

Abstract
Background
A fraction of coagulation factor VII circulates as an active protease (FVIIa). FVIIa also circulates as an inactivated complex with antithrombin (FVIIa-AT).
Objective
Evaluate associations of FVIIa and FVIIa-AT with genome-wide single nucleotide polymorphisms (SNPs) and incident coronary heart disease, ischemic stroke, and mortality.
Patients/Methods
We measured FVIIa and FVIIa-AT in 3,486 Cardiovascular Health Study (CHS) participants. We performed a genome-wide association scan for FVIIa and FVIIa-AT in European-Americans (n=2,410) and examined associations of FVII phenotypes with incident cardiovascular disease.
Results
In European-Americans, the most significant SNP for FVIIa and FVIIa-AT was rs1755685 in the F7 promoter region on chromosome 13 (FVIIa: β= −25.9 mU/mL per minor allele; FVIIa-AT: β= −26.6 pM per minor allele). Phenotypes were also associated with rs867186 located in PROCR on chromosome 20 (FVIIa: β= 7.8 mU/mL per minor allele; FVIIa-AT: β= 9.9 per minor allele). Adjusted for risk factors, a 1-standard deviation higher FVIIa was associated with increased ischemic stroke risk (hazard ratio (HR) = 1.12; 95% confidence interval (CI): 1.01, 1.23). Higher FVIIa-AT was associated with mortality from all-causes (HR: 1.08, 95% CI: 1.03, 1.12). Among European-American CHS participants the rs1755685 minor allele was associated with lower ischemic stroke (HR=0.69, 95% CI: 0.54, 0.88), but this association was not replicated in a larger multi-cohort analysis.
Conclusions
Results support the importance of the F7 and PROCR loci on variation in circulating FVIIa and FVIIa-AT. Findings suggest FVIIa is a risk factor for ischemic stroke in older adults while higher FVIIa-AT may reflect mortality risk.
Keywords: antithrombin, cardiovascular disease, coagulation factor VII, epidemiology, single nucleotide polymorphisms
Introduction
Coagulation factor VII (FVII) is a vitamin K-dependent zymogen cleaved to the activated serine protease FVIIa during hemostatic activation. A fraction (~1-2%) of the total FVII concentration circulates as FVIIa in human plasma and requires tissue factor (TF) for proteolytic activity [1]. FVIIa-TF initiates the extrinsic coagulation pathway by activating coagulation factors IX and X leading to thrombin generation and fibrin clot formation.
The FVIIa-TF complex is immediately inactivated by tissue factor pathway inhibitor- (TFPI) and antithrombin- (AT) dependent mechanisms. Inhibition by TFPI involves formation of a TF-FVIIa-TFPI-FXa complex that remains bound to the cell surface and does not dissociate into circulating blood [2]. AT binding to TF-FVIIa causes FVIIa dissociation and FVIIa-AT complex formation. This complex is released into circulation and levels are strongly related to FVIIa concentrations [3]. Emerging evidence from clinical studies have suggested FVIIa-AT as a potential biomarker of hemostatic activation [3, 4].
In epidemiological studies, evaluation of FVII has predominantly relied on FVII clotting activity (FVIIc) measurements, which are influenced by levels of FVIIa and FVII antigen. The Northwick Park Heart Study implicated FVIIc as a risk factor for fatal myocardial infarction (MI) [5]. Prospective associations of FVIIc with coronary heart disease (CHD) or stroke were inconsistent in subsequent population-based studies [6–11]. Inter-individual differences in circulating FVIIa and FVIIa-AT levels are under-evaluated in prospective studies and may be important for thrombotic risk [3].
Several common single nucleotide polymorphisms (SNPs) in the F7 gene are associated with FVII antigen and activity levels [12, 13]. Well-studied F7 polymorphisms, including the −323 decanucleotide insertion (−323 ins10) (rs5742910) and R353Q mutation (rs6046) were shown to be important for circulating FVIIa concentrations [14, 15]. A common polymorphism in the protein C receptor gene (PROCR) (rs867186), encoding a serine to glycine substitution (Ser219Gly) in endothelial cell protein C receptor (EPCR), was associated with higher FVIIa levels [16] and with increased risk of thrombosis [17]. Whether polymorphisms associated with FVIIa influence cardiovascular disease (CVD) risk remains uncertain [10, 18–22].
To date, no epidemiological studies have evaluated genome-wide SNP associations with FVIIa or FVIIa-AT, and very few prospective studies have assessed these coagulation factors’ relationships with incident CVD. We evaluated associations of circulating FVIIa and FVIIa-AT with CVD risk factors, genome-wide SNPs, incident CHD, ischemic stroke, and mortality in older adults of the Cardiovascular Health Study (CHS).
Methods
Study Population
The CHS is a prospective population-based cohort study of risk factors for CHD and stroke in adults aged 65-years and older [23]. The original cohort of 5,201 men and women were recruited in 1989-1990 from 4 U.S. field centers: Forsyth County, NC; Sacramento County, CA; Washington County, MD; and Pittsburgh, PA. In 1992-93 (CHS Year 5), an additional cohort of 687 primarily African-American (AA) participants were recruited. Participants answered standardized questionnaires assessing CVD risk factors, lifestyle, and medical histories [24]. The standardized clinical examination included fasting blood collection, resting electrocardiograms (ECGs), duplex carotid ultrasonography ultrasound [25], and echocardiographic examinations [26].
Between 1990 and 1999, cohort follow-up occurred semi-annually, alternating between clinical examinations and telephone contacts. Since 1999, potential events were identified by two phone calls per year to participants. Relatives were asked to report all hospitalizations to the CHS Field Center. All subjects gave written informed consent for participation in the study and all procedures were conducted under institutionally approved protocols at each center.
Laboratory Methods
Citrated plasma from the 1992-93 CHS examination was used for evaluation of FVIIa and FVIIa-AT (n=3,486). Technicians who completed centralized training programs collected blood using standardized protocols [27]. Plasma was processed and aliquoted at room temperature to limit the cold-activation of FVII [28] using standardized times for plasma processing (≤ 15 minutes) and aliquot storage at −70°C (≤ 10 minutes) [27].
FVIIa was measured using the Staclot VIIa-rTF assay run on a Stago STA-R Evolution according to the manufacturer’s instructions (Diagnostica Stago, Inc., Mount Olive, NJ); inter-assay coefficient of variation (CV) 8.7%. The assay used recombinant soluble tissue factor (rsTF), phospholipids, and Ca2+ to initiate coagulation; rsTF retained FVIIa cofactor function but was deficient in promoting activation of FVII to FVIIa [1, 29].
FVIIa-AT was measured using the Asserachrom VIIa-AT ELISA (Diagnostica Stago, Inc.). The assay captures FVIIa-AT with a mouse monoclonal anti-human FVIIa antibody and a peroxidase-coupled monoclonal anti-AT antibody. The assay’s lower detection limit is 40 pM. Samples were run in duplicate (CV 8.0%). FVIIa / FVIIa-AT assay standardization and quality control were performed using controls provided with the assays and pooled normal plasma samples (George King Bio-Medical, Inc., Overland Park, KS).
Unless noted otherwise, biomarkers of inflammation and coagulation were measured at the 1992-93 CHS examination as previously described [27, 30, 31]. FVIIc was measured in citrated plasma on a Coag-A-Mate X2 (Organon-Teknika, Durham, NC) (CV 5.9%) [6]. The coagulation inhibitor proteins, AT and TFPI, were measured at the CHS baseline examination (1989-1990) by in-house assays among a randomly selected subgroup (n=400) without CVD [32]. Soluble EPCR (sEPCR) was measured among a subset of participants (n=577) at baseline using an in-house ELISA (CV 3.1%) [33].
Genotyping
Genotyping was performed using the Illumina 370CNV platform on 3,208 European-American (EA) participants at baseline (1989-90) who had available DNA samples and provided informed consent for participation in DNA studies. Genotype data for 330,189 SNPs were used to impute to the 2.5 million SNPs from HapMap’s Phase 2 Centre d’Etude du Polymorphisme Humain (CEU) reference samples using MaCH [34]. SNPs were not included in the imputation target panel when R2 values were < 3, the minor allele frequency (MAF) was <0.5%, the missing rate across subjects >5%, or when the respective genotype distribution significantly departed from expected Hardy-Weinberg equilibrium proportions (p< 1.0×10−5).
Event Ascertainment
The primary clinical endpoints for this study were incident CVD events and mortality. Events included adjudicated fatal or non-fatal MI, CHD (defined as MI, coronary artery angioplasty or bypass grafting, or angina), and ischemic stroke. Mortality included from all-causes or from CVD, defined as adjudicated fatal events due to definite or probable MI, CHD, or stroke. Incident events were reported at clinical visits or during semiannual telephone contacts by participants or proxies. Events were adjudicated by a physician review panel using hospital and out-patient medical records, including when available copies of ECGs, CT scans, MRI scans, death certificates, autopsy reports, and coroner reports [35–37]. Censoring dates were defined as death, loss to follow-up, study drop out, or event date, occurring through December 31, 2011. Hypertension was defined as systolic blood pressure >140 mmHg, diastolic blood pressure >90 mmHg, or current use of antihypertensive medication. Diabetes was defined as the use of insulin, oral hypoglycemic medications, or a fasting glucose level ≥126 mg/dL.
Statistical Analysis
Cross-sectional associations of FVIIa and FVIIa-AT with demographic variables and cardiovascular-related biomarkers were evaluated using linear regression models adjusted for age, sex, and race; models stratified by age, sex, or race were adjusted only for the 2 remaining variables. Models evaluating diabetes were adjusted for age, sex, race, smoking, BMI, total-cholesterol, HDL-cholesterol, and triglycerides. Models evaluating carotid artery intima medial thickness (IMT) included these variables plus hypertension and systolic blood pressure. FVII phenotypes were modeled as outcome variables with independent variables modeled per standard deviation (SD) increment higher. Triglycerides, C-reactive protein (CRP), and IMT phenotypes had non-normal distributions and were natural log-transformed. To correct for multiple testing, results were considered significant if p<0.003 (using a Bonferonni correction for 20 tests). Pearson correlation was used to estimate associations among FVII phenotypes, AT, and TFPI.
We used linear regression models implemented in Mach2qtl [34] to test for genetic associations between FVII phenotypes and imputed SNPs (n=2,410 participants with genotype and phenotype measures), with imputation data analyzed as continuous dosage values (i.e., expected number of copies of the minor alleles). We adjusted for age, sex, and the first two principal components (PCs) estimated using Eigensoft to control for global ancestry [38]. The threshold for genome-wide statistical significance was set at 5.0×10−8. To assess for heterogeneity by disease status, we ran a sensitivity analysis evaluating SNP relationships with FVII phenotypes stratified by CVD status (i.e., no prevalent CVD, prevalent CHD, and prevalent stroke).
Targeted conditional analyses in the F7 and PROCR gene regions were performed to assess whether multiple SNPs within these regions independently contributed to the signals. We also assessed whether SNP associations were mediated by FVIIc, FVIIa, or sEPCR levels, respectively, by including these phenotypes as covariates in linear regression models.
Relationships between FVII phenotypes and incident CVD events and mortality were evaluated using Cox proportional hazards ratios, modeled per SD increment higher FVII or by tertiles of the distributions. To allow comparisons among the different FVII phenotypes, only participants with measures of FVIIa, FVIIa-AT, and FVIIc were included in the analyses (n=3,427). We also performed a sensitivity analyses that included participants with missing information for any one of the FVII phenotypes.
Participants with adjudicated CVD at the 1992-93 examination were excluded from analyses for the relevant outcome (n=382 participants with MI; n=762 with CHD; and, n=181 with stroke). Incident hemorrhagic strokes (n=67) and strokes of unknown subtype (n=35) were excluded from stroke analyses. Models were adjusted for age, sex, and race (demographic model), with subsequent adjustment for LDL-cholesterol, smoking status, systolic blood pressure, hypertension status, and diabetes status (risk factor model). Mortality analyses included adjustment for prevalent MI, CHD, and stroke at the 1992-93 examination. We evaluated genotype associations with incident ischemic stroke risk by including the most significant F7 or PROCR SNPs in risk factor-adjusted models.
Genetic Analyses in CHARGE
A candidate SNP look-up for rs1755685 with stroke risk was assessed within 18 community-based prospective cohort studies, including CHS, participating in the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium [39, 40]. Analyses included 75,508 white participants free of stroke at baseline; expert committees in the participating studies performed stroke adjudication and classification. We evaluated associations with ischemic stroke type and cardioembolic and non-cardioembolic ischemic stroke subtypes using Cox regression models with adjustment for age, sex, principal components accounting for population stratification, and study site [40]. We had >80% power to detect an ischemic stroke HR of approximately 1.11 per copy of the rs1755685 minor allele with an alpha of 0.05.
Results
Cross-sectional associations of FVIIa and FVIIa-AT
Characteristics of CHS participants with FVII measurements at the 1992-93 CHS clinical examination are presented in Table S1. Mean FVIIa (SD) was 52.7 mU/mL (25.2 mU/mL); mean FVIIa-AT (SD) was 141.1 pM (56.1 pM). Both phenotypes were approximately normally distributed (Fig S1). FVIIa was moderately correlated with FVIIa-AT (r2=0.44) and FVIIc (r2=0.44), and FVII-AT was less so with FVIIc (r2=0.26). AT weakly correlated with FVIIc (r2=0.06) and FVIIa-AT (r2=0.03) but not FVIIa (r2=0.01). FVII phenotypes were not correlated with TFPI (Table S2).
Table 1 presents age, sex, and race-adjusted associations of FVIIa and FVIIa-AT with CVD risk factors and inflammation biomarkers. The correlates of FVIIc in CHS were previously published [41]. Compared with men, women had significantly higher FVIIa (11.8 mU/mL higher) and FVIIa-AT (25.1 pM higher) levels. Compared with AAs, EAs had significantly higher FVIIa-AT (19.4 pM higher). Higher FVIIa was associated with moderately higher systolic blood pressure, total and HDL-cholesterol, triglycerides, CRP, and platelet count. Higher FVIIa-AT was strongly associated with current smoking and moderately associated with higher systolic blood pressure, hypertension, total and HDL-cholesterol, CRP, common carotid IMT, and lower BMI (Table 2).
Table 1.
Associations of Coagulation Factor VII Phenotypes with Cardiovascular Disease Risk Factors
| Variable (standard deviation) | FVIIa [mU/mL] β (SE) | P-Value | FVIIa-AT [pM] β (SE) | P-Value |
|---|---|---|---|---|
| Demographic | ||||
| Age (5.2 years) | −0.53 (0.42) | 0.21 | 0.79 (0.94) | 0.40 |
| Female sex | 11.8 (0.85) | <0.0001 | 25.1 (1.9) | <0.0001 |
| African-American race | −4.3 (2.1) | 0.04 | −19.4 (4.7) | <0.0001 |
| Former smoking | 2.1 (0.92) | 0.02 | 3.0 (2.1) | 0.14 |
| Current smoking | 3.4 (1.6) | 0.03 | 11.6 (3.5) | 0.0008 |
| BMI (4.5 kg/m2) | −1.0 (0.42) | 0.02 | −3.5 (0.94) | 0.0002 |
| Hypertension | 1.1 (0.46) | 0.02 | 3.7 (1.0) | 0.0003 |
| Systolic blood pressure (21 mmHg) | 1.8 (0.43) | <0.0001 | 4.8 (0.96) | <0.0001 |
| Diabetes* | −0.93 (1.2) | 0.44 | 4.2 (2.8) | 0.12 |
| Lipids | ||||
| Total-cholesterol (39 mg/dL) | 2.9 (0.43) | <0.0001 | 3.7 (0.97) | 0.0002 |
| HDL-cholesterol (14 mg/dL) | 3.9 (0.45) | <0.0001 | 6.4 (1.0) | <0.0001 |
| LDL-cholesterol (34 mg/dL) | 0.70 (0.42) | 0.10 | 0.74 (0.95) | 0.43 |
| Triglycerides (0.49) | 2.1 (0.42) | <0.0001 | 2.1 (0.94) | 0.03 |
| Inflammation and Coagulation | ||||
| CRP (1.13) | 2.0 (0.42) | <0.0001 | 3.0 (0.94) | 0.002 |
| Platelet count (69 × 103/mm3) | 1.6 (0.44) | 0.0003 | 1.9 (0.97) | 0.05 |
| Fibrinogen (68.6 mg/dL) | −0.12 (0.42) | 0.78 | 1.4 (0.93) | 0.14 |
| FVIIc (26.8 %) | 17.5 (0.34) | <0.0001 | 29.9 (0.87) | <0.0001 |
| FVIIa (25.2 mU/mL) | – | – | 37.3 (0.71) | <0.0001 |
| FVII-AT (56.1 pM) | 17.2 (0.31) | <0.0001 | – | – |
| Subclinical Atherosclerosis | ||||
| Common carotid intima medial thickness† (0.37) | 0.59 (0.47) | 0.21 | 2.9 (1.07) | 0.007 |
| Internal carotid intima medial thickness† (0.20) | −0.21 (0.45) | 0.64 | 2.2 (1.0) | 0.03 |
Associations between FVII phenotypes and biomarkers were estimated in separate linear regression models with FVII as the outcome variable, adjusted for age, sex, and race. Independent variables were modeled per 1-SD increase (values shown in parenthesis); β-coefficients (standard errors) and p-values are presented. Triglycerides, C-reactive protein (CRP), and intima medial thickness (IMT) phenotypes were natural log-transformed. In models of age, sex, and race, the estimates were adjusted only for the 2 remaining variables.
Diabetes models were adjusted for age, sex, race, smoking, BMI, total-cholesterol, HDL-cholesterol, and triglycerides.
IMT models were adjusted for age, sex, race, smoking, BMI, hypertension, systolic blood pressure, total-cholesterol, HDL-cholesterol, and triglycerides.
Table 2.
Top Genome-wide Association Results for FVIIa and FVIIa-AT in European-American Participants of the Cardiovascular Health Study
| Phenotype | Chromosome | Number of SNPs with P<5×10−8 | Candidate genes | Top SNP in region | Closest gene | Position | A1 | A2 | MAF | Beta (SE) | P-Value Top SNP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FVIIa [mU/mL] | |||||||||||
| 13 | 35 | F7, F10, MCF2L | rs1755685 | F7 | 112805193 | C | A | 0.12 | −25.9 (1.2) | 1.2×10−112 | |
| 20 | 29 | PROCR, EDEM2, TRPC4AP, MYH7B | rs867186 | PROCR | 33228215 | A | G | 0.10 | 7.8 (1.1) | 6.6×10−12 | |
| FVIIa-AT [pM] | |||||||||||
| 13 | 25 | F7, F10, MCF2L, | rs1755685 | F7 | 112805193 | C | A | 0.12 | −26.6 (1.7) | 6.2×10−55 | |
| 20 | 9 | PROCR, EDEM2, | rs867186 | PROCR | 33228215 | A | G | 0.10 | 9.9 (1.7) | 4.7×10−9 |
Positions are for build 36 (hg18). Beta values are presented for A2. SNPs were coded on the forward strand of the genome. A1, allele 1 (major allele); A2, allele 2 (minor allele) ; EDEM2, endoplasmic reticulum degradation-enhancing alpha-mannosidase-like 2; F7, coagulation factor 7; F10, coagulation factor 10; MAF, minor allele frequency; MCF2L, MCF.2 cell line-derived transforming sequence-like; MYH7B, myosin heavy chain 7B; PROCR, protein C receptor; TRPC4AP, transient receptor potential cation channel, subfamily C, member 4 associated protein.
Genome-wide associations of FVIIa and FVIIa-AT in European-Americans
In a GWAS of FVIIa performed among 2,391 EAs, 2 regions, located on chromosomes 13 and 20, reached genome-wide significance (p<5×10−8) (Table 2). On chromosome 13, 35 SNPs located within the F7/F10 and MCF2L (MCF.2 cell line derived transforming sequence-like) loci exceeded significance (Table 2; Figure 1A). The most significant genome-wide SNP was rs1755685 (p=1.2×10−122) located within the 5′ flanking/promoter region of F7. Each copy of the rs1755685 minor allele (MAF=0.12) was associated with 25.9 mU/mL (standard error (SE) = 1.2 mU/mL) lower FVIIa.
Figure 1.
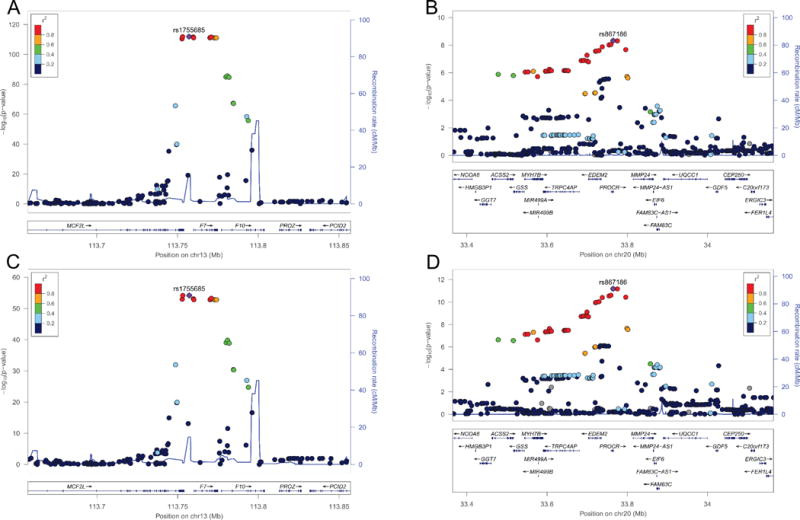
FVIIa and FVIIa-AT regional association plots for chromosome 13 and chromosome 20 in European-Americans. FVIIa (A & B) and FVIIa-AT (C & D) regional association plots for the chromosome 13q34 (A & C) and chromosome 20q11.2 (B & D) loci are presented. The colors of each single nucleotide polymorphism (SNP) indicate pairwise linkage disequilibrium (LD) relative to the lead SNP in the region. SNPs with missing LD information are shown in gray. LD was based on r-squared values calculated using 1000 Genomes (March 2012 EUR) LD for European-Americans. Plots were generated using LocusZoom [60].
On chromosome 20, 29 SNPs attained genome-wide significance (Table 2; Figure 1B). The most significant SNP was rs867186 (p=6.6×10−12) located in exon 4 of the PROCR gene. Each minor allele copy of rs867186 (MAF=0.096) was associated with 7.8 mU/mL (SE= 1.1 mU/mL) higher FVIIa. SNPs located near 3 additional genes on chromosome 20 also reached genome-wide significance: EDEM2 (endoplasmic reticulum degradation-enhancing alpha-mannosidase-like 2); MYH7B (myosin heavy chain 7B, cardiac muscle, beta); and, TRPC4AP (transient receptor potential cation channel, subfamily C, member 4 associated protein). These SNPs were in strong linkage disequilibrium (LD) with rs867186 [13].
In the FVIIa-AT GWAS (n=2,410 EAs), signals on chromosomes 13 and 20 again exceeded genome-wide significance (Table 2). The strongest signal was also rs1755685 (β= −26.6 pM, SE=1.7; p=6.2×10−55) in the F7 locus. The signal on chromosome 13 encompassed 24 additional SNPs located within the F7/F10 and MCF2L loci (Figure 1C). On chromosome 20, 9 SNPs located within the PROCR and EDEM2 genes exceeded significance; the most significant association was again rs867186 (β= 9.9 pM, SE=1.7, p=4.7×10−9) in PROCR (Figure 1D). SNP associations with both FVII phenotypes were essentially the same when stratified by prevalent CVD status.
Genome-wide significance for associations between FVIIc and variants in the F7 locus (lead variant rs488703) were previously reported by the CHARGE Consortium (which includes CHS) [13]. We assessed associations of rs1755685 and rs867186 with FVIIc. Each copy of the rs1755685 minor allele was associated with an estimated 20.9% (SE= 1.3%; p=6.7 ×10−56) lower FVIIc, and each minor allele copy of rs867186 was associated with an estimated 9.9% (SE= 1.3%; p=4.9 ×10−14) higher FVIIc.
Conditional genetic analysis
We performed an iterative, forward-selection analysis that conditioned on the top F7 (rs1755685) and PROCR (rs867186) SNPs. In FVIIa analyses, no SNPs reached genome-wide significance on chromosome 13 after conditioning on rs1755685 (Figure 2A), although nominal evidence for association with rs555212 remained (p=1.7×10−5). After conditioning on rs867186, no additional SNPs on chromosome 20 remained significantly associated with FVIIa (Figure 2B). Together, these 2 SNPs explained 24.2% of the variance in FVIIa levels (rs1755685, r2=0.220; rs867186, r2=0.022). Similarly, no additional SNPs remained significantly associated with FVIIa-AT after conditioning on rs1755685 or rs867186 (Figure 2C & D). Rs1755685 explained 7.2% of the variance in FVIIa-AT and rs867186 explained 1.3% of the variance.
Figure 2.
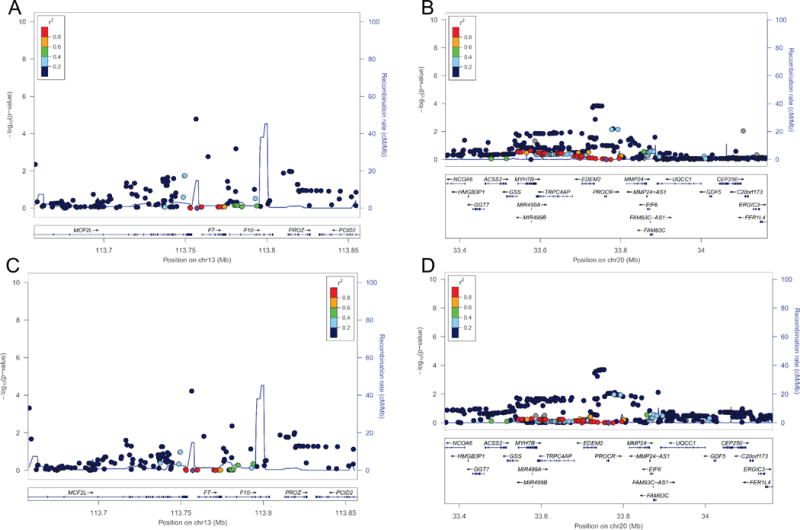
Conditional analysis regional association plots for chromosomes 13q34 and 20q11.2 in European-Americans. Shown are the FVIIa (A & B) and FVIIa-AT (C & D) regional association plots for the chromosome 13q34 locus adjusted by rs1755685 (A & C) and the chromosome 20q11.2 locus adjusted by rs867186 (B & D). The threshold used for defining statistical significance was set at p<5.0×10−8.
We evaluated whether relationships between rs1755685 or rs867186 and FVIIa were mediated by FVIIc or sEPCR levels, respectively, by adding these phenotypes as covariates in linear regression models that included age, sex, clinical site, and rs1755685 or rs867186. With FVIIc in the model the association of rs1755685 with FVIIa was moderately attenuated (β= −15.8 mU/mL; p=8.5×10−59). In participants with sEPCR measured (n=577), adjustment completely attenuated the association of rs867186 with FVIIa (without sEPCR adjustment: β=8.7 mU/mL, p=5.0×10−5; with sEPCR adjustment: β=1.3 mU/mL, p=0.54). We also assessed whether associations of rs1755685 with FVIIa-AT were mediated by FVIIa. In models adjusted for age, sex, clinical site, and FVIIa, the association between rs1755685 and FVIIa-AT was attenuated (β=3.7 (SE=2.2), p=0.10).
Relationships of FVII phenotypes with incident CVD events and mortality
Among the 3,427 CHS participants with measures of FVIIa, FVIIa-AT, and FVIIc at the 1992-93 CHS clinical examination, incident events comprised 576 MIs, 945 CHD events, and 470 ischemic strokes over a maximum possible 19.5-years of follow-up (median 11.5 years). Of the 2,035 deaths over this period from all-causes, 1,027 were attributed to cardiovascular causes. The relationships between FVII phenotypes, modeled per SD units, with events are shown in Table 3. Table S3 presents associations with CVD events stratified by FVII tertiles.
Table 3.
Hazards Ratios for Associations between 1-SD Unit Increases in FVII Phenotypes and Incident Cardiovascular Events and Mortality
| MI (n=576) HR (95% CI) | CHD (n=945) HR (95% CI) | Ischemic Stroke (n=470) HR (95% CI) | CVD-Mortality (n=1,027) HR (95% CI) | All Cause-Mortality (n=2,035) HR (95% CI) | |
|---|---|---|---|---|---|
| FVIIa | |||||
| Demographic Model | 0.95 (0.87, 1.04) | 1.02 (0.95, 1.09) | 1.12 (1.02, 1.24) | 0.97 (0.91, 1.04) | 1.01 (0.97, 1.05) |
| Risk Factor Model | 0.96 (0.88, 1.05) | 1.01 (0.94, 1.08) | 1.12 (1.01, 1.23) | 0.99 (0.92, 1.06) | 1.02 (0.98, 1.06) |
| FVIIa-AT | |||||
| Demographic Model | 1.03 (0.94, 1.12) | 1.04 (0.97, 1.11) | 1.10 (1.01, 1.20) | 1.09 (1.02, 1.17) | 1.08 (1.04, 1.12) |
| Risk Factor Model | 1.03 (0.94, 1.13 | 1.02 (0.95, 1.10) | 1.09 (0.99, 1.19) | 1.06 (0.99, 1.14) | 1.08 (1.03, 1.12) |
| FVIIc | |||||
| Demographic Model | 1.01 (0.92, 1.11) | 1.04 (0.96, 1.12) | 1.04 (0.94, 1.16) | 0.94 (0.87, 1.01) | 0.97 (0.92. 1.01) |
| Risk Factor Model | 0.97 (0.87, 1.07) | 1.00 (0.92, 1.08) | 1.00 (0.89, 1.12) | 0.92 (0.85, 0.99) | 0.96 (0.92, 1.01) |
Analyses using Cox proportional hazards ratios and 95% confidence intervals (CI) modeled per 1-SD higher FVII phenotype value among participants with each of the FVII phenotypes measured (n=3,427).
Demographic Model: Age, sex, and race.
Risk Factor Model: Age, sex, race, smoking status, diabetes status, hypertension, systolic blood pressure, and LDL-cholesterol; mortality models also included adjustment for prevalent MI, CHD, and stroke at the 1992-93 CHS examination.
CHD, coronary heart disease; CI, confidence interval; HR, hazard ratio; MI, myocardial infarction.
In demographic-adjusted models, higher FVIIa and FVIIa-AT were each associated with increased ischemic stroke risk. After adjustment for CVD risk factors, FVIIa remained associated with ischemic stroke (hazard ratio (HR): 1.12, 95% confidence interval (CI): 1.01, 1.23) and results for FVIIa-AT were slightly attenuated (HR: 1.09, 95% CI: 0.99, 1.19). FVIIa-AT was associated with cardiovascular- and all-cause-mortality in demographic-adjusted models. After adjustment for other risk factors, evidence for association with CVD-mortality was no longer observed while results for all-cause mortality were essentially unchanged (HR: 1.08, 95% CI: 1.03, 1.12). FVIIc was not associated with incident events or mortality (Table 3). In sensitivity analyses that evaluated participants with measurements for any one of the FVII phenotypes (n=3,486 for FVIIa and FVIIa-AT and n=4,669 for FVIIc) results were similar.
SNP associations with ischemic stroke
Among EA CHS participants (n=2,794), each additional copy of the rs1755685 minor allele was associated with a decrease in the estimated ischemic stroke HR of 0.69 (95% CI: 0.54, 0.88) in risk-factor-adjusted models. With the inclusion of FVIIa in the model (n=2,079), the relationship was attenuated towards the null (HR=0.73, 95% CI: 0.53, 1.02). Relationships were more robust to inclusion of FVIIa-AT in the model in place of FVIIa (HR=0.70, 95% CI: 0.51, 0.96). The rs867186 genotype was not associated with ischemic stroke (HR=0.99, 95% CI: 0.79, 1.23).
We performed a candidate SNP look-up in CHARGE [40]. Among 66,023 white participants with 2,842 ischemic stroke events, rs1755685 was not associated with ischemic stroke type (β= −0.018; SE=0.04; p=0.67) or cardioembolic (n total= 47,075; n event= 537) (β= 0.12; SE=0.09; p=0.19) and non-cardioembolic (n total= 55,070; n event= 1,735) (β= −0.016; SE=0.05; p=0.76) ischemic stroke subtypes.
Discussion
In a prospective cohort of older adults, we identified associations of higher FVIIa with increased risk of incident ischemic stroke and relationships of higher FVIIa-AT with mortality from all-causes. We also confirmed the importance of the F7 and PROCR loci on variation in FVIIa levels.
Associations of FVII with CVD events and mortality
In epidemiological studies, FVII has predominantly been evaluated by FVIIc assays and relationships with incident CHD or ischemic stroke have been inconsistent [5, 6, 8–10, 42, 43]. FVIIc assays are influenced by levels of both FVII antigen and FVIIa, as highlighted in our correlation analysis whereby FVIIa explained approximately half of the variation in FVIIc. FVIIc assay sensitivity to FVIIa is variable, influenced by the source of thromboplastin and presence or absence of plasma protein C [44].
Our study is among only a few to evaluate circulating FVIIa or FVIIa-AT with incident CVD events [45, 46]. We did not observe relationships of FVIIa or FVIIa-AT with MI or CHD, consistent with several reports [6, 9, 19, 47], but not all [21, 48, 49], for FVIIc or F7 SNPs. In contrast, we identified associations of higher FVIIa, but not FVIIc, with ischemic stroke after adjustment for risk factors. These results suggest FVIIa may be a more sensitive risk marker of ischemic stroke compared with FVIIc; the FVIIa and FVIIa-AT HRs and CIs for ischemic stroke were similar indicating similar sensitivity of the assays. An earlier study in CHS [10] reported higher FVIIc measured at the baseline examination (1989-90) was associated with increased ischemic stroke risk. Differences in follow-up times, or fewer FVIIc measurements available at the 1992-1993 CHS examination, may account for these differences.
The minor allele of rs1755685, associated with lower FVII levels, was associated with lower ischemic stroke risk in our study, and these associations were partly mediated by FVIIa and its correlated phenotype FVIIa-AT. Previous results in CHS demonstrated associations of rs6046, in strong LD with rs1755685, with lower ischemic stroke risk, consistent with our results [10]. Our analyses in the CHARGE Consortium, however, showed rs1755685 was not associated with the ischemic stroke type or its subtypes. This suggests relationship between FVIIa and stroke may not be causal or rs1755685 has only a small effect size. Several other studies also reported null associations of F7 polymorphisms with stroke (summarized in Table S4). CHARGE represents several heterogeneous cohorts whose susceptibility towards stroke risk may vary depending on the influence of additional factors, such as age [50]. Collectively, our results, along with prior findings in CHS [10], suggest the importance of FVII as a risk factor for ischemic stroke among older adults.
FVIIa-AT complexes may be a biomarker of activated coagulation [4] and may reflect vascular injury or underlying chronic inflammatory diseases, such as atherosclerosis [51]. Our results identified associations of FVIIa-AT with mortality attributed to all-causes. Findings were consistent with previous studies reporting higher FVIIa-AT levels predicted total- and cardiovascular-mortality in patients with clinically stable CAD [51] but not incident cardiovascular events [46].
Genetic associations of FVIIa and FVIIa-AT
To the best of our knowledge, our study is the first to evaluate genome-wide associations of FVIIa or FVIIa-AT. Our results confirmed the importance of the F7 and PROCR loci on variation in FVII levels [13–16, 52, 53]. The strongest association for both FVIIa and FVIIa-AT was with rs1755685 located within the 5′ flanking / promoter region of F7. These findings are consistent with a previous study reporting independent associations of rs1755685 with FVIIc [20]. Associations of rs1755685 with FVIIa-AT are consistent with the importance of FVIIa levels on FVIIa-AT [3]. After iterative conditional analyses, no additional SNPs in the chromosome 13 F7 locus remained significantly associated with either FVII phenotype.
Several known F7 promoter variants are in strong LD in European-ancestry populations, and whether rs1755685 plays a functional role is unclear. An in vitro site-directed mutagenesis study demonstrated allelic variants in rs1755685 increased F7 gene expression, but these effects were offset by the −323ins10 polymorphisms in LD with rs1755685 [54]. This study suggested −323ins10 was the major haplotype variant responsible for lower FVII levels in a Spanish population [54]. The −323ins10 variant is also in strong LD with the R353Q polymorphisms [12]. Whether the effects of R353Q are due to −323ins10 are controversial [55, 56]. The minor allele of the R353Q variant was associated with statistically significantly lower levels of FVIIa and FVIIa-AT in our study, with effect sizes comparable to rs1755685, but these associations were no longer significant after conditioning on rs1755685. We did not have information for −323ins10.
The rs867186 (Ser219Gly) polymorphism tags a common PROCR haplotype and is associated with higher sEPCR levels [17]. Approximately 70% of the variance in sEPCR levels are explained by the Ser219Gly mutation and increased EPCR shedding has been confirmed in carriers of the Gly allele [16, 57]. Associations of rs867186 with higher FVIIa, FVIIc, FVIIag and other coagulation biomarkers were reported previously [16] and EPCR-dependent endocytosis was proposed as a FVII clearance mechanism [58]. Our results did not indicate a role for rs867186 with ischemic stroke, consistent with previous results in CHS [33].
Physiological correlates of FVIIa and FVIIa-AT
Consistent with known relationships of FVII with hormonal status [59], FVIIa and FVIIa-AT were significantly higher among post-menopausal women than men. Modest relationships between lipids and FVIIa in our study and others [47, 59] suggest plasma lipid levels are more closely related to FVII protein production than activation. Our results showed the strongest correlate of FVIIa-AT was FVIIa, confirming previous findings [3, 51]. Cross-sectional relationships of higher FVIIa-AT with CRP, smoking status, and common carotid IMT are consistent with interactions between inflammatory pathways and TF expression / exposure.
Limitations and conclusions
Our study was limited by sample availability which precluded FVII measurements in the entire cohort and restricted power. Our genetic analyses were limited to EAs and a previous study demonstrated lower LD and greater haplotype diversity for FVII-associated SNPs among AAs compared with EAs [52]. We only evaluated common SNPs; rare and low-frequency variants were also associated with FVII [53]. FVII measurements were performed among a cohort of older adults and relationships of FVII phenotypes, or associated SNPs, with stroke may differ among younger populations. Although these FVII assays may be useful in evaluating hemostatic activation, their clinical utility is currently unclear.
In conclusion, our results support the importance of F7 and PROCR SNPs on variation in circulating FVIIa and FVIIa-AT. Findings suggest higher FVIIa is a risk factor for ischemic stroke in older adults while FVIIa-AT may reflect mortality risk.
Supplementary Material
Table S1. Characteristics of Cardiovascular Health Study (CHS) participants with FVII phenotypes measured at the CHS Year 5 examination (1992-1993)
Table S2. Pearson correlation coefficients among coagulation factor VII (FVII) phenotypes and anticoagulant proteins in CHS
Table S3. Cox proportional hazards ratios for associations of coagulation factor VII tertiles with incident cardiovascular disease, ischemic stroke, and mortality
Table S4. Summary of previous studies evaluating relationships of F7 SNPs with stroke risk
Fig. S1. Distributions of activated coagulation factor VII and FVIIa-antithrombin complex in participants of the Cardiovascular Health Study
Essentials.
A fraction of coagulation factor VII circulates in blood as an activated protease (FVIIa).
We evaluated FVIIa and FVIIa-antithrombin (FVIIa-AT) levels in the Cardiovascular Health Study.
Polymorphisms in the F7 and PROCR loci were associated with FVIIa and FVIIa-AT levels.
FVIIa may be an ischemic stroke risk factor in older adults and FVIIa-AT may assess mortality risk.
Acknowledgments
The authors acknowledge Diagnostica Stago, Inc. for proving the FVIIa and FVIIa-AT assays. Diagnostica Stago, Inc. had an opportunity to review the manuscript but did not have any role in the design or conduct of the study, the collection, management, analysis, or interpretation of the data, in preparation of the manuscript, or in the decision to publish.
The authors thank the staff and participants of each of the studies participating in the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium for their important contributions and all members of Neurology Working Group of the CHARGE Consortium.
This study was supported by an R01 (HL71862) from the National Heart, Blood, and Lung Institute (NHLBI) to A. P. Reiner. N C. Olsen was supported by the NHLBI post-doctoral training award 5T32HL007894 and NHLBI K99HL129045. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. G. Chauhan and S. Debette were supported by a grant from the Fondation Leducq and the Agence Nationale de la Recherche (Chaire d’Excellence).
Information about the funding for cohorts included in the CHARGE Consortium are provided in the Supplementary Material.
Footnotes
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Addendum
N. C. Olson, A. P. Reiner, and R. P. Tracy conceived the research study design; G. Chauhan, S. Debette, S. Seshadri, W. T. Longstreth Jr., A. P. Reiner, and R. P. Tracy generated data; N. C. Olson, L. M. Raffield, L. A. Lange, E. M. Lange, G. Chauhan, S. Debette, and S. Seshadri performed statistical analyses; N. C. Olson, L. M. Raffield, L. A. Lange, E. M. Lange, W. T. Longstreth, Jr., G. Chauhan, S. Debette, S. Seshadri, A. P. Reiner, and R. P. Tracy interpreted data; N. C. Olson drafted the manuscript; L. M Raffield., L. A. Lange, E. M. Lange, W. T. Longstreth Jr., A. P. Reiner, and R.P. Tracy critically revised the manuscript.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
References
- 1.Morrissey JH, Macik BG, Neuenschwander PF, Comp PC. Quantitation of activated factor VII levels in plasma using a tissue factor mutant selectively deficient in promoting factor VII activation. Blood. 1993;81:734–44. [PubMed] [Google Scholar]
- 2.Rao LV, Nordfang O, Hoang AD, Pendurthi UR. Mechanism of antithrombin III inhibition of factor VIIa/tissue factor activity on cell surfaces. Comparison with tissue factor pathway inhibitor/factor Xa-induced inhibition of factor VIIa/tissue factor activity. Blood. 1995;85:121–9. [PubMed] [Google Scholar]
- 3.Spiezia L, Rossetto V, Campello E, Gavasso S, Woodhams B, Tormene D, Simioni P. Factor VIIa-antithrombin complexes in patients with arterial and venous thrombosis. Thromb Haemost. 2010;103:1188–92. doi: 10.1160/TH09-08-0606. [DOI] [PubMed] [Google Scholar]
- 4.Spiezia L, Campello E, Valle FD, Woodhams B, Simioni P. Factor VIIa-antithrombin complex: a possible new biomarker for activated coagulation. Clin Chem Lab Med. 2017;55:484–8. doi: 10.1515/cclm-2016-0399. [DOI] [PubMed] [Google Scholar]
- 5.Meade TW, Mellows S, Brozovic M, Miller GJ, Chakrabarti RR, North WR, Haines AP, Stirling Y, Imeson JD, Thompson SG. Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet. 1986;2:533–7. doi: 10.1016/s0140-6736(86)90111-x. [DOI] [PubMed] [Google Scholar]
- 6.Tracy RP, Arnold AM, Ettinger W, Fried L, Meilahn E, Savage P. The relationship of fibrinogen and factors VII and VIII to incident cardiovascular disease and death in the elderly: results from the cardiovascular health study. Arterioscler Thromb Vasc Biol. 1999;19:1776–83. doi: 10.1161/01.atv.19.7.1776. [DOI] [PubMed] [Google Scholar]
- 7.Zakai NA, Katz R, Jenny NS, Psaty BM, Reiner AP, Schwartz SM, Cushman M. Inflammation and hemostasis biomarkers and cardiovascular risk in the elderly: the Cardiovascular Health Study. J Thromb Haemost. 2007;5:1128–35. doi: 10.1111/j.1538-7836.2007.02528.x. [DOI] [PubMed] [Google Scholar]
- 8.Folsom AR, Rosamond WD, Shahar E, Cooper LS, Aleksic N, Nieto FJ, Rasmussen ML, Wu KK. Prospective study of markers of hemostatic function with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Circulation. 1999;100:736–42. doi: 10.1161/01.cir.100.7.736. [DOI] [PubMed] [Google Scholar]
- 9.Folsom AR, Wu KK, Rosamond WD, Sharrett AR, Chambless LE. Prospective study of hemostatic factors and incidence of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 1997;96:1102–8. doi: 10.1161/01.cir.96.4.1102. [DOI] [PubMed] [Google Scholar]
- 10.Zakai NA, Lange L, Longstreth WT, Jr, O’Meara ES, Kelley JL, Fornage M, Nikerson D, Cushman M, Reiner AP. Association of coagulation-related and inflammation-related genes and factor VIIc levels with stroke: the Cardiovascular Health Study. J Thromb Haemost. 2011;9:267–74. doi: 10.1111/j.1538-7836.2010.04149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junker R, Heinrich J, Schulte H, van de Loo J, Assmann G. Coagulation factor VII and the risk of coronary heart disease in healthy men. Arterioscler Thromb Vasc Biol. 1997;17:1539–44. doi: 10.1161/01.atv.17.8.1539. [DOI] [PubMed] [Google Scholar]
- 12.Soria JM, Almasy L, Souto JC, Sabater-Lleal M, Fontcuberta J, Blangero J. The F7 gene and clotting factor VII levels: dissection of a human quantitative trait locus. Hum Biol. 2005;77:561–75. doi: 10.1353/hub.2006.0006. [DOI] [PubMed] [Google Scholar]
- 13.Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, Hayward C, Rudan I, Sabater-Lleal M, Bis JC, de Maat MP, Rumley A, Kong X, Yang Q, Williams FM, Vitart V, Campbell H, Malarstig A, Wiggins KL, Van Duijn CM, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121:1382–92. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moor E, Silveira A, van’t Hooft F, Suontaka AM, Eriksson P, Blomback M, Hamsten A. Coagulation factor VII mass and activity in young men with myocardial infarction at a young age. Role of plasma lipoproteins and factor VII genotype. Arterioscler Thromb Vasc Biol. 1995;15:655–64. doi: 10.1161/01.atv.15.5.655. [DOI] [PubMed] [Google Scholar]
- 15.Bernardi F, Arcieri P, Bertina RM, Chiarotti F, Corral J, Pinotti M, Prydz H, Samama M, Sandset PM, Strom R, Garcia VV, Mariani G. Contribution of factor VII genotype to activated FVII levels. Differences in genotype frequencies between northern and southern European populations. Arterioscler Thromb Vasc Biol. 1997;17:2548–53. doi: 10.1161/01.atv.17.11.2548. [DOI] [PubMed] [Google Scholar]
- 16.Ireland HA, Cooper JA, Drenos F, Acharya J, Mitchell JP, Bauer KA, Morrissey JH, Esnouf MP, Humphries SE. FVII, FVIIa, and downstream markers of extrinsic pathway activation differ by EPCR Ser219Gly variant in healthy men. Arterioscler Thromb Vasc Biol. 2009;29:1968–74. doi: 10.1161/ATVBAHA.109.191551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saposnik B, Reny JL, Gaussem P, Emmerich J, Aiach M, Gandrille S. A haplotype of the EPCR gene is associated with increased plasma levels of sEPCR and is a candidate risk factor for thrombosis. Blood. 2004;103:1311–8. doi: 10.1182/blood-2003-07-2520. [DOI] [PubMed] [Google Scholar]
- 18.Maguire JM, Thakkinstian A, Sturm J, Levi C, Lincz L, Parsons M, Whyte S, Attia J. Polymorphisms in platelet glycoprotein 1balpha and factor VII and risk of ischemic stroke: a meta-analysis. Stroke. 2008;39:1710–6. doi: 10.1161/STROKEAHA.107.507228. [DOI] [PubMed] [Google Scholar]
- 19.Ye Z, Liu EH, Higgins JP, Keavney BD, Lowe GD, Collins R, Danesh J. Seven haemostatic gene polymorphisms in coronary disease: meta-analysis of 66,155 cases and 91,307 controls. Lancet. 2006;367:651–8. doi: 10.1016/S0140-6736(06)68263-9. [DOI] [PubMed] [Google Scholar]
- 20.Kathiresan S, Yang Q, Larson MG, Camargo AL, Tofler GH, Hirschhorn JN, Gabriel SB, O’Donnell CJ. Common genetic variation in five thrombosis genes and relations to plasma hemostatic protein level and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. 2006;26:1405–12. doi: 10.1161/01.ATV.0000222011.13026.25. [DOI] [PubMed] [Google Scholar]
- 21.Ken-Dror G, Drenos F, Humphries SE, Talmud PJ, Hingorani AD, Kivimaki M, Kumari M, Bauer KA, Morrissey JH, Ireland HA. Haplotype and genotype effects of the F7 gene on circulating factor VII, coagulation activation markers and incident coronary heart disease in UK men. J Thromb Haemost. 2010;8:2394–403. doi: 10.1111/j.1538-7836.2010.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiner AP, Carlson CS, Rieder MJ, Siscovick DS, Liu K, Chandler WL, Green D, Schwartz SM, Nickerson DA. Coagulation factor VII gene haplotypes, obesity-related traits, and cardiovascular risk in young women. J Thromb Haemost. 2007;5:42–9. doi: 10.1111/j.1538-7836.2006.02279.x. [DOI] [PubMed] [Google Scholar]
- 23.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–76. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 24.Psaty BM, Lee M, Savage PJ, Rutan GH, German PS, Lyles M. Assessing the use of medications in the elderly: methods and initial experience in the Cardiovascular Health Study. The Cardiovascular Health Study Collaborative Research Group. J Clin Epidemiol. 1992;45:683–92. doi: 10.1016/0895-4356(92)90143-b. [DOI] [PubMed] [Google Scholar]
- 25.O’Leary DH, Polak JF, Wolfson SK, Jr, Bond MG, Bommer W, Sheth S, Psaty BM, Sharrett AR, Manolio TA. Use of sonography to evaluate carotid atherosclerosis in the elderly. The Cardiovascular Health Study. CHS Collaborative Research Group. Stroke. 1991;22:1155–63. doi: 10.1161/01.str.22.9.1155. [DOI] [PubMed] [Google Scholar]
- 26.Gardin JM, Wong ND, Bommer W, Klopfenstein HS, Smith VE, Tabatznik B, Siscovick D, Lobodzinski S, Anton-Culver H, Manolio TA. Echocardiographic design of a multicenter investigation of free-living elderly subjects: the Cardiovascular Health Study. J Am Soc Echocardiogr. 1992;5:63–72. doi: 10.1016/s0894-7317(14)80105-3. [DOI] [PubMed] [Google Scholar]
- 27.Cushman M, Cornell ES, Howard PR, Bovill EG, Tracy RP. Laboratory methods and quality assurance in the Cardiovascular Health Study. Clin Chem. 1995;41:264–70. [PubMed] [Google Scholar]
- 28.Seligsohn U, Osterud B, Griffin JH, Rapaport SI. Evidence for the participation of both activated factor XII and activated factor IX in cold-promoted activation of factor VII. Thromb Res. 1978;13:1049–56. doi: 10.1016/0049-3848(78)90233-5. [DOI] [PubMed] [Google Scholar]
- 29.Neuenschwander PF, Morrissey JH. Deletion of the membrane anchoring region of tissue factor abolishes autoactivation of factor VII but not cofactor function. Analysis of a mutant with a selective deficiency in activity. J Biol Chem. 1992;267:14477–82. [PubMed] [Google Scholar]
- 30.Tracy RP, Lemaitre RN, Psaty BM, Ives DG, Evans RW, Cushman M, Meilahn EN, Kuller LH. Relationship of C-reactive protein to risk of cardiovascular disease in the elderly. Results from the Cardiovascular Health Study and the Rural Health Promotion Project. Arterioscler Thromb Vasc Biol. 1997;17:1121–7. doi: 10.1161/01.atv.17.6.1121. [DOI] [PubMed] [Google Scholar]
- 31.Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH, Jr, Heimovitz H, Cohen HJ, Wallace R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506–12. doi: 10.1016/s0002-9343(99)00066-2. [DOI] [PubMed] [Google Scholar]
- 32.Sakkinen PA, Cushman M, Psaty BM, Kuller LH, Bajaj SP, Sabharwal AK, Boineau R, Macy E, Tracy RP. Correlates of antithrombin, protein C, protein S, and TFPI in a healthy elderly cohort. Thromb Haemost. 1998;80:134–9. [PubMed] [Google Scholar]
- 33.Reiner AP, Carty CL, Jenny NS, Nievergelt C, Cushman M, Stearns-Kurosawa DJ, Kurosawa S, Kuller LH, Lange LA. PROC, PROCR and PROS1 polymorphisms, plasma anticoagulant phenotypes, and risk of cardiovascular disease and mortality in older adults: the Cardiovascular Health Study. J Thromb Haemost. 2008;6:1625–32. doi: 10.1111/j.1538-7836.2008.03118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ives DG, Fitzpatrick AL, Bild DE, Psaty BM, Kuller LH, Crowley PM, Cruise RG, Theroux S. Surveillance and ascertainment of cardiovascular events. The Cardiovascular Health Study. Ann Epidemiol. 1995;5:278–85. doi: 10.1016/1047-2797(94)00093-9. [DOI] [PubMed] [Google Scholar]
- 36.Psaty BM, Kuller LH, Bild D, Burke GL, Kittner SJ, Mittelmark M, Price TR, Rautaharju PM, Robbins J. Methods of assessing prevalent cardiovascular disease in the Cardiovascular Health Study. Ann Epidemiol. 1995;5:270–7. doi: 10.1016/1047-2797(94)00092-8. [DOI] [PubMed] [Google Scholar]
- 37.Price TR, Psaty B, O’Leary D, Burke G, Gardin J. Assessment of cerebrovascular disease in the Cardiovascular Health Study. Ann Epidemiol. 1993;3:504–7. doi: 10.1016/1047-2797(93)90105-d. [DOI] [PubMed] [Google Scholar]
- 38.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 39.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, Uitterlinden AG, Harris TB, Witteman JC, Boerwinkle E. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Identification of additional risk loci for stroke and small vessel disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2016;15:695–707. doi: 10.1016/S1474-4422(16)00102-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cushman M, Yanez D, Psaty BM, Fried LP, Heiss G, Lee M, Polak JF, Savage PJ, Tracy RP. Association of fibrinogen and coagulation factors VII and VIII with cardiovascular risk factors in the elderly: the Cardiovascular Health Study. Cardiovascular Health Study Investigators. Am J Epidemiol. 1996;143:665–76. doi: 10.1093/oxfordjournals.aje.a008799. [DOI] [PubMed] [Google Scholar]
- 42.Smith FB, Lee AJ, Fowkes FG, Price JF, Rumley A, Lowe GD. Hemostatic factors as predictors of ischemic heart disease and stroke in the Edinburgh Artery Study. Arterioscler Thromb Vasc Biol. 1997;17:3321–5. doi: 10.1161/01.atv.17.11.3321. [DOI] [PubMed] [Google Scholar]
- 43.Smith A, Patterson C, Yarnell J, Rumley A, Ben-Shlomo Y, Lowe G. Which hemostatic markers add to the predictive value of conventional risk factors for coronary heart disease and ischemic stroke? The Caerphilly Study. Circulation. 2005;112:3080–7. doi: 10.1161/CIRCULATIONAHA.105.557132. [DOI] [PubMed] [Google Scholar]
- 44.Miller GJ, Stirling Y, Esnouf MP, Heinrich J, van de Loo J, Kienast J, Wu KK, Morrissey JH, Meade TW, Martin JC, et al. Factor VII-deficient substrate plasmas depleted of protein C raise the sensitivity of the factor VII bio-assay to activated factor VII: an international study. Thromb Haemost. 1994;71:38–48. [PubMed] [Google Scholar]
- 45.Cooper JA, Miller GJ, Bauer KA, Morrissey JH, Meade TW, Howarth DJ, Barzegar S, Mitchell JP, Rosenberg RD. Comparison of novel hemostatic factors and conventional risk factors for prediction of coronary heart disease. Circulation. 2000;102:2816–22. doi: 10.1161/01.cir.102.23.2816. [DOI] [PubMed] [Google Scholar]
- 46.Silveira A, Scanavini D, Boquist S, Ericsson CG, Hellenius ML, Leander K, de Faire U, Ohrvik J, Woodhams B, Morrissey JH, Hamsten A. Relationships of plasma factor VIIa-antithrombin complexes to manifest and future cardiovascular disease. Thromb Res. 2012;130:221–5. doi: 10.1016/j.thromres.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghaddar HM, Folsom AR, Aleksic N, Hearne LB, Chambless LE, Morrissey JH, Wu KK. Correlation of factor VIIa values with factor VII gene polymorphism, fasting and postprandial triglyceride levels, and subclinical carotid atherosclerosis. Circulation. 1998;98:2815–21. doi: 10.1161/01.cir.98.25.2815. [DOI] [PubMed] [Google Scholar]
- 48.Bozzini C, Girelli D, Bernardi F, Ferraresi P, Olivieri O, Pinotti M, Martinelli N, Manzato F, Friso S, Villa G, Pizzolo F, Beltrame F, Corrocher R. Influence of polymorphisms in the factor VII gene promoter on activated factor VII levels and on the risk of myocardial infarction in advanced coronary atherosclerosis. Thromb Haemost. 2004;92:541–9. doi: 10.1160/TH04-02-0130. [DOI] [PubMed] [Google Scholar]
- 49.Mo X, Hao Y, Yang X, Chen S, Lu X, Gu D. Association between polymorphisms in the coagulation factor VII gene and coronary heart disease risk in different ethnicities: a meta-analysis. BMC Med Genet. 2011;12:107. doi: 10.1186/1471-2350-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng YC, Stanne TM, Giese AK, Ho WK, Traylor M, Amouyel P, Holliday EG, Malik R, Xu H, Kittner SJ, Cole JW, O’Connell JR, Danesh J, Rasheed A, Zhao W, Engelter S, Grond-Ginsbach C, Kamatani Y, Lathrop M, Leys D, Thijs V, et al. Genome-Wide Association Analysis of Young-Onset Stroke Identifies a Locus on Chromosome 10q25 Near HABP2. Stroke. 2016;47:307–16. doi: 10.1161/STROKEAHA.115.011328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinelli N, Girelli D, Baroni M, Guarini P, Sandri M, Lunghi B, Tosi F, Branchini A, Sartori F, Woodhams B, Bernardi F, Olivieri O. Activated factor VII-antithrombin complex predicts mortality in patients with stable coronary artery disease: a cohort study. J Thromb Haemost. 2016;14:655–66. doi: 10.1111/jth.13274. [DOI] [PubMed] [Google Scholar]
- 52.Taylor KC, Lange LA, Zabaneh D, Lange E, Keating BJ, Tang W, Smith NL, Delaney JA, Kumari M, Hingorani A, North KE, Kivimaki M, Tracy RP, O’Donnell CJ, Folsom AR, Green D, Humphries SE, Reiner AP. A gene-centric association scan for Coagulation Factor VII levels in European and African Americans: the Candidate Gene Association Resource (CARe) Consortium. Hum Mol Genet. 2011;20:3525–34. doi: 10.1093/hmg/ddr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huffman JE, de Vries PS, Morrison AC, Sabater-Lleal M, Kacprowski T, Auer PL, Brody JA, Chasman DI, Chen MH, Guo X, Lin LA, Marioni RE, Muller-Nurasyid M, Yanek LR, Pankratz N, Grove ML, de Maat MP, Cushman M, Wiggins KL, Qi L, et al. Rare and low-frequency variants and their association with plasma levels of fibrinogen, FVII, FVIII, and vWF. Blood. 2015;126:e19–29. doi: 10.1182/blood-2015-02-624551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabater-Lleal M, Chillon M, Howard TE, Gil E, Almasy L, Blangero J, Fontcuberta J, Soria JM. Functional analysis of the genetic variability in the F7 gene promoter. Atherosclerosis. 2007;195:262–8. doi: 10.1016/j.atherosclerosis.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 55.Humphries S, Temple A, Lane A, Green F, Cooper J, Miller G. Low plasma levels of factor VIIc and antigen are more strongly associated with the 10 base pair promoter (-323) insertion than the glutamine 353 variant. Thromb Haemost. 1996;75:567–72. [PubMed] [Google Scholar]
- 56.Hunault M, Arbini AA, Lopaciuk S, Carew JA, Bauer KA. The Arg353Gln polymorphism reduces the level of coagulation factor VII. In vivo and in vitro studies. Arterioscler Thromb Vasc Biol. 1997;17:2825–9. doi: 10.1161/01.atv.17.11.2825. [DOI] [PubMed] [Google Scholar]
- 57.Qu D, Wang Y, Song Y, Esmon NL, Esmon CT. The Ser219–>Gly dimorphism of the endothelial protein C receptor contributes to the higher soluble protein levels observed in individuals with the A3 haplotype. J Thromb Haemost. 2006;4:229–35. doi: 10.1111/j.1538-7836.2005.01676.x. [DOI] [PubMed] [Google Scholar]
- 58.Nayak RC, Sen P, Ghosh S, Gopalakrishnan R, Esmon CT, Pendurthi UR, Rao LV. Endothelial cell protein C receptor cellular localization and trafficking: potential functional implications. Blood. 2009;114:1974–86. doi: 10.1182/blood-2009-03-208900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scarabin PY, Vissac AM, Kirzin JM, Bourgeat P, Amiral J, Agher R, Guize L. Population correlates of coagulation factor VII. Importance of age, sex, and menopausal status as determinants of activated factor VII. Arterioscler Thromb Vasc Biol. 1996;16:1170–6. doi: 10.1161/01.atv.16.9.1170. [DOI] [PubMed] [Google Scholar]
- 60.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of Cardiovascular Health Study (CHS) participants with FVII phenotypes measured at the CHS Year 5 examination (1992-1993)
Table S2. Pearson correlation coefficients among coagulation factor VII (FVII) phenotypes and anticoagulant proteins in CHS
Table S3. Cox proportional hazards ratios for associations of coagulation factor VII tertiles with incident cardiovascular disease, ischemic stroke, and mortality
Table S4. Summary of previous studies evaluating relationships of F7 SNPs with stroke risk
Fig. S1. Distributions of activated coagulation factor VII and FVIIa-antithrombin complex in participants of the Cardiovascular Health Study