

Abstract
Toll-like receptor (TLR) agonists are effective at treating superficial cancerous lesions but their use internally for other types of tumors remains challenging due to toxicity. Here we report murine and human naïve CD4+ T cells spontaneously sequester the TLR2 agonist Pam3Cys4 (CD4+ TPam3) and become primed for Th1 differentiation. Naïve CD4+ TPam3 cells encoding the ovalbumin-specific T cell receptor (OT2Pam3), when transferred into mice bearing established TGF-β-ovalbumin-expressing thymomas, produce high amounts of IFN-γ and sensitize tumors to PD-1/PD-L1 blockade-induced rejection. In contrast naïve OT2 cells without Pam3Cys4 cargo are prone to TGF-β-dependent iTreg conversion and accelerate tumor growth that is largely unaffected by PD-1/PD-L1 blockade. Ex vivo analysis reveal CD4+ TPam3 cells are resistant to TGF-β-mediated gene expression through Akt activation controlled by inputs from the T cell receptor and a TLR2-MyD88-dependent PI3 kinase-signaling pathway. These data show CD4+ TPam3 cells are capable of Th1 differentiation in the presence of TGF-β suggesting a novel approach to adoptive cell therapy.
INTRODUCTION
Toll-like receptors (TLR) promote host defense through recognizing pathogen-associated molecular patterns (PAMPs) released by microorganisms (1). TLR activation initiates potent inflammatory cytokine production and dendritic cell activation that drives the expansion and differentiation of antigen-specific T cells. These observations have led to the clinical use of TLR agonists to promote anti-tumor responses. These include the use of TLR7 agonist imiquimod and live preparations of Mycobacterium bovis bacillus of the Calmette–Guerin strain to treat superficial skin and bladder carcinomas, respectively (2, 3). However, TLR agonist therapy has been largely restricted to mucosal lesions due to potential systemic toxicity (4).
Although most studies have focused on TLR2 in antigen presenting cells (APCs) it has been recognized for over a decade that human and mouse T lymphocytes express TLR2 and directly respond to its agonists following T cell receptor stimulation (5). TLR2 on T lymphocytes is primarily thought to function as a costimulatory molecule that controls effector function (6). This activity has best been described in CD8+ T cells where TLR2 was shown to stimulate the clonal expansion of long-lived memory cells (5). The expression of Tbx21 (T-bet), a transcription factor that directs T helper 1 (Th1) lineage commitment (7), is upregulated by TLR2 agonist stimulation of CD8+ T cells (8). However, T-bet is not required for the regulation of IFN-γ expression in CD8+ T cells (9) and it remains unclear how TLR2 promotes T-bet expression or Th1 lineage development in CD4+ T cells.
Th1 development is strongly opposed by TGF-β, an immunosuppressive cytokine that is often found in the tumor microenvironment (10). TGF-β not only inhibits T-bet expression but also additionally limits effector cell expression of IFN-γ (11), a critical mediator of anti-tumor immunity (12). TGF-β also facilitates the conversion of peripheral naïve CD4+ T cells into inducible regulatory Foxp3+ CD4+ T cells (iTregs) (13), which in turn blunt CD8+ T cell effector cytotoxic activity (14). In T lymphocytes the transcription factors SMAD 2 and 3 play redundant roles in TGF-β–mediated inhibition of IFN-γ expression and iTreg development (15). Besides being inhibited by TGF-β Th1 cells may also become functionally impaired through the development of exhaustion due to chronic antigen exposure. In particular high expression of programmed cell death ligand 1 (PD-L1) by tumors, an immune checkpoint inhibitor, has been to be strongly linked to poor outcomes in solid tumors (16). PD-L1/PD-1 signaling can inhibit IFN-γ expression along with the exprssion of other Th1 effector molecules important in controlling tumor progression (17). These observations have led to the use of strategies to block PD-L1/PD-1 engagment although such approaches have not always proved successful due to the co-expression of other immune checkpoint inhibitors that promote T cell dysfunction such as TIM-3 (18).
Adoptive cell therapy (ACT) employing tumor-infiltrating T cells expanded ex vivo or with lymphocytes expressing engineered antigen receptors have been used to successfully treat metastasis (19). The majority of ACT reports have described the activity of ex vivo differentiated CD8+ T cells. However, CD8+ T cells require CD4+ T cell help to maintain functionality in vivo (20). This has been exemplified by ACT protocols rendered more effective with the addition of CD4+ T cells (21). Optimal priming and differentiation of CD4+ T cells is likely to occur within tumor draining lymph nodes (TDLN) as evident by the potent anti-tumor activity of TDLN-derived Th1 cells (22). Previous observations have shown that adoptively transferred naïve CD4+ T cells preferentially home to draining lymph nodes (23) suggesting in vivo priming of tumor-specific naïve CD4+ T cells may improve ACT protocols. Here we report in a model of TGF-β–mediated tumor immune evasion that naïve tumor-specific CD4+ T cells that carry the synthetic TLR2 agonist Pam3Cys4 differentiate into Th1 cells and control tumor growth. We also show that resistance to TGF-β directed effects on Th1 development is mediated through TLR2-MyD88 dependent Akt activation pathway that antagonizes Foxp3 expression.
MATERIAL AND METHODS
Mice and Humans
C57BL/6 (B6), CD45.1, TLR2−/−, CD14−/−, MyD88−/− T-bet−/−, Nur77EGFP and SBE luc mice were purchased from Jackson Labs (Bar Harbor, Maine USA). OT-II (OT2) and OT-I (OT1) mice were purchased from Charles River Labs (Wilmington, MA USA). CD45.1+ OT2 mice were generated by intercrossing with CD45.1 mice. Experiments were conducted with mice between 6 and 9 weeks age in accordance with an approved IACUC protocol. Human CD4+ T cells were isolated under an approved IRB protocol ID # 201012829
Preparation of naïve CD4+ T Pam3 cells
Spleen and lymph nodes were gently mechanically disrupted through a 70 μm filter, lysed for red blood cells with ACK Lysing buffer (Lonza) and stained with the following Abs (all obtained from ebiosciences): anti-NK-1.1-FITC (clone PK136), anti-CD45R-FITC (clone RA3-6B2), anti-CD11c-FITC (clone N418), anti-CD11b-FITC (clone M1/70), anti-CD69-FITC (clone H1.2F3), anti-CD4-PerCP-Cy 5.5 (clone RM4-5), anti-CD90.2-APC (clone 53-2.1), anti-CD62L-APC-Alexa 780 (clone MEL-14), anti-CD44-FITC (clone IM7), anti-Gr1-FITC (clone RB6-8C5) and anti-CD25-FITC (clone PCS1). Cells were double FACS-sorted through a CD90.2+ CD4+ CD62L+ CD25− CD69− CD44− CD11c− CD11b− NK1.1-CD45R-Gr1- gate using a Synergy 3200 BSC machine (Sony Biotechnology) into complete culture medium (CM: RPMI 1640, 1% Penicillin/Streptomycin (Life technologies), 10% FBS, 28.6 μM β-2-mercaptoethanol (Sigma). A human naïve CD4+ T cell isolation kit II (Miltenyi) were used to isolate CD4+ T cells from the peripheral blood of healthy human volunteers. CD4+ T cells were co-incubated with 10 μg/mL unconjugated, biotinylated or rhodmaine-conjugated Pam3CSK4 (Invivogen) at 37° C for 3 hrs in CM, centrifuged at 250 × g, and washed three times with 15 mls CM prior to an experiment. Plasma membrane staining was conducted with Cell Mask Deep Red (Thermo Fisher Scientific) in accordance with manufacturers recommendations.
APCs, CD4+ T cell activation and polarization
APCs were prepared from TLR2−/− disrupted spleens that were T lymphocyte depleted with anti-CD 90.2 beads and LS Magnetic Bead Columns (Miltenyi Biotec) and then gamma irradiated with 2000 rads. For polyclonal activation 0.3 μg/ml CD3ε Abs (clone 2C11, Biolegend) was used or 0.3 μM of OVA peptide (ISQAVHAAHAEINEAGR, Sigma) for OT2 cells. Plate bound stimulation was conducted in 96 well flat bottom plates (Corning) with 0.3 μg/ml CD3ε Abs and soluble 1 μg/ml CD28 Abs (clone CD28.2, Biolegend). Th1 polarization for mouse CD4+ T cells was conducted with 10 μg/ml mIL4 Abs (clone 11B11, Biolegend), 10 ng/ml mIFN-γ (Peprotech) and 10 ng/ml mIL-12 (Peprotech). Th1 polarization for human CD4+ T cells was accomplished with CellXVivo Human Th1 Cell Differentiation Kit (R&D Systems) in accordance with manufacturers recommendations. For iTreg development 0.5 ng/ml of mTGF-β1 (R&D) was used alone or in combination with Th1 polarizing medium.
Tumors, CD4+ T cell transfer and TGF-β & PD-L1 neutralization
EG.7-OVA, (ATCC) and was maintained in CM prior to subcutaneous injection into the left flank of B6 or TLR2−/− mice. Tumors were allowed to progress to approximately 250 mm3 in volume prior to the addition of intravenous administration of 106 indicated naïve CD4+ T cells. Tumors were evaluated for changes in tumor volume every three days with the formula 3.14 × [largest diameter × (perpendicular diameter) 2]/6. In some experiments mice were injected intraperitoneally with 1 mg/kg of a pan-specific neutralizing antibody against TGF-β1, -β2, and -β3 (R&D Systems) or control rabbit polyclonal IgG for one day before and every other day until the conclusion of the experiment. PD-L1 neutralization was conducted with intraperitoneal administration of 100 μg of the clone 10F.9G2 (Bio-X-Cell) or control mouse IgG every 3 days after tumor establishment until completion of the study. Tumors were minced with a razor, digested with DNAse I (Sigma) and Collagenase/Dispase (Roche) for one hour at 37 °C and cell suspensions were filtered through a 70 μm filter. Tumor cells and infiltrating lymphocytes were quantitated using fluorescent flow cytometric counting beads (BD Biosciences).
FACS, ELISA and CFSE assays
Cell suspensions were surface-stained with flourochrome-conjugated Abs specific mouse CD45.1 (Biolegend, clone A20), CD45.2 (Biolegend, clone 104), CD4 (eBioscience, clone RM4-5), CD90.2 (eBioscience, clone 53-2.1), PD-1 (eBioscience, clone RMP1-30), PD-L1 (BD Biosciences, clone MIH-5), TIM-3 (eBioscience, clone 88.2C12), and CD8α (Biolegend, clone 53-6.7). For intracellular IFNγ staining cell suspensions were stimulated for 4 hours with 20 ng/ml PMA (Sigma), and 1 μg/ml of Ionomycin (Sigma) in the presence of 1 μg/ml Golgi Plug (BD Biosciences) for 4 hours. Cells were fixed and permeabilized with a BD Cytofix/Cytoperm kit (BD Biosceinces) and then stained with IFN-γ Ab (eBioscience, clone XMG1.2), IL-17A Ab (Biolegend, clone TC11-18H10.1) or SMAD7 specific rabbit polyclonal Ab and alexflour 647 conjugated goat anti-rabbit IgG (ThermoFisher). For detection of Foxp3 and T-bet clone MF-14 (Biolegend) and clone 4B10 (Biolegend) were respectively used with a Foxp3/transcription factor staining kit (eBioscience). IFN-γ accumulation in supernatant was measured with a Ready-Set Go ELISA kit (eBiosciences). For Akt activation cells were treated with DMSO vehicle or indicated concentrations of Ly2924002 (Sigma) 30 minutes prior to the initiation of an assay. At the conclusion of an assay cells were fixed with 70% ethanol for 10 min and permeabilized with 0.4% Triton X-100, blocked 2.5 % BSA, incubated rabbit phospho-specific Ab to Serine 473 of Akt (ThermoFisher, clone 700392) for 5 hrs at room temp and then labelled with alexflour 647 conjugated goat anti-rabbit IgG.
Suppression Assay
Regulator CD45.1+ OT2 cells was isolated from primary cultures by positive selection with CD45.1 biotinylated Abs (Clone 820, Biolegend) and anti-biotin micro beads and LS columns (Miltenyi) in accordance with manufacturers instructions. 1.5 × 105 OT2 regulators were added to 96 well round bottom plates with 1.5 × 105 APCs, pulsed with SIINFEKL (0.1 μg/ml) and SQAVHAAHAEINEAGR (0.1 μg/ml) peptides and co-cultured in triplicate with CFSE labelled responder CD45.2+ OT1 cells at regulator to responder ratios of 0:1, 1:9, 1:3 and 1:1. Suppression was measured as percent retention of CFSE intensity after 5 days of culture.
Semiquantitative real time RTPCR
Cells were lysed and fractionated for total RNA with Trizol reagent (Invitrogen) in accordance with manufacturers recommendations. RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription kit (ThermoFisher). 400 ng of cDNA were equally distributed in quadruplicate wells and amplified 7900HT Fast Real Time PCR System (Applied Biosystems) and TaqMan Assays (ThermoFisher) in accordance with the manufacturers recommended cycling conditions and using these gene specific primers: bactin (Mn00607939_s1); tbx21 (Mn00450960_m1), ifng (Mn01168134_m1), gata3 (Mm00484683_m1), rorc (Mm01261022_m1), foxp3 (Mm00475162_m1), eomes (Mm01351984_m1), prdm1 (Mm00476128_m1), il12rb2 (Mm00434200_m1), icos (Mm00497600_m1), cxcr3 (Mm99999054_s1), cxcr4 (Mm01996749_s1), fasl (Mm00438864_m1), prfn (Mm00812512_m1), gzmb (Mm00442837_m1), and smad3 (Mm01170760_m1).
Software and statistical analysis
FACS analysis was conducted with FlowJo software version 9 (TreeStar). Statistics were performed with GraphPad Prims Software using one-way ANOVA and Tukey-Kramer multiple comparison posttest. Results were considered significant if P < 0.05.
RESULTS
Naïve CD4+ T cells sequester TLR2 ligands leading to enhanced IFN-γ expression
Myeloid cells can sequester TLR agonists (24) but it remains unclear if this occurs in lymphocytes. We incubated naïve C57BL/6 (B6) CD4+ T cells with biotin-labeled Pam3Cys4 for 3 hours at 37 °C. Cells were then washed extensively to remove unbound agonist (Fig. 1A). Streptavidin staining revealed that CD4+ T cells take up Pam3Cys4 intracellularly with about half remaining on the outer plasma membrane. As naïve CD4+ T cells are reported to express low levels of TLR2 (25) we also determined if Pam3Cys4 is sequestered by naïve TLR2−/− and CD14−/− CD4+ T cells as both receptors have been shown to promote ligand binding (26). Surprisingly, Pam3Cys4 uptake in TLR2−/− and CD14−/− CD4+ T cells was comparable to B6 wild type CD4+ T cells. Using a rhodamine conjugate of Pam3Cys4 we also visualized ligand binding in naïve mouse and human CD4+ T cells (Fig. 1B and C).
Figure 1. Naïve CD4+ T cells sequester Pam3Csy4 in a TLR2 independent manner.
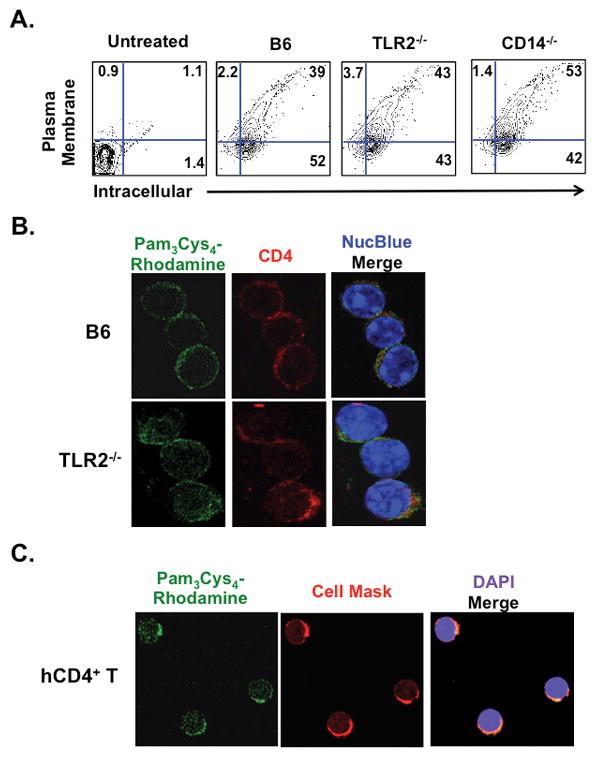
(A) Indicated naïve CD4+ T cells were co-incubated with 10 μg/ml of biotinylated Pam3Cys4 for 3 hours at 37 °C, washed 3 times, and the plasma membrane and intracellular compartments stained with fluorescent conjugates of streptavidin. (B) Naïve B6 and TLR2−/− CD4+ T cells were co-incubated 10 μg/ml of Pam3Cys4 conjugated to rhodamine for 3 hours, washed 3 times and then stained with CD4-PE antibodies and NucBlue. (C) Human naïve CD4+ T cells (hCD4+ T) were prepared with Pam3Cys4-rhodamine conjugate as described in (B) and stained with Cell Mask, a plasma membrane specific stain, and DAPI. (A–C) are a representative results from at least three independent experiments.
Based on previous reports that T lymphocytes co-incubated with Pam3Cys4 during activation enhance IFN-γ production (6, 8), we asked if we could detect similar responses in naïve CD4+ T cells that sequestered Pam3Cys4 (CD4+ TPam3). To this end TLR2−/− and B6 CD4+ TPam3 cells were polyclonally activated with CD3ε Abs and TLR2−/− APCs under Th1 polarizing conditions (Fig. 2A). B6 CD4+ TPam3 cell IFN-γ expression was markedly greater when compared to B6 CD4+ T cells without Pam3Cys4 cargo. Pam3Cys4-mediated IFN-γ expression was dependent on TLR2 irrespective of whether ligand was co-incubated or sequestered prior to activation Pam3Csy4 cargo also promoted naïve CD4+ T cells encoding the ovalbumin-specific T cell receptor (OT2Pam3) and naive human CD4+ T cells isolated from healthy human volunteers (hCD4+ TPam3) to produce high amounts of IFN-γ when activated under Th1 polarizing conditions (Figs. 2B and C). Moreover, two alternative TLR2-specific ligands, Pam2Cys4 and FSL-1, also enhanced IFN-γ expression demonstrating that augmented Th1 differentiation is not specific to Pam3Cys4. (Supplemental Fig. 1).
Figure 2. Pam3Cys4 cargo promotes IFN-γ expression.
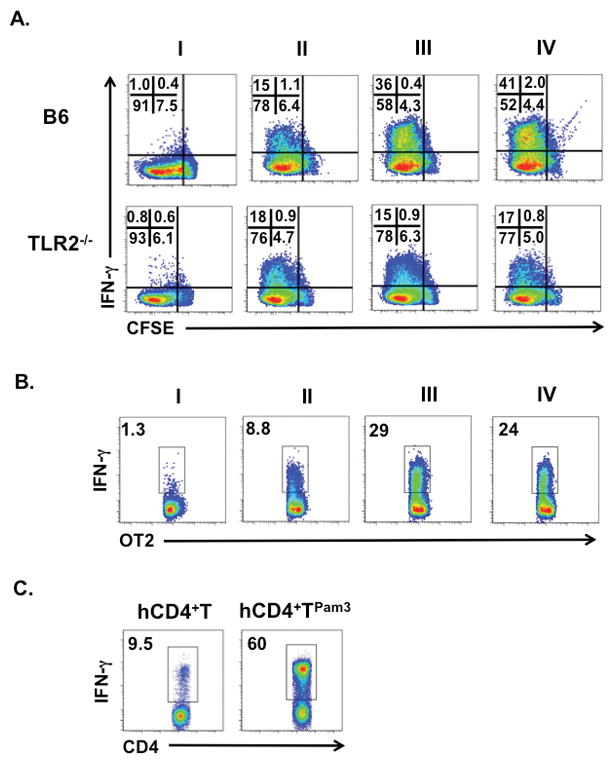
(A) Indicated naïve untreated CFSE labeled CD4+ T cells were co-cultured with 0.1 μg/ml CD3ε Abs and TLR2−/− APCs in the absence (I) or presence (II) of Th1 polarization or (III) with Th1 polarization and 10 μg/ml of Pam3Cys4 for 96 hours. (IV) Indicated naïve CD4+ TPam3 cells were stimulated as in (II). The corresponding CFSE dilution and IFN-γ accumulation are shown as a percent value in the inset. (B) Naïve untreated OT2 were co-cultured with OVA peptide (OVAp) bearing TLR2−/− APCs in the absence (I) or presence (II) of Th1 polarization or (III) with Th1 polarization and 10 μg/ml of Pam3Cys4 for 96 hours and assessed for IFN-γ expression. (IV) Naïve OT2Pam3 cells were stimulated as in (II). (C) Naïve human CD4+ T cells with or without Pam3Cys4 cargo were stimulated under Th1 polarizing conditions with CD3ε Ab-conjugated beads for 96 hours and assessed for IFN-γ expression. (A–C) are representative results from at least three independent experiments.
Naïve tumor specific CD4+ T Pam3 cells differentiate into Th1 cells in vivo
We next determined the fate of naïve CD4+ TPam3 cells in vivo. One million naïve CD45.1+ OT2 cells with or without Pam3Cys4 cargo were intravenously administered into CD45.2+ B6 or CD45.2+ TLR2−/− hosts bearing established ovalbumin-expressing EG.7 (EG.7-OVA) tumors (Fig. 3A). OT2 cells preferentially homed to tumor draining lymph nodes (TDLN) independent of Pam3Cys4 sequestration (Supplemental Fig. 2). However, only OT2Pam3 cells substantially controlled tumor growth when compared recipients that received untreated OT2 or B6 CD4+ TPam3 cells tumors. Quantitation of tumor-infiltrating OT2 cells showed comparable accumulation irrespective of treatment with Pam3Cys4 (Fig. 3B) but only hosts that received OT2Pam3 cells had high numbers of OT2 IFN-γ+ cells in their tumors (Fig. 3C). OT2Pam3 cell-treated hosts also had few tumor-infiltrating IL-17A+ OT2 cells (Supplemental Fig. 3A). In line with these observations tumor-infiltrating OT2Pam3 cells had significantly elevated gene transcripts linked to Th1 lineage determination including T-bet and its target genes that direct early steps in differentiation (il12rβ2)(27) and trafficking into inflammatory sites (cxcr3)(28). Additionally, we detected known T-bet-targeted genes that stimulate cytoxicity (fasl, Prfn and Gzmb) (29) suggesting that infiltrating OT2Pam3 cells acquire direct anti-tumor activity (Fig. 3D). In TDLN we observed elevated IFN-γ+ OT2 and IFN-γ+ CD8+ T cell accumulation of hosts that received OT2Pam3 cells (Fig. 3E & Supplemental 3B). TDLN CD4+ T cells from hosts that received OT2Pam3 cells also produced high amounts of IFN-γ when challenged with OVA peptide (OVAp) (Fig. 3F). In contrast we observed little OVAp-mediated IFN-γ expression from TDLN CD4+ T cells from hosts that received B6 or untreated OT2 cells. Taken collectively, these data demonstrate that tumor-specific CD4+ TPam3 cells differentiate into Th1 cells in vivo.
Figure 3. OT2Pam3 cells control the growth of EG.7-OVA tumors.
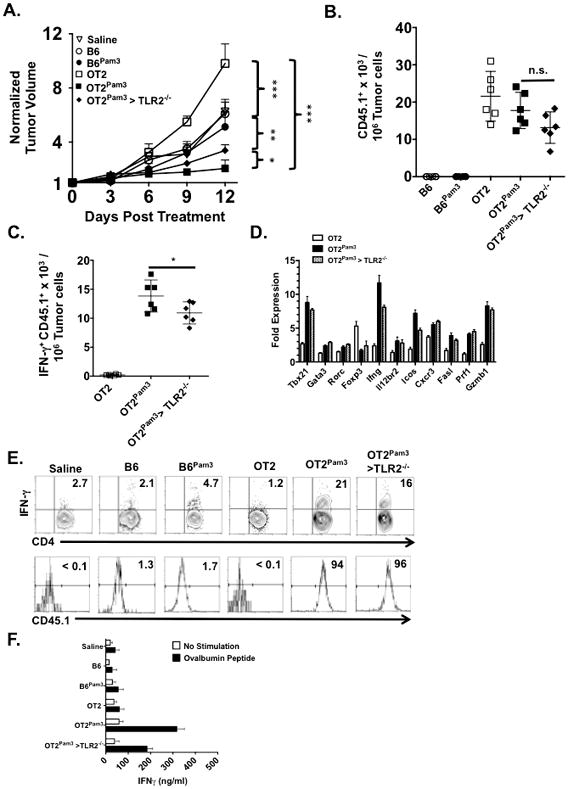
B6 and TLR2−/− mice (n=6/group) with established tumors received saline or 106 indicated naïve CD45.1+ CD4+ T cells intravenously and were assessed for (A) tumor size on three-day intervals for 12 days. The plot shows mean tumor size per group ± Standard Deviation (S.D.) Tumors from (A) were evaluated for numbers of infiltrating (B) CD45.1+ CD90.2+ CD4+ T cells and (C) CD45.1+ CD90.2+ IFN-γ+ CD4+ T cells. The plots (B, C) shows individual host tumor-infiltrating cell numbers, normalized to 107 tumor cells, along with a mean per group ± S.D. (D) Indicated day 12 host tumors represented in (A) were FACS sorted for infiltrating CD45.1+ cells, fractionated for RNA, and assessed for indicated transcripts by semiquantitive real time RT PCR. Transcript levels are normalized to freshly isolated naïve untreated OT2 cells (line) and is a representative result from 2 independent experiments. (E) Day 12 host TDLN in (A) were pooled for each group and analyzed for the percent abundance of (E) CD90.2+ IFN-γ+ CD4+ T cells and CD45.1 expression within this compartment. (F) Day 12 host spleens removed from (A), pooled for each group, fractionated for CD4+ T cells, co-cultured in quadruplicate with APCs with or without OVAp for 72 hours and supernatants were assessed for IFN-γ production by ELISA. Unless noted data shown for A–F are representative results from three independent experiments where n.s. p > 0.05, *p < 0.05, **p < 0.01, *** p< 0.005.
OT2 Pam3 cells induce PD-L1 expression in the tumor microenvironment
The absence of tumor rejection despite the high frequency of IFN-γ+ OT2Pam3 tumor-infiltrating cells suggested alterations within the tumor microenvironment could be inhibiting optimal Th1 effector function. To examine this possibility tumor-bearing hosts were analyzed for expression of programmed death ligand 1 (PD-L1), a well-established stimulator of T cell exhaustion that can be upregulated by IFN-γ (30) (Figs. 4A–C). PD-L1 expression on tumor cells at both 12 and 24 days after OT2Pam3 cell treatment was significantly higher than in hosts that received OT2 cells and was linked to an increase in late stage tumorigenesis. PD-L1 engagement of its cognate receptor, programmed death ligand (PD-1) has been shown to impede Th1 effector responses (31). We examined expression levels of PD-1 on tumor-infiltrating OT2 and OT2Pam3 cells along with another co-inhibitory receptor that negatively regulates Th1 cell survival, the T cell immunoglobulin mucin (TIM)-3 (32) (Figs. 4D and E). In tumor-infiltrating OT2Pam3 cells PD-1 and TIM-3 expression levels increased between 12 and 24 days after treatment. Interestingly, co-inhibitory receptor expression changes were coupled to the loss of T-bet suggesting the late stage development of T cell exhaustion. To explore this relationship further we analyzed IFN-γ expression in PD-1− TIM-3−, PD-1+ TIM-3− and PD-1+ TIM-3+ tumor-infiltrating OT2Pam3 cells at 12 and 24 days after treatment (Fig. 4F). Irrespective of PD-1 or TIM-3 expression patterns most OT2Pam3 tumor-infiltrating cells expressed IFN-γ twelve days after treatment. However, at the 24 day time point the bulk of tumor-infiltrating OT2Pam3 cells were either IFN-γ− PD-1+Tim-3+ or IFN-γ− PD-1+ Tim-3− while some IFN-γ expression could be still found in the PD-1− Tim-3− cell compartment. As the loss of IFN-γ expression could be explained by repolarization we assessed tumor-infiltrating PD-1+ and PD-1− OT2Pam3 cell mRNA for evidence of transcription factor expression changes indicative of alternative CD4+ T cell helper lineage determination (Fig. 4G). However, we failed to detect substantial changes in Rorc, Gata3 or Foxp3 transcript levels in PD-1+ or PD-1− cells indicating the absence the lack of repolarization into Th17, Th2 or iTreg fates. However, Ifng, Prf1 Gzmb1, fasl mRNA levels were all lower in the PD-1+ fraction at 24 days after treatment suggesting the progressive loss of a Th1 effector phenotype. Additionally, we observed the upregulation of transcripts encoding blimp-1 (Prdm1) and Eomes, transcription factors recently linked to PD-1-mediated CD4+ T cell exhaustion (33). Given these observations suggested the development of Th1 exhaustion was predominantly associated with high PD-1 expression we evaluated the effect of PD-L1 neutralizing Abs in tumor-bearing recipients that were left untreated or received either OT2 or OT2Pam3 cells (Fig. 4H). Although PD-1/PD-L1 blockade had a mild effect on tumor growth in untreated and OT2-treated recipients it led to the complete rejection of tumors in OT2Pam3 cell-treated hosts. Analysis of isolated tumor-infiltrating OT2Pam3 cells (Supplemental Fig. 4) demonstrated that IFN-γ expression was significantly higher in PD-L1 Ab-treated recipients when compared to hosts that received control Abs indicating that PD-1 engagement in the tumor microenvironment limits ACT-mediated Th1 effector responses.
Figure 4. OT2Pam3 cells synergize with PD-1/PD-L1 blockade to induce tumor rejection.
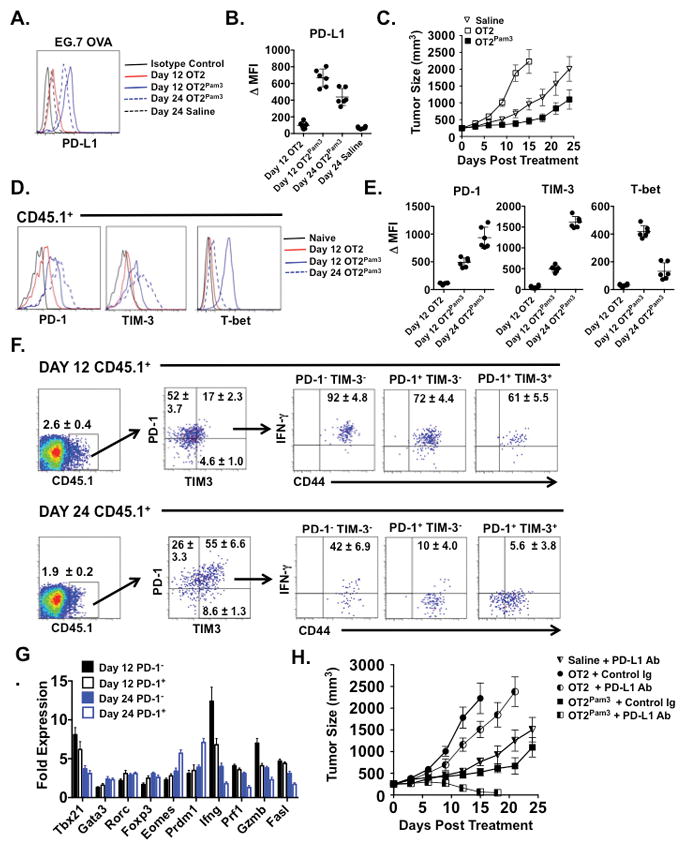
(A) B6 mice with established tumors (n = 6/group) received saline, 106 untreated OT2 or OT2Pam3 cells and EG.7 OVA cells assessed for PD-L1 expression at indicated times. Data is shown as a histogram or (B) Δ MFI ± S.D. relative to isotype control staining. (C) Tumor growth rates plotted as mean tumor size ± S.D. for indicated treatments. Tumor-infiltrating CD45.1+ cells were analyzed for either (D) PD-1, TIM-3 and T-bet expression, (E) IFN-γ and CD44 expression for indicated PD-1 and TIM-3 gates or (F) indicated mRNA levels for PD-1+ and PD-1− subsets from hosts treated as described in (A). Data is shown as representative histograms, Δ MFI ± S.E relative to naïve CD4+ T cells, indicated mean percent abundance ± S.D., or mean transcript levels ± S.E relative naïve CD4+ T cells. (G) Tumor bearing recipients received saline or an indicated OT2 cell treatment (n = 6/group) along with either neutralizing 100 μg PD-L1 Abs or Control Rabbit Ig every 3 days and assessed for tumor growth. Data is shown as mean tumor size ± S.D. Data shown in A–G are representative results from at least two independent experiments.
CD4+ T Pam3 cells are resistant to TGF-β–mediated iTreg development
In light of several reports that show EG.7 tumors produce TGF-β (34, 35) we compared the effects of administering TGF-β neutralizing Abs to tumor bearing hosts that received either untreated OT2 or OT2Pam3 cells (Fig. 5A). In hosts that received untreated OT2 cells TGF-β Ab treatment significantly slowed tumor growth and inhibited infiltration of Foxp3+ CD4+ T cells when compared to Control Ig-treated recipients that received the same cells (Fig. 5B). Surprisingly, hosts that received OT2Pam3 cells and TGF-β Abs did not exhibit significant increases tumor-infiltrating IFN-γ+ CD4+ T cells relative to OT2Pam3 and control Ig-treated hosts suggesting that CD4+ T cells carrying Pam3Cys4 cargo are resistant to the regulatory effects of TGF-β on Th1 development (Fig. 5C). A similar pattern was observed in the TDLN (Fig. 5D). Analysis of CD45.1 expression within the IFN-γ+ and Foxp3+ CD4+ T cell compartments demonstrated that conversion OT2Pam3 cells into Th1 cells was not markedly affected by treatment with TGF-β Abs while differentiation of OT2 cells into Foxp3+ CD4+ T cells was sharply attenuated by TGF-β neutralization.
Figure 5. OT2Pam3 cells are resistant to acquiring Foxp3 expression in tumor bearing hosts.
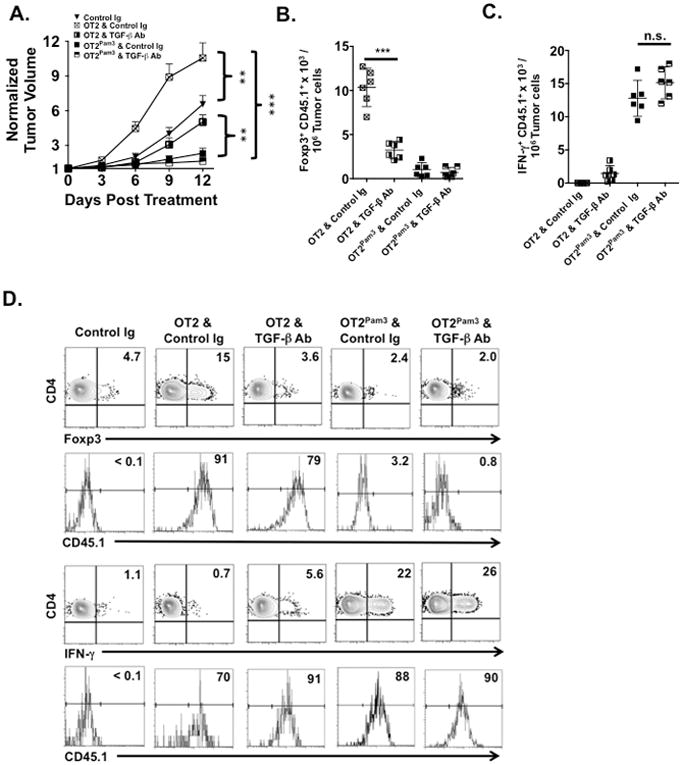
B6 mice with established tumors (n ≥ 5/group) received 106 untreated OT2 or OT2Pam3 cells with either Control Ig or TGF-β Abs and were assessed for (A) tumor size on three-day intervals for 12 days. The plot shows mean tumor size ± S.D. Tumors from (A) were evaluated for numbers of infiltrating (B) CD45.1+ CD90.2+ Foxp3+ CD4+ and (C) CD45.1+ CD90.2+ IFN-γ+ CD4+ T cells. The plots (B and C) show individual host infiltrating cell numbers normalized to 107 tumor cells along with a mean per group ± S.D. Host TDLN represented in (A) were pooled and evaluated for the percent abundance of (D) CD90.2+ Foxp3+ CD4+ and CD90.2+ IFN-γ+ CD4+ T cells and CD45.1 expression within each of the aforementioned compartments, respectively. Data from (A–D) are representative results from three independent experiments where n.s. p > 0.05, **p < 0.01, *** p< 0.005.
We next asked if resistance of CD4+ TPam3 cells to TGF-β–mediated attenuation of IFN-γ expression is mediated by TLR2 signaling pathways. B6, TLR2−/− and MyD88 −/− CD4+ T cells carrying Pam3Cys4 cargo were activated under Th1 polarizing conditions in the absence or presence of graded amounts of TGF-β1 (Fig. 6A). Consistent with previous reports TGF-β1 inhibited IFN-γ production in all cultures (11). However, at concentrations of up to 0.5 ng/ml of TGF-β1 IFN-γ production was significantly higher in B6 CD4+ TPam3 cells when compared to analogous cultures of untreated CD4+ T cells. A similar pattern of TGF-β1 resistance was also detected in OT2 cell cultures. In contrast, TLR2−/− and MyD88−/− CD4+ T cells showed no resistance to TGF-β1-mediated suppression of IFN-γ expression. We next analyzed the effects of TGF-β1 on T-bet and Foxp3 expression levels (Fig. 6B). In agreement with previous observations TGF-β1 upregulated Foxp3 and suppressed T-bet expression in cultures with untreated OT2 cells independent of the application of Th1 polarization (34). However, the addition of TGF-β1 to the Th1 polarization milieu only had a mild effect on T-bet suppression in OT2Pam3 cells. To test the functional significance of these observations we also performed suppression assays (Fig. 6C). Untreated OT2 cells differentiated with TGF-β1 alone or in combination with Th1 polarization were potent suppressors of responder ovalbumin specific CD8+ T cell proliferative responses while OT2Pam3 cells differentiated under the same conditions exhibited significantly less regulatory activity.
Figure 6. In the presence of TGF-β1 and Th1 polarizing conditions CD4+ TPam3 cells are biased towards a Th1 fate.
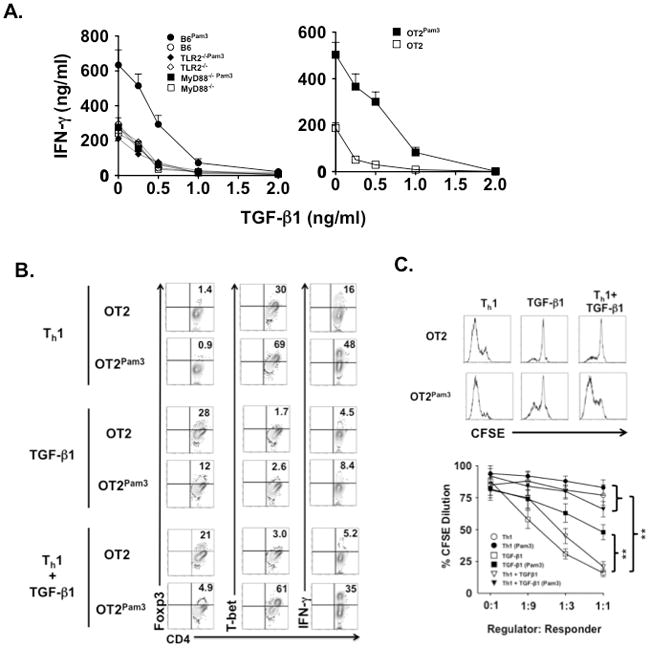
(A) Naïve B6 CD4+ T, TLR2−/− CD4+ T and OT2 cells with or without Pam3Cys4 cargo were stimulated with TLR2-deficient splenocyte-derived APCs bearing CD3ε Abs (B6 or TLR2−/− CD4+ T cells) or OVAp (OT2 cells) in the presence of IL-12 and indicated amounts of TGF-β1. After 72 hours culture supernatants were assessed for IFN-γ by ELISA. Data are representative of at least four independent experiments. (B) Naïve untreated OT2 and OT2Pam3 cells were stimulated with TLR2−/− APCs bearing OVAp cultured under Th1 polarization, 0.5 ng/ml TGF-β1 or Th1 polarization + 0.5 ng/ml TGF-β1. After 5 days of culture OT2 cells were evaluated for the percent abundance of Foxp3, T-bet and IFN-γ expression. Data shown is a representative result of three independent experiments. (C) Naïve untreated OT2 and OT2Pam3 cells were stimulated as in (A) and then fractionated from 72 hrs later, mixed at indicated ratios with responder CFSE labeled responder OT1 cells and re-cultured in triplicate wells with TLR2−/− APCs bearing peptides specific for OT2 and OT1. (Upper panel) Representative CFSE dilution plots of OT2 regulators were mixed with OT1 responders at a 1:1 ratio. (Lower panel) Mean CFSE dilution ± S.E of responders over indicated regulator to responder ratios (**p< 0.01). The result shown is representative of three independent experiments.
Pam3Cys4 antagonizes T cell receptor-mediated suppression of TGF-β signaling
Low levels of antigen stimulation or suboptimal costimulation favors naïve CD4+ T cell differentiation into iTregs. TGF-β is thought to mimic the effects of low antigen engagement by antagonizing T cell receptor (TCR) signaling (36). Given our observations of CD4+ TPam3 cell resistance to conversion into iTregs we asked if Pam3Cys4 cargo alters TCR signaling strength. To examine this possibility we utilized Nur77EGFP CD4+ T cells, which encode an orphan nuclear hormone receptor Nur77-EGFP transgene that is expressed in proportion to TCR signaling strength (37) (Fig. 7A). In the absence of TGF-β1 TCR signaling strength was moderately higher in CD4+ TPam3 cells relative to CD4+ T cells without Pam3Cys4 cargo and became more elevated with TGF-β1 concentrations ranging up to 0.5 ng/ml.
Figure 7. CD4+ TPam3 cells are refractory to TGF-β1-mediated signals following TCR engagement.
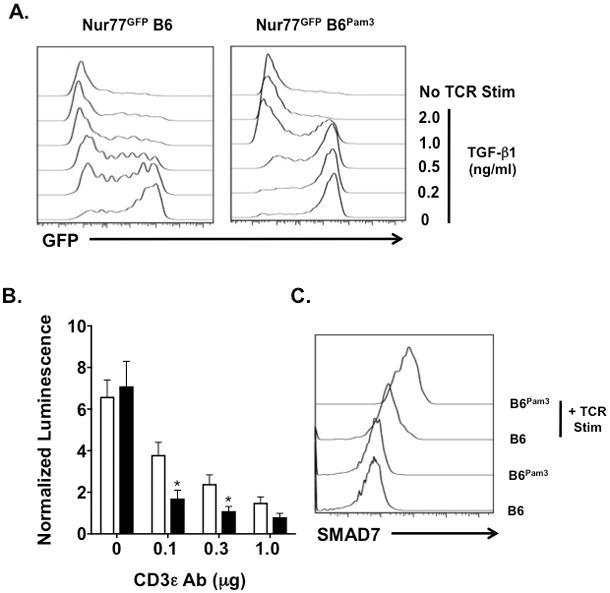
(A) Naïve untreated and naïve Pam3Cys4 cargo-loaded Nur77EGFP CD4+ T cells were cultured without or with 0.1μg/ml plate bound CD3ε Ab and 1 μg/ml soluble CD28 Abs in the absence or presence of indicated concentrations of TGF-β1. GFP expression was evaluated 24 hours after culture. (B) Naïve untreated (white bars) and naïve Pam3Cys4 cargo loaded (black bars) SMAD2/3 luciferase reporter (SBE luc) CD4+ T cells were cultured in triplicate with indicated amounts of plate bound CD3ε and 1 μg/ml soluble CD28 Abs for 3 hours and 0.5 ng/ml TGF-β1. Three hours later cultures were assessed for luciferase activity. Luminescence is represented as a normalized mean ± S.D. relative to corresponding cultures without TGF-β1 and TCR stimulation (*p < 0.05). (C) Untreated CD4+ T cells and CD4+ TPam3 cells were cultured with 0.5 ng/ml TGF-β1 with or without stimulation with 0.1μg/ml plate bound CD3ε Ab stimulated for 8 hrs and then stained intracellularly with SMAD7 specific Abs. Data (A–C) are representative of at least three independent experiments.
We next examined the effects of Pam3Cys4 cargo on TGF-β-mediated signaling. In CD4+ T cells the transcription factors SMAD 2 and 3 promote TGF-β–mediated iTreg development (15). We loaded Pam3Cys4 cargo into reporter CD4+ T cells that encode a SMAD2/3 promoter driven luciferase transgene (SBE-luc) (38) (Fig. 7B). TGF-β1-stimulated luciferase expression was significantly lower in SBE-luc CD4+ TPam3 cells when compared to untreated SBE-luc CD4+ T cells in response to low to moderate TCR engagement strength. Additionally, SMAD7, which antagonizes TGF-β signaling in part by promoting the degradation of internalized TGF-β receptor complexes (39), accumulated in higher amounts in CD4+ TPam3 cells (Fig. 7C). Taken together, these data show that TGF-β receptor signaling is blunted in CD4+ TPam3 cells.
TLR2-MyD88-dependent Akt activation pathway antagonizes TGF-β1-mediated suppression of Th1 development
T cell receptor signaling triggers PI3 kinase-mediated activation of Protein Kinase B (Akt). Several reports have shown that inhibiting Akt activation either by blocking PI3 kinase or premature cessation of TCR signaling promotes iTreg development (40–42). We measured the effects of Pam3Cys4 cargo on Akt activation in B6 wild type and CD4+ T cells deficient in TLR2 and MyD88 following TCR engagement (Figs. 8A and B). Relative to untreated CD4+ T cells CD4+ TPam3 cells had significantly higher Akt activation that could be largely abated by either TLR2 or MyD88 ablation or treatment with Ly2924002, a pharmacological inhibitor of PI3 kinase that prevents Akt activation. Blunting Akt activation also inhibited CD4+ TPam3 cell bias towards Th1 differentiation in favor of gene expression patterns consistent with iTreg development as denoted in the suppression of T-bet, IL-12β2 receptor and CXCR3 transcripts and the upregulation of Foxp3, CXCR4 and SMAD2 mRNA (Fig. 8C). Additionally, when compared to TLR2−/− or MyD88−/−CD4+ T cells with Pam3Cys4 cargo, B6 CD4+ TPam3 cells were more responsive to the inhibition of Akt activation. Relative to TLR2−/− and MyD88−/− CD4+ TPam3 cells Ly2924002 treatment induced larger changes in the upregulation of Foxp3 protein and the suppression of T-bet protein in B6 CD4+ TPam3 cells (Fig. 8D).
Figure 8. CD4+ TPam3 cell sensitivity to TGF-β1-stimulated gene expression patterns is dependent on a TLR2-MyD88-Akt pathway.
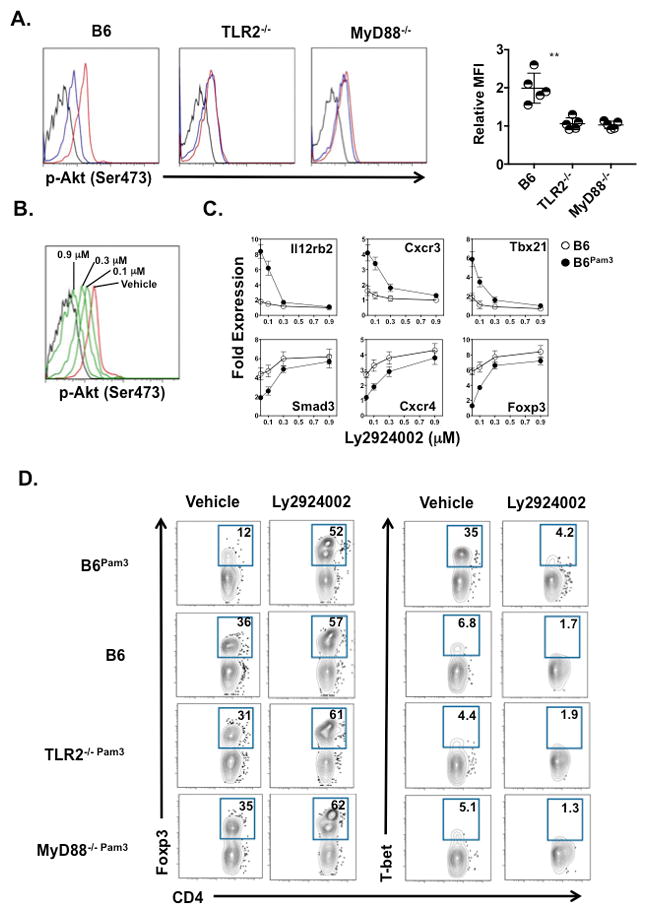
(A) Indicated naïve CD4+ T cells unactivated (black line) or activated for 10 mins with plate bound 0.1μg/ml CD3ε Ab and 1 μg/ml soluble CD28 Ab with (red line) or without (blue line) Pam3Cys4 cargo were evaluated for p-AktSer473. Left histograms are a representative result from 5 experiments. In the right panel data is shown as a individual and mean relative MFI calculated as the ratio of the MFI with Pam3Csy4 cargo to the MFI of untreated CD4+ cells ± S.D. where **p < 0.01. (B) B6 CD4+ TPam3 left untreated and unstimulated (black line) or pretreated for 30 mins with either indicated concentrations of Ly2924002 (green lines) or DMSO vehicle (red line) and then TCR stimulated as in (A) and assessed for p-AktSer473. The result is representative of 2 independent experiments. (C) B6 CD4+ T and B6 CD4+ TPam3 cells were pretreated Ly2924002 as in (B) and activated as in (A) under Th1 polarization & 0.5 ng/ml TGF-β1 and assessed for indicated transcripts 36 hrs after culture by semiquantitative real time RT-PCR. Data is normalized to transcript levels in freshly isolated untreated naïve B6 CD4+ T cells and the result shown is representative of 3 independent experiments. (D) Indicated naïve CD4+ T cells were pretreated with DMSO vehicle or 0.3 μM of Ly2924002 for 30 mins and stimulated as in (C) for 72 hours and assessed for percent abundance of Foxp3 and T-bet expression. Data are representative of 3 independent experiments.
DISCUSSION
To the best of our knowledge this is the first report of a TLR2 agonist taken up by T lymphocytes. TLRs can directly bind PAMPs but may require other pattern recognition receptors such as CD14 to induce optimal responses (43). Our data show that TLR2 and CD14 are not required for Pam3Cys4 uptake by CD4+ T cells. This is consistent with a similar observation made in dendritic cells (24). However, in contrast to this single report pharmacological inhibition of endocytosis or pinocytosis had only a negligible effect on Pam3Cys4 accumulation in naïve CD4+ T cells (data not shown). Given that Pam3Cys4, Pam2Csy4 and FSL-1 all contain lipid soluble moieties it is conceivable that agonist sequestration is first mediated by nonspecific hydrophobic interactions with the outer plasma membrane, which is then followed by translocation to the inner membrane leaflet as part of the normal homeostatic turnover of outer membrane lipids.
Our results show a direct role for TLR2 expression on CD4+ T cells in Th1 development. A potential complication in specifically dissecting TLR function on T cells is the indirect effects mediated by TLR expression on APCs. To minimize this possibility in ex vivo studies we FACS sorted through a negative selection gate that removes macrophages, dendritic cells, granulocytes, monocytes, NK cells and B cells. In vivo we assessed the contribution of TLR2 expression in host cells to anti-tumor responses. In TLR2−/− hosts that received OT2Pam3 cells there was moderate but significant late stage increase in tumor growth when compared to wildtype hosts treated with the same cells. We also observed a reduction of tumor-infiltrating IFN-γ+ OT2 cells and less OT2-mediated IFN-γ production in TLR2−/− hosts. These data suggest that some Pam3Cys4 cargo is delivered to TLR2 on host cells, possibly through agonist located on the outer plasma cell membrane. As TLR2 agonists promote APC IL-12 expression (44) priming by Pam3Cys4 carrying CD4+ T cells may boost Th1 differentiation and subsequent effector responses against tumors. This effect has been attributed to CD8+ T cell effector responses where Pam3Cys4 has been injected intravenously or around a tumor site (45, 46).
We chose to study naïve CD4+ T cells based on previous observations that in vivo activation of naïve lymphocytes induces differential responses that lead to better immunity against tumors. These include the maintenance of longer telomeres, less metabolic exhaustion and augmented resistance to TGF-β-mediated apoptosis (47). Although such factors are likely to have contributed to early OT2Pam3 cell-mediated tumor growth control there was a marked increase in tumorigenesis 18 days after treatment that correlated with high PD-L1 expression. This observation was linked to PD-1 expression on infiltrating OT2Pam3 cells, which additionally lacked IFN-γ production and mRNA transcripts that promote Th1 effector function. Nevertheless, PD-1 expression in itself may not be a reliable marker for exhaustion as previous observations suggest that PD-1 expression level or its co-expression with other immune checkpoint regulators may be more predictive of dysfunction (18). Indeed, we observed that PD-1+ OT2Pam3 cells maintained most of their IFN-γ expression relative to their PD-1− counterparts early after ACT. However, when OT2Pam3 cells expressed high levels of PD-1 and TIM-3 there was a sharp loss of IFN-γ expression linked to a sudden increase in late stage tumor growth. The functional relevance of these observations was revealed when we administered PD-L1 neutralizing Abs and assessed tumor growth. Blockade of PD-L1 led to the complete rejection of tumors in OT2Pam3 cell-treated hosts while it had only had a small effect on tumor growth in saline and OT2 cell-treated recipients. Additionally, analysis of IFN-γ production by tumor-infiltrating OT2Pam3 cells showed sharply higher production from recipients treated with PD-L1 Abs when compared to hosts that received control Abs. Therefore, these data suggest that the suppressive effects of the tumor microenvironment are likely progressive on CD4+ TPam3 cells but are likely reversed by PD-L1 blockade.
In line with previous findings TGF-β blockade suppressed Foxp3+ OT2 cell accumulation within tumors and the TDLN (35). Nevertheless, we found initially surprising that OT2Pam3 cells are substantially resistant to acquiring Foxp3 expression irrespective of TGF-β blockade. To better understand these results we conducted a series of ex vivo studies where we measured the impact of TGF-β on Th1 versus iTreg differentiation from naïve CD4+ TPam3 cells. Consistent with previous reports TGF-β1 also blocked Th1 polarization-mediated expression of T-bet and IFN-γ in untreated OT2 cells (11, 13). However, at TGF-β1 concentrations that normally induced untreated OT2 cell conversion into iTregs OT2Pam3 cells were largely able to differentiate into Th1 cells. As TCR stimulation is known to blunt TGF-β-mediated gene activity (39, 41) we also investigated if Pam3Csy4 cargo increases TCR signaling strength. Using CD4+ T cells that report TCR signaling strength we observed CD4+ TPam3 cells perceive more TCR signal for given amount of engagement when compared to untreated CD4+ T cells. Importantly, these differences were most pronounced for low strength TCR engagement, a point where iTreg generation would be most favored (41). Additionally, TGF-β1 stimulated CD4+ TPam3 cells had markedly higher levels of SMAD7. Thus our results suggest that Pam3Csy4 cargo blunts TGF-β signaling through controlling TCR-mediated regulation of SMAD activity.
Our observations of Akt activation in CD4+ TPam3 cells are consistent with several reports of MyD88-dependent PI3 kinase activation in T lymphocytes (8, 48). In this context MyD88 may play a role synonymous with CD28 where Akt activation is initiated by the recruitment of the PI3 kinase p85 regulatory subunit to an SH2 domain that has been reported on the intracellular domains of both molecules (48, 49). Akt activation promotes Th1 differentiation (50) through activating mTOR complex 1 (51, 52), which turn augments STAT4-mediated T-bet dependent transcriptional activity. In contrast, Akt activation antagonizes iTreg development (41, 42) through stimulating the nuclear export of Foxo3a and Foxo1, transcription factors critical to drive TGF-β-mediated Foxp3 expression (53). In agreement with our observations of decreased SMAD2/3 mediated-reporter activity in CD4+TPam3 cells Akt has been reported to directly interact with SMAD3 to prevent its activation and subsequent promotion of iTreg generation (54). These observations led us to that hypothesize that decisions between iTreg and Th1 lineage commitment are regulated by Akt activation levels driven by signaling inputs from TCR and TLR2 signaling pathways. We observed that when both fates were possible CD4+ TPam3 cells were significantly more sensitive to Akt activation inhibition when compared to TLR2−/− and MyD88−/− CD4+ TPam3 cells as evident by comparatively larger changes in lineage specific gene expression. Taken collectively, our findings support a model where TLR2 activation plays a pivotal role in negatively regulating TGF-β suppression on Th1 development through bolstering Akt activation (Fig. 9). In summary we report in a ACT model of TGF-β driven tumor immune evasion that naïve tumor-specific CD4+ T cells carrying a TLR2 agonist are able to differentiate into Th1 cells in vivo and control tumor growth. The sequestration of TLR ligands by engineered lymphocytes may be useful to enhance adoptive cell immunotherapies.
Figure 9. A proposed model for how TLR2 engagement promotes Th1 over iTreg differentiation from naïve CD4+ T cells.
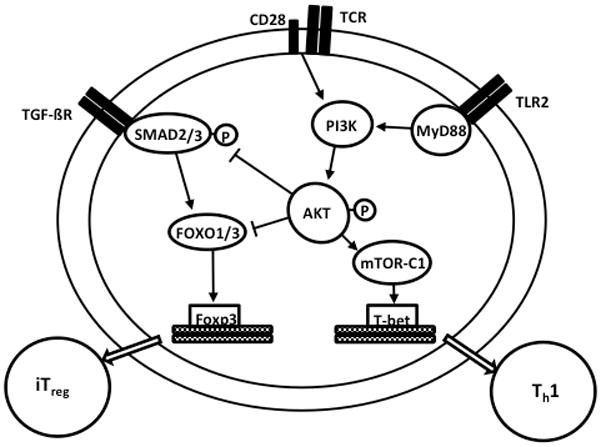
Both TCR/CD28 engagement and TLR2-MyD88 signaling stimulates PI3 kinase-mediated Akt activation as measured by phosphorylation at serine 473. Akt activation induces mTORC1 to promote T-bet activation to stimulate Th1 lineage determination. TGF-β–signaling leads to SMAD2/3 phosphorylation, which in turn drives Foxo1/Foxo3a mediated transcription of Foxp3 to specify iTreg development. SMAD3 and Foxo1/Foxo3a activity are antagonized by Akt activation. TCR/CD28 activated CD4+ TPam3 cells exposed to a TGF-β + Th1 cytokine environment (e.g., IL-12) favor Th1 over iTreg lineage determination due to additional Akt activation contributed by TLR2-MyD88 dependent PI3 kinase activation.
Supplementary Material
Acknowledgments
This work is supported by the Barnes Jewish Foundation, the Jacqueline G and William E Maritz Chair in Immunology & Oncology, and NIH grants P01AI116501-01, R01HL113436-01A1, and R01HL121218-01.
References
- 1.Adams S. Toll-like receptor agonists in cancer therapy. Immunotherapy. 2009;1:949–964. doi: 10.2217/imt.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulze HJ, Cribier B, Requena L, Reifenberger J, Ferrandiz C, Garcia Diez A, Tebbs V, McRae S. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from a randomized vehicle-controlled phase III study in Europe. Br J Dermatol. 2005;152:939–947. doi: 10.1111/j.1365-2133.2005.06486.x. [DOI] [PubMed] [Google Scholar]
- 3.Silverstein MJ, DeKernion J, Morton DL. Malignant melanoma metastatic to the bladder. Regression following intratumor injection of BCG vaccine. JAMA. 1974;229:688. [PubMed] [Google Scholar]
- 4.Baxevanis CN, I, Voutsas F, Tsitsilonis OE. Toll-like receptor agonists: current status and future perspective on their utility as adjuvants in improving anticancer vaccination strategies. Immunotherapy. 2013;5:497–511. doi: 10.2217/imt.13.24. [DOI] [PubMed] [Google Scholar]
- 5.Mercier BC, Cottalorda A, Coupet CA, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol. 2009;182:1860–1867. doi: 10.4049/jimmunol.0801167. [DOI] [PubMed] [Google Scholar]
- 6.Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029–3034. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 8.Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, Davila E. When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood. 2010;116:3494–3504. doi: 10.1182/blood-2010-02-268169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 10.Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2010;21:49–59. doi: 10.1016/j.cytogfr.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JT, Martin SL, Xia L, Gorham JD. TGF-beta 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: differential involvement of Stat4 and T-bet. J Immunol. 2005;174:5950–5958. doi: 10.4049/jimmunol.174.10.5950. [DOI] [PubMed] [Google Scholar]
- 12.Segal JG, Lee NC, Tsung YL, Norton JA, Tsung K. The role of IFN-gamma in rejection of established tumors by IL-12 : source of production and target. Cancer Res. 2002;62:4696–4703. [PubMed] [Google Scholar]
- 13.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, von Andrian UH. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–141. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Saika S, Hara T, Nomura M, Yoshimura A. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol. 2010;185:842–855. doi: 10.4049/jimmunol.0904100. [DOI] [PubMed] [Google Scholar]
- 16.Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, Sengupta S, Frank I, Parker AS, Zincke H, Blute ML, Sebo TJ, Cheville JC, Kwon ED. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66:3381–3385. doi: 10.1158/0008-5472.CAN-05-4303. [DOI] [PubMed] [Google Scholar]
- 17.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 18.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. Adoptive Cell Therapy--Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin Oncol. 2015;42:626–639. doi: 10.1053/j.seminoncol.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 21.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Yu B, Grover AC, Zeng X, Chang AE. Therapeutic effects of tumor reactive CD4+ cells generated from tumor-primed lymph nodes using anti-CD3/anti-CD28 monoclonal antibodies. J Immunother. 2002;25:304–313. doi: 10.1097/00002371-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Catron DM, Rusch LK, Hataye J, Itano AA, Jenkins MK. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J Exp Med. 2006;203:1045–1054. doi: 10.1084/jem.20051954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan S, Bijker MS, Weterings JJ, Tanke HJ, Adema GJ, van Hall T, Drijfhout JW, Melief CJ, Overkleeft HS, van der Marel GA, Filippov DV, van der Burg SH, Ossendorp F. Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J Biol Chem. 2007;282:21145–21159. doi: 10.1074/jbc.M701705200. [DOI] [PubMed] [Google Scholar]
- 25.Lee SM, Joo YD, Seo SK. Expression and Function of TLR2 on CD4 Versus CD8 T Cells. Immune Netw. 2009;9:127–132. doi: 10.4110/in.2009.9.4.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muroi M, Ohnishi T, Tanamoto K. Regions of the mouse CD14 molecule required for toll-like receptor 2- and 4-mediated activation of NF-kappa B. J Biol Chem. 2002;277:42372–42379. doi: 10.1074/jbc.M205966200. [DOI] [PubMed] [Google Scholar]
- 27.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 28.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eshima K, Chiba S, Suzuki H, Kokubo K, Kobayashi H, Iizuka M, Iwabuchi K, Shinohara N. Ectopic expression of a T-box transcription factor, eomesodermin, renders CD4(+) Th cells cytotoxic by activating both perforin- and FasL-pathways. Immunol Lett. 2012;144:7–15. doi: 10.1016/j.imlet.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Abiko K, Mandai M, Hamanishi J, Yoshioka Y, Matsumura N, Baba T, Yamaguchi K, Murakami R, Yamamoto A, Kharma B, Kosaka K, Konishi I. PD-L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clin Cancer Res. 2013;19:1363–1374. doi: 10.1158/1078-0432.CCR-12-2199. [DOI] [PubMed] [Google Scholar]
- 31.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 33.Hwang S, Cobb DA, Bhadra R, Youngblood B, Khan IA. Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J Exp Med. 2016;213:1799–1818. doi: 10.1084/jem.20151995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace A, Kapoor V, Sun J, Mrass P, Weninger W, Heitjan DF, June C, Kaiser LR, Ling LE, Albelda SM. Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res. 2008;14:3966–3974. doi: 10.1158/1078-0432.CCR-08-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gil-Guerrero L, Dotor J, Huibregtse IL, Casares N, Lopez-Vazquez AB, Rudilla F, Riezu-Boj JI, Lopez-Sagaseta J, Hermida J, Van Deventer S, Bezunartea J, Llopiz D, Sarobe P, Prieto J, Borras-Cuesta F, Lasarte JJ. In vitro and in vivo down-regulation of regulatory T cell activity with a peptide inhibitor of TGF-beta1. J Immunol. 2008;181:126–135. doi: 10.4049/jimmunol.181.1.126. [DOI] [PubMed] [Google Scholar]
- 36.Choudhry MA, Sir O, Sayeed MM. TGF-beta abrogates TCR-mediated signaling by upregulating tyrosine phosphatases in T cells. Shock. 2001;15:193–199. doi: 10.1097/00024382-200115030-00006. [DOI] [PubMed] [Google Scholar]
- 37.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin AH, Luo J, Mondshein LH, ten Dijke P, Vivien D, Contag CH, Wyss-Coray T. Global analysis of Smad2/3-dependent TGF-beta signaling in living mice reveals prominent tissue-specific responses to injury. J Immunol. 2005;175:547–554. doi: 10.4049/jimmunol.175.1.547. [DOI] [PubMed] [Google Scholar]
- 39.Giroux M, Delisle JS, O’Brien A, Hebert MJ, Perreault C. T cell activation leads to protein kinase C theta-dependent inhibition of TGF-beta signaling. J Immunol. 2010;185:1568–1576. doi: 10.4049/jimmunol.1000137. [DOI] [PubMed] [Google Scholar]
- 40.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, Shokat KM, Fisher AG, Merkenschlager M. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao G, Zhao Y, Li Z, Tang PQ, Langdon WY, Yang T, Zhang J. T cell activation threshold regulated by E3 ubiquitin ligase Cbl-b determines fate of inducible regulatory T cells. J Immunol. 2013;191:632–639. doi: 10.4049/jimmunol.1202068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Etemire E, Krull M, Hasenberg M, Reichardt P, Gunzer M. Transiently reduced PI3K/Akt activity drives the development of regulatory function in antigen-stimulated Naive T-cells. PLoS One. 2013;8:e68378. doi: 10.1371/journal.pone.0068378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teghanemt A, Prohinar P, Gioannini TL, Weiss JP. Transfer of monomeric endotoxin from MD-2 to CD14: characterization and functional consequences. J Biol Chem. 2007;282:36250–36256. doi: 10.1074/jbc.M705995200. [DOI] [PubMed] [Google Scholar]
- 44.Dabbagh K, Lewis DB. Toll-like receptors and T-helper-1/T-helper-2 responses. Curr Opin Infect Dis. 2003;16:199–204. doi: 10.1097/00001432-200306000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, Chu Y. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J Immunol. 2011;186:1963–1969. doi: 10.4049/jimmunol.1002320. [DOI] [PubMed] [Google Scholar]
- 46.Asprodites N, Zheng L, Geng D, Velasco-Gonzalez C, Sanchez-Perez L, Davila E. Engagement of Toll-like receptor-2 on cytotoxic T-lymphocytes occurs in vivo and augments antitumor activity. FASEB J. 2008;22:3628–3637. doi: 10.1096/fj.08-108274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen HH, Kim T, Song SY, Park S, Cho HH, Jung SH, Ahn JS, Kim HJ, Lee JJ, Kim HO, Cho JH, Yang DH. Naive CD8(+) T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-beta immunosuppression in the tumor microenvironment. Sci Rep. 2016;6:28208. doi: 10.1038/srep28208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pages F, Ragueneau M, Rottapel R, Truneh A, Nunes J, Imbert J, Olive D. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994;369:327–329. doi: 10.1038/369327a0. [DOI] [PubMed] [Google Scholar]
- 50.Arimura Y, Shiroki F, Kuwahara S, Kato H, Dianzani U, Uchiyama T, Yagi J. Akt is a neutral amplifier for Th cell differentiation. J Biol Chem. 2004;279:11408–11416. doi: 10.1074/jbc.M309063200. [DOI] [PubMed] [Google Scholar]
- 51.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-alpha impairs differentiation and function of TGF-beta-induced Treg cells in autoimmune diseases through Akt and Smad3 signaling pathway. J Mol Cell Biol. 2013;5:85–98. doi: 10.1093/jmcb/mjs063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.