

Abstract
The β-lactam antibiotics inhibit penicillin-binding proteins (PBPs) by forming a stable, covalent, acyl-enzyme complex. During the evolution from PBPs to Class A β-lactamases, the β-lactamases acquired Glu166 to activate a catalytic water and cleave the acyl-enzyme bond. Here we present three product complex crystal structures of CTX-M-14 Class A β-lactamase with a ruthenocene-conjugated penicillin — a 0.85 Å resolution structure of E166A mutant complexed with the penilloate product, a 1.30 Å resolution complex structure of the same mutant with the penicilloate product, and a 1.18 Å resolution complex structure of S70G mutant with a penicilloate product epimer — shedding light on the catalytic mechanisms and product inhibition of PBPs and Class A β-lactamases. The E166A-penilloate complex captured the hydrogen bonding network following the protonation of the leaving group and, for the first time, unambiguously show that the ring nitrogen donates a proton to Ser130, which in turn donates a proton to Lys73. These observations indicate that in the absence of Glu166, the equivalent lysine would be neutral in PBPs and therefore capable of serving as the general base to activate the catalytic serine. Together with previous results, this structure suggests a common proton relay network shared by Class A β-lactamases and PBPs, from the catalytic serine to the lysine, and ultimately to the ring nitrogen. Additionally, the E166A-penicilloate complex reveals previously unseen conformational changes of key catalytic residues during the release of the product, and is the first structure to capture the hydrolyzed product in the presence of an unmutated catalytic serine.
Keywords: X-ray crystallography, CTX-M, Penicillin-Binding Protein, Penilloate, Penicilloate
Graphical abstract
A sub-Angstrom resolution crystal structure of CTX-M-14 Class A β-lactamase in complex with a ruthenocene-conjugated penicillin product reveals that Lys73 is neutral in the absence of Glu166. Two additional crystal structures shed light on changes in the active site during product release. These results give new insight into the catalytic mechanism of both Class A β-lactamases and penicillin-binding proteins.
Introduction
The β-lactam antibiotics inhibit penicillin-binding proteins (PBPs), which are essential for bacterial cell wall synthesis [1, 2]. The marked rise in resistance to these antibiotics over the past several decades has been particularly facilitated by bacterial production of β-lactamases [3–8], which are divided into four classes, A-D, with Classes A and C being the most commonly observed types in the clinic [9, 10]. While Class B is comprised of metallo-enzymes that require catalytic Zn ions to function and do not involve a covalent intermediate, Classes A, C, and D all share a common active site serine residue that leads to the formation of an acyl-enzyme intermediate, similar to those found in PBPs, from which serine β-lactamases are hypothesized to have evolved [10–12]. However, unlike PBPs whose acyl-enzyme adduct is stable, serine β-lactamases can catalyze the removal of the hydrolyzed β-lactams from their active site by deacylating the acyl-enzyme intermediate using a catalytic water [5, 10, 11, 13, 14]. This allows serine β-lactamases to continuously “turn over” β-lactam antibiotics, thereby conferring resistance to β-lactam antibiotics [10, 11, 13, 14].
Resistance to early β-lactam antibiotics led to the development of newer generations of β-lactams that were less susceptible to β-lactamase hydrolysis, but overuse of these drugs has led to the emergence of extended spectrum β-lactamases (ESBLs) [15, 16]. These ESBLs have the ability to hydrolyze third-generation cephalosporins such as cefotaxime, and are mostly comprised of Class A and D enzymes [15, 17]. CTX-M Class A β-lactamase is the most frequently occurring ESBL worldwide [17, 18]. Like other Class A enzymes, CTX-M contains a catalytic Ser70 residue which attacks and forms a covalent bond with the β-lactam ring (Fig. 1). The reaction pathway for the acylation half of β-lactam hydrolysis begins with the ground-state Michaelis complex and then proceeds through a high-energy acylation transition state, before forming the acyl-enzyme complex [19]. The deacylation half of the reaction proceeds through a high-energy deacylation transition state formed after a catalytic water attacks the acyl-enzyme complex, resulting in the release of the hydrolyzed β-lactam product [20]. For Class A β-lactamases, the catalytic water is activated by Glu166, the general base for the deacylation reaction [21, 22]. The E166A mutation has been shown to severely impair the deacylation rate and is often used to trap the complex at the acyl-enzyme stage in X-ray crystallographic analysis [23–26].
Figure 1. Hydrolysis mechanism of Class A β-lactamase.

The reaction pathway for the acylation half of β-lactam hydrolysis begins with the ground-state Michaelis complex (I), and then proceeds through a high-energy acylation transition state (II), before forming the acyl-enzyme complex (III). The deacylation half of the reaction begins when a high-energy deacylation transition state is formed after a catalytic water attacks the acyl-enzyme complex (IV), resulting in the release of the hydrolyzed β-lactam product (V). A proton is transferred to the ring nitrogen during the collapse of the acylation TS (II), although the ultimate source of the proton has been debated. The identities of the general bases and acids have been debated for the acylation and deacylation reactions, and they may not necessarily be the same for the two steps.
Although the catalytic mechanisms of PBPs and Class A β-lactamases have been intensely studied, several key questions persist. One such question is the protonation state of the purported general base, a lysine residue (Lys73 in Class A β-lactamases). The pKa of this lysine has been hypothesized to increase by two units due to the introduction of Glu166 in the active site during the evolution from PBPs to Class A β-lactamases [27]. Whereas the protonation state of Lys73 has been extensively analyzed in Class A β-lactamases in the presence of Glu166, similar studies on the equivalent lysine in PBPs have been limited. Direct structural analysis probing the lysine protonation state in the absence of Glu166 has yielded ambiguous results in Toho-1 E166A mutant, where neutron diffraction suggests a positively charged Lys73 incapable of carrying out the general base function in the apo enzyme [28]. Another remaining question concerning β-lactam hydrolysis is in regards to the general acid catalysis in the acylation reaction of Class A β-lactamase. During catalysis, the ring nitrogen of the substrate must accept a proton for scission of the C-N bond to occur and allow for the formation of the acyl-enzyme [27]. The role of Ser130 as this proton donor is generally accepted, but the original source of the proton that Ser130 donates is unknown, with both Lys73 and Lys234 suggested to fulfill this role [19, 21, 27, 29–31]. In addition, to date, all the product complexes of serine β-lactamases have been obtained by mutating the catalytic serine to a smaller residue such as glycine, which prevents the acylation reaction from occurring [32–34]. These mutations significantly alter the center of the catalysis in the enzyme active site. It remains unknown how the product would actually interact with the catalytic serine and other residues during its release.
To answer this question and others related to PBP and Class A β-lactamase catalysis, we have determined the X-ray crystal structures of multiple ruthenocene conjugated 6-aminopenicillinic acid (6-APA-Ru) (1, Figure 2a) product complexes using both E166A and S70G mutants of CTX-M-14 Class A β-lactamase. The 6-APA-Ru compound was used as part of our broader efforts to investigate the antibacterial properties of the ruthenocene moiety on β-lactam compounds [35]. These product complexes not only represent an important stage of the reaction pathway, but also capture the active site in a state bound by a non-covalent ligand, offering valuable information about the catalytic mechanisms of β-lactamases and PBPs, as well as the protein microenvironment. As non-covalent complexes, they also provide valuable insights into inhibitor discovery against two important antibiotic targets: β-lactamases and PBPs.
Figure 2. Structures of ruthenocene conjugated 6-aminopenicillinic acid and hydrolyzed products.

a) Intact β-lactam substrate 1. b) Hydrolyzed product of 1, ruthenocene conjugated penicilloate. c) Decarboxylated and hydrolyzed product of 1, ruthenocene conjugated penilloate. d) Penicilloate with alternative chirality at C5. Compounds 2 and 3 are termed as penicilloate and penilloate products respectively in the text.
Results
We determined three crystal structures (Table 1) of CTX-M-14 in complex with hydrolyzed products of our novel 6-APA-Ru β-lactam compound (1, Figure 2a) [35]. All three crystals belong to the P21 space group, and have two copies of CTX-M in the asymmetric unit. Initially, two different CTX-M mutants were used in an attempt to capture the 6-APA-Ru compound at different points in the reaction pathway. An E166A mutant was used to capture the 6-APA-Ru acyl-enzyme complex, as the E166A mutation impairs the ability of CTX-M to deacylate the acyl-enzyme complex, essentially trapping the compound in the acyl-enzyme state. An S70G mutation was used in the hopes of capturing the intact, unhydrolyzed, 6-APA-Ru compound in the active site, as the S70G mutation renders CTX-M catalytically inactive. Instead, both mutants captured interesting hydrolyzed products in the CTX-M active site, likely a result of the long soaking time for complex crystal preparation, particularly for the S70G mutant, as well as the remaining deacylating activity of the E166A mutant. In addition, compared to other common β-lactamase crystal growth conditions with acidic pH (e.g., pH 4–5), our crystallization buffer, at pH 7.9, is more alkaline, which can promote auto-hydrolysis of β-lactam compounds [36].
Table 1.
X-ray Data Collection and Refinement Statistics
| Data Collection | |||
| Structure (PDB ID) | 5VLE | 5TOP | 5TOY |
| Space Group | P21 | P21 | P21 |
| Cell Dimensions | |||
| a, b, c (Å) | 44.745 | 45.093 | 45.012 |
| 106.418 | 106.821 | 106.787 | |
| 47.611 | 47.933 | 47.759 | |
| α, β, γ (°) | 90 | 90 | 90 |
| 102.38 | 101.66 | 101.757 | |
| 90 | 90 | 90 | |
| Resolution (Å) | 50–0.85 | 50–1.18 | 50–1.30 |
| No. Reflections | 378347 | 133593 | 103057 |
| Rmerge (%) | 4.1 | 3.6 | 9.5 |
| I / σ(I) | 45.8 (2.3)* | 25.8 (4.3)* | 15.8 (2.9) |
| Completeness (%) | 99.6 | 97.7 | 100.0 |
| Redundancy | 6.6 (5.3) | 3.7 (3.1) | 3.4 (2.7) |
| Refinement | |||
| Resolution (Å) | 50–0.85 | 50–1.18 | 50–1.30 |
| Rwork/Rfree (%) | 11.8/12.7 | 12.9/14.7 | 14.3/16.4 |
| No. Heavy Atoms | |||
| Protein | 4456 | 4430 | 4430 |
| Ligand/Ion | 170 | 144 | 214 |
| Water | 668 | 578 | 600 |
| B-Factors (Å2) | |||
| Protein | 9.66 | 14.88 | 13.84 |
| Ligand/Ion | 9.97 | 10.65 | 9.04 |
| Water | 24.11 | 27.28 | 24.52 |
| Ramanchandran Plot | |||
| Most Favored Region(%) | 91.5 | 91.3 | 91.6 |
| Additionally Allowed (%) | 7.8 | 7.8 | 7.6 |
| Generously Allowed (%) | 0.7 | 0.9 | 0.9 |
Values in parentheses represent highest resolution shells
Ultrahigh resolution crystal structure of a penilloate product complex with E166A mutant
The first structure was solved at 0.85 Å resolution, and contained the decarboxylated product (3, Fig. 2c), a ruthenocene-conjugated penilloate, in the active site of CTX-M-14 E166A mutant for each of the two monomers in the asymmetric unit (Fig. 3a), while capturing two copies of the carboxylated product (2, Fig. 2b), a ruthenocene-conjugated penicilloate, and an additional copy of the penilloate product bound outside the active site and at the crystal packing interface. We had previously determined this structure to 1.18 Å resolution (PDB ID: 4XXR) [35]. We sought a sub-Angstrom resolution structure of the same complex in an attempt to elucidate the protonation state of Lys73 and the source of the proton donated to the substrate ring nitrogen. We found in the previous structure that the penilloate product of the ruthenocene-conjugated β-lactam compound bound readily to the active site in a non-covalent fashion [35], and that the position of the substrate five-membered ring ends up in the same position as in the covalent acyl-enzyme complex [23]. This provided an excellent model system to gain insight into the hydrogen bonding network after the general acid proton transfer of the acylation reaction has taken place. Extensive optimization of the crystallization process (e.g., soaking conditions, seeding) and sampling a large number of crystals resulted in significant improvement of the data quality. This was reflected by the low R-factors after anisotropic refinement with SHELXH (Table 1).
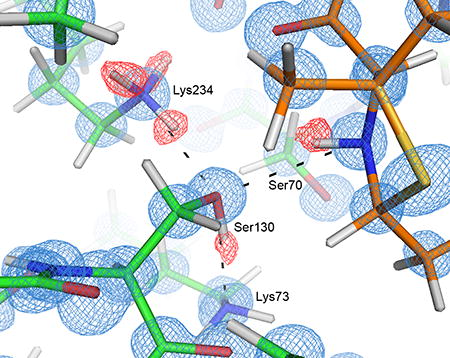
Figure 3. Hydrogen bonding network at 0.85 Å resolution in the active site of CTX-M in complex with the penilloate product.

The carbon atoms of the protein and the 6-APA-Ru penilloate product are colored in green and orange respectively. Hydrogen bonds between the product and the protein are represented by dashed lines. a) Unbiased Fo-Fc electron density map (3σ, green) showing the product molecule in the active site. b) Hydrogen bonding network involving the substrate ring nitrogen, Ser130, Lys234 and Lys73. 2Fo-Fc electron density map (blue) is contoured at 1.5σ. Unbiased Fo-Fc electron density map (2σ, red) is calculated using a hydrogen-omitted model to identify hydrogen atom positions.
The penilloate product bound in the active site in an identical fashion to our previously published 1.18 Å structure, with the 6-APA portion of the compound forming many favorable interactions with several active site residues. Away from the active site however, there were several differences between the 0.85 Å structure and our previously published structure. Most notably, whereas two copies of the penicilloate product were observed outside the active site in the previous structure, only one of them is recaptured in the current complex. The other copy is replaced by a penicilloate product of a different conformation overlapping with a penilloate product bound in the same pocket. These differences have likely resulted from the long soaking time used for the higher resolution data.
While the 0.85 Å and 1.18 Å structures are otherwise nearly identical, the ultrahigh resolution of the 0.85 Å structure allowed us to visualize almost all the riding hydrogen atoms in the Fo-Fc density maps at 1.5σ or higher contour levels for both monomers in the asymmetric unit (chain A and chain B), while many of the highly ordered hydrogen atoms on the polar side chains were visible at 1.5σ or slightly lower contour levels. Most importantly, the hydrogen atoms on the side chains for Ser130 and Lys234 were clearly visible in chain A, as was the hydrogen on the substrate ring nitrogen. These electron density peaks clearly show that the substrate ring nitrogen is protonated and serves as a donor in its HB (hydrogen bond) with Ser130, which in turn donates its proton to Lys73 in a second HB (Fig. 3b). Ser130 also functions as an acceptor in a third HB with Lys234. The hydrogen atoms on the Ser70 and Lys73 side chains in chain A were not observed in the electron density maps, probably due to the increased mobility of these two residues as reflected by their temperature factors (7.78 and 8.27 Å2 respectively), in comparison to the lower values of Ser130 and Lys234 (6.67 and 6.09 Å2 respectively). However, because it accepts a proton in its HB with Ser130, Lys73 is clearly neutral in the product complex. Lys73 also serves as donor in two other HBs with Ser130O and Asn132Oδ1 (Fig. 3a). The HB between Ser70 and Lys73 observed in many previous structures [20, 37] appears to be absent, with a distance of 3.3 Å and unfavorable orientations between the two polar groups. Meanwhile, the densities for the hydrogen atoms in the chain B active site were generally weaker than those found in chain A, and only a few side chain hydrogen atoms in the active site were observed.
An intact product complex with E166A mutant
The second product complex structure was solved at 1.30 Å resolution and captured a mixture of both the penicilloate and penilloate products in the active site of CTX-M-14 E166A, with the two overlapping perfectly except for the newly generated carboxylate group. The products can be unambiguously identified in the unbiased 2Fo-Fc electron density map at 3σ (Fig. 4a). In the 2Fo-Fc map, the penilloate product is clearly visible in the active site for both chain A and chain B at 1.5σ contour level, but density for the carboxylate group becomes apparent at contour levels below 1σ. Both monomers also show two conformations for Lys73 visible at 1.5σ, with a second conformation of Ser70 becoming apparent at 1σ and lower. Comparing the two product complex structures, and the weaker density for the newly generated carboxylate group, suggests that the penicilloate and penilloate products are both present in the current structure and bind to the protein in exactly the same fashion except for the carboxylate group, resulting in perfect superimposition with one another in the electron density maps. The two conformations of Ser70 and Lys73 correspond to the two populations of the different product complexes (Fig. 4b), and the relative occupancies for these different conformations (Conf. 1 = 0.70, and Conf. 2 = 0.30) are in agreement with the relative occupancies of the different product complexes (Penilloate = 0.75, and Penicilloate = 0.25). In particular, the newly generated carboxylate group on the product would clash with the conformation of Ser70 (conformation 1) that was observed in the 0.85 Å complex structure with the penilloate product. In comparison, conformation 2 places the serine hydroxyl group 3.2 Å away from the carboxylate group, and in the oxyanion hole formed by the backbone amide groups of Ser70 and Ser237, replacing the water molecule that usually occupies it. This results in a new HB between Ser70 and the substrate ring nitrogen. Lys73 also adopts two conformations in the 1.30 Å structure, with the terminal amine group pointing towards Ser130 when the penilloate product is present (conformation 1, Fig. 4b), and away from Ser130, towards Ser70 and forming a HB with the newly generated carboxylate group when the penicilloate product is present (albeit without any hydrogen bonding contact with Ser70 due to the conformational change of Ser70) (conformation 2, Fig. 4b). Interestingly, the penicilloate product was not found to bind away from the active site at the crystal-packing interface as was observed in our 0.85 Å structure, likely due to the shorter soaking time for the current structure (24 hrs vs. 5 days). Instead, sucrose, which was used as a cryoprotectant, appears in the structure, as well as several phosphate ions that were not present in the 0.85 Å structure.
Figure 4. Binding of the penicilloate product in the 1.3 Å resolution complex crystal structure.

The carbon atoms of the protein, the penicilloate product, and the penilloate product are colored in green, orange, and cyan respectively. Hydrogen bonds between the product and the protein are represented by black dashed lines. Hydrogen bonds between the oxyanion hole water and the protein are represented by red dashed lines. a) Unbiased Fo-Fc electron density map (3σ, green) showing the product molecules in the active site. The penilloate product (cyan) overlaps perfectly with the penicilloate product (orange). As a result, only the newly generated carboxylate group is shown in orange. b) Interactions between the newly generated carboxylate group and active site residues. 2Fo-Fc electron density map (blue) is contoured at 1.5 σ. Only the penicilloate product is shown. Two conformations are observed for Ser70 and Lys73, reflecting the mixture of products in the active site. Ser70 and Lys73 adopt conformation 1 when the penilloate product is present, and conformation 2 when the penicilloate product is present. The water is only present when the protein adopts conformation 1.
A product complex with S70G mutant
The third product complex was obtained with S70G mutant and determined to 1.18 Å resolution. The electron density for the small molecule is well defined and it appears to have only one species, the penicilloate product, at full occupancy (Fig. 5a). One oxygen atom of the carboxylate group is placed inside the oxyanion hole formed by the backbone amide groups of Gly70 and Ser237, and corresponds to the ring carbonyl oxygen from the substrate (Fig. 5b). The other carboxylate oxygen atom forms HBs with Lys73 and Ser130, and originated from the attack of a water molecule on the ring carbonyl group. At first glance, this molecule seems to be the original hydrolyzed product, identical to the one partially occupying the active site of the E166A mutant as described above (Fig. 4a). However, a closer examination revealed that this product molecule has a different chirality at C5; the carbon atom bound to the ring nitrogen, sulfur, and the newly generated carboxylate group (4, Fig. 2d). It should be mentioned that a similar product molecule with alternative chirality at C5 (S in contrast to R in the intact substrate) was previously observed in another CTX-M S70G product complex structure with penicillin, as well as a product complex with PBP3 [33, 38]. For PBP3, the conversion from 5R to 5S stereochemistry in the product was found to occur over time after the initial hydrolysis and was hypothesized to follow a non-enzymatic pathway. Additionally, one copy of the hydrolyzed product with the expected chirality (5R) was found in our structure at the crystal-packing interface. This suggests that the alternative epimer (5S) may bind preferentially over the original product with the expected chirality (5R). Another unique feature of the S70G product complex is that the ring nitrogen forms a HB with Ser237, rather than Ser130 as observed in the previous two product complexes. Interactions between the protein and the different product molecules are largely similar except for the region surrounding the newly generated carboxylate group (Fig. 5c).
Figure 5. Crystal structure of the S70G mutant in complex with the penicilloate product.

The carbon atoms of the protein and the penicilloate product are colored in orange and cyan respectively. Hydrogen bonds between the product and the protein are represented by dashed lines. a) Unbiased Fo-Fc electron density map (3σ, green) showing the product molecule in the active site. b) Interactions between the newly generated carboxylate group and active site residues. c) The penicilloate product from the E166A complex (magenta) superimposed onto the S70G complex, showing a different chirality for the C5 atom on the five-membered ring.
Discussion
Lys73 protonation state in β-lactamases and PBPs
The acylation reaction of Class A β-lactamases and PBPs involve the extraction of a proton from the catalytic serine (Ser70 in Class A β-lactamases) and the addition of a proton to the β-lactam ring nitrogen [20, 27]. The fate of the first proton, and the origin of the second, has been the subject of many experimental and computational studies [19, 21, 27, 29–31, 37, 39]. Lying at the center of these questions is the protonation state of the purported general base, Lys73 in Class A β-lactamases. Whereas recent ultrahigh resolution X-ray crystallography and neutron diffraction analysis has shed light on Lys73’s protonation state in Class A β-lactamases by revealing the hydrogen atoms [19, 37, 40–42], similar structural analysis has proven challenging for PBPs. In our current study, the use of the E166A mutant mimics the protein microenvironment in the PBP active site. Glu166 was acquired by Class A β-lactamases during their evolution from PBPs, and the catalytic machineries of these two groups of enzymes are otherwise nearly identical (Fig. 6) [7, 12, 43]. Our 0.85 Å resolution structure suggests that in the absence of Glu166, Lys73 and its counterpart in PBP would be neutral, at least in the non-covalent Michaelis complex, allowing it to serve as the general base to deprotonate Ser70. There are significant similarities between the penilloate product complex and the Michaelis complex. Both are non-covalent complexes with the key catalytic machinery, Ser70 and Lys73, isolated from bulk solvent [34]. The ligands in both complexes have one single negative charge placed in the same subpocket of the active site. The key difference is the hydrogen atom on the β-lactam ring nitrogen, which will undoubtedly alter the hydrogen bonding network involving the nitrogen, Ser130, Lys73 and Ser70. However, we deem it unlikely that this change in HB pattern will result in a different protonation state of Lys73 in the Michaelis complex, which would lead to a positively charged Lys73 and different net charges in the active sites of two similar non-covalent complexes. The penilloate complex structure supports a neutral Lys73 in the Michaelis complex, even though it does not directly prove this. Our observation is consistent with previous biochemical and QM/MM analysis of Class A β-lactamase E166A mutants [27, 44], and DD-carboxypeptidases PBP5 and 6 [45, 46], while providing the first direct structural support. This result has important implications for computational modeling of the PBP active site in enzyme analysis and inhibitor discovery.
Figure 6. Comparison of active site configurations in β-lactamase and PBP.

Structural similarities between key active site residues are shown by CTX-M-14 Class A β-lactamase (magenta) and Pseudomonas aeruginosa PBP5 (4K91, green). Residues are labeled based upon their numbering in CTX-M-14. Glu166 is labeled in red to denote the key difference between the two proteins.
The observation of a neutral Lys73 in our E166A mutant complex is reminiscent of the neutral Lys73 in a previous non-covalent complex of CTX-M-14 WT using a tetrazole-based inhibitor (Fig. 7a) [37], and underscores the evolutionary connection between PBPs and Class A β-lactamases. The introduction of Glu166 in the active site of Class A β-lactamase increases the pKa of Lys73 from ~6 to 8–8.5, making this lysine more likely to be positively charged at physiological pH and hindering its role as general base [44, 45, 47–49]. A positively charged Lys73 was indeed observed in WT CTX-M-14 by ultrahigh resolution X-ray crystallography [19, 37]. This observation, together with other experimental and computational results [39, 40], led to a hypothesis that Glu166, instead of Lys73, is the general base to deprotonate Ser70 during acylation in Class A β-lactamases. However, QM/MM calculations have suggested that upon substrate binding, a series of protonation state changes occur in the β-lactamase active site, leading to a neutral Glu166 and neutral Lys73 [27, 37, 44]. This hypothesis is supported by the aforementioned non-covalent tetrazole-based inhibitor complex with WT CTX-M-14, determined at 0.89 Å. In particular, the previous structure showed a neutral Lys73 forming a low barrier HB (LBHB) with Ser70, with a HB length of 2.53 Å and the hydrogen at equal distance to Lys73Nζ and Ser70Oγ (Fig. 7a), making Lys73 partially positively charged and Ser70 partially negatively charged. A LBHB is a special type of hydrogen bond that occurs when two functional groups equally share a proton, resulting in an unusually short (~2.5 Å) HB, with a strength several times stronger than a standard HB [50]. The observation of a Lys73-Ser70 LBHB is consistent with a previous hypothesis that such a strong HB between the general base and the nucleophilic Ser70 would stabilize the activated serine during its attack on the substrate [37, 50].
Figure 7. Influence of ligand binding on the active site hydrogen bonding network.

a) Experimentally observed hydrogen atoms in a previously determined 0.89 Å resolution non-covalent inhibitor complex with CTX-M-14. Carbon atoms of the protein and inhibitor are shown in green and cyan respectively. Hydrogen bonds are indicated by black dashed lines. Compared with the penicilloate product complex, the HB donor/acceptor roles are reversed in this complex for the ligand nitrogen, Ser130, and Lys73. Note the hydrogen atom between Ser70 and Lys73 is equally shared with a HB length of 2.53 Å. b) The penicilloate product complex of E166A mutant (carbon atoms shown in orange; HBs in purple) superimposed onto the non-covalent inhibitor complex (carbon atoms shown in green; HBs in black). Note the significant movement of Lys73. The distance between Ser70 and Lys73 is 3.3 Å in the product complex.
Residue protonation states in crystal structures are influenced by the pH of the crystallization buffer, which is ~7.5 for the current structures (after the addition of 30% sucrose as cryoprotectant to the crystallization buffer at pH 7.9) and close to the physiological pH, but high compared to the other common β-lactamase crystal growth pH range (pH 4–5). In an apo Toho-1 Class A β-lactamase E166A neutron structure, Lys73 was shown to be positively charged [28]. This has most likely resulted from the acidic crystallization pH, approximately pH 5, which is below the calculated pKa of ~6 for Lys73. At the physiological pH, Lys73 would be in its free-base form in the apo E166A mutant. Similarly, the equivalent lysine in PBP5 and 6 has been suggested to be neutral at the optimal pH of these enzymes, which is approximately pH 9 [45, 46]. Therefore, unlike the WT Class A β-lactamases, there would be no substrate/ligand-induced protonation state change for Lys73 in the E166A mutant and its counterpart in PBPs during catalysis. Taken together, our new structures and previous studies demonstrate that a neutral lysine would serve as the general base to deprotonate the catalytic serine both in the presence and absence of Glu166.
Mechanism of proton relay during acylation of Class A β-lactamase
Despite the similarities between the previous 0.89 Å resolution non-covalent tetrazole-based inhibitor complex and the current 0.85 Å resolution product complex, these structures capture two different states of the active site HB network, i.e., before and after the proton transfer of the acylation process respectively. This, together with the previously determined sub-Angstrom apo (PDB ID: 4UA6) and acylation TS analog structures (PDB ID: 4UA9) [37], has allowed us to visualize all the active site hydrogen atoms and follow their movement during the entire acylation reaction (Fig. 8). It has enabled us to investigate the origin of the proton donated to the substrate leaving group, i.e., the ring nitrogen of β-lactam compounds. For Class A β-lactamases, Lys73 and Lys234 have been alternatively proposed to be the ultimate source of this proton [27, 30, 31].
Figure 8. Comparison of active site configurations in β-lactamase complexes and PBP.

a) Superimposition of the apo protein (PDB ID: 4UA6, magenta, state 1), non-covalent complex mimicking the pre-covalent Michaelis complex (PDB ID: 4UA7, yellow, state 2), covalent acylation TS analog complex (PDB ID: 4UA9, cyan, state 3) of CTX-M-14 and the new penicilloate product complex (green, state 4) of CTX-M-14 E166A mutant. The catalytic water is represented by a red sphere. Arrows indicate the movement of active site residues from state 1 to 2 and 3. Due to the E166A mutation in the product complex, the catalytic water (not shown) and the conformation changes for Asn170 may not reflect the real movement in the actual reaction. However, Lys73 in the product complex (4) overlaps nearly perfectly with that of the acylation TS (3). b) Superposition of penicilloate/penilloate product complex with Toho-1 E166A and benzylpenicillin acyl-enzyme complex. The penicilloate/penilloate complex is shown in green, and the benzylpenicillin acyl-enzyme complex is shown in cyan. c) Experimentally determined hydrogen bonding network of the CTX-M-14 active site when (1) in the apo state, (2) bound to a non-covalent inhibitor, (3) bound covalently to a boronic acid inhibitor which mimics the acylation TS, and (4) in complex with the penilloate products.
In the apo enzyme of CTX-M-14, both Lys73 and Lys234 are positively charged with three protons on the amine group (Fig. 8c, state 1). Lys73 donates a proton in its HB with Ser70 and Ser70 serves as a HB donor to the catalytic water, which in turn donates its proton to a negatively charged Glu166. This HB network changes upon ligand binding as described above, leading to a neutral Glu166, and a neutral Lys73 forming a LBHB with Ser70 (Fig. 8c, state 2).
In this structure mimicking the pre-transfer Michaelis complex [37], a neutral Lys73 and a positively charged Lys234 both serve as a donor in their respective HB with Ser130, while Ser130’s proton is pointed towards a ring nitrogen in the tetrazole group of the inhibitor. This ring nitrogen has no proton associated with it and can only serve as a HB acceptor, and thus mimics the substrate ring nitrogen in the pre-covalent Michaelis complex. In comparison, in a 0.84 Å acylation TS analog structure using a boronic acid covalent inhibitor, Lys73 becomes positively charged following the extraction of the proton on Ser70, and moves closer to Ser130 (Fig. 8c, state 3) [37]. Again, both Lys73 and a positively charged Lys234 serve as donors in their respective HB with Ser130, which in turn donates a proton to the boronic acid oxygen mimicking the ring nitrogen. Interestingly, in our current 0.85 Å resolution product complex (Fig. 8c, state 4), Lys73 adopts a nearly identical conformation as the acylation TS but with the HB network involving the compound, Ser130, and Lys73 reversed. In this reversed state, Lys73 is now neutral, and the substrate ring nitrogen donates a proton to Ser130, which then donates a proton to Lys73.
We hypothesize that the HB network observed in the current product complex has captured the active site state immediately after the proton transfer of the acylation reaction in Class A β-lactamases, with or without Glu166. First, for the E166A mutant, the positions of the substrate ring nitrogen, Ser130, Lys73, and nearby protein/substrate atoms are nearly identical between our penilloate product complex and an acyl-enzyme complex of Toho-1 E166A and benzylpenicillin (Fig. 8b) [23]. Although the Ser70 side chain is in completely different states between the two structures, it does not form a HB with the substrate nitrogen, Ser130, or Lys73, and has minimal influence on the protonation state and HB network involving these three functional groups. The only charged functional group with potential long-range influence in both the penilloate and acyl-enzyme complex is the C3 carboxylate group, which is placed in the same area of active site. These observations suggest that the HB network in the penilloate product complex involving the substrate ring nitrogen, Ser130, and Lys73 would be the same as the acyl-enzyme complex of E166A mutant due to nearly identical configuration and environment. Second, the ligand-bound active site microenvironment is highly similar between the WT and the E166A mutant during the proton transfer from Ser70 to Lys73 and to the ring nitrogen. Our previous studies have demonstrated that the binding of a non-covalent inhibitor would induce protonation state changes in both Lys73 and Glu166, converting both from a charged state to being neutral. A neutral Lys73 is reminiscent of our observation in the penilloate complex, whereas a neutral Glu166 has diminished influence on the active site microenvironment, particularly in comparison to a negatively charged glutamate. In addition, Glu166 remains neutral in the WT enzyme during the acylation process [37]. Although Lys73 is in close proximity to Glu166 in the WT apo enzyme, it moves away from Glu166 upon ligand binding, particularly in the acylation TS. Therefore, Glu166 may not significantly influence the general acid catalysis involving Lys73, Ser130, and the substrate. The similarities between the WT and E166A active site microenvironment is further highlighted by the observations that the same non-covalent inhibitor induces a very similar Ser70-Lys73 short HB in the presence and absence of Glu166 (unpublished data), despite the different Lys73 conformations in the apo enzymes of the WT and E166A mutant.
Key differences, however, do exist between the reaction in the presence and absence of Glu166. For Class A β-lactamases, whereas the acyl-enzyme intermediate with a neutral lysine is stable in E166A mutant, it is only a transient species in the WT protein. QM/MM calculations have shown that a neutral Lys73 would take a proton from a neutral Glu166, following the formation of the acyl-enzyme intermediate [27]. This would activate a negatively charged Glu166 for the deacylation process of the reaction. For PBPs, based on the calculations using the peptide substrate that also undergoes acylation, the equivalent of Ser130 may also play a less important role during the proton-transfer process, and the Lys73 counterpart may transfer the proton extracted from Ser70 to the substrate leaving group directly or through a water molecule, without involving additional residues [45, 46].
Product Expulsion from the Active Site
It has been hypothesized that during catalysis when the β-lactam ring is opened and the penicilloate product formed, steric clashes and electrostatic repulsion between active site residues and the newly generated carboxylate group would help to expel the product from the active site [32, 33]. However, the only published structures of serine β-lactamases in complex with the penicilloate product have had the catalytic serine mutated to a smaller residue such as glycine [32, 33]. This has prevented the observation of any interactions the catalytic serine has with the newly generated carboxylate group, and may also have distorted other important interactions between the product and the enzyme.
Our 1.30 Å resolution penicilloate product complex represents the first time a β-lactam product has been captured in complex with a serine β-lactamase where the catalytic serine remains unreacted. The structure revealed that the newly generated substrate carboxylate group pushed Ser70 up and away to adopt an unusual, and potentially high energy, conformation in order to avoid clashes (Fig. 4b). Lys73 also formed a favorable HB with the carboxylate group through a new conformation (Fig. 4b), playing a direct role in shuffling the hydrolyzed product outside the active site. However, in this new configuration, Lys73 establishes fewer interactions with surrounding residues, including the loss of its HBs with Ser70 and Ser130, further suggesting that the active site bound by the penicilloate product may be an overall energetically unfavorable state, particularly compared with the substrate. This is also underscored by the 0.85 Å resolution penilloate product complex, where only the penilloate product is observed in the active site, even though both product molecules are present inside the crystal [32]. In addition, the E166A mutation used in the current study most likely helped us obtain the product complexes by eliminating one source of potential electrostatic repulsion in Class A β-lactamases.
Compared with the E166A penicilloate product complex, the S70G product complex captures the C5 atom of the five-membered substrate ring in a different chirality (S in comparison to R in the substrate and other product complexes), similar to a previous study (4, Fig. 2d) [33]. Superimposition of the two complex structures illustrates the reason: the product with the alternative C5 chirality allows the newly generated carboxylate group to establish more interactions with the oxyanion hole and Ser237. These observations suggest caution of using the Ser→Gly mutation in obtaining product complex structures as commonly done for serine β-lactamases.
Potential Product Inhibition and New Scaffold for Drug Discovery
The findings from our E166A mutant product complex structures raise questions about the potential of penilloate products to serve as β-lactamase inhibitors. Product inhibition by penicilloates and penilloates were previous reported, with some experiments showing that penilloates bind to the β-lactamases with significantly higher affinities than penicilloates, albeit both exhibiting Ki values in the mM range [51]. These results are consistent with our observations that the penilloate product is preferred by the protein active site and suggest that the penilloate product may provide a more suitable scaffold for lead optimization. The penilloate scaffold may also be useful for PBP inhibition as well. Based upon the similarity of the CTX-M active site with PBPs (Fig. 6), it can be expected that pencilloates would form many of the same favorable interactions with the PBP active site. A 5S-penicilloate molecule, similar to what is observed in our S70G product complex, has been shown to bind to the active site of PBP3 and serve as an inhibitor, further demonstrating the potential of different β-lactam products in novel antibiotic development targeting PBPs [38].
In summary, the acylation reaction of Class A β-lactamases and PBPs by β-lactam antibiotics has been intensely studied due to its importance in antibiotic mechanism and resistance. The 0.85 Å resolution penilloate product complex structure of a ruthenocene-conjugated penicillin has captured the HB network immediately after the acylation general acid catalysis, and for the first time, identified the hydrogen atoms on the substrate ring nitrogen and Ser130 in this state. Together with three previously determined sub-Angstrom resolution complex structures, we can now track the movement of each proton during the acylation reaction, including deprotonation of Ser70 and protonation of the substrate ring nitrogen. Meanwhile, the 1.30 Å resolution product complex structure illustrates the first original product complex with an intact catalytic serine, and demonstrates that alternative conformations of Ser70 and Lys73 aid the release of the product from the active site. Finally, the product complex of the S70G mutant highlights the potential misleading artifacts that can occur when studying catalysis using mutants, and provides valuable information for future analysis of serine β-lactamases.
Materials and Methods
Protein Purification
The CTX-M-14 E166A gene sequence was cloned into the pET9a expression vector and transformed into BL21 (DE3) competent cells for protein expression. The cells were grown in 1 liter 2XYT media containing 16 g tryptone, 10 g yeast extract, and 5 g NaCl. Before use, sorbitol and betaine were added to the media to a final concentration of 0.2 M and 5 mM respectively. Cultures were grown in the presence of 50 µg/ml kanamycin. The cultures were incubated at 37° C with shaking at 225 rpm until the OD600 reached 0.6–0.8. Protein expression was induced by the addition of 0.5 mM IPTG, and then cultures were incubated overnight at 20° C with shaking at 225 rpm. The total induction time was around 24 hrs. After overnight incubation, cells were centrifuged at 4000g for 10 min at 4° C, and then the supernatant was discarded.
The cell pellet was then resuspended in lysis buffer (10 mM MES pH 6.0, 2 mM EDTA). A protease inhibitor tablet was then crushed and dissolved in the resuspension. Cells were then lysed by sonication using 10 seconds ON and 15 seconds OFF at amplitude 10, for a total of 15 min. Lysed cells were then centrifuged at 35,000 rpm for 35 min at 4° C. The supernatant was filtered and loaded onto a 60 ml CM-sepharose ion exchange column using Buffer A (50 mM MES pH 6.0, 2 mM EDTA). After loading, the column was washed with two column volumes of Buffer A. The protein was eluted from the column by using an increasing concentration of Buffer B (50 mM MES pH 6.0, 2 mM EDTA, 150 mM NaCl) from 0–100%. SDS-PAGE was used to verify protein containing fractions. The fractions were pooled and prepared for loading onto a gel filtration column.
The pooled fractions were loaded onto a SuperDex75 16/60 gel filtration column using Buffer C (10 mM Tris pH 7.0, 50 mM NaCl, 2 mM EDTA). SDS-PAGE was used to verify protein containing fractions, and protein purity. The protein containing fractions were pooled, and concentrated to approximately 20 mg/ml. The protein was aliquoted and then flash frozen and stored at −80° C.
Protein Crystallization
CTX-M-14 E166A (20 mg/ml) was crystallized in 1.0 M potassium phosphate pH 7.9 at 20° C using hanging drops in a 1:1 ratio (protein:buffer). Single crystals formed in 3–4 days. 10 mM compound 1 dissolved in the crystallization buffer was then added to the crystal drops and allowed to soak into the crystals. Soaking times were as follows: 24 hrs for the 1.30 Å E166A structure; 4 days for the 1.18 Å S70G structure; 5 days for the 0.85 Å E166A structure. After soaking, the crystals were collected and cryo-cooled using a solution of the crystallization buffer with 30% sucrose added.
X-ray Data Collection and Refinement
Sub-Angstrom (< 1.0 Å) resolution data were collected using the 23ID-D beamline of GM/CA@APS at the Advanced Photon Source, Argonne, Illinois. Remaining data were collected using the 22-ID beamline of SER-CAT at the Advanced Photon Source, Argonne, Illinois. All data were processed using HKL2000 [52]. Sub-Angstrom resolution data were refined using SHELXL [53], and the CCP4 [54] suite was used to refine all other data. Coot [55] was used to complete the model building and refinement for all data. Figures of protein structures were made with PYMOL.
Acknowledgments
We thank Orville Pemberton for reading the manuscript. The Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory, is supported by the U.S. Department of Energy (Contract No. W-31-109-Eng-38). GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Y.C. has been supported by the NIH (AI103158). K.K and J.S. thank the National Science Centre (Poland) for their financial support, grant no. DEC-2013/11/B/ST5/00997.
Abbreviations
- PBP
penicillin-binding protein
- ESBL
extended spectrum β-lactamase
- 6-APA
6-aminopenicillinic acid
- HB
hydrogen bond
- LBHB
low barrier hydrogen bond
- QM/MM
quantum mechanics/molecular mechanics
- TS
transition state
Footnotes
Database:
Structural data are available in the PDB database under the accession numbers 5TOP, 5TOY, and 5VLE.
Conflict of Interest:
The authors declare no conflicts of interest.
Author Contributions:
E.L. and Y.C. designed the experiments. E.L. crystallized proteins, collected crystal data, and solved all structures. K.L. crystallized proteins. R.S. collected ultrahigh crystal data. J.S. and K.K. synthesized all compounds. E.L., J.S., K.K., and Y.C. wrote the manuscript.
References
- 1.Tipper DJ, Strominger JL. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci U S A. 1965;54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lovering AL, Safadi SS, Strynadka NC. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem. 2012;81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- 3.Taubes G. The Bacteria Fight Back. Science. 2008;321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- 4.Bush K, Fisher JF. Epidemiological expansion, structural studies, and clinical challenges of new beta-lactamases from gram-negative bacteria. Annu Rev Microbiol. 2011;65:455–478. doi: 10.1146/annurev-micro-090110-102911. [DOI] [PubMed] [Google Scholar]
- 5.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikolaidis I, Favini-Stabile S, Dessen A. Resistance to antibiotics targeted to the bacterial cell wall. Protein Sci. 2014;23:243–259. doi: 10.1002/pro.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith JD, Kumarasiri M, Zhang W, Hesek D, Lee M, Toth M, Vakulenko S, Fisher JF, Mobashery S, Chen Y. Structural analysis of the role of Pseudomonas aeruginosa penicillin-binding protein 5 in beta-lactam resistance. Antimicrob Agents Chemother. 2013;57:3137–3146. doi: 10.1128/AAC.00505-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Therrien C, Levesque RC. Molecular basis of antibiotic resistance and beta-lactamase inhibition by mechanism-based inactivators: perspectives and future directions. FEMS Microbiol Rev. 2000;24:251–262. doi: 10.1111/j.1574-6976.2000.tb00541.x. [DOI] [PubMed] [Google Scholar]
- 9.Livermore DM. beta-Lactamases in laboratory and clinical resistance. Clin Microbiol Rev. 1995;8:557–584. doi: 10.1128/cmr.8.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher JF, Mobashery S. Three decades of the class A beta-lactamase acyl-enzyme. Curr Protein Pept Sci. 2009;10:401–407. doi: 10.2174/138920309789351967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massova I, Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and beta-lactamases. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adediran SA, Deraniyagala SA, Xu Y, Pratt RF. Beta-secondary and solvent deuterium kinetic isotope effects on beta-lactamase catalysis. Biochemistry. 1996;35:3604–3613. doi: 10.1021/bi952107i. [DOI] [PubMed] [Google Scholar]
- 14.Frere JM. Beta-lactamases and bacterial resistance to antibiotics. Mol Microbiol. 1995;16:385–395. doi: 10.1111/j.1365-2958.1995.tb02404.x. [DOI] [PubMed] [Google Scholar]
- 15.Paterson DL, Bonomo RA. Extended-Spectrum β-Lactamases: a Clinical Update. Clin Microbiol Rev. 2005;18:657–686. doi: 10.1128/CMR.18.4.657-686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford PA. Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev. 2001;14:933–951. doi: 10.1128/CMR.14.4.933-951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonnet R. Growing Group of Extended-Spectrum β-Lactamases: the CTX-M Enzymes. Antimicrobial Agents and Chemotherapy. 2004;48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J Mol Biol. 2005;348:349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Bonnet R, Shoichet BK. The acylation mechanism of CTX-M beta-lactamase at 0.88 Å resolution. J Am Chem Soc. 2007;129:5378–5380. doi: 10.1021/ja0712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Shoichet B, Bonnet R. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J Am Chem Soc. 2005;127:5423–5434. doi: 10.1021/ja042850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 A resolution. Nature. 1992;359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 22.Escobar WA, Tan AK, Lewis ER, Fink AL. Site-directed mutagenesis of glutamate-166 in beta-lactamase leads to a branched path mechanism. Biochemistry. 1994;33:7619–7626. doi: 10.1021/bi00190a015. [DOI] [PubMed] [Google Scholar]
- 23.Shimamura T, Ibuka A, Fushinobu S, Wakagi T, Ishiguro M, Ishii Y, Matsuzawa H. Acyl-intermediate Structures of the Extended-spectrum Class A β-Lactamase, Toho-1, in Complex with Cefotaxime, Cephalothin, and Benzylpenicillin. J Biol Chem. 2002;277:46601–46608. doi: 10.1074/jbc.M207884200. [DOI] [PubMed] [Google Scholar]
- 24.Tremblay LW, Xu H, Blanchard JS. Structures of the Michaelis Complex (1.2 Å) and the Covalent Acyl Intermediate (2.0 Å) of Cefamandole Bound in the Active Sites of the Mycobacterium tuberculosis β-Lactamase K73A and E166A Mutants. Biochemistry. 2010;49:9685–9687. doi: 10.1021/bi1015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonseca F, Chudyk EI, van der Kamp MW, Correia A, Mulholland AJ, Spencer J. The Basis for Carbapenem Hydrolysis by Class A β-Lactamases: A Combined Investigation using Crystallography and Simulations. J Am Chem Soc. 2012;134:18275–18285. doi: 10.1021/ja304460j. [DOI] [PubMed] [Google Scholar]
- 26.Padayatti PS, Helfand MS, Totir MA, Carey MP, Hujer AM, Carey PR, Bonomo RA, van den Akker F. Tazobactam Forms a Stoichiometric trans-Enamine Intermediate in the E166A Variant of SHV-1 β-Lactamase: 1.63 Å Crystal Structure. Biochemistry. 2004;43:843–848. doi: 10.1021/bi035985m. [DOI] [PubMed] [Google Scholar]
- 27.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. Ab initio QM/MM study of class A beta-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc. 2005;127:15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 28.Tomanicek SJ, Blakeley MP, Cooper J, Chen Y, Afonine PV, Coates L. Neutron diffraction studies of a class A beta-lactamase Toho-1 E166A/R274N/R276N triple mutant. J Mol Biol. 2010;396:1070–1080. doi: 10.1016/j.jmb.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 29.Wladkowski BD, Chenoweth SA, Sanders JN, Krauss M, Stevens WJ. Acylation of β-Lactams by Class A β-Lactamase: An ab Initio Theoretical Study on the Effects of the Oxy-Anion Hole. J Am Chem Soc. 1997;119:6423–6431. [Google Scholar]
- 30.Atanasov BP, Mustafi D, Makinen MW. Protonation of the β-lactam nitrogen is the trigger event in the catalytic action of class A β-lactamases. Proc Natl Acad Sci U S A. 2000;97:3160–3165. doi: 10.1073/pnas.060027897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandavasi VG, Weiss KL, Cooper JB, Erskine PT, Tomanicek SJ, Ostermann A, Schrader TE, Ginell SL, Coates L. Exploring the Mechanism of beta-Lactam Ring Protonation in the Class A beta-lactamase Acylation Mechanism Using Neutron and X-ray Crystallography. J Med Chem. 2016;59:474–479. doi: 10.1021/acs.jmedchem.5b01215. [DOI] [PubMed] [Google Scholar]
- 32.Beadle BM, Trehan I, Focia PJ, Shoichet BK. Structural milestones in the reaction pathway of an amide hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC beta-lactamase. Structure. 2002;10:413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]
- 33.Delmas J, Leyssene D, Dubois D, Birck C, Vazeille E, Robin F, Bonnet R. Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J Mol Biol. 2010;400:108–120. doi: 10.1016/j.jmb.2010.04.062. [DOI] [PubMed] [Google Scholar]
- 34.Leyssene D, Delmas J, Robin F, Cougnoux A, Gibold L, Bonnet R. Noncovalent complexes of an inactive mutant of CTX-M-9 with the substrate piperacillin and the corresponding product. Antimicrob Agents Chemother. 2011;55:5660–5665. doi: 10.1128/AAC.00245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewandowski EM, Skiba J, Torelli NJ, Rajnisz A, Solecka J, Kowalski K, Chen Y. Antibacterial properties and atomic resolution X-ray complex crystal structure of a ruthenocene conjugated β-lactam antibiotic. Chem Commun. 2015;51:6186–6189. doi: 10.1039/c5cc00904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshpande AD, Baheti KG, Chatterjee NR. Degradation of β-lactam antibiotics. Curr Sci. 2004;87:1684–1695. [Google Scholar]
- 37.Nichols DA, Hargis JC, Sanishvili R, Jaishankar P, Defrees K, Smith EW, Wang KK, Prati F, Renslo AR, Woodcock HL, Chen Y. Ligand-Induced Proton Transfer and Low-Barrier Hydrogen Bond Revealed by X-ray Crystallography. J Am Chem Soc. 2015;137:8086–8095. doi: 10.1021/jacs.5b00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Berkel SS, Nettleship JE, Leung IK, Brem J, Choi H, Stuart DI, Claridge TD, McDonough MA, Owens RJ, Ren J, Schofield CJ. Binding of (5S)-penicilloic acid to penicillin binding protein 3. ACS Chem Biol. 2013;8:2112–2116. doi: 10.1021/cb400200h. [DOI] [PubMed] [Google Scholar]
- 39.Hermann JC, Hensen C, Ridder L, Mulholland AJ, Holtje HD. Mechanisms of antibiotic resistance: QM/MM modeling of the acylation reaction of a class A beta-lactamase with benzylpenicillin. J Am Chem Soc. 2005;127:4454–4465. doi: 10.1021/ja044210d. [DOI] [PubMed] [Google Scholar]
- 40.Minasov G, Wang X, Shoichet BK. An ultrahigh resolution structure of TEM-1 beta-lactamase suggests a role for Glu166 as the general base in acylation. J Am Chem Soc. 2002;124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 41.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR. Ultrahigh resolution structure of a class A beta-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J Mol Biol. 2003;328:289–301. doi: 10.1016/s0022-2836(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 42.Tomanicek SJ, Standaert RF, Weiss KL, Ostermann A, Schrader TE, Ng JD, Coates L. Neutron and X-ray crystal structures of a perdeuterated enzyme inhibitor complex reveal the catalytic proton network of the Toho-1 beta-lactamase for the acylation reaction. J Biol Chem. 2013;288:4715–4722. doi: 10.1074/jbc.M112.436238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meroueh SO, Minasov G, Lee W, Shoichet BK, Mobashery S. Structural aspects for evolution of beta-lactamases from penicillin-binding proteins. J Am Chem Soc. 2003;125:9612–9618. doi: 10.1021/ja034861u. [DOI] [PubMed] [Google Scholar]
- 44.Swaren P, Maveyraud L, Guillet V, Masson JM, Mourey L, Samama JP. Electrostatic analysis of TEM1 beta-lactamase: effect of substrate binding, steep potential gradients and consequences of site-directed mutations. Structure. 1995;3:603–613. doi: 10.1016/s0969-2126(01)00194-0. [DOI] [PubMed] [Google Scholar]
- 45.Shi Q, Meroueh SO, Fisher JF, Mobashery S. Investigation of the mechanism of the cell wall DD-carboxypeptidase reaction of penicillin-binding protein 5 of Escherichia coli by quantum mechanics/molecular mechanics calculations. J Am Chem Soc. 2008;130:9293–9303. doi: 10.1021/ja801727k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumarasiri M, Zhang W, Shi Q, Fisher JF, Mobashery S. Protonation States of Active-Site Lysines of Penicillin-Binding Protein 6 from Escherichia coli and the Mechanistic Implications. Proteins. 2014;82:1348–1358. doi: 10.1002/prot.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang W, Shi Q, Meroueh SO, Vakulenko SB, Mobashery S. Catalytic mechanism of penicillin-binding protein 5 of Escherichia coli. Biochemistry. 2007;46:10113–10121. doi: 10.1021/bi700777x. [DOI] [PubMed] [Google Scholar]
- 48.Chow C, Xu H, Blanchard JS. Kinetic Characterization of Hydrolysis of Nitrocefin, Cefoxitin, and Meropenem by β-Lactamase from Mycobacterium tuberculosis. Biochemistry. 2013;52:4097–4104. doi: 10.1021/bi400177y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Golemi-Kotra D, Meroueh SO, Kim C, Vakulenko SB, Bulychev A, Stemmler AJ, Stemmler TL, Mobashery S. The Importance of a Critical Protonation State and the Fate of the Catalytic Steps in Class A β-Lactamases and Penicillin-binding Proteins. J Biol Chem. 2004;279:34665–34673. doi: 10.1074/jbc.M313143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cleland W, Kreevoy M. Low-barrier hydrogen bonds and enzymic catalysis. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 51.Kiener PA, Waley SG. Reversible inhibitors of penicillinases. Biochem J. 1978;169:197–204. doi: 10.1042/bj1690197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Elsevier; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 53.Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 54.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 55.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]