

Abstract
Current post-operative standard of care for surgical procedures, including device implantations, dictates prophylactic antimicrobial therapy, but a percentage of patients still develop infections. Systemic antimicrobial therapy needed to treat such infections can lead to downstream tissue toxicities and generate drug-resistant bacteria. To overcome issues associated with systemic drug administration, a polymer incorporating specific drug affinity has been developed with the potential to be filled or refilled with antimicrobials, post-implantation, even in the presence of bacterial biofilm. This polymer can be used as an implant coating or stand-alone drug delivery device, and can be translated to a variety of applications, such as implanted or indwelling medical devices, and/or surgical site infections. The filling of empty affinity-based drug delivery polymer was analyzed in an in vitro filling/refilling model mimicking post-implantation tissue conditions. Filling in the absence of bacteria was compared to filling in the presence of bacterial biofilms of varying maturity to demonstrate proof-of-concept necessary prior to in vivo experiments. Antibiotic filling into biofilm-coated affinity polymers was comparable to drug filling seen in same affinity polymers without biofilm demonstrating that affinity polymers retain ability to fill with antibiotic even in the presence of biofilm. Additionally, post-implantation filled antibiotics showed sustained bactericidal activity in a zone of inhibition assay demonstrating post-implantation capacity to deliver filled antibiotics in a timeframe necessary to eradicate bacteria in biofilms. This work shows affinity polymers can fill high levels of antibiotics post-implantation independent of biofilm presence potentially enabling device rescue, rather than removal, in case of infection.
Keywords: Cyclodextrin, Biofilm, In situ filling, Antibiotic, Affinity-based release
1. Introduction
Infection is a major complication that can result following all surgical procedures including implanted medical devices [1]. Orthopedic joint and soft tissue implants are of particular concern because infection can often lead to removal of the implanted device and potentially serious complications. Staphylococcus aureus (S. aureus) infections are one of the most frequent types of infections that can develop following these procedures and in severe cases can lead to osteomyelitis, abscesses or fistulas [2–5]. Staphylococcus epidermidis is less pathogenic than S. aureus, but both can form biofilms on the surface of implanted materials [6–11]. Biofilms are complex clusters of bacteria composed of live and dead bacteria, proteins, polysaccharides, and an extracellular matrix formed by the bacteria [7,8]. A biofilm begins developing when bacteria adhere to proteins that have adsorbed to the surface of the material. Over time, several layers of bacteria cluster together on the surface of the material. A dense polysaccharide layer surrounds a mature biofilm forming a diffusional barrier that combined with a metabolic reduction in bacteria means traditional antibiotic treatments are often unsuccessful and can lead to resistant bacterial populations [12–14]. The only treatment for the biofilm-coated device and surrounding necrotic tissue is surgical removal [14]. This current work demonstrates the ability of a drug delivery polymer to load drug in situ and potentially treat infected devices even when coated with a biofilm thereby rescuing the device and possibly avoiding additional surgical procedures.
Prophylactic antimicrobial therapy before and after surgery has led to rates of infection that are low compared to the total number of procedures: 1–4% of 1 million knee and hip joint replacements completed in 2010 in the US [15,16] and ~10% of more than 1 million soft tissue repair procedures performed annually in the US [17]; however, these infections still add up to nearly 2 million total cases of hospital-acquired infections costing the US >$11 billion annually [1]. Systemic prophylactic antimicrobial therapy can also potentially drive drug resistance in the bacteria [18].
For those patients who develop a chronic infection such as osteomyelitis, treatment is very difficult often leading to osteolysis and significantly increasing treatment costs and tissue morbidity [19]. To address these concerns, engineers have developed a variety of localized delivery approaches ranging from antibiotic-filled bone cement [19] to soft tissue meshes coated with chlorhexidine and silver [20]; however, all patients receiving these antimicrobial devices still receive antimicrobials independent of whether they develop an infection.
Ideally only patients who develop infections would be exposed to post-operative antimicrobials. Therefore, in this work, a customizable affinity polymer delivery system has been developed that allows the physician to fill with antimicrobials only when an infection presents. Briefly, the physician would perform the surgical procedure as normal and implant a small amount of affinity polymer without any drug at the end of the procedure prior to closing. Only if a patient develops an infection, the physician can then inject a small dosage of antimicrobial into the tissue near the location of the implanted affinity polymer. Some of the injected drug would immediately act on the planktonic bacteria while the remaining drug would be able to fill into the affinity “pockets” of the polymerized cyclodextrin (pCD) affinity polymer system. For patients at high risk of infection, the affinity polymer could be filled with drug prior to implantation and then refilled with either additional drug or a different drug if an infection presents. The insoluble pCD affinity polymer exploits the affinity binding interaction between drug and CD resulting in high drug filling and prolonged, controlled affinity-based drug release for a range of hydrophobic and hydrophilic drugs and is capable of eradicating bacteria [21–24]. Specifically, the affinity interaction between pCD and the antibiotics is driven by the structure of pCD that has hydrophobic inner “pockets” that enable the drug to form a reversible inclusion complex. The strength of the affinity interaction is governed both by the structure of the drug and the size of the “pockets” in pCD. Previous in vivo work has demonstrated that the unfilled pCD-based affinity polymer system can fill with doxorubicin post-implantation and deliver therapeutic levels of post-implantation filled drug sufficient to eradicate subcutaneous tumors following resection [25]. Doxorubicin is poorly soluble in water (~2 mg/ml) compared to many antibiotics that are readily soluble (>50 mg/ml). In addition, the presence of established biofilm is believed to present a significant diffusional barrier that may prevent post-implantation loading.
In this work, the ability to refill antibiotics into affinity-based polymers across the diffusional barrier of mature biofilms is reported for the first time and builds upon the previous work of refilling affinity-based polymers with the chemotherapeutic doxorubicin. This work demonstrates the versatility of the affinity-based delivery system by refilling rifampicin, which has similar water solubility to doxorubicin (~2 mg/ml) and minocycline, which is significantly more water soluble (~50 mg/ml). Specifically, in this paper, empty affinity polymer disks have been filled with antibiotics in an in vitro post-implantation “tissue-mimicking” agarose phantom. For the “non-affinity” control a linear glucose polymer (polymerized dextran; pDEX) was selected since it represents a chemistry most similar to pCD but lacks the affinity interaction of the drug with the CD pocket (Fig. 1). Previous work with other polymers such as poly(L-lactic acid-co-glycolic acid) (PLGA) shows that, as expected, these polymers do not refill with drug either in vitro or in vivo (data not shown). Post-implantation drug filling into both non-affinity and affinity polymers was quantified, and the bioactivity of filled drug was shown via zone of inhibition assay. Since many patients do not present with infection immediately following surgery, the effect of bacteria and biofilm development on post-implantation filling of affinity polymer disks was also investigated. Briefly, pCD affinity polymer disks were exposed to bacteria prior to filling using a modified protocol for developing mature biofilm [26,27]. pCD affinity polymer disks were also preconditioned with serum proteins prior to bacterial exposure to evaluate the robustness of non-preconditioned biofilms and to validate the efficacy of non-preconditioned biofilms in the filling model studies.
Fig. 1.
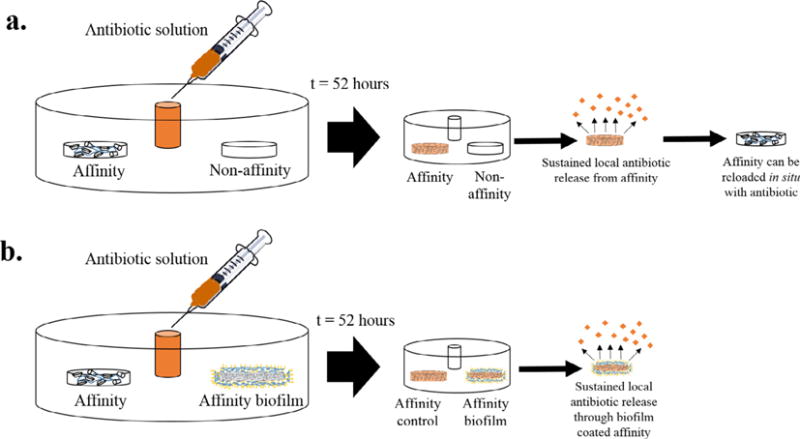
In situ “tissue-mimicking” antibiotic filling models with and without biofilm. (a) Validation of filling model with affinity and non-affinity (control) polymer disks. After 52 h of in situ filling, more antibiotic is filled into the affinity polymer disk. The filled affinity polymer disk can release filled antibiotic locally in a sustained manner and be refilled with more drug (in situ) when emptied. (b) Filling model with affinity polymer without biofilm (biofilm control) and affinity polymer disk with either an immature or mature biofilm. After 52 h of in situ filling, a similar amount of drug is filled into both the disks with and without the biofilm. Following this in situ filling, the antibiotic can be released from the disks with and without the biofilm locally in a sustained manner. The model also supports the filling of polymers coated with serum preconditioned biofilms where the polymers are exposed to FBS prior to bacterial exposure and are subsequently implanted into the model.
2. Experimental details
2.1. Materials
Lightly epichlorohydrin crosslinked β-cyclodextrin prepolymer was purchased from CycloLab, Budapest, Hungary (previously characterized in cross-linked form [22]). Low molecular weight dextran prepolymer was from Polysciences Inc., Warrington, PA. Hexamethylene diisocyanate (HDI) was from Sigma-Aldrich (St. Louis, MO). Rifampicin (RMP) and Minocycline (MC) were purchased from Research Products International (Mt. Prospect, IL). All other reagents, solvent, and chemicals were purchased from Fisher Scientific in the highest grade available. Green fluorescent protein (GFP)-labeled Staphylococcus aureus culture was kindly provided by Dr. Ed Greenfield (Case Western Reserve University).
2.2. Synthesis of affinity and non-affinity polymer disks
Affinity polymer disks comprised of polymerized cyclodextrin (pCD) and non-affinity polymer disks of polymerized dextran (pDEX) were synthesized according to a previously published protocol and cross-linked with HDI in a molar ratio of 1:0.16 (glucose residue:HDI) [21,22,28]. Briefly, 1 g of each β-cyclodextrin prepolymer (CD) and dextran were dissolved in 4 mL dimethylformamide (DMF; CD) or dimethylsulfoxide (DMSO; dextran). The affinity polymers were cured at 70 °C for 45 min and non-affinity polymers for 120 min and punched into 5 mm disks. Disks were thoroughly washed over a period of several days in 100% solvent, 50:50 solvent:water, and 100% water prior to use in order to remove unreacted residual polymer and solvent from pCD and pDEX.
2.3. Preparation of agarose ‘tissue phantom’
An agarose gel was created by dissolving 0.075% weight agarose in phosphate buffered saline (PBS) and bringing the solution to a boil. Briefly, 10 mL of the hot solution was placed in each well of a Costar 6-well tissue culture plate and allowed to gel at room temperature. Two 5 mm disks (affinity or non-affinity) were placed on top of the agarose and an additional 5 mL of hot agarose solution was placed on top. Polymer disks were aligned in the agarose to the center far left and right. The entire agarose solution was allowed to solidify at room temperature and a 3 mm biopsy punch was used to form a well in the center of each agarose gel to inject the drug solution (Fig. 1A). Agarose “tissue phantoms” are most commonly used for imaging (i.e. ultrasound and MRI) applications to mimic soft tissue properties (mechanical and acoustic) while providing good optical properties to enable easy imaging and analysis [29–33]. In this application agarose was selected due to the diffusional resistance that it provides, similar to that found in soft tissue, and its low background absorbance signal.
2.4. Biofilm formation on polymer disks
A 5 mm affinity or non-affinity polymer disk was placed in a 5 mL solution of 2× Trypticase soy broth (BD, BBL Trypticase soy broth) and 50 μL of freshly cultured GFP-labeled S. aureus was added. Each solution was incubated at 37 °C for 24 or 72 h to form an immature or mature biofilm, respectively. The biofilm-coated polymer disks were then implanted into agarose as above.
Additional preconditioned biofilms were formed on affinity polymer disks where 5 mm affinity polymer disks were placed in 1 mL of fetal bovine serum (FBS) for 24 h at 37 °C. Disks were then removed from the FBS, blotted on a Kimwipe, and placed in a solution of 5 mL 2× Trypticase soy broth and 50 μL of freshly cultured GFP-labeled S. aureus. Solutions were incubated at 37 °C for 72 h to form a mature biofilm prior to implantation in agarose.
2.5. In situ biofilm filling model
Biofilm coated disks were carefully removed from the Trypticase soy broth and blotted on a Kimwipe to remove culture media and non-adherent bacteria. After the initial layer (10 mL) of 0.075% agarose had solidified, a control affinity polymer disk without biofilm was placed on one side of the agarose and the biofilm coated affinity polymer disk was placed opposite. The final hot agarose layer (5 mL) was added and allowed to set at room temperature. A 3 mm well was punched in the center of each agarose gel for the drug filling solution (Fig. 1B).
2.6. In situ drug filling
A Biotek™ 96 well plate reader (H1; Winooski, VT) was used to record the absorbance of each drug diffusing through the agarose at set time points using an area scan with 19 × 19 data points Background absorbance scans were collected for each polymer disk at wavelengths of 485 nm (RMP) and 390 nm (MC) prior to injection of the drug and were later subtracted from absorbance scans following the drug injection (See Supplementary Fig. 1). Drug solutions of RMP dissolved in methanol (1 mg/mL; 50 μL) and MC dissolved in PBS (11 mg/μL; 100 μL) were injected into each agarose gel and the absorbance signal was recorded after the initial background scans were collected. Methanol was used to solubilize water-insoluble RMP and despite its inherent toxicity in large quantities, the amount used in the filling model (~0.03% of the total agarose volume) does not impact bacterial viability for in vitro work. Microsoft Excel 2013 was used to analyze the data and conditional formatting was used to color scale the absorbance signals. Microsoft PowerPoint 2013 was used to construct the MP4 video files of each drug diffusing through the agarose at each time point where an absorbance scan was collected following the antibiotic injection (RMP: t = 0, 2, 4, 7, 23, 25, 27, 29, 46, 48, 52 h; MC: t = 0, 16, 18, 20, 22, 39, 41, 45 h) (Supplementary Fig. 1; Supplementary Videos S1–S2).
2.7. Quantification of antibiotic filling
The mass of RMP filled into the implanted polymers was quantified by leaching antibiotic into a dimethylformamide (DMF) solvent sink. Every couple of hours, the entire volume of release media was removed from the sample and replaced with fresh DMF to maintain infinite sink conditions. The concentration of drug in the solvent was quantified by scanning small (200 μL) samples of release media with UV absorbance spectroscopy (485 nm, RMP) and a calibration curve (RMP in DMF). Table 1A displays the averaged filling data for each in situ filling condition.
Table 1.
Mass of antibiotics filled in situ into affinity and non-affinity polymer disks. Mass of RMP filled into implanted polymers from in situ filling studies with and without bacteria (a) and mass of MC filled into implanted polymers from in situ filling studies with and without bacteria (b) averaged (n = 3) with standard deviation.
| a. | ||
|---|---|---|
|
| ||
| Rifampicin | Average loaded drug mass (n = 3) (μg) | |
| No biofilm | pCD | 2.8 ± 0.6 |
| pDEX | 1.0 ± 0.4 | |
| 24 h biofilm | pCD (no biofilm) | 3.1 ± 0.6 |
| pCD (biofilm) | 3.3 ± 0.4 | |
| 72 h biofilm | pCD (no biofilm) | 2.9 ± 0.2 |
| pCD (biofilm) | 1.8 ± 0.2 | |
| b. | ||
|
| ||
| Minocycline | Average loaded drug mass (n = 3) (μg) | |
|
| ||
| No biofilm | pCD | 38.1 ± 15.6 |
| pDEX | 13.6 ± 0.9 | |
| 24 h biofilm | pCD (no biofilm) | 50.5 ± 1.2 |
| pCD (biofilm) | 40.4 ± 2.0 | |
| 72 h biofilm | pCD (no biofilm) | 39.6 ± 9.0 |
| pCD (biofilm) | 50.0 ± 7.3 | |
Due to the challenge of MC oxidizing once filled into the disks, the mass of MC filled into the implanted polymers was quantified using UV absorbance spectroscopy using a 9 × 9 data point area scan of both empty polymer disks and MC filled disks (390 nm). An initial calibration curve was created of the absorbance of known quantities of MC filled into disks with the background disk absorbance subtracted. This calibration curve was used to correlate the absorbance of the in situ MC filled disks to the mass of MC. Table 1B displays the averaged filling data for each in situ filling condition.
2.8. In vitro zone of inhibition assay
The ability of in situ filled antibiotic to release from affinity polymer disks and eradicate bacteria was evaluated against S. aureus in a zone of inhibition assay as done previously (Fig. 3) [22]. S. aureus was cultured overnight and 70 μL was then spread on a Trypticase soy agar plate. Affinity polymer disks were removed from the agarose following in situ antibiotic filling and then placed on a fresh S. aureus lawn and incubated at 35 °C overnight. After 24 h, the zone of inhibition around each disk was measured with calipers and averaged. Each disk was transferred onto a freshly seeded S. aureus plate and placed in the incubator. This process was repeated until the zone was no longer visible (<0.5 mm).
Fig. 3.
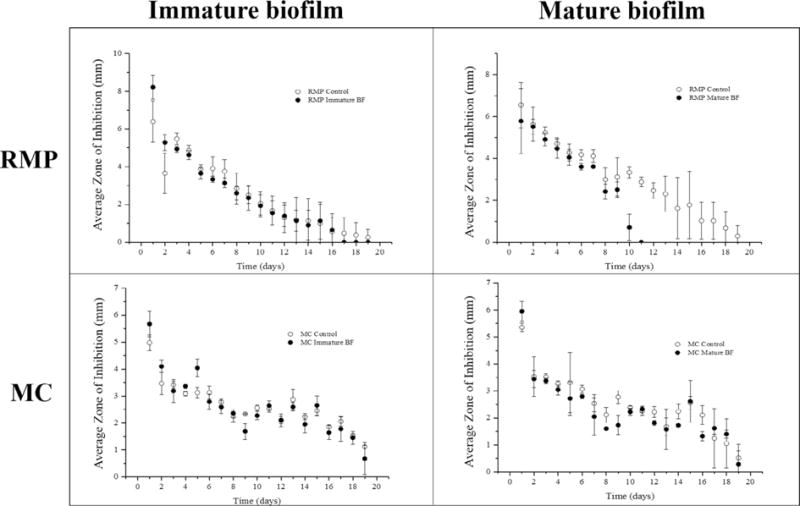
Zone of inhibition study of post-implantation filled RMP and MC filled affinity polymer disks with immature or mature biofilm (t = 19 days) averaged (n = 3) with standard deviation. Control affinity polymer disks without biofilm were filled at the same time. Post-implantation filling was carried out for 52 h. RMP filled affinity polymer disks with immature (top left) and mature (top right) and MC filled affinity polymer disks with immature (bottom left) and mature biofilm (bottom right) demonstrated antibacterial activity against S. aureus for approximately 20 days.
2.9. Bacterial quantification of biofilms
The bacterial load present on biofilm coated affinity or non-affinity polymer disks was quantified at the start of in situ filling. Briefly, biofilm coated disks were implanted in agarose and removed once the agarose solidified. Each disk was placed in a tube with 4 mL of Trypticase soy broth and homogenized for 30 s with an Omni TH homogenizer. 70 μL aliquots of the homogenized solution were then spread on Trypticase soy agar plates in duplicate and incubated overnight. Bacterial colonies were counted and averaged. Bacteria in biofilms was also quantified after 1 and 2 days during in situ filling of RMP carried out as detailed above. After 1 or 2 days the disks were removed from the agarose, homogenized and bacteria plated and counted 24 h later. Further experiments were conducted with mature biofilm coated affinity polymer disks incubated for 1, 2, 3, 7, 14, and 21 days in in situ conditions without any drug in order to detect changes in bacterial colony viability after different periods of incubation.
Similar experiments were conducted to quantify the bacteria present in serum preconditioned mature biofilm coated affinity polymer disks after 2 days of in situ filling with RMP and MC.
2.10. Statistical analysis
All data is displayed as the mean of each condition tested in triplicate with the standard deviation as the error bars. One-way ANOVA statistics was performed on the antibiotic filling data (Table 1) with Microsoft Excel 2013. A two-tailed Student’s t-test assuming unequal variances was performed on the antibiotic filling data (n = 3) and bacterial quantification data (n = 6) (Table 2). A t-test value <0.01 was determined to be statistically significant.
Table 2.
Bacterial quantification of affinity and non-affinity polymer disks with mature biofilms. Affinity polymer disks and non-affinity polymer disks with mature biofilm (n = 6) after t = 1 and 2 days of RMP filling as a percent of control disks incubated for 1 or 2 days without antibiotic filling. All data shown is reported as the average percentage of bacterial colonies remaining with the standard deviation.
| % Remaining control colonies after drug exposure (n = 6) t = 24 h |
% Remaining control colonies after drug exposure (n = 6) t = 48 h |
|
|---|---|---|
| pCD | 63.2 ± 50.0 | 8.9 ± 4.4 |
| pDEX | 39.8 ± 31.2 | 95.1 ± 82.3 |
3. Results and discussion
3.1. Affinity-based in situ filling and quantification
The efficacy of the post-implantation antibiotic filling was evaluated with affinity and non-affinity polymer disks. The diffusion of RMP and MC through the agarose and subsequent filling into the implanted polymer disks was tracked using colorimetric absorbance spectroscopy (Supplementary Fig. 1; Supplementary Videos S1–S2). Fig. 2 (top) shows the disks embedded in agarose after 52 h (RMP) and 45 h (MC) of antibiotic filling as well as the removed implants (Fig. 2 bottom). For both antibiotics tested, affinity polymer disks filled more drug compared to non-affinity polymer disks as qualitatively indicated by the dark orange color of RMP and yellow/green color of MC and quantitatively indicated by the results of Table 1. Table 1A shows that over three times more RMP was filled into the affinity polymer compared to the non-affinity polymer (p = 0.0035). Similar results were observed for MC filling into affinity versus non-affinity polymers where nearly three times more MC was filled into the affinity polymer compared to non-affinity control (p = 0.056) (Table 1B). The increased antibiotic filling into affinity polymers is attributed to the affinity “pockets” present in the affinity polymer that are not present in the conventional, non-affinity polymer. The hypothesis is that the CD “pockets” in the affinity polymer create a sink for drug to stick into thereby maintaining a steady concentration gradient from the bulk agarose leading drug to concentrate into the affinity polymer relative to that in the agarose system. Further, the increased filling of antibiotics into affinity-based pCD can be attributed to the relatively high binding energies of RMP (−4.39 kcal/mol) [34] and MC for β-cyclodextrin.
Fig. 2.
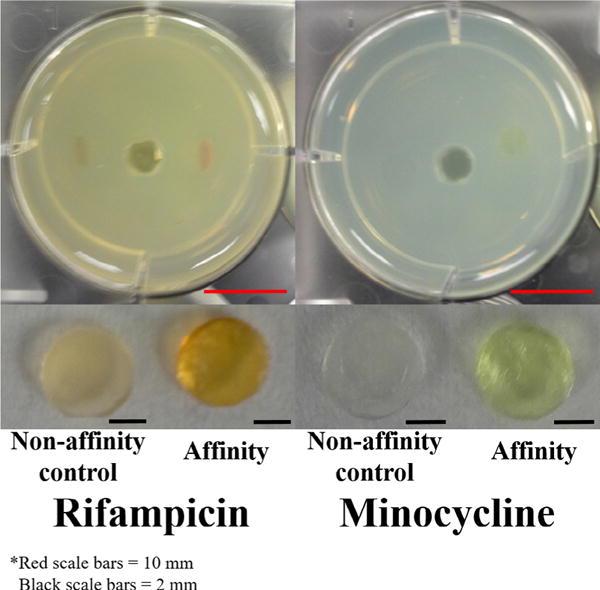
(Top images) In situ filling model with antibiotic filled disks after 52 h (RMP) and 45 h (MC) of filling. (Bottom images) Removed polymer disks after 52 h (RMP) and 45 h (MC) of filling. The dark orange (RMP) and yellow (MC) colors indicate the presence of the drug. The diameter of each well is 34.8 mm and all disks are 5 mm in diameter. Scale bars (bottom right corner of images) for wells are 10 mm (red) and scale bars for disks are 2 mm (black). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Previous RMP release data demonstrates that under physiological release conditions (pH 7.4) RMP is gradually released from pCD over 1200 h [24] and over 384 h for MC (manuscript in preparation). Regarding drug release through tissue, such as the agarose filling model, it is hypothesized that the pCD delivery system will be capable of demonstrating the same, slow and consistent release profile for RMP and MC over >1 month to treat infections over an extended period of time. Conversely, release from the pDEX polymer typically exhibits a “burst” release profile with the majority of the release occurring over only several hours or days [35]. Therefore, with the pCD delivery system a more consistent and prolonged release profile of the antibiotics can be obtained.
Further, the affinity-based pCD polymer system has been shown to exhibit low degradation and be mechanically robust under in vivo physiological conditions [25]. Low degradation is desirable for the treatment of long-term severe infections and ensures that the polymer has the mechanical integrity to withstand repeated filling and drug release cycles without significant changes to release kinetics. The mechanical integrity and viscoelastic properties of pCD polymer systems have been extensively analyzed using thermo-gravimetric analysis, differential scanning calorimetry, and rheology with and without sterilization [24,28]. These studies confirm that the moduli of pCD polymers is within the range of native tissue [24] and that neither heat nor ethylene oxide sterilization techniques significantly alter the mechanical integrity of the polymer [28]. Future work can include synthesizing hydrolytically cleavable linkages onto pCD to develop more biodegradable forms of pCD to increase the versatility of pCD for both short-term and long-term therapeutic applications.
3.2. Post-implantation filling in the presence of biofilm
After demonstrating that affinity interactions drive higher filling of both RMP and MC into affinity polymer disks, the effect of bacterial biofilm’s diffusional resistance on filling polymers with drug was investigated. Affinity polymer disks were exposed to S. aureus for 24 h to form an immature biofilm or 72 h to form a mature biofilm [27,36] prior to embedding in the agarose system. RMP and MC filling were then tracked via colorimetric absorbance. Supplementary Fig. 2 shows the affinity polymer disks after 52 h of post-implantation antibiotic filling. There was no difference in RMP filling into affinity polymer disks with the immature biofilm (3.3 ± 0.4 μg) compared to control affinity polymer disks without any bacterial exposure (3.1 ± 0.6 μg; p = 0.6).
For affinity polymer disks with mature biofilms, more RMP was present in the control disk without bacterial exposure (2.9 ± 0.2 μg) compared to the disk with the biofilm (1.8 ± 0.2 μg), but the difference was not statistically significant (p = 0.15). Two-way ANOVA showed that the affinity polymer disks with the mature biofilm filled less RMP than the disks with a less mature biofilm (p < 0.005). No statistically significant differences were observed for MC filling into affinity polymer disks with mature, immature, or no biofilms. Overall, the presence of bacteria did not significantly affect the post-implantation filling of RMP or MC into the pCD affinity polymer disks.
3.3. Bacterial quantification of biofilms
Bacteria present in the mature biofilm coated affinity polymer disks were quantified in an effort to correlate drug filling to the number of bacteria present in the biofilm. Each condition was tested with n = 6 replicates and averaged. Table 2 shows quantification of bacteria from affinity polymer disks with mature biofilms after 1 and 2 days of RMP filling time expressed as a percentage of bacteria on control affinity polymer disks incubated in the agarose model for the respective time but without any antibiotic filling. The bacterial count for RMP filled affinity polymer disks decreased as the filling time increased, and after two days the majority of bacteria in the biofilm had been eradicated. Bacteria in mature biofilms on the surface of affinity polymers were quantified after 1, 2, and 3 days of incubation in the filling model without antibiotics and showed that there was no change in bacterial counts when antibiotics are not injected to day 3.
Further, the effect of RMP filling into non-affinity control polymers with mature biofilms was evaluated. The non-affinity polymer disks had significantly more bacteria in the mature biofilm (p = 0.008) at the time of implantation into the agarose. Both affinity and non-affinity polymer disks with mature biofilms showed a decrease in bacteria at day 1 of RMP filling. At day 2 the non-affinity polymer disks with the mature biofilm showed similar bacteria counts as day 1 compared to the affinity polymer disks, which showed a decrease.
Due to a large degree of variation in bacteria present in less mature biofilms (consistent with previous observations), the primary focus was on quantifying the bacteria present in mature biofilms [27,36]. While the mature biofilm did decrease the amount of drug present in the affinity polymer disk slightly (p = 0.15; Table 1A), quantitative culturing revealed a lower bacterial load on these disks. The hypothesis is that some of the initial antibiotic in the bolus injection was immediately used to eradicate the planktonic S. aureus around the biofilm and the rest of the antibiotic was filled into the pCD polymers to be gradually released to eradicate the less metabolically active bacteria present in the biofilm, rather than the biofilm resisting drug diffusion into the pCD disk. This hypothesis was further validated by the observation that the agarose background absorbance signal of the immature (t = 24 h) was comparable to the signal of the mature (t = 72 h) biofilm coated pCD disk. Since the signal was similar and the more mature biofilm coated pCD disk contained less drug, some of the initial injected drug must have been used to eradicate some of the bacteria in the mature biofilm. The presence of bacteria still remaining on the affinity polymer surface after the post-implantation filling underscores the importance of sustained antibiotic delivery where the drug released back into the biofilm over the period of days or weeks is needed to continue eradicating the bacteria present on the polymer or implant surface. Additional studies quantifying bacteria from mature biofilms on affinity polymers after 1–3 days of incubation verified that RMP and not unfavorable survival conditions in the agarose model was responsible for eradicating the majority of bacteria in the biofilm. Further, the post-implantation filling model used in this work supports bacterial biofilm viability for at least 7 days (13.3% of initial biofilm colonies) with minimal viability after 14 days. Future work will optimize culture conditions to support long-term biofilm viability so that the bacterial eradicating due to post-implantation drug filling release from affinity polymers can be tracked.
In the bacterial quantification studies of the mature biofilm on non-affinity control polymers, it is hypothesized that the initial decrease in bacteria counts for affinity and non-affinity polymers from t = 0 to day 1 is driven by the antibiotic bolus whereas the decrease from day 1 to 2 seen only for affinity polymers with mature biofilms is driven by the affinity filling (Tables 1–2). The lack of decrease in bacteria counts (over 2 days) for non-affinity polymers with mature biofilms supports this hypothesis.
3.4. Bacterial quantification of serum-preconditioned biofilms
One further concern was that in a biological setting the “pockets” present in the affinity polymer might be obstructed by other affinity moieties that are present in a more robust biofilm such as proteins that adsorb to the surface prior to bacterial adhesion [7,8]. Affinity polymer disks were pre-conditioned in fetal bovine serum for 24 h prior to incubation in S. aureus to form a more robust biofilm. The presence of adsorbed serum components (e.g. proteins) did not significantly impact biofilm formation on the affinity polymer disks. Initial bacterial counts for serum-preconditioned affinity polymer disks with mature biofilms was 102% of the bacterial count of the non-conditioned samples, and as observed in the non-conditioned samples, negligible bacteria were present after 2 days of post-implantation filling RMP or MC. The serum proteins represent a slightly more physiologically relevant biofilm formation model, which is utilized by several other groups to model a more robust biofilm [37–39]. A whole blood biofilm model was not used in an effort to model the best possible conditions for biofilm formation (i.e. without leukocytes, etc.).
3.5. Zone of inhibition study
To demonstrate that post-implantation filled antibiotic was active and capable of continued bacterial eradicating, post-implantation filled RMP and MC affinity polymer disks were removed from the agarose filling model, and evaluated with a zone of inhibition assay against S. aureus. Four separate conditions were tested in triplicate. Fig. 3 shows that post-implantation filled RMP from affinity polymer disks without biofilm was able to eradicate S. aureus for 19 days. Affinity polymer disks with immature biofilms were also able to clear S. aureus out to 19 days, but affinity polymer disks with mature biofilms only cleared bacteria for 11 days, which is consistent with lower drug filling (Table 1A). This difference in bacterial eradicating between RMP from immature and mature biofilms demonstrated that increased drug filling into affinity polymer (Table 1A) results in an extended antimicrobial timeframe. Fig. 3 also shows that the filled MC was active for 19 days independent of the presence or maturity of biofilm. Overall, the results of this study confirmed that post-implantation filled affinity polymers are capable of releasing a therapeutic dose of active antibiotic sufficient to inhibit the growth of bacteria for nearly 3 weeks.
As an example in hernia repair, current clinical treatment for hernia mesh infection can include intraperitoneal injection of antibiotics [40], which results in a spike in local antibiotic concentration on the scale of hours. Any bacteria that remain on or in the mesh post-antibiotic bolus can continue growing to form a biofilm, and the one-time antibiotic dose can also contribute to the development of drug-resistant bacteria. In this work the decreased bacterial load observed on the polymer disk surface following bolus injection even on the diffusion controls demonstrates how this one-time antimicrobial injection is insufficient to eradicate the infection (Table 2). The result is usually revision surgery and removal of the infected mesh significantly increasing overall cost and putting the patient at risk from a second surgical procedure. The work presented here could improve the outcome by rescuing the infected mesh, or any infected device, by providing a localized drug sink capable of delivering a steady concentration of antibiotic that can eradicate bacteria even with the decreased metabolic rates observed when a biofilm is present. Previously biofilms had been observed to pose a diffusion barrier that would prevent lethal concentrations of drug to reach the bacteria. This work shows that drug does freely diffuse through a biofilm; however, the capacity to trap that drug and re-administer it at therapeutic doses had previously been missing. For the bacteria in the biofilm which are less metabolically active than planktonic, or rapidly growing bacteria, a re-administration of the drug on the scale of days to weeks are needed to eradicate the bacteria and eradicate the infection.
The affinity interactions of the pCD system have previously shown antibiotic release for ≥ 1200 h [24]. This work demonstrates that these affinity interactions act like a localized sink for free drug in an in vitro model of post-implantation loading enabling drug to concentrate in the unloaded polymer disk (Fig. 2; Supplementary Videos S1 and S2). The chemically similar control polymer disks without affinity moieties did not load drug. Once free drug has concentrated in the affinity pCD the polymer will begin releasing the drug in sustained manner that is pharmacokinetically more suitable to eradicating the infection than a one-time bolus dose.
The presence of biofilm was believed to pose a diffusional barrier. This work shows that biofilm did not significantly decrease antibiotic post-implantation loading even when serum proteins were present (Table 1). These proteins had the potential to form a more robust biofilm or even block the affinity groups, but the data show that neither scenario impacted drug filling.
Polymer disks were used in this work for ease of handling, but this affinity polymer can be formulated as a device coating or formed into microparticles for independent use with a variety of implants or incorporated into the bulk device. The ultimate goal of this and future work is to treat infections ranging from soft tissue hernia infections to hard tissue bone infections.
4. Conclusions
Prophylactic antimicrobial treatment is not clinically sufficient since patients may still present with infection up to a year after surgery. In these cases, the infection will likely be well established and potentially have formed a biofilm. The in vitro evaluation of post-implantation drug filling presented in this work indicates neither bacteria nor biofilm will impair antibiotic filling or the affinity polymer’s ability to eradicate bacteria. The delivery system in this work utilizes two components to treat established infections. A bolus antibiotic injection serves to eradicate the planktonic bacteria surrounding the initial infection, while the implanted affinity polymer serves as an antibiotic depot slowly re-administer drug to eradicate the less metabolically active bacteria that are present in biofilm. Affinity polymer filled with vancomycin prior to implantation has already been shown to prevent device infection in rodents [41,42] and has also prevented Methicillin-resistant S. aureus (MRSA) hernia defect infection in pigs (manuscript submitted). The versatility of affinity polymers makes them amenable for use in both hard and soft tissue applications. Future work will focus on evaluating the in vivo ability to fill affinity polymers with antimicrobials post-implantation, the impact the host foreign body response has on in vivo filling, and refilling affinity polymers that have been implanted prefilled with antibiotic for patients at high risk of device infection. The work presented here demonstrates for the first time that antimicrobials can be loaded into affinity polymers independent of the presence of a biofilm and opens the door to potentially rescuing infected devices rather than removing them.
Supplementary Material
Statement of Significance.
Post-operative prophylactic antimicrobial therapy greatly reduces risk of infection, such as on biomedical implants, but does not totally eliminate infections, and the healthcare cost of these remaining infections remains a major concern. Systemic antimicrobial therapy to treat these infections can lead to tissue toxicity and drug-resistant bacteria. In order to treat only those patients who have developed infections, a customizable antimicrobial delivery system made of cyclodextrin-based affinity polymer has been developed that is capable of filling post-implantation and delivering the filled antibiotic in a sustained manner even when the delivery device covered in bacterial biofilm. These observations have the potential to be translated to a wide variety of applications, such as implanted or indwelling medical devices, and/or surgical site infections.
Acknowledgments
The authors would like to thank Christopher Hernandez (Case Western Reserve University) for assistance in the development of the reproducible in situ agarose filling model. This work was funded by NIH NIGMS SBIR 1R43GM100525 and NSF CAREER Award CBET-0954489.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.actbio.2017.04.015.
Footnotes
Conflict of interest/Disclosure statement
STZ is employed by Affinity Therapeutics who is commercializing the technology developed by HvR’s lab. HvR is a co-founder of Affinity Therapeutics but does not receive salary. JNK is a co-founder & employee of Affinity Therapeutics.
References
- 1.Schierholz JM, Beuth J. Implant infections: a haven for opportunistic bacteria. J Hosp Infect. 2001;49:87–93. doi: 10.1053/jhin.2001.1052. [DOI] [PubMed] [Google Scholar]
- 2.Olson ME, Horswill AR. Staphylococcus aureus osteomyelitis: bad to the bone. Cell Host Microbe. 2013;13:629–631. doi: 10.1016/j.chom.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green BN, Johnson CD, Egan JT, Rosenthal M, Griffith EA, Evans MW. Methicillin-resistant Staphylococcus aureus: an overview for manual therapists. J Chiropr Med. 2012;11:64–76. doi: 10.1016/j.jcm.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Troidle L, Eisen T, Pacelli L, Finkelstein F. Complications associated with the development of bacteremia with Staphylococcus aureus. Hemodial Int. 2007;11:72–75. doi: 10.1111/j.1542-4758.2007.00156.x. [DOI] [PubMed] [Google Scholar]
- 5.Falagas ME, Kasiakou SK. Mesh-related infections after hernia repair surgery. Clin Microbiol Infect. 2005;11:3–8. doi: 10.1111/j.1469-0691.2004.01014.x. [DOI] [PubMed] [Google Scholar]
- 6.Naber CK. Staphylococcus aureus bacteremia: epidemiology, pathophysiology, and management strategies. Clin Infect Dis. 2009;48:S231–S237. doi: 10.1086/598189. [DOI] [PubMed] [Google Scholar]
- 7.Arciola CR, Campoccia D, Speziale P, Montanaro L, Costerton JW. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials. 2012;33:5967–5982. doi: 10.1016/j.biomaterials.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 8.Cha JO, Yoo JI, Yoo JS, Chung HS, Park SH, Kim HS, Lee YS, Chung GT. Investigation of biofilm formation and its association with the molecular and clinical characteristics of methicillin-resistant Staphylococcus aureus. Osong Pub Health Res Perspect. 2013;4:225–232. doi: 10.1016/j.phrp.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iordache F, Grumezescu V, Grumezescu AM, Curutiu C, Ditu LM, Socol G, Ficai A, Trusca R, Holban AM. Gamma-cyclodextrin/usnic acid thin film fabricated by MAPLE for improving the resistance of medical surfaces to Staphylococcus aureus colonization. Appl Surf Sci. 2015;336:407–412. [Google Scholar]
- 10.Francolini I, Donelli G, Stoodley P. Polymer designs to control biofilm growth on medical devices. Rev Environ Sci Biotechnol. 2003;2:307–319. [Google Scholar]
- 11.Desrousseux C, Sautou V, Descamps S, Traore O. Modification of the surfaces of medical devices to prevent microbial adhesion and biofilm formation. J Hosp Infect. 2013;85:87–93. doi: 10.1016/j.jhin.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 12.Cohen NR, Lobritz MA, Collins JJ. Microbial persistence and the road to drug resistance. Cell Host Microbe. 2013;13:632–642. doi: 10.1016/j.chom.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campoccia D, Montanaro L, Arciola CR. The significance of infection related to orthopedic devices and issues of antibiotic resistance. Biomaterials. 2006;27:2331–2339. doi: 10.1016/j.biomaterials.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 14.Davies D. Understanding biofilm resistance to antibacterial agents. Nat Rev. 2003;2:114–122. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- 15.Tande AJ, Patel R. Prosthetic joint infection. Clin Microbiol Rev. 2014;27:302–345. doi: 10.1128/CMR.00111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laudermilch DJ, Fedora CJ, Heyl A, Rao N, McGough RL. Outcomes of revision total knee arthroplasty after methicillin-resistant Staphylococcus aureus infection. Clin Orthop Relat Res. 2010;468:2067–2073. doi: 10.1007/s11999-010-1304-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mavros MN, Athanasiou S, Alexiou VG, Mitsikos PK, Peppas G, Falagas ME. Risk factors for mesh-related infections after hernia repair surgery: a meta-analysis of Cohort studies. World J Surg. 2011;35:2389–2398. doi: 10.1007/s00268-011-1266-5. [DOI] [PubMed] [Google Scholar]
- 18.Cavanaugh DL, Berry J, Yarboro SR, Dahners LE. Better prophylaxis against surgical site infection with local as well as systemic antibiotics. J Bone Joint Surg Am. 2009;91:1907–1912. doi: 10.2106/JBJS.G.01237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inzana JA, Schwarz EM, Kates SL, Awad HA. Biomaterials approaches to treating implant-associated osteomyelitis. Biomater. 2016;81:58–71. doi: 10.1016/j.biomaterials.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Kohler B, Garcia-Moreno F, Brune T, Pascual G, Bellon JM. Preclinical bioassay of a polypropylene mesh for hernia repair pretreated with antibacterial solutions of chlorohexidine and allicin: an in vivo study. PLoS ONE. 2015;10:e0142768. doi: 10.1371/journal.pone.0142768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang NX, von Recum HA. Affinity-based drug delivery. Macromol Biosci. 2011;11:321–332. doi: 10.1002/mabi.201000206. [DOI] [PubMed] [Google Scholar]
- 22.Thatiparti TR, von Recum HA. Cyclodextrin complexation for affinity-based antibiotic delivery. Macromol Biosci. 2010;10:82–90. doi: 10.1002/mabi.200900204. [DOI] [PubMed] [Google Scholar]
- 23.Fu AS, Thatiparti TR, Saidel GM, von Recum HA. Experimental studies and modeling of drug release from a tunable affinity-based drug delivery platform. Ann Biomed Eng. 2011;39:2466–2475. doi: 10.1007/s10439-011-0336-z. [DOI] [PubMed] [Google Scholar]
- 24.Halpern JM, Gormley CA, Keech M, von Recum HA. Thermomechanical properties, antibiotic release, and bioactivity of a sterilized cyclodextrin drug delivery system. J Mater Chem. 2014;2:2764–2772. doi: 10.1039/C4TB00083H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu AS, von Recum HA. Affinity-Based Delivery and Refilling of Doxorubicin for Treatment of Glioblastoma Multiforme. Case Western Reserve University; Cleveland: 2013. [Google Scholar]
- 26.Zhao G, Ye L, Huang Y. In vitro model of bacterial biofilm formation on polyvinyl chloride biomaterial. Cell Biochem Biophys. 2011;61:371–376. doi: 10.1007/s12013-011-9220-6. [DOI] [PubMed] [Google Scholar]
- 27.Orgaz B, Lobete MM, Puga CH, San Jose C. Effectiveness of chitosan against mature biofilms formed by food related bacteria. Int J Mol Sci. 2011;12:817–828. doi: 10.3390/ijms12010817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thatiparti TR, Shoffstall AJ, von Recum HA. Cyclodextrin-based device coatings for affinity-based release of antibiotics. Biomater. 2010;31:2335–2347. doi: 10.1016/j.biomaterials.2009.11.087. [DOI] [PubMed] [Google Scholar]
- 29.Maxwell AD, Wang TY, Yuan L, Duryea AP, Xu Z, Cain CA. A tissue phantom for visualization and measurement of ultrasound-induced cavitation damage. Ultrasound Med Biol. 2010;36:2132–2143. doi: 10.1016/j.ultrasmedbio.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Culjat MO, Goldenberg D, Tewari P, Singh RS. A review of tissue substitutes for ultrasound imaging. Ultrasound Med Biol. 2010;36:861–873. doi: 10.1016/j.ultrasmedbio.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Chen ZJ, Gillies GT, Broaddus WC, Prabhu SS, Fillmore H, Mitchell RM, Corwin FD, Fatouros PP. A realistic brain tissue phantom for intraparenchymal infusion studies. J Neurosurg. 2004;101:314–322. doi: 10.3171/jns.2004.101.2.0314. [DOI] [PubMed] [Google Scholar]
- 32.Chmarra MK, Hansen R, Mårvik R, Langø T. Multimodal phantom of liver tissue. PLoS One. 2013;8:e64180. doi: 10.1371/journal.pone.0064180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vassiliou VS, Heng EL, Gatehouse PD, Donovan J, Raphael CE, Giri S, Babu-Narayan SV, Gatzoulis MA, Pennell DJ, Prasad SK, Firmin DN. Magnetic resonance imaging phantoms for quality-control of myocardial T1 and ECV mapping: specific formulation, long-term stability and variation with heart rate and temperature. J Cardiovasc Magn Reson. 2016;18 doi: 10.1186/s12968-016-0275-9. http://dx.doi.org/10.1186/s12968-016-0275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chadha R, Saini A, Gupta S, Arora P, Thakur D, Jain DVS. Encapsulation of rifampicin by natural and modified β-cyclodextrins: characterization and thermodynamic parameters. J Incl Phenom Macrocycl Chem. 2010;67:109–116. [Google Scholar]
- 35.Thatiparti TR, Averell N, Overstreet D, von Recum HA. Multiplexing interactions to control antibiotic release from cyclodextrin hydrogels. Macromol Biosci. 2011;11:1544–1552. [PubMed] [Google Scholar]
- 36.Leid JG, Shirtliff ME, Costerton JW, Stoodley P. Human leukocytes adhere to penetrate, and respond to Staphylococcus aureus biofilms. Infect Immun. 2002;70:6339–6345. doi: 10.1128/IAI.70.11.6339-6345.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramage G, Vande Walle K, Wickes BL, Lopez-Ribot JL. Biofilm formation by Candida dubliniensis. J Clin Microbiol. 2001;39:3234–3240. doi: 10.1128/JCM.39.9.3234-3240.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKintosh EE, Patel JD, Marchant RE, Anderson JM. Effects of biomaterial surface chemistry on the adhesion and biofilm formation of Staphylococcus epidermidis in vitro. J Biomed Mater Res A. 2006;78:836–842. doi: 10.1002/jbm.a.30905. [DOI] [PubMed] [Google Scholar]
- 39.Patel JD, Ebert M, Ward R, Anderson JM. S. epidermidis biofilm formation: effects of biomaterial surface chemistry and serum proteins. J Biomed Mater Res A. 2007;80:742–751. doi: 10.1002/jbm.a.31103. [DOI] [PubMed] [Google Scholar]
- 40.Tzovaras G, Delikoukos S, Christodoulides G, Spyridakis M, Mantzos F, Tepetes K, Athanassiou E, Hatzitheofilou C. The role of antibiotic prophylaxis in elective tension-free mesh inguinal hernia repair: results of a single-center prospective randomized trial. Int J Clin Pract. 2007;61:236–239. doi: 10.1111/j.1742-1241.2006.00977.x. [DOI] [PubMed] [Google Scholar]
- 41.Harth KC, Rosen MJ, Thatiparti TR, Jacobs MR, Halaweish I, Bajaksouzian S, Furlan J, von Recum HA. Antibiotic-releasing mesh coating to reduce prosthetic sepsis: an in vivo study. J Surg Res. 2010;163:337–343. doi: 10.1016/j.jss.2010.03.065. [DOI] [PubMed] [Google Scholar]
- 42.Grafmiller KT, Zuckerman ST, Petro C, Liu L, von Recum HA, Rosen MJ, Korley JN. Antibiotic-releasing microspheres prevent mesh infection in vivo. J Surg Res. 2016;206:41–47. doi: 10.1016/j.jss.2016.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.