization energy accounts for almost the entire catalytic power of enzymes”). This point is clear from repeated parts of Ref. 1 (e.g.,. the statement that “Solvent reorganization, as opposed to molecular interaction, has been dogmatically set forth by others as the sole source of enzymatic accelerations”). Furthermore, misunderstandings of the preorganization idea (e.g. the problematic definition in Ref. 6) and how to evaluate it (e.g. 7 (also see discussion in Ref. 8)) are quiet common. Furthermore, there is a common tendency to assume that enzymes work by preorganization of the substrate9, 10 which is, in fact, a combination of the near attack conformation (NAC) idea,11 the entropy idea,12 the strain idea,13, 14 and different forms of many of the ground state destabilization ideas. A recent example of the problems with a substrate preorganization proposal was pointed out in a careful study by Hilvert and Gelman showing that the dipeptide (which was suggested to be sufficient to catalyze amide bond hydrolysis15) is incapable of catalyzing the hydrolysis of several ester and amide substrates16 and that without the preorganized protein environment, achieving catalysis is extremely difficult. Another example is the work of Ref. 1. Interestingly, despite the major problems in many of the substrate preorganization proposals (see below) they are much simpler to accept, unless one actually checks their validity by computational energy considerations.
1.2 Conceptual Clarifications of the Enzyme Preorganization Idea
In clarifying possible difficulties in understanding the preorganization idea it is important to repeat this concept, following the guide of Fig.1. The figure illustrates the nature of the catalytic effect in a typical SN2 enzymatic reaction (e.g. 17). In such a case the catalysis is defined (see Ref. 2) as the difference between the SN2 step in the enzyme active site and in the corresponding reference reaction in water solution. In water the change of the charge distribution of the substrate, upon moving from the reactant state (RS) to the transition state (TS), leads to rotation of the water molecules (drawn as dipoles) so that they point towards the charges of the TS. The rotation of the water costs energy, which is referred to as the reorganization energy. In the enzyme on the other hand, the active site polar (or charged) groups that are also drawn as dipole do not have to undergo significant rotation to stabilize the TS charge distribution since they are already pointing in the correct direction. The energy that holds the protein dipole reorganized is invested by the protein folding forces. Quite surprisingly, the strong interaction between the protein dipoles and the TS charges is very similar to the corresponding interaction between the water molecules and the TS charge in the reaction in water (see right of Fig 1). Since the interaction energy between the environment and the TS charges is very similar in the water and the protein (and thus the contribution of the interaction to catalysis cancels), the main difference is that in the reference water reaction we have to invest significant reorganization free energy, while in the protein active site the investment of energy is very small.
Figure 1.

Schematic representation of the reorganization effect in water and protein for an SN2 reaction.
In other words, in water the free energy of moving from the ground state to the TS is larger than in the enzyme since the water dipoles have to reorganize and pay for the reorganization energy, whereas in the enzyme the polar groups or the catalytic groups are preorganized in the correct direction and thus do not have to pay large preorganization energy. The difference between the large reorganization energy in water and the small reorganization of the protein groups is the reason for catalysis.
Obviously, the preorganization concept has never suggested that the protein does not have catalytic groups. The confusion may be due to the major inability to realize that interaction energy at a specific point along the reaction coordinate is not equal to free energy of moving from the RS to the TS.
Of course, there are key catalytic groups, whose effect has been explored in details in all of our studies (e.g. 2), but such groups lead to catalysis not by just being in the active site and interacting strongly with the substrate TS, but by not having to pay for reaching the configuration that leads to such interaction. In a clear contrast to the presumption of Ref. 1 (where it was stated that “Our work here shows that catalytic groups, carboxy groups in this case, need not be mere idle spectators. Thus, only two carboxy groups, properly placed, can lead to enzymatic rates”), we never suggested that the enzyme catalytic groups are just some “idle spectators” groups, but rather critical groups that provide crucial preorganization effect.
The statement in Ref. 5 that “enzyme catalysis is not due to the interaction between the enzyme and substrate (which is what was believed by most people) meant, for those who followed the preorganization idea and the paper, that the interactions at the TS (which are never zero) cancel out and not that the catalytic groups are not needed for catalysis.
2. Computational Methods
We calculated the activation energies of systems I-V given in the main text using our empirical valence bond (EVB)18-20 and free energy perturbation/umbrella sampling (FEP/US) approach.21 The calculations were performed using MOLARIS software with the ENZYMIX force field.22 Further details on the methods can be found in the Supporting Information.
3. Results and Discussion
3.1 Considering the Problems with Using Model Compounds in Promoting the Substrate Preorganization Idea
The idea that the substrate preorganization is the origin of enzyme catalysis, is a seemingly reasonable idea, although it does not pass a careful examinations. In clarifying the problems with the misunderstanding about the presumed role of substrate preorganization in enzyme catalysis, it is useful to consider the difficulties in deducing the effectiveness of substrate preorganization from experiment with model compounds. Here we will focus on the studies presented in Ref. 1, using a model system, I (Fig. 2) to mimic aspartic proteases. The problems start with the simple fact that the model for a peptide cleavage reaction is coupled by delocalized π electrons to the naphthalene skeleton. This coupling moves a very large fraction of the oxyanion TS charges to the naphthalene and such delocalization effect does not exist in proteases. This brings us to the conceptually much more important problem of not defining the relevant reference state. More specifically, in considering catalysis it is crucial to address the issue “catalysis relative to what”? The best reference state is the same reaction in a water cage,2 but Ref. 1 does not consider any references state. Thus, we must try to consider the most logical reference state.
Figure 2.

Different systems that are studied using the EVB method. The EVB region is enclosed within dotted lines in compound I.
The most obvious selection is to consider the reference state for the reaction of aspartic proteases.23, 24 Unfortunately, this reference state does not correspond to our model compound and does tell us what is the origin of the observed rate of the model compound (since the model compound has delocalized charges, which make it very different than the catalytic system in aspartic proteases). To gain some understating of the model compound, we used the EVB approach, which is described extensively elsewhere,18 and in the SI but with the following twist; since we do not have a proper reference state with delocalized charges, we consider the effect of going from the original model compound (where the corresponding parameters and charge distribution were obtaining by ab initio calculations) to the butadiene type system (II in Fig. 2) and to the systems where an acids were disconnected from the napthalene skeleton (III, IV and V in Fig. 2). In all cases we kept the charge distribution obtained in system I. Since in all cases we kept the same type of transition state we consider the TS as shown in Fig. 3 as an intermediate (see our study of Chorismate Mutase25 and the corresponding EVB parameters are given in the SI). This EVB approach allows us to explore the catalytic effect obtained going from system I to V of Fig. 2. The calculated activation barriers are given in Table 1 and Fig. 3 and can be attributed to different effects. The change from I to II reflects mainly release of ground state strain. The change from I to III reflects some strain release and the regular cage effect of about 2.5 kcal mol which is not a part of the rigorously defined enzyme catalysis.2 The change from I to IV to reflects the cage effect and the strain plus NAC effect. The change from I to V probably reflects the above effects.
Figure 3.

Concerted type transition state for the hydrolysis of I.
Table 1. Calculated and Observed Activation Barriers (kcal/mol) for Different Systems.
| System | ΔG‡ (cal) | ΔG‡ (obs) |
|---|---|---|
| I | 20.2 | 19.8 |
| II | 25.6 | - |
| III | 27.3 | - |
| IV | 32.6 | - |
| V | 37.6 | - |
The trend in Fig. 4 may suggest that we have a model for an enzyme active site, since the magnitude of the decrease in the activation energy upon going from systems V to I are relatively close to the effect of a typical enzyme catalysis. However, such an idea encounters several problems.
Figure 4.
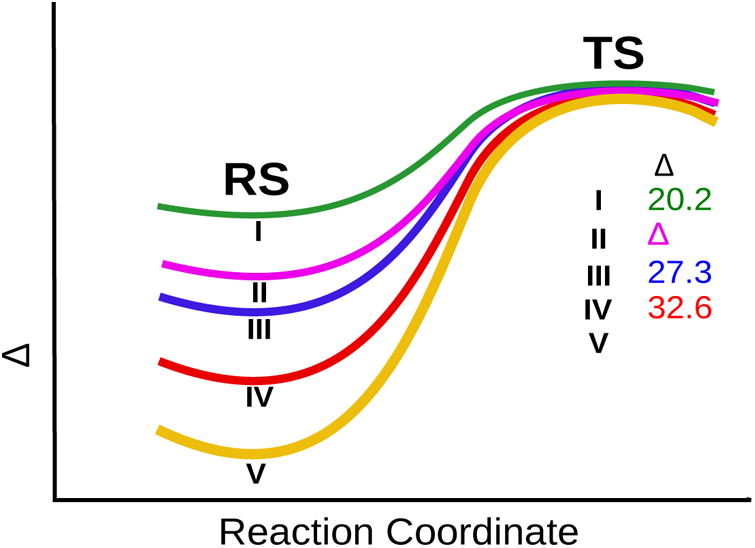
The EVB profiles for the motion to the TS in different systems. The TS energies are arbitrarily placed at the same height.
First, if we try for the sake of argument to take III or IV as a reference for I we may still try to claim that we have here a NAC or another type of substrate GS destabilization. However, the conceptual error here is in the assumption that if we have a rate acceleration in the enzyme, it must be due to the same NAC effect. In this respect, it is not useful to assert incorrectly (as done in Ref. 1) that theoretical analysis of aspartic proteases have shown that the catalysis is due to proximity effects (stating that “this mechanism, supported by both computational and experimental investigations, achieves its huge rate via “interaction” rather than “reorganization”). In fact, Refs.23 and 24 clearly stated that the origin of catalysis lies in the protein preorganization as opposed to the misunderstanding by Ref. 1. Obviously, the model compound I does not use the key factor of preorganization of the environment (as it works in solution). Furthermore, aspartic proteases use water attack rather than attack by an acidic group.
However, regardless of the missing active site preorganization effect in I, what about the use of the substrate preorganization in some other related enzymes? Can we extrapolate from the problematic conclusions obtained from the study of I to the assumption that the enzyme catalysis must include some type of substrate reorganization and in particular a NAC effect of compressing the O…C nucleophilic attack distance. Here it is essential to clarify that as shown in countless studies, enzymes do not use the NAC for ground state destabilization (see the very careful analysis of Ref. 25). In fact, a specific closely related study of a nucleophilic attack by an acidic group in haloalkane dehalogenase26 (which is much closer to the case in I than Asp proteases) has shown that the enzyme does not use the NAC in catalysis.
4. Conclusion
This work addressed the conceptual problems associated with the misunderstanding of the enzyme preorganization idea. Special attention was given to the misunderstanding promoted by Ref. 1 about our preorganization idea, presuming that it somehow overlooks the role of the protein catalytic groups. This confusion might have stemmed from misunderstanding that catalysis is defined relative to a well-defined references state (namely the reaction in a solvent cage). Thus if the interaction energy between the enzyme and the substrate TS is similar to that of the substrate TS and the surrounding solvent this interaction does not contribute to the catalytic effect. Ref. 1 and other works seem to overlook the key role of the protein environment, except in providing chemically active groups (this includes overlooking the role of the preorganization of the oxyanion holes). Ref. 1 attempted to support its view by using a model compound, where forcing catalytic groups are forced to strained interactions and thus can lead to fast reactions. What was missing has been a proper comparison of the model compounds to enzyme active site. Unfortunately, such a comparison can only be done consistently by computational approaches (that properly dissects the different energy contributions) and no such comparison has been done.
The analysis here established that although the model compound I can be considered as a catalytic system relative to some reference states, this effect is not related to the presumably similar catalytic effect in aspartic proteases. Such an assumption is similar to related assumptions in other works that suggested that strain in model compounds must mean that such strain exists in enzyme (see above). However, what is new in Ref. 1 is the major misunderstanding of the preorganization proposal and our related finding that the interaction energy at the TS is not the origin of the catalytic effect. This misunderstanding, as well as the general issue of using model compounds without clear reference state, are the main subjects of the present work.
Supplementary Material
Acknowledgments
This work was supported by Grant GM024492 from the National Institutes of Health (NIH). We also gratefully acknowledge the University of Southern California's High Performance Computing and Communication Center (HPCC) for computer time.
References
- 1.Souza BS, Mora JR, Wanderlind EH, Clementin RM, Gesser JC, Fiedler HD, et al. Transforming a Stable Amide into a Highly Reactive One: Capturing the Essence of Enzymatic Catalysis. Angew Chem Int Ed. 2017;56:5345–5348. doi: 10.1002/anie.201701306. [DOI] [PubMed] [Google Scholar]
- 2.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 3.Warshel A. Energetics of Enzyme Catalysis. Proc Natl Acad Sci USA. 1978;75:5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 5.Warshel A. Multiscale Modeling of Biological Functions: From Enzymes to Molecular Machines (Nobel Lecture) Angew Chem Int Ed. 2014;53:10020–10031. doi: 10.1002/anie.201403689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagel ZD, Klinman JP. A 21st century revisionist's view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 7.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, et al. A Dynamic Knockout Reveals That Conformational Fluctuations Influence the Chemical Step of Enzyme Catalysis. Science. 2011;332:234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adamczyk AJ, Cao J, Kamerlin SC, Warshel A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc Natl Acad Sci USA. 2011;108:14115–14120. doi: 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menger FM. An alternative view of enzyme catalysis. Pure Appl Chem. 2005;77:1873–1886. [Google Scholar]
- 10.Anslyn EV, Dougherty DA. Modern physical organic chemistry. University Science Books; 2006. [Google Scholar]
- 11.Bruice TC. A view at the millennium: the efficiency of enzymatic catalysis. Acc Chem Res. 2002;35:139–148. doi: 10.1021/ar0001665. [DOI] [PubMed] [Google Scholar]
- 12.Page MI, Jencks WP. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc Natl Acad Sci USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khanjin NA, Snyder JP, Menger F. Mechanism of chorismate mutase: Contribution of conformational restriction to catalysis in the Claisen rearrangement. J Am Chem Soc. 1999;121:11831–11846. [Google Scholar]
- 14.Blake CCF, Mair GA, North ACT, Phillips DC, Sarma VR. On Conformation of Hen Egg-White Lysozyme Molecule. Proc R Soc London, Ser B. 1967;167:365–377. doi: 10.1098/rspb.1967.0034. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Zhao Y, Hatfield S, Wan R, Zhu Q, Li X, et al. Dipeptide seryl-histidine and related oligopeptides cleave DNA, protein, and a carboxyl ester. Biorg Med Chem. 2000;8:2675–2680. doi: 10.1016/s0968-0896(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 16.MacDonald MJ, Lavis LD, Hilvert D, Gellman SH. Evaluation of the Ser-His Dipeptide, a Putative Catalyst of Amide and Ester Hydrolysis. Org Lett. 2016;18:3518–3521. doi: 10.1021/acs.orglett.6b01279. [DOI] [PubMed] [Google Scholar]
- 17.Olsson MHM, Warshel A. Solute solvent dynamics and energetics in enzyme catalysis: The SN2 reaction of dehalogenase as a general benchmark. J Am Chem Soc. 2004;126:15167–15179. doi: 10.1021/ja047151c. [DOI] [PubMed] [Google Scholar]
- 18.Kamerlin SC, Warshel A. The EVB as a quantitative tool for formulating simulations and analyzing biological and chemical reactions. Faraday Discuss. 2010;145:71–106. doi: 10.1039/B907354J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamerlin SC, Warshel A. The empirical valence bond model: theory and applications. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2011;1:30–45. [Google Scholar]
- 20.Warshel A, Weiss RM. An empirical valence bond approach for comparing reactions in solutions and in enzymes. J Am Chem Soc. 1980;102:6218–6226. [Google Scholar]
- 21.Zwanzig RW. High-temperature equation of state by a perturbation method. I. nonpolar gases. J Chem Phys. 1954;22:1420–1426. [Google Scholar]
- 22.Warshel A, Chu ZT, Villa J, Strajbl M, Schutz CN, Shurki A, Vicatos S, Plotnikov NV, Schopf P, Molaris XG. v 9.15. Los Angeles: University of Southern California; 2012. [Google Scholar]
- 23.Krzemińska A, Moliner V, Świderek K. Dynamic and Electrostatic Effects on the Reaction Catalyzed by HIV-1 Protease. J Am Chem Soc. 2016;138:16283–16298. doi: 10.1021/jacs.6b06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bjelic S, Åqvist J. Catalysis and linear free energy relationships in aspartic proteases. Biochemistry. 2006;45:7709–7723. doi: 10.1021/bi060131y. [DOI] [PubMed] [Google Scholar]
- 25.Strajbl M, Shurki A, Kato M, Warshel A. Apparent NAC effect in chorismate mutase reflects electrostatic transition state stabilization. J Am Chem Soc. 2003;125:10228–10237. doi: 10.1021/ja0356481. [DOI] [PubMed] [Google Scholar]
- 26.Shurki A, Strajbl M, Villa J, Warshel A. How much do enzymes really gain by restraining their reacting fragments? J Am Chem Soc. 2002;124:4097–4107. doi: 10.1021/ja012230z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.