

Abstract
Reverse translational research takes a bedside to bench approach, using sophisticated basic research to explain the biological mechanisms behind observed clinical data. For transporters, which play a role in human disease and drug response, this approach offers a distinct advantage over the typical translational research, which often falters due to inadequate in vitro and preclinical animal models. Research on ABCG2, which encodes the Breast Cancer Resistance Protein (BCRP), has benefited immensely from a reverse translational approach due to its broad implications to disease susceptibility and both therapeutic and adverse drug response. In this review, we describe the success of reverse translational research for ABCG2 and opportunities for further studies.
Keywords: Pharmacogenomics, GWAS, transporter polymorphism, pharmacokinetics, pharmacodynamics, drug development
Translational research generally takes a bench to bedside approach, in which laboratory research is translated to address pertinent problems in the clinic. This approach has led to a wealth of new information about human physiology and pathophysiology. However, the approach falters in one key area: the discovery and development of new drug therapies. What is often described as the “valley of death,” refers to the large gap between the number of potential drug targets identified through laboratory research each year and the number of new therapies on the market. Notably, budgets of the top 10 pharmaceutical companies totaled $70.5 billion in 2016, with only 22 new drugs being approved by the FDA in the same year (1, 2).
So why has translational research failed to result in more approved prescription drugs? The leading cause for failure of drugs in phase II and III clinical trials is lack of efficacy, followed by lack of safety and market need (3). Lack of efficacy can be attributed to the failure of methods used in drug development to reliably predict therapeutic drug response. For example, cell based research fails to fully recapitulate the complexity of the human body as a whole system whereas large differences in pharmacologic action between humans and preclinical animal species may result in false predictions of drug response. Further, differences in drug metabolism and transport among species may result in poor predictions of pharmacokinetics, which in turn may lead to lack of efficacy or safety in clinical trials. Because of these species differences, drug developers are increasingly relying on human genetic data as well as in vitro assays from human cells. In vitro drug metabolism studies are typically performed in primary or cryopreserved hepatocyte cultures and microsomes derived from human liver. However, hepatocytes lose much of their canalicular drug transporters during culture and undergo substantial de-differentiation when plated (4, 5). The artificial environment of the system also has no flow or shear stress, which has been shown to affect transporter expression in mice (6).
Increasingly, a vast amount of human data has been extracted and stored in publicly accessible databases, providing an enabling resource for reverse translation. Reverse translational research offers a complementary approach to traditional translational research. In particular, in reverse translation, the challenge for the researcher is to explain the observational data through detailed and in depth mechanistic studies, reproducing clinical findings in preclinical in vitro or in vivo models that can then be used for further translational research. This kind of research is especially powerful in the context of drug development, as it not only improves the safety and efficacy of newly developed drugs but also identifies potential new targets or clinical subtypes of disease that can inform future drug development. With the increasing availability of computational tools and decreasing costs of high-throughput genetic screens, reverse translational research has become a mainstay of human genetic studies, and is becoming an important tool in discovering the endogenous role of proteins as well.
Against this backdrop reverse translational research in the area of membrane transporters has advanced rapidly. Traditionally, membrane transporters have been of particular interest to drug developers because of their multiple roles in drug toxicity and pharmacokinetics. However, more recently, transporters have increasingly been recognized as enticing drug targets, as human genetic studies have revealed their roles in the pathophysiology of both rare and common disease (7). For example, genome-wide association studies focused on serum uric acid levels have revealed an essential role for URAT1 (SLC22A12) in hyperuricemia, and supported the development of a new drug targeted to the transporter (lesinurad) in the treatment of gout (8). Another transporter revealed in genome-wide association studies for its association with uric acid levels is the ATP-Binding Cassette protein, ABCG2, which encodes Breast Cancer Resistance Protein (BCRP). This efflux transporter, which contributes to the disposition of various drugs and endogenous solutes such as uric acid, estrone sulfates, and folic acid, is of broad interest in drug development. Here, we describe the successes of reverse translational research for this transporter, and include future directions for the potential of translating research on BCRP into diagnostic and therapeutic products.
The Discovery and Characterization of ABCG2
Because of its wide expression, broad substrate specificity, and complex regulation, BCRP plays an important role in a number of endogenous pathways as well as xenobiotic metabolism and excretion. Reverse translational research has made a significant impact on the understanding the diverse roles of BCRP. Importantly, the discovery of BCRP itself has its own roots in reverse translation. Unlike the discovery of many other proteins including transporters, which typically occurred in rodents followed by identification of human orthologs, BCRP was cloned directly from a human breast cancer cell line selected for multi-drug resistance, as described below.
In the late 1970s, the discoveries of P-glycoprotein (P-gp, ABCB1) and Multi-drug Resistance Associated Protein (MRP, ABCC) introduced the idea of broad-spectrum xenobiotic efflux proteins that could induce multi-drug resistance, and sparked a new wave of research focused on improving current chemotherapies. However, these transporters alone could not account for the spectrum of multi-drug resistant cancers seen in the clinic (9). One trial dosed 41 EPOCH-resistant Non-Hodgkin's lymphoma patients with both dexverapamil, a potent P-gp inhibitor, and EPOCH chemotherapy. The results showed only a moderate response in 3 patients (7%), and concluded that P-gp inhibition was insufficient to reverse drug resistance in Non-Hodgkin's lymphoma (10). Another study of verapamil, another P-gp inhibitor, and chemotherapy in previously untreated small cell lung cancer yielded similar results, with no significant difference between the control and verapamil treatment groups (11).
These results, taken with many other instances of P-gp inhibitors falling short in the clinic, indicated other mechanisms by which tumors induced multi-drug resistance. Using MCF-7 cells that exhibited resistance to both adriamycin and verapamil, Doyle, et al used RNA fingerprinting to identify a novel 72 kDa protein, thus the discovery of Breast Cancer Resistance Protein (12). This finding was quickly replicated in other human cell lines, including colon carcinoma S1, gastric carcinoma EPG-85-257, and fibrosarcoma EPF86-079 cells (9). In fact, two of these replications were so soon after the discovery of BCRP that they were thought to be novel findings and the protein was given the names ABCP (ATP-Binding Cassette Placenta) and MXR (Mitoxantrone Resistance) (13). Today, we refer to the protein by its original name, BCRP.
BCRP has since been characterized as a half-transporter, requiring the formation of a homodimer by disulfide bridges to form a fully functional transporter. Similar to other human ABC transporters, BCRP functions as an efflux transporter, using ATP-hydrolysis to pump its substrates against the concentration gradient and protect tissues from harmful metabolites and xenobiotics. In addition to variable expression in tumors, BCRP is expressed on the apical cell membranes of the placenta, brain, prostate, gastrointestinal tract, testes, ovaries, hepatocytes, renal tubules, stem cells, adrenal gland, uterus, bile ducts, gallbladder, central nervous system, and endothelium of veins and capillaries. Many factors are involved in the regulation of BCRP expression and activity, such as cholesterol content, promoter methylation, and estrogen response elements (13, 14).
ABCG2 Polymorphisms
Though a number of single nucleotide polymorphisms (SNPs) are present in the ABCG2 gene, one SNP in particular has garnered much interest because of its high prevalence in Japanese and Chinese populations and its reduced function. ABCG2 c.421C>A (BCRP Q141K) encodes a change from glutamine to lysine at the 141st amino acid of the BCRP protein, and has an allele frequency ranging from 22 to 34% in East Asian populations (15). This missense codon does not alter mRNA levels of BCRP; rather, it increases the lysosomal breakdown of the protein from the endoplasmic reticulum. Studies suggest that this is due to instability in the nucleotide-binding domain, which disrupts protein folding and increases ubiquitination and degradation (16, 17). Other variants, such as F489L and N590Y have also been shown to affect BCRP expression and function, but are much less common and therefore are not as clinically relevant (18, 19).
ABCG2 and Drug Development
ABCG2 as a Target for Multi-Drug Resistance
Once BCRP had been identified as a possible mechanism for the multi-drug resistance phenotype in cancer, many studies were performed to assess the impact of BCRP expression on clinical outcomes and chemotherapy treatment response. Interestingly, expression of BCRP in breast cancer, the namesake of the protein, was found to be highly variable in clinical tumor samples and was not a significant predictor of prognosis or progression-free survival after treatment (20). However in other cancers such as acute myeloid leukemia, non-small cell lung cancer, ovarian cancer, chronic myeloid leukemia and esophageal squamous cell carcinoma, tumor expression level of BCRP was found to have a significant association with survival and response to therapy with higher expression being linked to worse prognosis (21-24). In a panel of 150 untreated solid tumors, BCRP expression was evident in all of them, but especially high in cancers of the digestive tract, endometrium, and lung, and in melanoma (25). These findings made BCRP a potential target for inhibition in chemotherapy-resistant cancers, especially for concomitant use with BCRP substrates (FIGURE 1).
Figure 1.
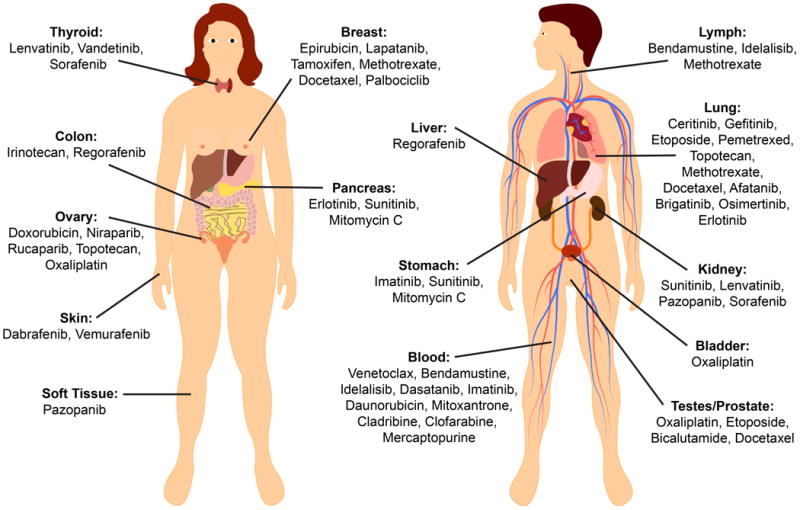
Chemotherapeutics that have shown to be BCRP substrates are summarized by their approved indications. These compounds are at risk for multi-drug resistance and may benefit from the concomitant use of BCRP inhibitors in the clinic. Sources for each chemotherapy can be found in the Supplemental Table S1. Adapted and printed with permission from Designua/Shutterstock.com.
Although the identification of BCRP as a target in the treatment of cancer was exciting in principle, thus far BCRP inhibitors have fallen prey to the “valley of death” in drug development (26). Many potent and selective BCRP inhibitors have been identified via high-throughput screens and other methods, described in detail in other reviews(9, 27, 28). Many of these inhibitors are able to re-sensitize multi-drug resistant cells to the cytotoxic effects of chemotherapeutics (9, 16). However, only a few BCRP inhibitors have made it to the clinic. As of 2015, only 5 inhibitors had been evaluated in the clinic, usually in a phase I study. Multiple clinical trials investigated lapatanib and topotecan as a method for overcoming BCRP mediated drug-resistant cancers, but showed no clinical benefit in platinum-refractory/resistant ovarian/peritoneal cancer and had increased toxicities in HER2 positive breast cancer that led to the discontinuation of the trial (26, 29, 30).
To further complicate clinical trials of BCRP inhibitors, a complex relationship was described between chemotherapies and BCRP regulation. Many studies found increased BCRP expression after treatment with chemotherapy, theoretically due to upregulation in the surviving tumor cells (31). The tyrosine kinase inhibitor gefitinib was found to decrease BCRP expression via inhibition of the EGFR pathway and re-sensitize cells to SN-38, the active metabolite of irinotecan, when given in short term (32). After long-term exposure, however, the increased SN-38 levels reactivated EGFR and actually increased BCRP expression to again reduce the levels and cytotoxic effects of SN-38 (27). These in vitro studies complicate the use of TKIs as re-sensitizing agents in the clinic, where patients would have long-term exposure to both drugs. Further, the co-administration of topotecan and sulfasalazine was shown to induce BCRP expression by promoter demethylation, suggesting a number of different mechanisms by which cancer cells upregulate BCRP expression after exposure to chemotherapeutics (33).
Thus, while reverse translational research led to the identification of BCRP as a target and even some early phase clinical trials, these trials have not resulted in an approved therapeutic to improve efficacy of anti-cancer drugs. A more complete understanding of the systems and proteins involved in the activity and regulation of BCRP may make the clinical administration of BCRP inhibitors as chemosensitizers more feasible. Further, complete characterization of the binding sites and oligomerization of BCRP may offer even more insight into the development of more specific and less toxic inhibitors.
Though the development of BCRP inhibitors has not been successful to date, some interesting studies have shown that BCRP expression can still serve as a marker of prognosis on treatment, even for drugs that are not substrates for the transporter. One study in acute myeloid leukemia showed that patients treated with cytarabine, which is not a substrate of BCRP, were much less likely to achieve complete remission if they had high BCRP expression. Two other studies confirmed these findings, showing associations between BCRP mRNA and either survival or complete response rate (34). Thus, expression of BCRP may be a biomarker for poor drug response irrespective of whether or not the chemotherapeutic agents are BCRP substrates.
Statin Exposure
The list of BCRP substrates is large and diverse. Because of its diverse substrate specificity and high expression in the intestine, kidney, and liver, BCRP plays an important role in the absorption, distribution, and excretion of many prescription drugs. While new drugs are now routinely screened as substrates/inhibitors of BCRP, this was not always the case, and thus, many older drugs including many statins are substrates of BCRP, and its variation in expression and activity can have a major impact on the efficacy and safety of these medications. In fact, there is perhaps no better example of the triumph and swift nature of reverse translational research than that of rosuvastatin and BCRP. While the average time to develop a drug is 10-12 years, the time it took to change the drug product insert based on observed ethnic differences in exposure to rosuvastatin was only two years (35). Perhaps more impressively, it would seem as though these observed ethnic differences could be almost entirely attributed to genetic polymorphisms in two membrane transporters: OATP1B1 (SLCO1B1) and BCRP.
HMG-CoA reductase inhibitors, also known as statins, were introduced in 1987 with the development of lovastatin for lowering low-density lipoprotein cholesterol (LDL-C). HMG-CoA reductase catalyzes the rate-limiting step in the cholesterol biosynthetic pathway (36). After lovastatin, synthetic statins such as atorvastatin and rosuvastatin were approved for the treatment of dyslipidemia (37). Rosuvastatin, marketed as Crestor, was approved by the FDA in August 2003 as the sixth statin on the market, with a maximum dose of 40 mg and a recommended starting dose of 10 mg daily for patients without renal impairment (38). Despite being the sixth statin on the market, Crestor is widely used and was the top statin on the market in 2015 according to revenue and sales (39).
Clinical trials for the development of rosuvastatin were done primarily in subjects of European descent, and a dose-limiting muscle toxicity of myopathy and rhabdomyolysis was observed at a rate of 0.4 percent in subjects taking 80 mg daily (40). However, in an assessment of a multinational phase II/III program, no rhabdomyolysis was attributed to rosuvastatin at doses <40 mg daily (41). Patients with conditions that increase exposure such as renal impairment or concomitant medications were found to have an increased risk of the dose-dependent toxicities of rosuvastatin (38, 40). When similar trials were performed in Japanese cohorts, a large increase in rosuvastatin exposure was observed compared to corresponding doses in Caucasian subjects (40, 42). This increase in exposure mixed with the exposure-dependent toxicities prompted AstraZeneca to release a revised drug insert suggesting a decreased starting dose for Asian subjects in March 2005, just two years after initial approval (38).
Up to this time, the differences in exposure had only been observed in the clinic but could not be fully explained. SLCO1B1 (OATP1B1) was known to take up rosuvastatin in the liver, and reduced-function variants had been previously associated with differences in other statin pharmacokinetics (43). However, many speculated that differences could also be attributed to diet and environmental factors. To best solve this problem, Lee, et al. studied rosuvastatin exposure in various ethnic groups all living within the metropolitan area of Singapore. They showed that Japanese subjects had up to twice the exposure as European subjects given the same dose, and that SLCO1B1 genotypes could not account for the observed differences (44).
This study provided strong evidence that there was a genetic factor causing the difference in exposure between the two ethnic groups, and encouraged further research into the mechanism. Unlike other statins, rosuvastatin did not undergo extensive metabolism and was excreted mostly as unchanged drug in the bile (37, 38). Among the efflux transporters that could potentially be determinants of rosuvastatin exposure, BCRP, MDR1, or MRP2, BCRP indeed transported rosuvastatin (45). Thus, further studies were done to determine the effect of BCRP genetic polymorphisms on rosuvastatin exposure. One study, in healthy Chinese males, showed that when controlling for CYP2C9 and SLCO1B1 variation, BCRP Q141K significantly increased the AUC and decreased the CL/F for rosuvastatin (46). Another study in healthy Finnish volunteers showed that even in European subjects, the Q141K polymorphism markedly affected rosuvastatin exposure (47). Recent studies have suggested that when controlling for both ABCG2 and SLCO1B1 genotype, the difference in rosuvastatin exposure between Asian and Caucasian volunteers is nullified, although the study was not powered to detect modest differences. Previous studies have shown that with larger sample sizes, there are still intrinsic ethnic differences between the two population's rosuvastatin pharmacokinetics that are not explained by BCRP and SLCO1B1 variants (48, 49).
Though the FDA and other regulatory bodies do not require BCRP genotyping before dosing rosuvastatin, and the product label suggests giving a lower dose to individuals of East Asian ancestry, the data point to genotypes being the driving safety concern for dosing rather than ethnicity. Many individuals of non-East Asian ancestries harbor the BCRP Q141K variant, and these individuals should potentially be started on lower doses. Further, East Asians without the BCRP Q141K variant or reduced OATP1B1 function may benefit from higher doses, although larger studies are still needed to verify this benefit. Clearly, studies of the BCRP polymorphism and its effect on rosuvastatin exposure and adverse drug reactions have underscored the importance of BCRP in drug disposition, especially for drugs with narrow therapeutic indices. Due to the high prescription rate of statins and their potential for rare, but life-threatening adverse effects, the FDA now recommends screening new molecular entities for their inhibition or transport by BCRP to avoid drug-drug interactions. The International Transporter Consortium has issued recommendations for the design and implementation of these drug-drug interaction studies, and suggests a pharmacogenetic approach to any new molecular entity in which BCRP plays a large role in the disposition or response due to the high allele frequencies of the reduced function BCRP Q141K variant (50, 51), especially considering the number of BCRP substrates on the World Health Organization list of essential medicines (FIGURE 2). The change in regulatory guidance for BCRP represents a true outcome of reverse translational research focused on BCRP. Beginning with the observation that Asians have increased exposure and consequently more adverse events, the mechanisms were then identified and related to genetic polymorphisms in ABCG2. Dosing recommendations were changed for Asians, and extrapolated to guidances focused on BCRP-mediated drug-drug interactions, which can phenocopy the effects of genetic polymorphisms.
Figure 2.

The allele frequencies for the reduced-function ABCG2 c.421C>A (BCRP Q141K) variant are shown for world populations (actual percentages in Supplemental Table S2) (15). This variant is common among Hispanic, Asian, and Caucasian populations and could cause complications in the administration of BCRP substrates and inhibitors, which are numerous. For example, there are 42 compounds that interact with BCRP on the World Health Organization List of Essential Medicines, listed here. An asterisk (*) indicates substrates for which BCRP activity has been shown to significantly affect pharmacokinetics in vivo. Sources for each medication can be found in Supplemental Table S4.
Sunitinib and Irinotecan Toxicities
While pre-emptive screening helps identify BCRP-mediated drug-drug interactions that can lead to issues with safety and efficacy, a number of drugs that were approved before the discovery of BCRP have toxicities that have been since attributed to variation in BCRP activity and expression. Irinotecan, used for the treatment of colorectal and lung cancers, was identified as a BCRP substrate; that is, high BCRP expression in tumors associated with non-response to the drug (52). However, a number of patients also experienced myelosuppression and early-onset diarrhea, dose-limiting toxicities of the drug. These toxicities were associated with high steady-state concentrations of irinotecan and its active metabolite, SN-38 (53). One study, screening 170 SNPs in 14 candidate genes, showed that rs2622604, a SNP in the first intron of ABCG2 correlated significantly with severe myelosuppression in patients taking irinotecan (54). Because this SNP is intronic, its relation to irinotecan pharmacokinetics is unknown, but these results suggest that the SNP may increase irinotecan exposure, perhaps by modification of BCRP expression or activity.
Similar studies were conducted for other substrates of ABCG2 with dose-limiting toxicities, such as sunitinib and gefitinib. Sunitinib, a tyrosine kinase inhibitor known to interact with BCRP, has a number of toxicities including thrombocytopenia, leukopenia, mucosal inflammation, and hand-foot syndrome (55). Risk for any of these toxicities at a grade of 2 or higher was significantly increased by reduced function BCRP variants, and specifically the BCRP Q141K variant was linked to an increased risk for grade 3 or 4 thrombocytopenia even after correcting for non-genetic risk factors. This could be due to higher exposure, as one group attributed an increased AUC of sunitinib to the BCRP Q141K variant and replicated these findings in Abcg2(-/-) mice (55, 56). Inclusion of BCRP Q141K in the population PK model for sunitinib was found to significantly improve the estimation of clearance (57).
BCRP Q141K was also linked to an increased risk for developing grade 1 or 2 diarrhea following gefitinib treatment in non-small cell lung cancer (58). These findings exemplify the importance of pharmacogenetic studies for BCRP substrates for more precise dosing of drugs with narrow therapeutic indices or dose-limiting toxicities. Though currently, there are no expert guidelines for use of BCRP genotype information in clinical dosing, such as a Clinical Pharmacogenetics Implementation Consortium paper, continued research and validation of the association of BCRP variants with toxicities to anti-cancer agents, may eventually result in clinical guidelines and genotype-driven dosing of various drugs.
The Endogenous Role of ABCG2
Due to its high levels of expression in tissues vital to the absorption, distribution, and excretion of exogenous compounds, BCRP functions as a barrier between vital organs and toxic compounds. This is especially true in some of the body's most sensitive tissues, the placenta and brain. BCRP is highly expressed in the Blood-Brain Barrier (BBB), and has been shown to protect the brain from toxic pharmaceuticals as well as endogenous compounds such as porphyrins that can be toxic at high levels and under hypoxic conditions (59).
Some of the endogenous roles and regulation of BCRP have been uncovered via in vitro studies and studies in knockout mice, which have then been translated to the clinic. Large, high-throughput screens offer a quick way to screen compounds for their ability to inhibit BCRP, but these screens tend to generate a number of false positives. Further, these large-scale screens give no information about the importance of a compound's disposition in vivo. As a prime example, almost all of the statins are substrates for the transporter, but only rosuvastatin and atorvastatin exposure seem to be altered by BCRP genotype, likely due to intrinsic differences in intestinal bioavailability and the contribution of BCRP to both the absorption and excretion of each statin (60).
Though translational studies from mice to humans have revealed much about the pharmacological role of BCRP, genome-wide association studies (GWAS) in the reverse translational direction, have revealed the endogenous roles of BCRP, and in particular, its role in uric acid disposition.
Uric Acid Levels and Gout
Gout is a painful arthritic disease that manifests as uric acid crystals in the joints of the body. Hyperuricemia or high uric acid levels in the serum (>6.8 mg/dL) is the central risk factor for developing gout, which has been associated with fatal comorbidities such as chronic kidney disease, stroke, and myocardial infarctions (61). In 2008, the first genome-wide association study (GWAS) on uric acid levels and dyslipidemia was performed, identifying genetic variants in SLC2A9 as a risk for high uric acid levels. Replication cohorts and further studies expanded on these findings, and with increasing numbers, an association between ABCG2 variants and uric acid levels was uncovered (62). Specifically, the BCRP Q141K variant was linked to high uric acid levels.
These GWAS led to the hypothesis that BCRP was a uric acid transporter and played a role in uric acid handling in the kidney and/or intestine, a function that was not previously known. Indeed, in vitro studies confirmed that BCRP is a high capacity uric acid transporter with a Km and Vmax of 8.24 ± 1.44 mM and 6.96 ± 0.89 nmol/min/mg protein, respectively (63). Many elegant studies following the initial GWAS have identified BCRP as not only an important factor in the kidney “urate transportersome” but also as a vital transporter for uric acid excretion in the intestine (64).
Since then, researchers have categorized gout and hyperuricemia into two clinical subtypes: renal under-excretion and renal overload. Renal under-excretion results from reduced excretion of urate in the kidney, potentially due to kidney disease or genetic polymorphisms in renal urate transporters. Renal overload, on the other hand, results from reduced extra-renal excretion or overproduction of uric acid (64, 65). Both subtypes have been linked to BCRP dysfunction, but recent studies in chronic kidney disease patients suggest that BCRP may be the most important factor for extra-renal excretion of uric acid, as it is the top gene that associates with uric acid levels in patients with deteriorated kidney function (66).
This distinction, stemming from a better understanding of the underlying mechanisms in urate handling, has generated many hypotheses on more precise treatment for hyperuricemia and gout. One GWAS in particular found that splitting subjects by their clinical subtype of gout revealed unique associations between genetic risk factors and clinical parameters such as fractional excretion of uric acid and urinary urate excretion, suggesting that these genes play different roles depending on the clinical manifestation of gout (67). Future studies are needed to determine the most effective treatment for each clinical subtype.
ABCG2 variation has also proven to be a useful probe in deciphering the relationship between uric acid levels and other risk factors for comorbidities. Uric acid levels have been shown to associate with comorbidities of gout such as cardiovascular disease, type 2 diabetes, and chronic kidney disease. However, the cause and effect relationship between these disease states is largely unknown. The genetic architecture of urate levels and gout has been used to create a genetic urate score that can be used to predict uric acid levels and risk for gout. These factors were not good predictors for cardiovascular disease or chronic kidney disease, suggesting that uric acid levels may not be the sole determinant of these conditions (68).
Recently, ABCG2 has also been implicated in response to the gout treatment allopurinol through GWAS. Allopurinol is a xanthine oxidase inhibitor and closely resembles uric acid and its precursor, xanthine. A GWAS in 2,027 multi-ethnic subjects linked the BCRP Q141K variant with a reduced response to allopurinol, with an independent study replicating these results, confirming adherence to the drug by accounting for plasma drug levels in subjects (69, 70). Further studies are needed to confirm the role of BCRP variation on the pharmacokinetics and pharmacodynamics of allopurinol and other xanthine oxidase inhibitors.
Other Genome-Wide Association Studies
Through GWAS, genetic variants in ABCG2 have been associated with a number of other phenotypes highlighted in Table 1. The wide array of BCRP substrates and high allele frequencies of reduced-function variants make it easier to detect BCRP-related changes in endogenous and exogenous substrate levels through GWAS. Beyond its associations with gout, uric acid levels, allopurinol response and LDL cholesterol lowering in patients taking statins, BCRP has also been associated at suggestive p-values with caffeine intake, sitting height ratio, Lipoprotein-associated phospholipase A(2) change, and dental caries. Many of these findings still require replication and functional studies to determine the mechanism by which BCRP variation plays a role, but offer further opportunities for reverse translational research (71).
Table 1. Associations Between ABCG2 Variants and Various Phenotypes at Genome-wide Level Significance.
Genetic variants within ABCG2 associate with a number of phenotypes, primarily uric acid and statin related phenotypes. Any association reaching genome-wide significance (5 × 10-8) for association studies is shown. Number of subjects represents the number of subjects in the largest meta-analysis per publication, if applicable (71). Sources for each study can be found in Supplemental Table S3.
| Phenotype | Subjects | Ethnicities | Top SNP | P-Value | Direction of Association |
|---|---|---|---|---|---|
| Allopurinol Response: SUA | 1,492 | Caucasian | rs10011796 | 2.0 × 10-8 | Worse response |
| Gout | 22,871 | Caucasian | rs2231142 | 3.3 × 10-15 | Increased Risk |
| Gout | 28,283 | Caucasian | rs2199936 | 2.6 × 10-23 | Increased Risk |
| Gout | 40,968 | Icelanders | rs2231142 | 2.8 × 10-12 | Increased Risk |
| Gout | 69,374 | Caucasians | rs1481012 | 2.0 × 10-32 | Increased Risk |
| Gout* | 19,427 | Multi-Ethnic | rs2231142 | 1.1 × 10-12 | Increased Risk |
| Gout | 4,540 | Japanese | rs2728125 | 7.2 × 10-54 | Increased Risk |
| Gout | 4,822 | Japanese | rs3114020 | 8.7 × 10-35 | Increased Risk |
| Gout | 10,902 | Caucasian | rs2231142 | 1.0 × 10-30 | Increased Risk |
| Gout | 3,103 | Han Chinese | rs1481012 | 9.0 × 10-11 | Increased Risk |
| Rosuvastatin Response: Lp-PLA2 | 2,673 | Caucasians | rs2199936 | 1.6 × 10-10 | Better Response |
| ROL gout* | 1,709 | Japanese | rs2231142 | 2.8 × 10-32 | Increased Risk |
| ROL gout | 3,659 | Japanese | rs2728104 | 5.1 × 10-33 | Increased Risk |
| RUE gout* | 1,843 | Japanese | rs2231142 | 2.5 × 10-12 | Increased Risk |
| RUE gout | 3,796 | Japanese | rs1871744 | 2.5 × 10-22 | Increased Risk |
| Statin Response: Absolute LDL-C | 6,989 | Caucasians | rs2199936 | 2.1 × 10-12 | Better Response |
| Statin Response: Fractional LDL-C | 6,989 | Caucasians | rs1481012 | 1.7 × 10-15 | Better Response |
| SUA | 22,871 | Caucasian | rs2231142 | 2.5 × 10-60 | Increased Levels |
| SUA | 28,141 | Caucasian | rs2231142 | 3.1 × 10-26 | Increased Levels |
| SUA | 8,868 | Japanese | rs4148155 | 7.1 × 10-24 | Increased Levels |
| SUA | 28,283 | Caucasian | rs2199936 | 1.2 × 10-75 | Increased Levels |
| SUA | 15,506 | Icelanders | rs2231142 | 2.3 × 10-20 | Increased Levels |
| SUA | 110,347 | Caucasians | rs2231142 | 1.0 × 10-134 | Increased Levels |
| SUA* | 39,853 | Multi-Ethnic | rs2231142 | 1.4 × 10-80 | Increased Levels |
| SUA | 12,281 | Chinese | rs2231142 | 3.3 × 10-42 | Increased Levels |
| SUA | 5,911 | Indian | rs2231142 | 2.7 × 10-10 | Increased Levels |
| SUA | 33,074 | East Asians | rs2725220 | 4.2 × 10-30 | Increased Levels |
| SUA (lean subjects) | 14,504 | Caucasian | rs2231142 | 1.6 × 10-29 | Increased Levels |
| SUA (overweight subjects) | 17,078 | Caucasian | rs2231142 | 1.6 × 10-30 | Increased Levels |
Performed as a candidate gene study, not as a genome-wide association study
Alzheimer's Disease and Amyloid Beta Deposition
GWAS represent a useful tool for probing how genetic variation can impact a phenotype, but gene expression studies are an even more direct method of determining the role of protein expression in metabolite levels and disease risk. The link between BCRP and Alzheimer's Disease (AD) was discovered via gene expression screening in human brain tissue, and has since been explored as a potential biomarker of the devastating disease.
The prevalence of AD, the most common form of dementia, is on the rise. An estimated 24 million people suffer from this disease globally, with approximately 95% of those cases being late onset. In comparison to early onset disease, which can be attributed to highly penetrant variants in genes for Amyloid Precursor Protein (APP) breakdown such as APOE and amyloid-beta (A-beta) generation, the genes responsible for late-onset AD are much more complex and therefore still being investigated (72).
AD is characterized by A-beta peptides deposited extracellularly in diffuse and neuritic plaques. However, the mechanism by which these A-beta peptides cross the BBB was unknown until 2009. Using gene expression analysis in brains from AD patients as well as Abcg2 null mice, Xiong, et al found that BCRP played an important role in preventing the deposition of these peptides in the brain (73). In particular, BCRP expression was upregulated in brain tissue from AD patients. Conversely, Abcg2 null mice showed an increased accumulation of labeled A-beta peptides compared to wild-type mice as well as an increase in NF-KB activation. Cell studies showed that A-beta peptide activated microglia could actually stimulate the expression of BCRP, further supporting the idea that BCRP protects the brain from A-beta peptides through efflux mechanisms (73, 74). Some studies have failed to replicate the original upregulation of BCRP in AD brains, although this may be attributed to which regions of the brain are studied (74).
Because of its proposed role in A-beta peptide deposition, BCRP Q141K was investigated for an association with Alzheimer's risk. Although it does associate with a small increase in risk, this association is most pronounced in those carrying the APOE4 risk variant (75). However, replication and validation are still necessary.
Summary and Conclusions
BCRP was originally discovered via reverse translational research as a possible mechanism for multi-drug resistance in cancer treatment because of its efficient efflux of a number of chemotherapeutics. Since then, it has been described as a broad-spectrum efflux transporter that regulates circulating levels of its substrates, both xenobiotic compounds and endogenous metabolites. Reduced-function variants in ABCG2, specifically BCRP Q141K, have been associated with changes in pharmacokinetics and pharmacodynamics of drugs that have resulted in reduced efficacy and increased toxicities in patients carrying these variants. Rosuvastatin is perhaps the most widely used and well known of these drugs, and differences in PK/PD were originally attributed to ethnic differences. Since then, reverse translational research has shown that genetic variation in SLCO1B1 and ABCG2 in both Asian and Caucasian patients contribute to the large variation in drug exposure and response, regardless of ethnicity.
Of the many endogenous metabolites of BCRP, uric acid has garnered quite a bit of interest in the field because of its discovery via GWAS. Genetic variants in ABCG2 associated with SUA levels in various ethnic groups, and further functional studies confirmed that BCRP and its reduced-function variants play a role in uric acid homeostasis and risk for gout and hyperuricemia. These variants, due to their strong predictive value, have since been used as probes for testing the associations between uric acid levels and comorbidities of high uric acid as well as discerning between clinical subtypes of gout. While uric acid is the major success story of discovering BCRP's endogenous role, ABCG2 variants have been associated with other phenotypes in GWAS including response to the anti-gout medication, allopurinol. These studies suggest the potential for future reverse translational studies to further elucidate the role of BCRP in the disposition of other drugs, beyond statins, methotrexate and allopurinol, which are substrates of the transporter.
BCRP's endogenous role has also been identified via gene expression studies, regardless of genotypic variation. Expression levels in brain tissues with Alzheimer's disease identified the upregulation of BCRP as a potential protective function from the disease. These studies showed that a build up of A-beta peptides could stimulate the upregulation of the transporter to protect precious brain tissue from the harmful peptide. Further studies will be needed to identify whether BCRP upregulation could be a druggable target for the prevention of Alzheimer's Disease.
Reverse translational research improves upon translational research by incorporating observed clinical data into preclinical models that not only validate these models but characterize the mechanisms behind the observed clinical phenotype. Here we have described a number of tools and studies that have successfully determined the role of BCRP as a drug target, modulator of pharmacokinetics and pharmacodynamics, and mediator of endogenous metabolite levels. This efflux transporter is of particular importance in drug development because of its role in drug disposition and drug-drug interactions, but recently has also been shown to be important in the pathogenesis of disease states. Great focus should be placed on elucidating the complex regulation and endogenous role of BCRP to better understand the dosing of its substrates as well as the potential of drugging it as a therapeutic target.
Supplementary Material
Table S1 References for Chemotherapies as BCRP Substrates: References identifying each chemotherapy as a BCRP substrate. References are given as PubMed ID number or Package Insert (PI), accessed via the FDA archives before August 28, 2017.
Table S2 Allele Frequencies of ABCG2 c.421C>A by Sub-Population: Allele frequencies of reference ABCG2 and ABCG2 c.421C>A for the subpopulations described in Figure 2 (15).
Table S3 References for Genome-Wide Associations with ABCG2 Variants: PubMed ID numbers for each study described in Table 1.
Table S4 PubMed ID Numbers for BCRP Substrates or Inhibitors on the WHO List of Essential Medications: PubMed ID numbers identifying WHO essential medications as a BCRP substrate or inhibitor. Additional PubMed ID numbers are given for medications with evidence of change in pharmacokinetics in knockout models or clinical studies.
Acknowledgments
This manuscript was funded by NIH grant R01DK103729.
Funding: This manuscript was funded by NIH grant R01DK103729. Deanna Brackman is partially funded by a UCSF Discovery Fellowship.
Footnotes
Disclaimers: None
Author Contributions: DJ Brackman and KM Giacomini wrote the manuscript.
Conflicts of Interest: Dr. Giacomini is co-founder of Apricity Therapeutics, a transporter based biotechnology company.
References
- 1.FDA. [Accessed August 28, 2017];Novel Drug Approvals for 2016. 2017 < https://www.fda.gov/drugs/developmentapprovalprocess/druginnovation/ucm483775.htm>.
- 2.Adams B. [Accessed August 28, 2017];The Top 10 Pharma R&D Budgets in 2016. 2017 < http://www.fiercebiotech.com/special-report/top-10-pharma-r-d-budgets-2016>.
- 3.Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- 4.Ramboer E, Vanhaecke T, Rogiers V, Vinken M. Primary hepatocyte cultures as prominent in vitro tools to study hepatic drug transporters. Drug metabolism reviews. 2013;45:196–217. doi: 10.3109/03602532.2012.756010. [DOI] [PubMed] [Google Scholar]
- 5.Feng B, Varma MV, Costales C, Zhang H, Tremaine L. In vitro and in vivo approaches to characterize transporter-mediated disposition in drug discovery. Expert Opin Drug Discov. 2014;9:873–90. doi: 10.1517/17460441.2014.922540. [DOI] [PubMed] [Google Scholar]
- 6.Duan Y, Weinstein AM, Weinbaum S, Wang T. Shear stress-induced changes of membrane transporter localization and expression in mouse proximal tubule cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21860–5. doi: 10.1073/pnas.1015751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov. 2015;14:543–60. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoy SM. Lesinurad: First Global Approval. Drugs. 2016;76:509–16. doi: 10.1007/s40265-016-0550-y. [DOI] [PubMed] [Google Scholar]
- 9.Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 10.Wilson WH, et al. Controlled trial of dexverapamil, a modulator of multidrug resistance, in lymphomas refractory to EPOCH chemotherapy. J Clin Oncol. 1995;13:1995–2004. doi: 10.1200/JCO.1995.13.8.1995. [DOI] [PubMed] [Google Scholar]
- 11.Milroy R. A randomised clinical study of verapamil in addition to combination chemotherapy in small cell lung cancer. West of Scotland Lung Cancer Research Group, and the Aberdeen Oncology Group. Br J Cancer. 1993;68:813–8. doi: 10.1038/bjc.1993.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doyle LA, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jani M, et al. Structure and function of BCRP, a broad specificity transporter of xenobiotics and endobiotics. Arch Toxicol. 2014;88:1205–48. doi: 10.1007/s00204-014-1224-8. [DOI] [PubMed] [Google Scholar]
- 14.Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chinese journal of cancer. 2012;31:73–99. doi: 10.5732/cjc.011.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yates A, et al. Ensembl 2016-Release 90. Nucleic Acids Res. 2016;44:D710–6. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mao Q, Unadkat JD. Role of the Breast Cancer Resistance Protein (BCRP/ABCG2) in Drug Transport-an Update. The AAPS journal. 2015;17:65–82. doi: 10.1208/s12248-014-9668-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furukawa T, et al. Major SNP (Q141K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteasomal degradations. Pharmaceutical research. 2009;26:469–79. doi: 10.1007/s11095-008-9752-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura A, et al. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 2007;98:231–9. doi: 10.1111/j.1349-7006.2006.00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vethanayagam RR, et al. Functional analysis of the human variants of breast cancer resistance protein: I206L, N590Y, and D620N. Drug metabolism and disposition: the biological fate of chemicals. 2005;33:697–705. doi: 10.1124/dmd.105.003657. [DOI] [PubMed] [Google Scholar]
- 20.Faneyte IF, et al. Expression of the breast cancer resistance protein in breast cancer. Clin Cancer Res. 2002;8:1068–74. [PubMed] [Google Scholar]
- 21.Yoh K, et al. Breast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer. Clin Cancer Res. 2004;10:1691–7. doi: 10.1158/1078-0432.ccr-0937-3. [DOI] [PubMed] [Google Scholar]
- 22.Tsunoda S, et al. ABCG2 expression is an independent unfavorable prognostic factor in esophageal squamous cell carcinoma. Oncology. 2006;71:251–8. doi: 10.1159/000106787. [DOI] [PubMed] [Google Scholar]
- 23.van den Heuvel-Eibrink MM, et al. Increased expression of the breast cancer resistance protein (BCRP) in relapsed or refractory acute myeloid leukemia (AML) Leukemia. 2002;16:833–9. doi: 10.1038/sj.leu.2402496. [DOI] [PubMed] [Google Scholar]
- 24.Tian C, et al. Common variants in ABCB1, ABCC2 and ABCG2 genes and clinical outcomes among women with advanced stage ovarian cancer treated with platinum and taxane-based chemotherapy: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;124:575–81. doi: 10.1016/j.ygyno.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diestra JE, et al. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol. 2002;198:213–9. doi: 10.1002/path.1203. [DOI] [PubMed] [Google Scholar]
- 26.Ricci JW, Lovato D, Larson RS. ABCG2 Inhibitors: Will They Find Clinical Relevance? J Develop Drugs. 2015;4:138. [Google Scholar]
- 27.Azzariti A, et al. Tyrosine kinase inhibitors and multidrug resistance proteins: interactions and biological consequences. Cancer Chemother Pharmacol. 2010;65:335–46. doi: 10.1007/s00280-009-1039-0. [DOI] [PubMed] [Google Scholar]
- 28.Hasanabady MH, Kalalinia F. ABCG2 inhibition as a therapeutic approach for overcoming multidrug resistance in cancer. J Biosci. 2016;41:313–24. doi: 10.1007/s12038-016-9601-5. [DOI] [PubMed] [Google Scholar]
- 29.Weroha SJ, et al. Phase II trial of lapatinib and topotecan (LapTop) in patients with platinum-refractory/resistant ovarian and primary peritoneal carcinoma. Gynecol Oncol. 2011;122:116–20. doi: 10.1016/j.ygyno.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin NU, et al. Randomized phase II study of lapatinib plus capecitabine or lapatinib plus topotecan for patients with HER2-positive breast cancer brain metastases. J Neurooncol. 2011;105:613–20. doi: 10.1007/s11060-011-0629-y. [DOI] [PubMed] [Google Scholar]
- 31.Stacy AE, Jansson PJ, Richardson DR. Molecular pharmacology of ABCG2 and its role in chemoresistance. Molecular pharmacology. 2013;84:655–69. doi: 10.1124/mol.113.088609. [DOI] [PubMed] [Google Scholar]
- 32.Pick A, Wiese M. Tyrosine kinase inhibitors influence ABCG2 expression in EGFR-positive MDCK BCRP cells via the PI3K/Akt signaling pathway. ChemMedChem. 2012;7:650–62. doi: 10.1002/cmdc.201100543. [DOI] [PubMed] [Google Scholar]
- 33.Bram EE, Stark M, Raz S, Assaraf YG. Chemotherapeutic drug-induced ABCG2 promoter demethylation as a novel mechanism of acquired multidrug resistance. Neoplasia. 2009;11:1359–70. doi: 10.1593/neo.91314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochemical pharmacology. 2012;83:1084–103. doi: 10.1016/j.bcp.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lipsky MS, Sharp LK. From idea to market: the drug approval process. J Am Board Fam Pract. 2001;14:362–7. [PubMed] [Google Scholar]
- 36.Endo A. A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:484–93. doi: 10.2183/pjab.86.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19:117–25. doi: 10.1111/j.1472-8206.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- 38.Crestor (R) [package insert] AstraZeneca Pharmaceuticals LP Gaithersburg, MD; 2010. < https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021366s016lbl.pdf>.
- 39.Aitken M, Kleinrock M, Pennente K, Lyle J, Nass D, Caskey L. Medicines Use and Spending in the U.S: A Review of 2015 and Outlook to 2020. QuintilesIMS, 2016; 2016. [Google Scholar]
- 40. [Accessed August 28, 2017];Drug Approval Package: Crestor (R) Application No: 021366. 2003 < https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-366_Crestor.cfm>.
- 41.Shepherd J, et al. Safety of rosuvastatin. Am J Cardiol. 2004;94:882–8. doi: 10.1016/j.amjcard.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto A, et al. Clinical effects of rosuvastatin, a new HMG-CoA reductase inhibitor, in Japanese patients with primary hypercholesterolemia: an early phase II study. J Atheroscler Thromb. 2002;9:48–56. doi: 10.5551/jat.9.48. [DOI] [PubMed] [Google Scholar]
- 43.Lee HH, Ho RH. Interindividual and interethnic variability in drug disposition: polymorphisms in organic anion transporting polypeptide 1B1 (OATP1B1; SLCO1B1) British journal of clinical pharmacology. 2017;83:1176–84. doi: 10.1111/bcp.13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee E, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clinical pharmacology and therapeutics. 2005;78:330–41. doi: 10.1016/j.clpt.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 45.Huang L, Wang Y, Grimm S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:738–42. doi: 10.1124/dmd.105.007534. [DOI] [PubMed] [Google Scholar]
- 46.Zhang W, et al. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006;373:99–103. doi: 10.1016/j.cca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clinical pharmacology and therapeutics. 2009;86:197–203. doi: 10.1038/clpt.2009.79. [DOI] [PubMed] [Google Scholar]
- 48.Wu HF, et al. Rosuvastatin Pharmacokinetics in Asian and White Subjects Wild Type for Both OATP1B1 and BCRP Under Control and Inhibited Conditions. Journal of pharmaceutical sciences. 2017;106:2751–7. doi: 10.1016/j.xphs.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugiyama Y, Maeda K, Toshimoto K. Is Ethnic Variability in the Exposure to Rosuvastatin Explained Only by Genetic Polymorphisms in OATP1B1 and BCRP or Should the Contribution of Intrinsic Ethnic Differences in OATP1B1 Be Considered? Journal of pharmaceutical sciences. 2017;106:2227–30. doi: 10.1016/j.xphs.2017.04.074. [DOI] [PubMed] [Google Scholar]
- 50.Giacomini KM, et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clinical pharmacology and therapeutics. 2013;94:23–6. doi: 10.1038/clpt.2013.12. [DOI] [PubMed] [Google Scholar]
- 51.International Transporter C, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagashima S, et al. BCRP/ABCG2 levels account for the resistance to topoisomerase I inhibitors and reversal effects by gefitinib in non-small cell lung cancer. Cancer Chemother Pharmacol. 2006;58:594–600. doi: 10.1007/s00280-006-0212-y. [DOI] [PubMed] [Google Scholar]
- 53.Innocenti F, et al. Dose-finding and pharmacokinetic study to optimize the dosing of irinotecan according to the UGT1A1 genotype of patients with cancer. J Clin Oncol. 2014;32:2328–34. doi: 10.1200/JCO.2014.55.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cha PC, et al. Single nucleotide polymorphism in ABCG2 is associated with irinotecan-induced severe myelosuppression. J Hum Genet. 2009;54:572–80. doi: 10.1038/jhg.2009.80. [DOI] [PubMed] [Google Scholar]
- 55.Mizuno T, et al. Impact of genetic variation in breast cancer resistance protein (BCRP/ABCG2) on sunitinib pharmacokinetics. Drug Metab Pharmacokinet. 2012;27:631–9. doi: 10.2133/dmpk.dmpk-12-rg-026. [DOI] [PubMed] [Google Scholar]
- 56.Low SK, et al. Association Study of a Functional Variant on ABCG2 Gene with Sunitinib-Induced Severe Adverse Drug Reaction. PloS one. 2016;11:e0148177. doi: 10.1371/journal.pone.0148177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mizuno T, et al. The effect of ABCG2 genotype on the population pharmacokinetics of sunitinib in patients with renal cell carcinoma. Ther Drug Monit. 2014;36:310–6. doi: 10.1097/FTD.0000000000000025. [DOI] [PubMed] [Google Scholar]
- 58.Cusatis G, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–42. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- 59.Krishnamurthy P, Schuetz JD. The ABC transporter Abcg2/Bcrp: role in hypoxia mediated survival. Biometals : an international journal on the role of metal ions in biology, biochemistry, and medicine. 2005;18:349–58. doi: 10.1007/s10534-005-3709-7. [DOI] [PubMed] [Google Scholar]
- 60.Ieiri I, Higuchi S, Sugiyama Y. Genetic polymorphisms of uptake (OATP1B1, 1B3) and efflux (MRP2, BCRP) transporters: implications for inter-individual differences in the pharmacokinetics and pharmacodynamics of statins and other clinically relevant drugs. Expert Opin Drug Metab Toxicol. 2009;5:703–29. doi: 10.1517/17425250902976854. [DOI] [PubMed] [Google Scholar]
- 61.Baker JF, Schumacher HR. Update on gout and hyperuricemia. International journal of clinical practice. 2010;64:371–7. doi: 10.1111/j.1742-1241.2009.02188.x. [DOI] [PubMed] [Google Scholar]
- 62.Dalbeth N, Stamp LK, Merriman TR. The genetics of gout: towards personalised medicine? BMC Med. 2017;15:108. doi: 10.1186/s12916-017-0878-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakayama A, et al. ABCG2 is a high-capacity urate transporter and its genetic impairment increases serum uric acid levels in humans. Nucleosides, nucleotides & nucleic acids. 2011;30:1091–7. doi: 10.1080/15257770.2011.633953. [DOI] [PubMed] [Google Scholar]
- 64.Cleophas MC, Joosten LA, Stamp LK, Dalbeth N, Woodward OM, Merriman TR. ABCG2 polymorphisms in gout: insights into disease susceptibility and treatment approaches. Pharmgenomics Pers Med. 2017;10:129–42. doi: 10.2147/PGPM.S105854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsuo H, et al. ABCG2 dysfunction increases the risk of renal overload hyperuricemia. Nucleosides, nucleotides & nucleic acids. 2014;33:266–74. doi: 10.1080/15257770.2013.866679. [DOI] [PubMed] [Google Scholar]
- 66.Bhatnagar V, et al. Analysis of ABCG2 and other urate transporters in uric acid homeostasis in chronic kidney disease: potential role of remote sensing and signaling. Clin Kidney J. 2016;9:444–53. doi: 10.1093/ckj/sfw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsuo H, et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Annals of the rheumatic diseases. 2016;75:652–9. doi: 10.1136/annrheumdis-2014-206191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Q, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet. 2010;3:523–30. doi: 10.1161/CIRCGENETICS.109.934455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wen CC, et al. Genome-wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clinical pharmacology and therapeutics. 2015 doi: 10.1002/cpt.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roberts RL, et al. ABCG2 loss-of-function polymorphism predicts poor response to allopurinol in patients with gout. Pharmacogenomics J. 2017;17:201–3. doi: 10.1038/tpj.2015.101. [DOI] [PubMed] [Google Scholar]
- 71.T B, et al. [Accessed August 28, 2017];The NHGRI-EBI Catalog of Published Genome-Wide Association Studies. < http://www.ebi.ac.uk/gwas>. v1.0.
- 72.Reitz C, Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochemical pharmacology. 2014;88:640–51. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiong H, et al. ABCG2 is upregulated in Alzheimer's brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J Neurosci. 2009;29:5463–75. doi: 10.1523/JNEUROSCI.5103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abuznait AH, Kaddoumi A. Role of ABC transporters in the pathogenesis of Alzheimer's disease. ACS Chem Neurosci. 2012;3:820–31. doi: 10.1021/cn300077c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feher A, Juhasz A, Laszlo A, Pakaski M, Kalman J, Janka Z. Association between the ABCG2 C421A polymorphism and Alzheimer's disease. Neurosci Lett. 2013;550:51–4. doi: 10.1016/j.neulet.2013.06.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 References for Chemotherapies as BCRP Substrates: References identifying each chemotherapy as a BCRP substrate. References are given as PubMed ID number or Package Insert (PI), accessed via the FDA archives before August 28, 2017.
Table S2 Allele Frequencies of ABCG2 c.421C>A by Sub-Population: Allele frequencies of reference ABCG2 and ABCG2 c.421C>A for the subpopulations described in Figure 2 (15).
Table S3 References for Genome-Wide Associations with ABCG2 Variants: PubMed ID numbers for each study described in Table 1.
Table S4 PubMed ID Numbers for BCRP Substrates or Inhibitors on the WHO List of Essential Medications: PubMed ID numbers identifying WHO essential medications as a BCRP substrate or inhibitor. Additional PubMed ID numbers are given for medications with evidence of change in pharmacokinetics in knockout models or clinical studies.