

Abstract
Thyroid hormone (TH) is critical for the maintenance of cellular homeostasis during stress responses, but its role in lung fibrosis is unknown. Here, we found that the activity and expression of iodothyronine deiodinase 2 (DIO2), an enzyme that activates TH, was higher in lungs of patients with idiopathic pulmonary fibrosis compared to control individuals and correlated with disease severity. We also found that Dio2 knockout mice exhibited enhanced bleomycin-induced lung fibrosis. Aerosolized TH delivery increased survival and resolved fibrosis in two models of pulmonary fibrosis in mice (intratracheal bleomycin and inducible TGF-β1). Sobetirome, a TH mimetic, also blunted bleomycin-induced lung fibrosis. Given after bleomycin-induced injury, TH promoted mitochondrial biogenesis, improved mitochondrial bioenergetics and attenuated mitochondria-regulated apoptosis in alveolar epithelial cells both in vivo and in vitro. TH did not blunt fibrosis in Ppargc1a or Pink1 knockout mice suggesting dependence on these pathways. We conclude that the TH anti-fibrotic properties are associated with protection of alveolar epithelial cells and restoration of mitochondrial function and thus may represent an effective therapy for pulmonary fibrosis.
Keywords: Pulmonary Fibrosis, Mitochondria, Thyroid Hormone, Sobiterome, Alveolar epithelial Cells
Pulmonary fibrosis (PF) describes a condition in which the normal lung anatomy is replaced by a process of active remodeling, deposition of extracellular matrix and dramatic changes in the phenotype of both fibroblasts and alveolar epithelial cells. This condition can be idiopathic, as in idiopathic pulmonary fibrosis (IPF), or secondary to genetic disorders, autoimmune disorders, or exposure to environmental toxins, chemical warfare, drugs, foreign antigens, or radiation1. IPF represents the most common idiopathic form of PF and is frequently pathologically indistinguishable from the other forms, especially at the later stages of the disease. IPF affects approximately 120,000 people in the US with a steady increase in both incidence and mortality. The median survival without transplant of IPF is approximately 3 years, making it the non-cancer-related lung disease with the gravest prognosis2.
IPF is the result of multiple cycles of epithelial cell injury and activation that provoke the formation of myofibroblastic foci, accumulation of extracellular matrix and abnormal wound repair3. Multiple pathways including sustained transforming growth factor-β1 (TGF-β1) activation, abnormal matrix deposition, epithelial injury and apoptosis, fibroblast activation and myofibroblast transdifferentiation, aberrant recapitulation of developmental pathways, endoplasmic reticulum stress, telomerase mutations, oxidative injury, metalloprotease activation and signaling4, as well as extensive changes in mRNA5 and microRNA6 expression, have been implicated in IPF. More recently it has been proposed that many of these features could be explained by the role of aging and indeed many of the hallmarks of aging including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication can be considered characteristics of the fibrotic lung7,8.
In the context of this study, the emerging evidence for mitochondrial dysfunction, and metabolic aberrations in PF are very compelling8. Metabolic reprogramming is a characteristic of myofibroblast differentiation9,10, alveolar epithelial cells in IPF lungs exhibit large numbers of damaged mitochondria11 and increased levels of free mitochondrial DNA are found in the plasma and bronchoalveolar lavage of subjects with IPF9. PTEN-induced putative kinase 1 (PINK1), a regulator of mitophagy is lower in the lungs of subjects with IPF compared to healthy controls and Pink1 knockout mice exhibit deformed mitochondria and are more susceptible to PF than wildtypes11. Considering that the original observation of PINK1 downregulation in IPF was derived from a public dataset generated by us (http://www.lung-genomics.org - GEO accession no. GSE47460)12, we looked for other changes in gene expression indicative of abnormalities in lung bioenergetics or metabolism. We discovered that DIO2, the iodothyronine deiodinase that converts the prohormone thyroxine (T4) to the active molecule 3,5,3′-triiodothyronine (T3) is among the most significantly increased genes in the lungs of individuals with IPF.
The purpose of the current study is to follow up on this finding and investigate the role of thyroid hormone (TH) signaling in the pathogenesis of experimental and human PF and its potential effects on mitochondrial function in IPF. In brief, we discovered that upregulation of DIO2 in the lungs of patients with IPF most likely represents an effort to boost local conversion of T4 to T3 to improve the metabolic state of alveolar epithelial cells in the stressed environment of the fibrotic lung. Dio2 knockout mice exhibited enhanced bleomycin-induced pulmonary fibrosis, whereas aerosolized delivery of TH blunted fibrosis in two experimental models. The beneficial effects of TH in mouse pulmonary fibrosis were associated with normalization of mitochondrial function and morphology in alveolar type II epithelial cells (AECIIs). T3-mediated restoration of bleomycin-induced mitochondrial abnormalities was associated with inhibition of mitochondria-regulated apoptosis in lung epithelial cells in vivo and in vitro and dependent on intact expression of regulators of mitochondrial biogenesis and mitophagy, including peroxisome proliferator-activated receptor gamma coactivator alpha (Ppargc1a) and Pink1. These effects may be potentially mediated through interaction of T3 with thyroid hormone receptors (THRs) given that we found that a THR-selective thyromimetic compound had anti-fibrotic properties in a bleomycin-model of lung fibrosis.
RESULTS
DIO2 is higher in IPF and its absence augments PF in mice
Reanalysis of the Lung Genomic Research Consortium (LGRC) dataset revealed that DIO2 was among the significantly highest expressed genes in lungs of patients with IPF compared to controls (Fig. 1a and Supplementary Table 1). DIO2 gene expression was negatively correlated with disease severity as reflected by diffusion capacity for carbon monoxide (DLCO % predicted) (Fig. 1b). DIO2 mRNA (Fig. 1c) and protein (Fig. 1d) were significantly higher (8- and 3-fold, respectively) in the lungs of patients with IPF compared to normal histology controls (P < 0.001). DIO2 enzymatic activity, assessed by in vitro T4 to T3 conversion, was also higher (3-fold, P < 0.05) in lung extracts from IPF patients compared to controls (Fig. 1e). DIO2 was mainly expressed in the alveolar epithelium surrounding the fibrotic interstitium in lungs of patients with IPF, and weakly expressed in the normal alveolar epithelium (Fig. 1f). In line with human data, Dio2 expression (Fig. 1g) and activity (Fig. 1h) were also significantly induced in the bleomycin model of lung fibrosis in mice.
Figure 1.
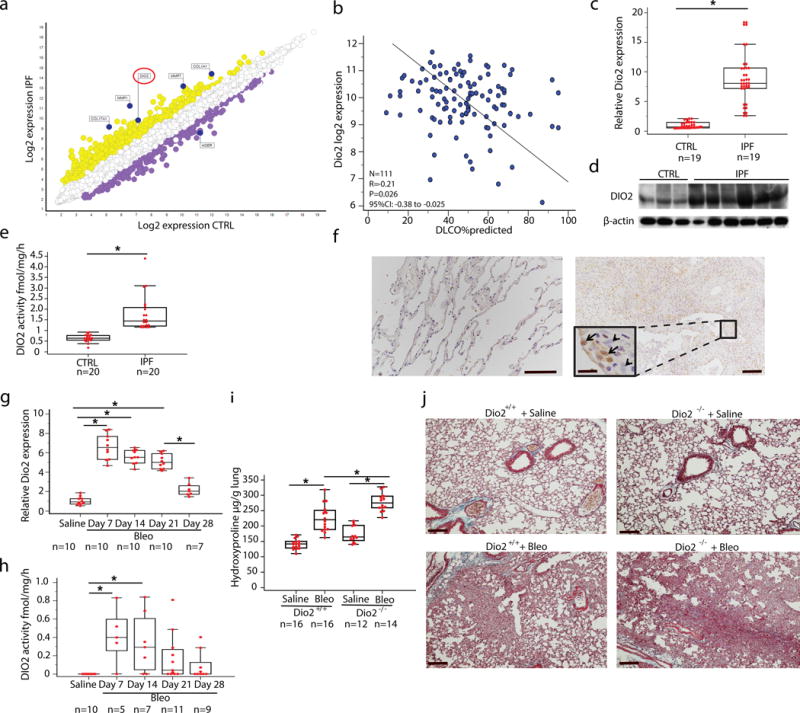
DIO2 is higher in lungs of patients with IPF compared to normal histology controls and its inhibition enhances bleomycin induced lung fibrosis. (a) Microarray gene expression scatterplot comparing the log2 expression values of all genes in subjects with IPF (n = 123, y axis) and control (n = 96, x axis). Yellow indicates upregulated genes whereas purple indicates downregulated genes, between IPF and controls. (b) Correlation of tissue DIO2 log2 gene expression with diffusion capacity for carbon monoxide, Pearson Correlation, *P = 0.026. (c) Quantitative RT-PCR analysis of the DIO2 mRNA expression (means+ SEM) in lungs of patients with IPF (n = 17) and normal histology controls (n = 17), *P < 0.001 (d) Immunoblot of IPF whole lung lysates (n = 6) showing DIO2 protein levels compared to control lungs (n = 3). Immunoblot gels were cropped. Uncropped images of the immunoblot gels are in Supplementary Fig 4. (e) DIO2 activity in lung homogenates from subjects with IPF compared to controls. Data are presented as box-and-whisker plots with horizontal bars representing means+ SEM of DIO2 activity fmol/mg/h, *P < 0.0001, (f) Immunohistochemistry analysis of representative lung tissue samples (n = 4) showing DIO2 expression in IPF lungs (right panel) compared to controls (left panel). Black arrows point to alveolar epithelial cells surrounding the fibrotic interstitium, black arrowheads indicate fibroblast-like cells. Boxed region is shown enlarged as inset on the left. Scale bars, 100 μm, inset, 20 μm. (g) Quantitative RT-PCR analysis of DIO2 relative expression (means+ SEM) at different time-points following disease progression in the bleomycin model of lung fibrosis, *P < 0.001 (h) DIO2 activity at different time-points following challenge with bleomycin. Data are presented as box-and-whisker plots with horizontal bars representing means of DIO2 activity fmol/mg/h, *P = 0.006. (i) Hydroxyproline content in 9–12 weeks-old, C57/BL6 female, Dio2-knockout mice (Dio2−/−) compared to wild-type (Dio2+/+) littermates, 14 days after intratracheal challenge with bleomycin (Bleo) (1.5U/kg) or equivalent volume of normal saline 0.9%. Data presented as box-and-whisker plots are from one of two independent experiments with similar results with horizontal bars representing mean hydroxyproline content per lung set (μg/gr) + SEM, *P < 0.001. (j) Masson’s Trichrome staining of representative lung sections (n = 3) from each group of treated mice. Scale bars, 100 μm. The statistical tests used were Mann-Whitney U-test for independent samples (d) (z=7.5), (f) (z=5.4) and one-way ANOVA with Student–Newman-Keuls post-hoc test for pairwise comparisons, (h) (F=75.5, df=46) (i) (F=4.3, df=41) (j) (F=55.1, df=57).
To determine the role of DIO2 increases in human lung fibrosis we administered bleomycin to Dio2 knockout (Dio2−/−) mice that are systemically euthyroid but exhibit localized hypothyroidism in Dio2-expressing tissues13,14. Both Dio2−/− mice and wild-type littermates developed pulmonary fibrosis 14 days after intratracheal bleomycin with significant increases in hydroxyproline levels (1.7- and 1.6-fold, respectively) (Fig. 1i) compared to saline-treated animals. However, after bleomycin Dio2−/− mice exhibited significantly higher hydroxyproline levels (1.3-fold) (Fig. 1i) and increased histological evidence of fibrosis in the lung (Fig. 1j) compared to wild-type littermates, but no increases in collagen mRNA (data not shown).
TH blunts lung fibrosis in two murine models
Systemic administration of T4 (100μg/kg) at days 10, 12, 14, 16, after bleomycin administration significantly blunted fibrosis but caused significant increases in serum T3 levels (Supplementary Fig. 1). To avoid the side effects of systemic administration of T4 and maximize therapeutic efficacy we focused on the pulmonary delivery of the physiologically active hormone T3 by aerosol. Aerosolized T3 (40 μg/Kg) therapy, given every other day starting at the established fibrosis stage (days 10–20 after bleomycin), caused significant decreases in hydroxyproline levels (2.25-fold, P < 0.05) (Fig. 2a), and histologic evidence of fibrosis (Fig. 2b) without affecting serum T3 levels (Fig. 2b). The effects were comparable to those observed with oral administration of pirfenidone (100 mg/kg) or nintedanib (60 mg/kg), the two drugs currently approved by the US FDA for the treatment of human IPF15 (Fig. 2a,b).
Figure 2.
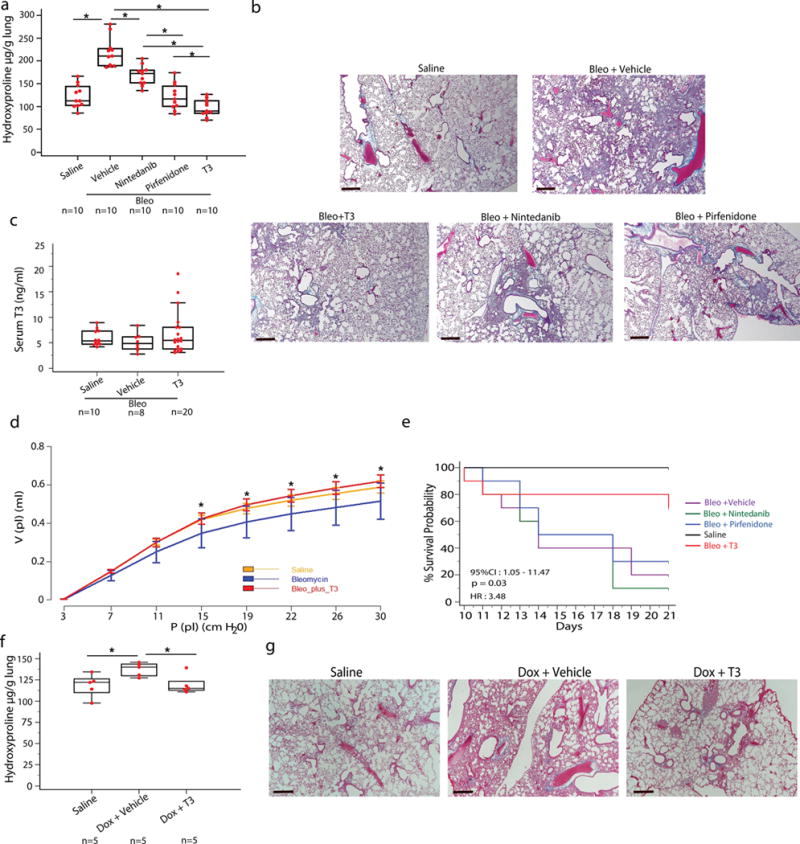
Aerosolized T3 blunts established fibrosis in two murine models of lung fibrosis. (a) Effects of aerosolized T3, nintedanib or pirfenidone on lung hydroxyproline content. Data presented as box-and-whisker plots are from one of two independent experiments with similar results with horizontal bars representing mean hydroxyproline content per lung set (μg/gr) + SEM, *P < 0,001. (b) Masson’s Trichrome staining of representative lung sections (n = 5) from each group of treated mice. Scale bars, 50 μm. (c) Aerosolized T3 administration effects on T3 serum levels. Data are presented as box-and-whisker plots with horizontal bars representing mean T3 serum levels (ng/ml) + SEM, P = 0.7. (d) Pressure–volume-loops (PV-loops) of mice (n = 5/group) treated with aerosolized T3 following intratracheal challenge with bleomycin or normal saline, P < 0.001. (e) Kaplan-Meier plot survival plots of mice treated with aerosolized T3, pirfenidone, nintedanib or equivalent volume of vehicle following intratracheal challenge with double dose of bleomycin or normal saline, n = 10 mice/group. (f) T3 effects on hydroxyproline content in mice in the following groups: Saline – transgene not induced, Dox+vehicle - transgene induced animals treated with vehicle, DOX+T3 – transgene induced and animals treated with aerosolized T3. Data are presented as box-and-whisker plots with horizontal bars representing mean hydroxyproline content per lung set (μg/gr) + SEM, *P = 0.035. (g) Masson’s Trichrome staining of representative lung sections (n = 5) in the triple transgenic (CC10-rtTA-tTS-TGF-β1) in the groups from f. Scale bars, 50 μm. The statistical test used was one-way ANOVA with Student-Newman-Keuls post-hoc test for pairwise comparisons, (a) (F=38.6, df=54), (c) (F=0.6, df=56), (d) (F=11.9, df=44), (f) (F=4.5, df=14)
Aerosolized T3 therapy reversed the significant reduction in static lung compliance induced by bleomycin, reflecting a functional improvement in respiratory mechanics and reduction in fibrosis (Fig. 2d). Similar functional benefits were observed with pirfenidone and nintedanib treatment (data not shown). To observe the effects of aerosolized T3 on survival we used a higher dose of bleomycin (3.0 U/kg). In contrast to pirfenidone and nintedanib, aerosolized T3 caused a substantial survival benefit with significant reduction in mortality from 90% in the untreated group to 20% in the T3 treated arm (Fig. 2e). This survival benefit suggested that T3 potentially limited the extent of acute lung injury caused by high dose bleomycin in addition to its anti-fibrotic effects. To assess the effects of aerosolized T3 in another animal model of experimental lung fibrosis, we used the triple transgenic mice in which a CC10 promoter drives the expression of active TGFβ1 in response to doxycycline treatment16. Aerosolized T3 therapy, given every other day, at days 10–20 after doxycycline treatment resulted in significantly lower pulmonary fibrosis as assessed by lower hydroxyproline levels (1.15-fold, P < 0.05) and Masson Trichrome staining compared to mice treated with doxycycline and vehicle (Fig. 2f,g).
TH reverses bleomycin-induced mitochondrial abnormalities
As previously observed11 AECIIs in bleomycin-treated mice exhibited swollen mitochondria, with severely damaged electron-lucent cristae and disrupted membranes and significantly reduced numbers of normal looking mitochondria, while aerosolized T3 reversed these changes (Fig. 3a,b). Aerosolized T3 also reversed many of the functional abnormalities induced by bleomycin in AECIIs. Specifically, primary AECIIs isolated on day 21 from the lungs of bleomycin-treated animals (AECBs) exhibited significantly lower mitochondrial membrane potential than AECIIs from mice treated with aerosolized T3 after bleomycin (AECBT3s) or AECIIs isolated from uninjured lungs (AECTRLs) (1.4- and 1.5-fold, respectively) (Fig. 3c). AECBs exhibited significantly decreased oxygen consumption rate (OCR) compared to AECTRLs (3-fold) that was restored to baseline levels in AECBT3s (Fig. 3d). In vitro, bleomycin treatment caused significant declines in the mitochondrial bioenergetic profile of primary human small airway epithelial cells (SAECs), primary mouse lung AECIIs and mouse lung epithelial cells (MLE12), but T3, added 4 hours after bleomycin, reversed this decline. SAECs, AECIIs and MLE12 that were treated with T3 after bleomycin exhibited significantly higher mitochondrial membrane potential (MMP, 1.2-, 1.3-, and 1.3-fold, respectively) (Fig 3e–g) and oxygen consumption rate (OCR, 2.1-, 1.5- and 6.9-fold, respectively) (Fig. 3h–j) compared to cells treated with vehicle after bleomycin. A detailed analysis of the mitochondrial bioenergetic profiles in lung epithelial cells, both in vivo (isolated AECIIs) and in vitro (SAECs, AECIIs, MLE12) showed similar differences between T3-treated groups and controls (Supplementary Fig 2).
Figure 3.
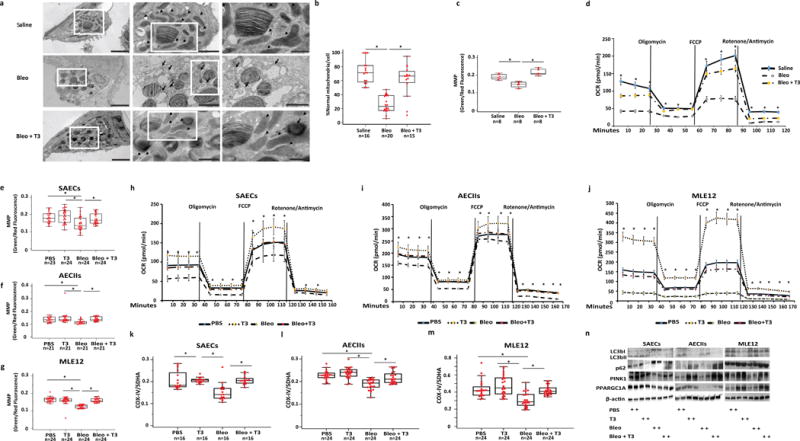
TH treatment restores bleomycin-induced mitochondrial abnormalities in alveolar epithelial cells. (a) Representative transmission electron microscopy (TEM) images (n = 16, 19 and 13, respectively) of AECIIs from mice treated with saline (upper panel), bleomycin (3.0U/kg) (middle panel) or bleomycin + aerosolized T3 (40μg/kg) (lower panel). Black arrows indicate damaged and swollen mitochondria with severely disrupted electron-lucent cristae, arrow heads indicate normal appearing mitochondria. Boxed regions are shown enlarged in the next column. Scale bars, 2 μM (left panels), 1 um (middle panels), 500 nm (right panels). (b) Quantitative analysis of the percentage of normal mitochondria per cell/group. Bars represent mean score + SEM, *P < 0,001. (c) Mitochondrial function in AECIIs cultured from animals treated with saline, bleomycin or bleomycin + T3. Data are presented as box-and-whisker plots with horizontal bars representing mean MMP levels (green/red fluorescence ratio) + SEM, *P < 0,001. (d) Oxygen consumption rate (OCR, pmol/min) was measured under basal conditions followed by addition of oligomycin, FCCP, rotenone and antimycin as indicated, *P < 0.001). (e–g) Green/red fluorescence ratio as a readout of MMP in primary human small airway epithelial cells (SAECs) (e), primary mouse AECIIs (f) and mouse lung epithelial cells (MLE12) (g) exposed to bleomycin or PBS and then treated with T3 or vehicle. Data are presented as box-and-whisker plots with horizontal bars representing mean MMP levels + SEM, *P < 0.001, P = 0.003 and P < 0.001, respectively). (h–j) Oxygen consumption rate (OCR, pmol/min) of the category of cells indicated in e–g as measured under basal conditions followed by addition of oligomycin (0.25μM), FCCP (1 μM), as well as rotenone and antimycin (1 μM), as indicated. (k–m) Effects of in vitro T3 treatment on mitochondrial biogenesis in the same category cells indicated in e–g as measured by the levels of Cytochrome c oxidase subunit IV (COX-IV) and Succinate Dehydrogenase Complex Flavoprotein subunit A (SDHA). Data are presented as box-and-whisker plots with horizontal bars representing mean COX-IV/SDHA ratio + SEM, *P < 0.001. (n) Immunoblot analysis of markers of autophagy (LC3BI, II, p62, PINK1) and mitochondrial biogenesis (PPARGCA1) in the same category of cells indicated in e–g. Immunoblot gels were cropped and uncropped images of the immunoblot gels are in Supplementary Figure 4. The statistical test used was One-way ANOVA with Student-Newman-Keuls post-hoc test for pairwise comparisons (b) (F=34.5, df=50), (c) (F=38.6, df=23) (d) (F=91.4, df=42), (e) (F=7.04, df=94) (f) (F=5.15, df=83) (g), (F=35.3, df=95), (k) (F=39.6, df=88), (l) (F=30.1, df=74), (m) (F= 74.6, df= 87).
To assess the effects of T3 treatment on mitogenesis we measured the Cytochrome c oxidase subunit IV (COX-IV) and Succinate Dehydrogenase Complex Flavoprotein subunit A (SDHA) ratio as previously described17. Epithelial cells treated with bleomycin exhibited a significantly lower COX-IV/SDHA ratio compared to vehicle treatment. Bleomycin treated cells that were also treated with T3 exhibited ratios similar to controls. This effect was observed in the three cell types (Fig. 3k–m, 1.4-, 1.2-, 1.4-fold, respectively). Epithelial cells treated with bleomycin exhibited significantly lower protein expression of PPARGC1A, a transcriptional co-activator of mitochondrial metabolism and biogenesis, compared to vehicle treated controls, but in cells treated with T3 following bleomycin PPARGC1A expression was similar to controls (Fig. 3n). T3 treatment after bleomycin was associated with significant higher levels of PINK1. Changes in autophagic flux, as indicated by altered autophagy protein light chain 3 (LC3B) and p62 protein levels were consistently seen in SAECs but not in AECIIs or MLE12 (Fig. 3n).
TH suppresses mitochondria-regulated apoptosis in AECIIs
After showing that TH opposes bleomycin-induced mitochondrial function abnormalities in primary mouse epithelial cells in vivo and in both human and mouse epithelial cells in vitro, we aimed to determine the effects of TH treatment on mitochondria-regulated apoptotic pathways. Immunoblot analyses revealed that exposure of AECIIs to bleomycin led to higher protein expression of the pro-apoptotic mediator BCL-2 associated X protein (BAX) and lower expression of the anti-apoptotic mediator BCL-xL (Fig. 4a). Importantly, bleomycin promoted the intracellular movement of BAX from the cytosol to mitochondria in SAECs, as indicated by co-localization of BAX with mito-tracker in the immunofluorescence analysis (Fig. 4b). BAX upregulation and mitochondrial translocation were associated with enhanced apoptotic activity as revealed by increased number of TUNEL-positive cells (Fig. 4c,d). This observation was verified in vivo by immunohistochemistry analysis showing increased TUNEL staining in the alveolar epithelium in mouse lungs challenged with bleomycin (Fig. 4e,f). T3 treatment reversed the BAX/BCL-xL ratio (Fig. 4a), attenuated BAX translocation to mitochondria (Fig. 4b) and consequently diminished bleomycin-induced apoptotic injury in AECs, both in vitro and in vivo, as indicated by significant reductions in TUNEL-positive cells (Fig. 4c–e).
Figure 4.
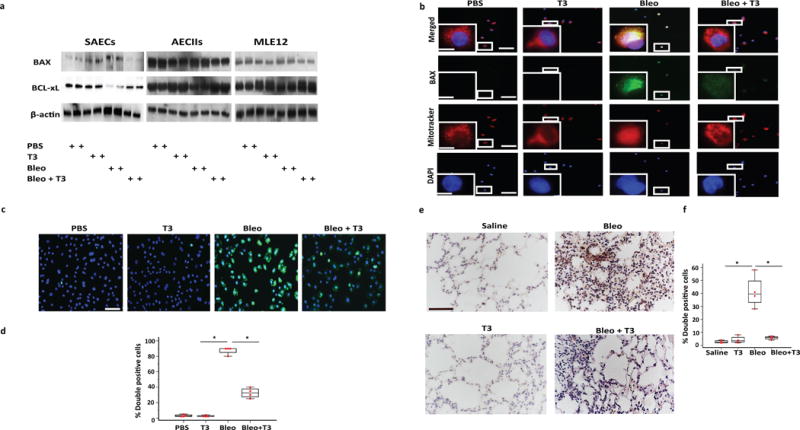
TH attenuates mitochondria-regulated apoptosis in lung epithelial cells. (a) Immunoblot analysis of markers of mitochondria-cell apoptosis (BAX, BCL-xL). Each lane represents a biological replicate and 2 experiments were done in each case. Immunoblot gels were cropped, and uncropped images of the immunoblot gels are in Supplementary Fig 4. (b) Immunofluorescence analysis for Mito-Tracker (red cationic dye that stains active mitochondria) and BAX (green) in SAECs after bleomycin or PBS exposure and treatment with or without T3. Localization of BAX with mito-tracker is indicated in yellow. Boxed regions are shown enlarged at lower left panels. Scale bars, 50μm, insets: 10 μm. (c,d) Immunofluorescence (c) and quantitative analysis (d) of double positive SAECs (TUNEL/DAPI). Data are presented as box-and-whisker plots with horizontal bars representing mean % percentage of double positive cells + SEM. One-way ANOVA (F=366.7, df=15) with Student-Newman-Keuls post-hoc test for pairwise comparisons, *P < 0.001. (e,f) Immunohistochemistry (e) and quantitative analysis (f) of TUNEL-positive cells in lung samples derived from mice challenged bleomycin or saline and then treated with T3 or vehicle at days 10–20. Data are presented as box-and-whisker plots with horizontal bars representing mean % percentage of double positive cells + SEM, *P < 0.001. Scale bars, 100μm. The statistical test used was one-way ANOVA (F=33.3, df=15) with Student-Newman-Keuls post-hoc test for pairwise comparisons.
Anti-fibrotic effects of TH are dependent on PPARGC1A and PINK1
Expression of PPARGC1A was lower following bleomycin in wild-type, Dio2−/− mice as well as in human IPF lungs (Supplementary Fig. 3). Mice treated with aerosolized T3 after bleomycin mice exhibited significantly higher (1.5-fold) expression of PPARGC1A mRNA in their lungs to mice treated with vehicle after bleomycin (Fig. 5a). To determine whether the anti-fibrotic effects of T3 were mediated through PPARGC1A we used Ppargc1a knockout mice (Ppargc1a−/−). After bleomycin treatment Ppargc1a−/− mice demonstrated significantly higher hydroxyproline levels (1.35-fold) compared to wild-type littermates but similar changes in Col1a1, Col3a1 mRNA levels (Fig. 5b–d). Unlike wild-type littermates, aerosolized T3 did not blunt fibrosis in Ppargc1a−/− mice and hydroxyproline content, Col1a1, Col3a1 mRNA levels and Masson Trichrome staining were unchanged (Fig. 5a–e). Consistent with previous reports18, in vitro treatment of alveolar epithelial cells (A549) induced higher expression and a shift in the cellular localization of PPARGC1A from nuclear, to both nuclear and mitochondrial (Fig. 4f,g). Dronedarone, an inhibitor of Thyroid-Hormone Receptors (THR)19 inhibited the T3-mediated induction and cellular redistribution of PPARGC1A (Fig. 5f,g). THR inhibition by dronedarone also prevented the anti-apoptotic effects of T3, as indicated by lower protein levels of BCL-xL and increased TUNEL-positive cells in the immunofluorescence analysis (Fig. 5g,h). Gold immunostaining for THRA1 and THRB in uninjured mouse lungs localized both receptors to nuclei and mitochondria of murine AECIIs (Fig. 5i). To further support our hypothesis that T3 anti-fibrotic properties are mediated through thyroid hormone receptors we investigated the therapeutic effects of sobetirome, a well-characterized thyroid hormone receptor agonist20,21. Oral administration of sobetirome started 10 days after bleomycin was associated with enhanced resolution of pulmonary fibrosis, as assessed by significantly decreased hydroxyproline content (P < 0.05) and Masson Trichrome staining (Fig 5j,k).
Figure 5.
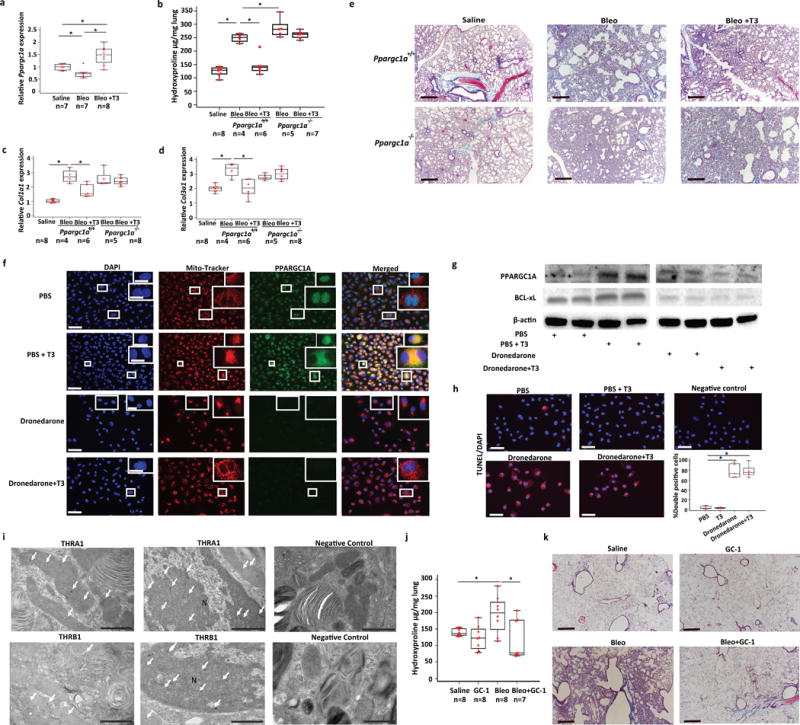
Anti-fibrotic effects of TH are mediated through PPARGC1A. (a) Quantitative RT-PCR analysis for Ppargc1a mRNA levels in the indicated treatment groups (means+ SEM), *P < 0.001. (b) Lung hydroxyproline content, and quantitative RT-PCR analysis of collagen type 1, alpha 1 (Col1a1) (c) and type 3, alpha 1 (Col3a1) (d) mRNA levels in Ppargc1a-deficient (Ppargc1a−/−) mice or wild-type littermates (Ppargc1a +/+) treated with aerosolized T3 following intratracheal challenge with bleomycin or equivalent volume of normal saline. Data presented are from one of two independent experiments with similar results and are expressed as mean hydroxyproline content per lung (μg/gr lung) set + SEM, *P < 0.001. (e) Masson’s Trichrome staining of representative lung sections (n = 3) from each group of mice indicated. Scale bars, 50 μm. (f) Immunofluorescence analysis for Mito-Tracker (red cationic dye that stains active mitochondria) and PPARGC1A (green) in A549 cells after pre-incubation with dronedarone for 24 hr or saline and treatment with or without T3. Colocalization of PPARGC1A with Mito-Tracker is denoted in yellow. Boxed regions are shown enlarged at upper right panels. Scale bars, 50μm, insets 10 μm. (g) Immunoblot of PPARGC1A in A549 cell lysates from cells treated as described for f (n = 2/group). Immunoblot gels were cropped and uncropped images of the immunoblot gels are in Supplementary Fig 4. (h) Immunofluorescence (left and upper right) and quantitative analysis (lower right) of double positive A549 cells (TUNEL/DAPI) after treatment as described in f. Quantitative data are presented as box-and-whisker plots with horizontal bars representing mean %percentage of double positive cells + SEM, *P < 0.001. (i) Representative TEM images of mouse lung sections (n = 5) stained with immunogold technique for expression of THRA1 (black dots-white arrows-upper panel) and THRB (black dots-white arrows-lower panel) inside morphologically normal mitochondria as well as nuclei (N) of primary AECIIs. Negative control was stained with secondary antibody only. (j) Lung hydroxyproline content in mice challenged with saline, bleomycin or bleomycin + sobiterome. Data are presented as box-and-whisker plots with horizontal bars representing mean hydroxyproline content per lung set (μg/gr) + SEM, *P = 0.01. (k) Masson’s Trichrome staining of representative lung sections (n = 3) from each group of mice indicated. The statistical test used was one-way ANOVA with Student-Newman-Keuls post-hoc test for pairwise comparisons (a) (F=14.5, df=21), (b) (F=54.8, df=29), (c) (F=25.2, df=30), (d) (F=12.5, df=30), (f) (F=96.7, df=19), (j) (F=4.3, df=23).
PINK1 is a known positive regulator of PPARGC1A-dependent mitochondrial metabolism22 and its absence renders mice susceptible to lung fibrosis due to inability to process dysfunctional mitochondria through mitophagy11. After demonstrating that the anti-fibrotic effects of TH were dependent on PPARGC1A we assessed the role of PINK1. In wild type mice aerosolized T3-treatment partially restored bleomycin-induced PINK1 downregulation (1.8-fold) (Fig. 6a). PINK1-knockout mice (Pink1−/−) exhibited higher collagen deposition measured by hydroxyproline levels (1.28-fold) (Fig. 6b), Col1a1, (1.5-fold) (Fig. 6c,d) mRNA levels and Masson Trichrome staining (Fig. 6e) compared to wild-types, indicating augmented fibrosis as previously reported11. Aerosolized T3 administration did not blunt fibrosis in Pink1−/− mice (Fig. 5b–e).
Figure 6.
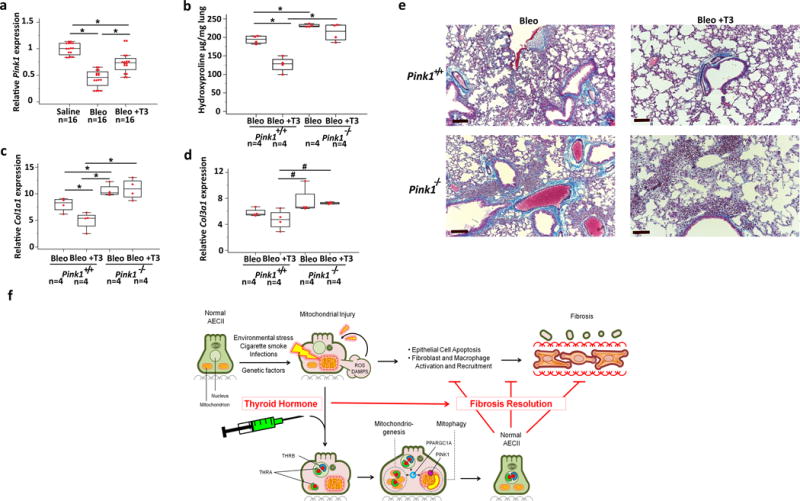
PINK1 is required for the antifibrotic effects of TH. (a) Quantitative RT-PCR analysis for Pink1 mRNA levels in wild-type littermates intratracheally challenged with saline, bleomycin or aerosolized T3 following bleomycin. (means+ SEM), *P < 0.001. (b) Collagen deposition assessed by hydroxyproline content in Pink1-deficient (Pink1−/−) mice or wild-type littermates (Pink1+/+) treated with aerosolized T3 following intratracheal challenge with bleomycin vs. controls. Data presented are from one of two independent experiments with similar results and are expressed as mean hydroxyproline content per lung set (μg/gr lung) + SEM, n = 4 mice/group, *P < 0.001. (c,d) Quantitative RT-PCR analysis of collagen type 1, alpha 1 (Col1a1) and type 3, alpha 1 (Col3a1) mRNA levels in a similar group of mice indicated in b (means+ SEM), *P < 0.001 and #P = 0.03. (e) Masson’s Trichrome staining of representative lung sections (n = 3) from each group of treated mice indicated. Scale bars, 100μm. The statistical test used was one-way ANOVA with Student-Newman-Keuls post-hoc test for pairwise comparisons (a) (F=46, df=47), (b) (F=29.9, df=15), (c) (F=12.9, df=15), (d) (F=4.2, df=15) (f) Schematic diagram of a model of the anti-fibrotic effect of TH via its restoration of mitochondrial homeostasis and function in alveolar type II epithelial cells (AECIIs). Injury of AECIIs leads to mitochondrial dysfunction and release of reactive oxygen species (ROS) and damage-associated molecular patterns (DAMPs) including mitochondrial DNA. TH supplementation modifies fibrosis through an epithelial protective effect via its binding to its receptor (THRA1, THRB) and its promotion of the expression of positive regulators of mitochondrial metabolism (PPARGC1A) and mitophagy (PINK1), resulting in restoration of normal mitochondrial function, rescue from mitochondria-regulated apoptosis and fibrosis resolution.
DISCUSSION
Our results here suggest that mitochondrial dysfunction, increasingly recognized as a hallmark of epithelial cell injury in pulmonary fibrosis11, can be attenuated using localized thyroid hormone delivery, leading to resolution of fibrosis in mouse models. Following the observation that both the expression and activity of DIO2 were substantially increased in lung from patients with IPF and correlated with disease severity, we studied the role of DIO2 and TH signaling in experimental pulmonary fibrosis in mice. Genetic deletion of DIO2 enhanced bleomycin-induced lung fibrosis. TH delivery to the lung blunted established pulmonary fibrosis in two experimental models as did oral therapy with sobiterome, a well characterized thyromimetic drug. TH restored bleomycin-induced mitochondrial derangements in epithelial cells in vivo and in vitro through improved mitochondrial function and homeostasis. These beneficial effects were associated with suppression of mitochondria-regulated death pathways, and dependent on intact PPARGC1A and PINK1 pathways.
TH regulates fundamental biological functions including bioenergetics and post-stress injury repair of every vertebrate tissue. Tissue-specific TH metabolism and availability are tightly regulated by DIO1-3. Among them DIO2 is mainly responsible for TH activation by converting T4 to active T323. Increased DIO2 expression and activity may either reflect TH deficiency or increased metabolic requirements23, whereas decreases reflect excess of TH in the body23. These changes are required to protect vital tissues and organs from unexpected fluctuations in TH levels. In the context of lung disease, DIO2 immunoreactivity and enzymatic activity were rapidly induced by ventilation-induced lung injury and DIO2 knockout mice exhibit increased susceptibility to acute lung injury24. TH therapy significantly attenuated experimental lung injury24 and produced similar therapeutic results in animal models of cardiac25 and renal injury26. Clinically, hypothyroidism has been associated with unfavorable prognosis in critically ill patients27, with myocardial infarction28, diabetic nephropathy29 and most recently IPF30. However, evidence for impaired TH signaling in the human IPF lung or experimental evidence in support of its therapeutic utility in IPF were not available. Our study provides novel observations that together establish the case for aerosolized TH therapy as an antifibrotic therapy. Firstly, we provide evidence that DIO2 expression and activity are increased in the lung of patients with IPF, correlate with disease severity and localize mainly to AECs, the highly metabolically active cell type thought to be at the center of the vicious cycle of injury and repair characteristic of IPF. Secondly, we establish a role for TH in experimental lung fibrosis; TH therapy effectively blunts experimental lung fibrosis in two mouse models in a manner that is replicated by the thyroid hormone mimetic sobiterome. Thirdly, the beneficial effect is observed when TH therapy is initiated during the established fibrosis phase, suggesting enhanced resolution of fibrosis. Fourthly, the antifibrotic effects are obtained with aerosolized T3 without an increase in T3 serum levels, suggesting that aerosolized delivery may be effective without the toxicity observed previously with systemic delivery31. Although some systemic deposition of TH following aerosolized delivery could not be entirely excluded as mice may clean their fur following mist exposure; yet, this appears to be non-significant, as assessed by minimal changes in T3 serum levels. Lastly, aerosolized T3 showed comparable or superior effects on lung fibrosis or survival compared to the two US FDA-approved therapeutic compounds for the treatment of IPF, pirfenidone and nintedanib. Taken together these observations establish a novel role for TH as a potential therapeutic agent in pulmonary fibrosis.
Mechanistically we established that TH exerted its antifibrotic effects in bleomycin induced pulmonary fibrosis by restoring mitochondrial function through induction of mitochondrial biogenesis and mitophagy. T3-mediated beneficial effects on mitochondrial homeostasis attenuated bleomycin-induced mitochondria-regulated cell death in alveolar epithelial cells. Recently, there has been significant interest in mitochondrial function in chronic lung diseases9,11,32. Bueno and colleagues discovered that PINK1, a regulator of clearance of dysfunctional mitochondria, was decreased in subjects with IPF11. They convincingly demonstrated abundant mitochondrial abnormalities in AECIIs derived from lungs from patients with IPF, as well as in mice treated with bleomycin11. Based on their findings we hypothesized that the effects of TH depletion or augmentation on severity of bleomycin induced pulmonary fibrosis were associated with changes in mitochondrial function. Indeed, when we isolated primary mouse AECIIs from lungs treated with bleomycin we discovered that they exhibited significant derangements in mitochondrial function including diminished ATP production, ineffective energy metabolism and mitochondrial membrane depolarization, an index of mitochondrial integrity and proton gradient across the mitochondrial inner membrane, and increased proton leak. Aerosolized TH treatment significantly improved all of the parameters of mitochondrial function, as well as mitochondrial morphology in AECIIs. TH did induce increases in proton leak but those correlated with increases in coupling efficiency and metabolic rates as has previously been observed33. This phenomenon may represent restoration of the basal mitochondrial adaptive mechanism to link higher proton conductance to higher metabolic rates resulting in protection against excessive reactive oxygen species production34.
In the context of critical illness, TH exerts cytoprotective and regenerative properties through effects on target genes leading to maintenance of cellular homeostasis, energy expenditure and modulation of stress responses to injury35. We propose a similar but more detailed model in pulmonary fibrosis; specifically, that repeated epithelial injury and increased metabolic demand lead to mitochondrial damage, increased reactive oxygen species (ROS) generation and epithelial cell apoptosis that eventually lead to downstream fibroblast activation and initiation of the fibrotic vicious cycle. TH opposes these processes by mediating mitochondrial biogenesis and effective mitophagy through induction of major transcription coactivators, PPARGC1A and PINK1. The role of PPARGC1A and PINK1 in the beneficial effect of TH is of particular interest. PPARGC1A controls mitochondrial remodeling and mitochondrial biogenesis36. PINK1 is downstream of PPARCG1A and is thought to have an important role in regulating PPARGC1A-dependent mitochondrial biogenesis through selective degradation of damaged mitochondria by mitophagy22. Both PPARGC1A and PINK1 were decreased in the lungs of animals treated with bleomycin, and TH treatment restored their levels. Aerosolized TH delivery failed to rescue the bleomycin-induced fibrotic phenotype in mice with either PPARGC1A or PINK1 deficiency suggesting that TH-mediated antifibrotic effects required intact PPARGC1A and PINK1. The relevance of this finding to human disease is enhanced because the expression of both molecules is decreased in IPF lungs, however it is unclear whether directly overexpressing either of the molecules will have any antifibrotic effect. In addition to in vivo observations, we demonstrated in vitro that T3 caused THR-dependent induction and redistribution of PPARGC1A in alveolar epithelial cells consistent with previous reports in other cells37. THR inhibition with dronedarone resulted in enhanced apoptotic activity and diminished TH-mediated PPARGC1A induction and translocation, whereas treatment with TH restored mitochondrial bioenergetics and reduced apoptotic activity in lung epithelial cells.
Epithelial cell apoptosis and impaired autophagy are increasingly considered hallmarks of pulmonary fibrosis16,38 and they are believed to reciprocally regulate each other39. Caspase-mediated apoptosis has been proposed to inhibit autophagy through cleavage of several autophagy-related proteins including LC3B40. On the other hand, mitophagy reduces the propensity of cells to undergo apoptosis and functions as an early cytoprotective response favoring adaptation to stress and injury by removing damaged mitochondria. PINK1 acts as a key cytoprotective kinase that activates anti-apoptotic members of the BCL-2 family41, which are known as positive regulators of mitochondrial bioenergetics by enhancing coupling efficiency and ATP production42. In line with these studies, we have demonstrated that alveolar epithelial cells exposed to bleomycin exhibited enhanced BAX mitochondrial translocation and diminished BCL-xL expression, mitochondrial membrane depolarization, and downregulation of PINK1 and PPARGC1A leading to impairment of mitophagy and mitochondrial biogenesis, as assessed by changes in the COX-IV/SDHA ratio. These in vitro observations were also supported by in vivo findings showing impaired mitochondrial homeostasis and enhanced apoptosis in AECIIs derived from mice challenged with bleomycin. Impressively, we observed that T3 mediated a sequence of cytoprotective anti-apoptotic events including: attenuation of mitochondrial membrane depolarization, enhancement of mitochondrial biogenesis, induction of effective mitophagy, and finally suppression of the mitochondria-regulated death pathway. These observations are in line with previous studies demonstrating that T3 protected ovarian cells from chemotherapy-induced apoptosis through downregulation of BAX and upregulation of BCL-xL levels43. Similarly, TH has been demonstrated to induce mitochondrial biogenesis and activity through positive regulation of autophagy in skeletal muscle cells44. Taken together, we propose that by restoring mitochondrial function, TH therapy allows functional recovery of alveolar epithelial cells including normalization of mitochondrial bioenergetics and attenuation of mitochondria-regulated apoptosis, which in turn stops the activation of fibroblasts by ROS and damage-associated molecular patterns (DAMPs), and thus allows resolution of fibrosis (Fig. 6f).
In this study we did not address several mechanistically relevant questions of interest. For instance, we did not establish the cause for induction of DIO2 in lung fibrosis. Demonstrating a link between mitochondrial dysfunction and upregulation of DIO2 would strengthen the evidence that the mechanism we propose is indeed also relevant in humans. We also did not study in detail the interaction between the antifibrotic effect of TH and the TGF-β signaling pathway, although we demonstrated that aerosolized T3 therapy was effective in the inducible TGF-β1 model of lung fibrosis. A recent study demonstrated antifibrotic TH effects in models of liver and skin fibrosis45. The study proposed that T3 acted as a modulator of the TGF-β signaling machinery45. We did not test this mechanism directly, and it could still be relevant in our models. Similarly, while our study focused on the beneficial effects of TH on epithelial cells, we did not directly evaluate its role in other cells. Metabolic aberrations, senescence and impaired autophagy have been reported in fibroblasts and macrophages in IPF38,46–50, and it is possible that improved mitochondrial function may also affect these cells. However, we believe that the impressive changes in mitochondrial morphology and function and the dependence of the antifibrotic effects of T3 on intact PPARGC1A and PINK1 pathways in vivo, and the impressive cytoprotective effects in vitro, support the notion that the antifibrotic effects of TH on mitochondrial homeostasis and function in epithelial cells were key mechanisms in our models, but does not rule out the role of additional mechanisms or cell types.
We believe that the observations in subjects with IPF, as well as the detailed and convincing mechanistic experiments we performed in vitro and in animal models of disease do establish a novel and significantly important observation; that TH augmentation, by localized T3 or systemic sobiterome therapy, enhances resolution of fibrosis in mouse models of pulmonary fibrosis, probably through an epithelial cytoprotective effect. Taken together, our results strongly suggest that strategies utilizing lung-targeted TH therapy should be further studied in IPF.
Online Methods
Microarray assays
For gene expression analysis we used the publically available (gene expression omnibus (GEO) GSE47460 and http://www.lung-genomics.org/) LGRC gene expression dataset, generated by our study group12,51. 123 subjects with IPF and 96 control subjects were included. Supplementary Table 1 summarizes the clinical characteristics of the subjects.
IPF Tissues
Lung tissue samples were obtained through the University of Pittsburgh Health Sciences Tissue Bank and Yale university Pathology Tissue service, which were surgical remnants of biopsies or lungs explanted from subjects with IPF who underwent pulmonary transplantation.
Human subject approval
All studies have been approved by the Institutional Review Board at the University of Pittsburgh and Yale School of Medicine.
RNA extraction
Total RNA was extracted from 30 – 50 mg of frozen lung tissue in 700 uL of Qiazol (Lysis buffer, Qiagen, Valencia, CA) according to the manufacturer’s instructions. The purity of the RNA was verified using NanoDrop at 260 nm and the quality of the RNA was assessed using the Agilent 2100 Bioanalyzer (Agilent, Technologies, Santa Clara, CA).
Quantitative Real-Time PCR
Gene expression was determined by TaqMan® (Life Technologies, Thermo Scientific Inc. Rockford IL, USA) according to manufacturer’s instruction. β-glucuronidase (Gusb) was employed as an internal standard control and the specific primers and probes were all obtained from Life Technologies (Thermo Scientific Inc. Rockford IL, USA). Each reaction was performed in triplicate. Relative gene expression was normalized to a value of 1.0 for the unstimulated control group. Control reactions without cDNA or RNA were done as a negative control. Fold change was calculated taking the mean of the controls as the baseline.
Protein extraction and western blot analysis
Proteins from tissues or cultured cells were extracted, transferred onto PVD-membranes, hybridized overnight with appropriate primary antibodies (PPARGC1A - sc-13067 and ab-54481, DIO2-sc-98716, BCL-xL- ab32124, PINK1-ab23707, LC3B-ab51520, p62/SQSTM1-ab56416, BAX-ab32503 and ab-53154, β-actin-sc-47778) obtained from Santa Cruz Biotechnology, CA (sc), or Abcam, Cambridge, UK (ab) and visualized using the Gel Doc XR+ System (Biorad, Lab. Inc. Life Science, USA), according to manufacturer’s protocol.
DIO2 activity
The DIO2 assay was established previously based on the release of radioiodide from the 125I- labelled substrate (100000 cpm-5′-125I of T4-Sigma, MO) as previously described52. Briefly, the mixture containing 25 ml tissue homogenate in 0·1mM-potassium phosphate buffer (pH 7·0) and 1mM-EDTA, and 650 ml of substrate for a final concentration of 1 nM-T4 (Sigma St Louis, MO), 20mM-cofactor diothiothreitol and 1mM-propylthiouracil (pH 7.0) was incubated at 37°C for 1 h. The reactions were stopped by the addition of 50 ml ice-cold 5% bovine serum albumin followed by 350 ml 10% ice-cold TCA, and mixtures were centrifuged at 4000 g for 20 min. The supernatant was further purified by cation exchange chromatography using 1·6ml Dowex 50 W-X2 (100–200 mesh; Sigma). The iodide was then eluted twice with 1ml 10% glacial acetic acid and counted in a g-counter. Enzymatic activity was expressed in fmol125I-released/h per mg protein. Enzymatic activity was expressed in fmol125I-released/h per mg protein.
Animal studies approval
All animal studies were conducted in accordance with the NIH guidelines for humane treatment of animals and were approved by the Institutional Animal Care and Use Committee (IACUC), Yale University (2014-11592).
Experimental models of lung fibrosis
All animal models were performed twice and data was jointly analyzed. All animals treated were included in the analysis. Interventions were not blinded, but analysis of animal samples was. a) Bleomycin-model:C57Bl/6, 9–12 weeks-old, female mice were purchased (Taconic Biosciences, Hudson, NY). Knockout mice (Dio2−/−), Ppargc1a−/− and Pink1−/− of C57/BL6 background were obtained from Jackson Lab (Bar-Harbor, ME). As previously published11,53 genetic deletion of either PINK1 or PPARGC1A does not produce any endogenous lung phenotype and is not associated with derangements of lung physiology. Ppargc1a−/− and Pink1−/− mice are not embryonically lethal, they produce litters of appropriate Mendelian size, are fertile and healthy and present with no gross morphology abnormalities. Mice were anesthetized by placing them in a chamber having paper towels soaked with 40% isoflurane solution diluted with 1,2-propanediol. Mice were randomly assigned to either intratracheal 1.5 U/kg of bleomycin (Hospira, IL) or equivalent volume (50 μL) of 0.9% normal saline was administered intratracheally as previously described54,55. To test therapeutic efficacy of TH in bleomycin-induced established fibrosis we used intraperitoneal T4 (T2376-Sigma Aldrich, 100μg/kg), or aerosolized T3 (T2877-Sigma Aldrich, 40 μg/kg). T4 was administered systemically at days 10, 12, 14, 16 and mice were sacrificed on day 19, following bleomycin-challenge. Aerosolized T3 was administered every other day at days 10–20 and mice were sacrificed on day 21. Dose regimens were based on previously published protocols24,56. Equivalent volumes of normal saline 0.9% were used as controls in all approaches. For survival analysis we used a double dose of bleomycin (3.0 U/kg) and administered aerosolized T3 (40 μg/kg) or normal saline 0.9% at days at days 10–20 (established fibrosis) following challenge and survival data was collected at day 21, respectively. Mice were randomly assigned to receive either pirfenidone (100 mg/kg) or nintedanib (60 mg/kg) or vehicle (0.9% saline) via oral gavage, as previously described57, at days 10–20 (on a daily basis) following bleomycin administration and mice were sacrificed at day 21. Aerosolized delivery of T3 was performed following a standardized protocol. Briefly: T3 was diluted to a final concentration of 40 μg/kg in 6 ml of PBS and suspension was aerosolized using a conventional aerosol nebulizer (Omron) throughout a chamber that allowed simultaneous exposure of 8 mice for 30 minutes until mist stopped forming in the nebulizer chamber. b) TGF-β1-induced lung fibrosis: Inducible lung targeted TGF-β1-overexpressing triple transgenic mice (CC10-rtTA-tTS-TGF-β1) generated as previously described16 were used. Briefly: A triple transgenic system based on the tetracycline-controlled transcriptional suppressor (tTS) and the reverse tetracycline transactivator (rtTA) was constructed. In this system, the CC10 promoter constitutively drives the expression of rtTA and tTS in a lung-specific fashion. In the absence of dox, tTS binds to and actively suppresses the expression of the tet-O–regulated TGF-β1 transgene. In the presence of dox, tTS is released allowing the activating, dox binding rtTA to bind to the tet-O and activate transgene expression. As expected induced TGF-β1 – overexpression upon addition of doxycycline water on a daily basis (days 0–20) caused airway and parenchymal fibrotic response as assessed by increased lung collagen deposition indicated by hydroxyproline levels and Masson Trichrome staining. Aerosolized T3 (40 μg/kg) or normal saline 0.9% was administered every other day on days 10–20 following addition of doxycycline and mice where sacrificed on day 21. c) Sobiterome therapeutic protocol: Sobiterome was provided by the laboratory of Dr. Scanlan as previously described21,58. C57Bl/6, 9–12 weeks-old, female mice were randomly assigned to be challenged with 1.5 U/kg of bleomycin or equivalent volume of normal saline 0.9% at day 0. Mice were then randomly assigned to treatment with 5 mg/kg of sobiterome diluted in 50 μL of normal saline 0.9% or vehicle (equivalent volume of normal saline 0.9%) administered by oral gavage at days 10, 12, 14, 16 and 18 following bleomycin administration and mice were sacrificed on day 21.
In-vivo respiratory mechanics (Flexi-Vent system-SCIREQ)
To evaluate in-vivo respiratory mechanics as an assessment tool for therapeutic efficacy we applied the Flexi-Vent computer-controlled piston-ventilator system according to manufacturer’s protocol. Briefly: Mice were anesthetized with ketamine and then mice tracheotomized and ventilated using the Flevi-Vent system with an average breathing frequency of 150 breaths/min. During the maximal vital capacity (MVC) perturbation, the lungs were inflated to a standard pressure of +30 cmH2O and then deflated to determine MVC. Maximal pressure–volume-loops (PV-loops) between TLC (+30 cmH2O) and FRC (2.8 cmH20) were finally generated to obtain static compliance (Cst) of the respiratory system. In every animal, each manoeuver was repeated until three acceptable measurements (coefficient of determination ≥0.95) were recorded. The average of three acceptable measurements was calculated.
Histology and immunohistochemistry
Tissue sections (4 μm) were stained with Masson Trichrome (collagen/connective tissue), two slices per animal, and two animals per group, as previously described. Immune staining for DIO2 (ab77481, Abcam, Cambridge, UK) or PPARGC1A (ab54481, Abcam, Cambridge, UK) was performed after paraffin removal, hydration, and blocking, following the recommendation of the manufacturer and described by us59. Sections were incubated overnight at 4°C with the primary antibody (diluted 1:100 in PBS) and during 1 hour at room temperature with the secondary antibodies (Sigma, USA). The sections were counterstained with hematoxylin. The primary antibody was replaced by non-immune serum for negative controls.
Hydroxyproline assay
Lung hydroxyproline was analyzed with hydroxyproline colorimetric assay kit from Biovision (Milpitas, CA) following manufacturer’s instruction, as previously described60. Data are expressed as μg of hydroxyproline/right lung.
Measurement of T3 serum levels
Serum from mouse peripheral blood was collected by BD vacutainers, and serum total total triiodothyronine T3 levels were measured T3 ELISA kits (ABIN2685558, Antibodies-Online, Atlanta, GA), following the manufacturer’s instructions.
Isolation of primary alveolar epithelial cells (AECIIs)
Primary AECIIs were isolated from mice treated with 0.9% saline, or bleomycin (1.5U/kg), or bleomycin + aerosolized T3 (40μg/kg), using a modified protocol of a previously published method61. AECIIs were identified as population with CD45/CD32/CD16/CD11b-ve,SSchi cells. Using this protocol, a purity exceeding 80% and viability of 90% was obtained. Isolated cells were used for functional assays.
Cell culture experiments, immunofluorescence staining and assays of apoptosis
Human small airway epithelial cells (SAECs) were purchased from Lonza, Allendale, NJ (LOT: 000470903). Primary mouse AECIIs were isolated from the lungs of unchallenged mice. Mouse lung epithelial cell line (MLE12) was purchased from ATCC (CRL-2110). Cells were seeded into 96-well plates (40.000/well), exposed to bleomycin (15 mU/ml) or PBS for 4 hours and then treated with T3 (15 ng/ml) or vehicle control for 8 hours. Immunofluorescence staining was performed using Mito-Tracker (Thermo Fisher Scientific, Waltham, MA), a cationic dye that stains active mitochondria and PPARGC1A (ab54481, Abcam, Cambridge, UK), according to manufacturer’s instructions. Detection of apoptotic cells was performed with TUNEL-assay using the in situ Cell Death Detection Kit, Fluorescein (Roche, Indianapolis, IA, USA, Catalog No 11684795910). DAPI staining was used to determine the number of nuclei and to assess gross cell morphology. The human lung adenocarcinoma cell line A549 (ATCC® CCL-185™, ATCC, Manassas, VA), negatively tested for mycoplasma, was cultured in DMEM supplemented with 10% fetal bovine serum. Cells were pre-incubated with either dronedarone 10 μM diluted in 0.04% DMSO or vehicle control (0.04% DMSO) for 24 hours and then treated with T3 (15 ng/ml) or vehicle (normal saline 0.9%) for 24 hrs.
Extracellular Flux Technology
The oxygen consumption rates (OCR) of primary mouse AECIIs, SAECs, and MLE-12 cells were measured by using a Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA), as previously described62. All assays were performed using a seeding density of 60,000 cells/well in 200μl of DMEM in a XF96 cell culture microplate (Seahorse Bioscience). After the cells were switched to unbuffered DMEM supplemented with 2 mM sodium pyruvate and 20mM carnosine 1h prior to the beginning of the assay and maintained at 37 °C. OCR was measured after sequentially adding to each well 25μl of oligomycin (an ATP-synthase inhibitor), FCCP (a protonophore) and rotenone (inhibitors of complex I and III), to reach working concentrations of 1μg/ml, 1μM and 0.5μM respectively. OCR is reported in picomoles/minute/60,000 cells.
Determination of ATP production
Total ATP was determined using an ATP Fluorometric Assay Kit (BioVision, San Francisco, CA) as previously described62. Briefly, harvested cultured cells were lysed with a lysis buffer followed by centrifugation at 10.000g for 2 min, at 4°C. The level of ATP was determined by mixing 20 μl of the supernatant with 100 μl of luciferase reagent which catalyzed the light production from ATP and luciferin. Reactions were performed in 6-well plates at a seeding density of 106 cells/well. Luminance was measured by a monochromator microplate reader. Total ATP levels were expressed as nmol/mg protein.
Mitochondrial membrane potential (MMP)
The changes in relative MMP (ΔΨm) were measured using the JC-10 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzamidazolocarbocyanine iodide) molecular probe, as previously described62.
Transmission Electron microscopy (TEM)
For TEM, mice were perfused with 4% Paraformaldehyde (PFA) and lung samples were dissected out in 2% PFA. These were fixed in 2.5% gluteraldehyde/2% PFA in 0.1M sodium cacodylate buffer pH7.4 for 1 hour, followed by rinsing in PBS buffer then post fixed in 1% osmium tetroxide. They were en-bloc stained in 2% aqueous uranyl acetate then rinsed and dehydrated in an ethanol series followed by resin infiltration Embed 812 (Electron Microscopy Sciences) and baked overnight at 60 C. Hardened blocks were cut using a Leica UltraCut UC7. 60nm sections were collected on formvar/carbon coated grids and contrast stained using 2% uranyl acetate and lead citrate. Grids were viewed FEI TencaiBiotwin TEM at 80Kv. Images were taken using Morada CCD and iTEM (Olympus) software. At least 10 cells from low- and high-magnification images (×11,500, ×26,000) were used to count the number of mitochondria per AECII (identified by the presence of lamellar bodies).
Mitobiogenesis assay
Human SAECs, primary (AECIIs) isolated from the mouse lungs and MLE-12 were seeded in 96-well microplates containing 100 μL of culture medium. Cells were first exposed to bleomycin (10mU/ml) diluted in PBS or vehicle control (PBS) for 4 hours and then were treated with T3 (15ng/ml) or vehicle control for 8 hours. The levels of two mitochondrial proteins were measured simultaneously in each well by using a colorimetric ELISA-kit, according to manufacturer’s protocol (MitoBiogenesis™ In-Cell ELISA Kit- Abcam-ab110217)17. The two proteins are each subunits of a different oxidative phosphorylation enzyme complex, one protein being subunit I of Complex IV (Cytochrome c oxidase subunit-COX-IV), which is mitochondrial-DNA-encoded, and Succinate Dehydrogenase Complex Flavoprotein subunit A-SDHA) which is a 70kDa subunit of Complex II and nuclear-DNA-encoded.
Immunogold staining protocol
The grids containing tissue samples were placed section side down on drops of 1% hydrogen peroxide for 5 minutes, rinsed and blocked for nonspecific binding on 3% bovine serum albumin in Tris buffered saline containing 1% Triton-X for 30 minutes. Grids were incubated with a primary antibody anti-rabbit thyroid hormone receptor alpha-1 and beta (PA1-211A and PA1-213A, respectively Thermo Fisher, Pierce) 1:500 overnight, rinsed in TBS then incubated with the secondary antibody 10nm protein A gold (University of Utrecht, Cell Microscopy Core, Netherlands) for 30 minutes. The grids were well rinsed in PBS, fixed using 1% gluteraldehyde for 5mins, rinsed again, dried and heavy metal contrasted using 2% aqueous uranyl acetate and lead citrate.
Statistical analysis
Data were statistically analyzed using MedCalc version 14. D’Agostino-Pearson test was used to test normal distribution. The results were analyzed by Mann-Whitney U test for comparisons between two groups when sample data were not normally distributed, by unpaired Student’s t-test for comparisons between two groups with normal distribution or by one-way ANOVA with Student-Newman-Keuls post-hoc test for pairwise comparisons, between three or more groups. Efficacy experiments were designed for 10 animals in control and treated groups, to allow for 82% power to detect a difference of 20% between the two groups at the 0.05 statistical significance level, but the actual size of groups differed because of mortality or availability of knockout mice. Data are presented as mean ± SEM, and were considered statistically significant at P < 0.05. Statistical analysis of microarray experiments was performed using BRB array tools as previously described by us63. Multiple comparisons were addressed using the FDR method.
Supplementary Material
Acknowledgments
The work was in part supported by US National Institute of Health (NIH) grants R01HL095397, R01HL127349 (N.K.), R01HL1109233, R01HL125250 (E.L.H.), RO1DK65055 (A.C.B.), RO1DK52798 (T.S.S.), Flight Attendant Medical Research Institute (P.J.L.), American Lung Association Award RT 350419, Marie Sklodowska/Curie, ERS/EU-RESPIRE 2-8860-2015 grant (A.T.). American Lung Association Award RG-415350 (A.S.), Pulmonary Fibrosis Foundation (PPF) Albert Rose Established Investigator Award 415245 (G.Y.)
A.T., G.Y., and N.K., are inventors on a pending patent on use of thyroid hormone as an antifibrotic agent entitled: “Novel Methods of Treating or Preventing Fibrotic Lung Diseases” - OCR 6368 - 047162-7029P1 (00219). N.K., consulted Biogen Idec, Boehringer Ingelheim, Numedii, MMI, and Pliant and has an ongoing collaboration with MiRagen, all outside the submitted work. E.L.H., consulted for Boehringer Ingelheim and held grant funding from Sanofi and Promedior, all outside the submitted work.
Footnotes
Authors contributions
G.Y., and A.T., performed most of the experiments with the assistance of R.W., J.D.H.-M., J.P.W.d.C., R.A.e.D., F.A., G.D., NA., T.W., J.D., Y.G., R.D., G.H.I., and A.C.B.,. A.S., and P.M., performed the mitochondrial function experiments. M.G., and X.L., performed the electron microscopy experiments and analysis. G.Y., A.T., and N.K performed the histologic analyses with R.J.H., T.S.S., G.H.I., assisted on the GC-1 animal experiments. G.Y., A.T., and N.K., conceived the project, designed the experiments and analyzed and interpreted the results. A.T., G.Y., and N.K., drafted and revised the manuscript. E.L.H., P.J.L., and A.C.B., interpreted results and revised the manuscript.
Competing financial interests
All other authors declare no additional competing interests.
Data Availability
All high throughput data mentioned in the paper are publicly available (gene expression omnibus (GEO) GSE47460 and http://www.lung-genomics.org/). All other data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Travis WD, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American journal of respiratory and critical care medicine. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology. 2016;21:427–437. doi: 10.1111/resp.12683. [DOI] [PubMed] [Google Scholar]
- 3.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 4.Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. 2015;24:102–114. doi: 10.1183/09059180.00003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kusko RL, et al. Integrated Genomics Reveals Convergent Transcriptomic Networks Underlying Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. American journal of respiratory and critical care medicine. 2016;194:948–960. doi: 10.1164/rccm.201510-2026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandit KV, Milosevic J. MicroRNA regulatory networks in idiopathic pulmonary fibrosis. Biochem Cell Biol. 2015;93:129–137. doi: 10.1139/bcb-2014-0101. [DOI] [PubMed] [Google Scholar]
- 7.Thannickal VJ. Mechanistic links between aging and lung fibrosis. Biogerontology. 2013;14:609–615. doi: 10.1007/s10522-013-9451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest. 2017;127:405–414. doi: 10.1172/JCI87440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryu C, et al. Extracellular Mitochondrial DNA is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. American journal of respiratory and critical care medicine Epub ahead of print. 2017 doi: 10.1164/rccm.201612-2480OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie N, et al. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. American journal of respiratory and critical care medicine. 2015;192:1462–1474. doi: 10.1164/rccm.201504-0780OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bueno M, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer Y, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. American journal of respiratory cell and molecular biology. 2015;52:217–231. doi: 10.1165/rcmb.2013-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christoffolete MA, et al. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. 2004;53:577–584. doi: 10.2337/diabetes.53.3.577. [DOI] [PubMed] [Google Scholar]
- 14.de Jesus LA, et al. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. 2001;108:1379–1385. doi: 10.1172/JCI13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raghu G, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2015;192:e3–19. doi: 10.1164/rccm.201506-1063ST. [DOI] [PubMed] [Google Scholar]
- 16.Lee CG, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Sikhry HE, Alsaleh N, Dakarapu R, Falck JR, Seubert JM. Novel Roles of Epoxyeicosanoids in Regulating Cardiac Mitochondria. PLoS One. 2016;11:e0160380. doi: 10.1371/journal.pone.0160380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorner T, Egerer K, Feist E, Burmester GR. Rheumatoid factor revisited. Current opinion in rheumatology. 2004;16:246–253. doi: 10.1097/00002281-200405000-00013. [DOI] [PubMed] [Google Scholar]
- 19.Van Beeren HC, et al. Dronerarone acts as a selective inhibitor of 3,5,3′-triiodothyronine binding to thyroid hormone receptor-alpha1: in vitro and in vivo evidence. Endocrinology. 2003;144:552–558. doi: 10.1210/en.2002-220604. [DOI] [PubMed] [Google Scholar]
- 20.Chiellini G, et al. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem Biol. 1998;5:299–306. doi: 10.1016/s1074-5521(98)90168-5. [DOI] [PubMed] [Google Scholar]
- 21.Scanlan TS. Sobetirome: a case history of bench-to-clinic drug discovery and development. Heart Fail Rev. 2010;15:177–182. doi: 10.1007/s10741-008-9122-x. [DOI] [PubMed] [Google Scholar]
- 22.Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation. 2004;110:2869–2874. doi: 10.1161/01.CIR.0000146889.46519.27. [DOI] [PubMed] [Google Scholar]
- 23.Williams GR, Bassett JH. Deiodinases: the balance of thyroid hormone: local control of thyroid hormone action: role of type 2 deiodinase. The Journal of endocrinology. 2011;209:261–272. doi: 10.1530/JOE-10-0448. [DOI] [PubMed] [Google Scholar]
- 24.Barca-Mayo O, et al. Role of type 2 deiodinase in response to acute lung injury (ALI) in mice. Proc Natl Acad Sci U S A. 2011;108:E1321–1329. doi: 10.1073/pnas.1109926108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forini F, et al. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology. 2014;155:4581–4590. doi: 10.1210/en.2014-1106. [DOI] [PubMed] [Google Scholar]
- 26.Lu X, et al. Thyroid hormone inhibits TGFbeta1 induced renal tubular epithelial to mesenchymal transition by increasing miR34a expression. Cellular signalling. 2013;25:1949–1954. doi: 10.1016/j.cellsig.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Fliers E, Bianco AC, Langouche L, Boelen A. Thyroid function in critically ill patients. Lancet Diabetes Endocrinol. 2015;3:816–825. doi: 10.1016/S2213-8587(15)00225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friberg L, Werner S, Eggertsen G, Ahnve S. Rapid down-regulation of thyroid hormones in acute myocardial infarction: is it cardioprotective in patients with angina? Archives of internal medicine. 2002;162:1388–1394. doi: 10.1001/archinte.162.12.1388. [DOI] [PubMed] [Google Scholar]
- 29.Han C, et al. Subclinical Hypothyroidism and Type 2 Diabetes: A Systematic Review and Meta-Analysis. PLoS One. 2015;10:e0135233. doi: 10.1371/journal.pone.0135233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oldham JM, et al. Thyroid Disease Is Prevalent and Predicts Survival in Patients with Idiopathic Pulmonary Fibrosis. Chest. 2015 doi: 10.1378/chest.14-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brent GA, Hershman JM. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration. J Clin Endocrinol Metab. 1986;63:1–8. doi: 10.1210/jcem-63-1-1. [DOI] [PubMed] [Google Scholar]
- 32.Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest. 2016;126:809–820. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porter RK, et al. Indirect measurement of mitochondrial proton leak and its application. Int J Obes Relat Metab Disord. 1999;23(Suppl 6):S12–18. doi: 10.1038/sj.ijo.0800937. [DOI] [PubMed] [Google Scholar]
- 34.Wrutniak-Cabello C, Casas F, Cabello G. Thyroid hormone action in mitochondria. J Mol Endocrinol. 2001;26:67–77. doi: 10.1677/jme.0.0260067. [DOI] [PubMed] [Google Scholar]
- 35.Salvatore D, Simonides WS, Dentice M, Zavacki AM, Larsen PR. Thyroid hormones and skeletal muscle–new insights and potential implications. Nature reviews. Endocrinology. 2014;10:206–214. doi: 10.1038/nrendo.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Austin S, St-Pierre J. PGC1alpha and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. Journal of cell science. 2012;125:4963–4971. doi: 10.1242/jcs.113662. [DOI] [PubMed] [Google Scholar]
- 37.Safdar A, et al. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. The Journal of biological chemistry. 2011;286:10605–10617. doi: 10.1074/jbc.M110.211466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Araya J, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L56–69. doi: 10.1152/ajplung.00213.2012. [DOI] [PubMed] [Google Scholar]
- 39.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oral O, et al. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis. 2012;17:810–820. doi: 10.1007/s10495-012-0735-0. [DOI] [PubMed] [Google Scholar]
- 41.Arena G, et al. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013;20:920–930. doi: 10.1038/cdd.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alavian KN, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verga Falzacappa C, et al. T(3) preserves ovarian granulosa cells from chemotherapy-induced apoptosis. The Journal of endocrinology. 2012;215:281–289. doi: 10.1530/JOE-12-0153. [DOI] [PubMed] [Google Scholar]
- 44.Lesmana R, et al. Thyroid Hormone Stimulation of Autophagy Is Essential for Mitochondrial Biogenesis and Activity in Skeletal Muscle. Endocrinology. 2016;157:23–38. doi: 10.1210/en.2015-1632. [DOI] [PubMed] [Google Scholar]
- 45.Alonso-Merino E, et al. Thyroid hormones inhibit TGF-beta signaling and attenuate fibrotic responses. Proc Natl Acad Sci U S A. 2016;113:E3451–3460. doi: 10.1073/pnas.1506113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel AS, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e41394. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernard K, et al. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. The Journal of biological chemistry. 2017;292:3029–3038. doi: 10.1074/jbc.M116.752261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernard K, et al. Metabolic Reprogramming Is Required for Myofibroblast Contractility and Differentiation. The Journal of biological chemistry. 2015;290:25427–25438. doi: 10.1074/jbc.M115.646984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu L, Larson-Casey JL, Carter AB. Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB J. 2017;31:3072–3083. doi: 10.1096/fj.201601371R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity. 2016;44:582–596. doi: 10.1016/j.immuni.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S, et al. Integrative phenotyping framework (iPF): integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genomics. 2015;16:924. doi: 10.1186/s12864-015-2170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buettner C, Harney JW, Larsen PR. The role of selenocysteine 133 in catalysis by the human type 2 iodothyronine deiodinase. Endocrinology. 2000;141:4606–4612. doi: 10.1210/endo.141.12.7831. [DOI] [PubMed] [Google Scholar]
- 53.Kleiner S, et al. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc Natl Acad Sci U S A. 2012;109:9635–9640. doi: 10.1073/pnas.1207287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montgomery RL, et al. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol Med. 2014;6:1347–1356. doi: 10.15252/emmm.201303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tzouvelekis A, et al. SH2 Domain-Containing Phosphatase-2 Is a Novel Antifibrotic Regulator in Pulmonary Fibrosis. American journal of respiratory and critical care medicine. 2017;195:500–514. doi: 10.1164/rccm.201602-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhargava M, et al. Triiodo-L-thyronine rapidly stimulates alveolar fluid clearance in normal and hyperoxia-injured lungs. American journal of respiratory and critical care medicine. 2008;178:506–512. doi: 10.1164/rccm.200709-1429OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myllarniemi M, Kaarteenaho R. Pharmacological treatment of idiopathic pulmonary fibrosis - preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine. European clinical respiratory journal. 2015;2 doi: 10.3402/ecrj.v2.26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiellini G, Nguyen NH, Yoshihara HA, Scanlan TS. Improved synthesis of the iodine-free thyromimetic GC-1. Bioorg Med Chem Lett. 2000;10:2607–2611. doi: 10.1016/s0960-894x(00)00531-x. [DOI] [PubMed] [Google Scholar]
- 59.Tzouvelekis A, et al. SH2 Domain-containing Phosphatase-SHP-2 is a Novel Anti-fibrotic Regulator in Pulmonary Fibrosis. American journal of respiratory and critical care medicine. 2016 doi: 10.1164/rccm.201602-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G, et al. Matrix metalloproteinase-19 is a key regulator of lung fibrosis in mice and humans. American journal of respiratory and critical care medicine. 2012;186:752–762. doi: 10.1164/rccm.201202-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gereke M, et al. Flow cytometric isolation of primary murine type II alveolar epithelial cells for functional and molecular studies. Journal of visualized experiments : JoVE. 2012 doi: 10.3791/4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mannam P, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L604–619. doi: 10.1152/ajplung.00272.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Herazo-Maya JD, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5:205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.