

Abstract
Membrane potential is a principal regulator of arterial contractility. Arterial smooth muscle cells express several different types of ion channel that control membrane potential, including voltage-gated K+ (KV) channels. KV channel activation leads to membrane hyperpolarization, resulting in inhibition of voltage-dependent calcium (Ca2+) channels, a reduction in intracellular Ca2+ concentration ([Ca2+]i) and vasodilation. In contrast, KV channel inhibition leads to membrane depolarization and vasoconstriction. The ability of KV channels to regulate arterial contractility is dependent upon the number of plasma membrane-resident channels and their open probability. Here, we will discuss mechanisms that alter the surface abundance of KV channel proteins in arterial smooth muscle cells and the functional consequences of such regulation. Cellular processes that will be described include those that modulate KV channel transcription, retrograde and anterograde trafficking and protein degradation.
Keywords: smooth muscle, ion channel, vasoconstriction, trafficking, voltage-dependent K+
Introduction
Membrane potential is a key regulator of arterial contractility. 1,2 Arterial smooth muscle cells express several different types of ion channel that control membrane potential, including voltage-gated K+ (KV) channels. 3–5 KV channel activation leads to membrane hyperpolarization, which reduces the activity of voltage-dependent calcium (Ca2+) channels, leading to a reduction in [Ca2+]i and vasodilation. 5–11 In contrast, KV channel inhibition leads to membrane depolarization and vasoconstriction. Physiological stimuli, including intravascular pressure and receptor ligands regulate KV currents in arterial smooth muscle cells to modify vascular contractility. Mechanisms by which these stimuli alter KV currents may involve modification of channel expression, retrograde and anterograde trafficking, post-translational modification, degradation and activity. 7–9,12–17
The current (I) generated by a plasma membrane population of ion channels is determined by the number of channels (N), single channel open probability (Po) and single channel current (i), such that I=N.Po.i. Previous studies primarily investigated mechanisms that regulate the gating (Po) of plasma membrane ion channels. In contrast, processes that control the surface abundance (N) of ion channels and functional significance are poorly understood. The focus of this review is to summarize studies that have described mechanisms that can regulate the surface abundance (N) of KV channels in arterial smooth muscle cells.
Expression and distribution of KV channels in arterial smooth muscle cells
KV channels are tetramers of four pore-forming α subunits that can associate with accessory β-subunits. 18,19 Each KV α subunit is a six transmembrane domain (S1–S6) protein in which S5 and S6 form the central pore and S4 acts as the voltage sensor. 18,19 KV channels are a family of ~40 proteins classified into 12 subtypes (KV1-12) that can assemble as homo- or hetero-tetramers. This diversity generates a vast array of KV current phenotypes that can differ in their kinetics, amplitude and responses to different modulators. 20–23 Tissue- and species-specific expression of KV channel isoforms has also been described, which further contributes to KV current heterogeneity. 24,25
Arterial smooth muscle cells express multiple different KV channel isoforms. 9,10,24–29 Using PCR, Western blotting and immunofluorescence, KV1.2, KV1.3, KV1.5 and KV2.1 channels were identified in rat mesenteric artery smooth muscle cells, whereas KV3.2 was absent. 24 Message for KV1.1, KV1.2, KV1.4, KV1.5, KV1.6, KV2.1 and KV9.3 was detected in cultured rat pulmonary arterial smooth muscle cells. 25 Nishijima et al. reported the expression of KV1.3, 1.4, 1.5 and 1.6 channels in whole human adipose arteries, with KV1.5 a principal isoform in smooth muscle cells isolated from these vessels. 17 KV7.1, KV7.4 and KV9.3 channel message was detected when using RT-PCR in rat cerebral arteries. 30,31 Thus, arterial smooth muscle cells express multiple different KV channel isoforms, with diversity that may depend upon the anatomical location of the vascular bed.
A recent study by Kidd and co-authors (2015) demonstrated that when several different KV channel isoforms were quantified in fresh-isolated rat mesenteric artery smooth muscle cells, Kv1.5 accounted for ~60% of mRNA transcripts, with KV2.1 and KV2.2 each ~15%. 9 Using arterial biotinylation, the abundance and cellular distribution of KV1.5 and KV2.1 proteins were measured in rat resistance-size mesenteric arteries. ~50% of total KV1.5 and ~80% of total KV2.1 proteins were located in the plasma membrane. 9 Evidence obtained using pharmacological blockers indicates that these surface KV channel proteins are functional and generate K+ currents in arterial smooth muscle cells. 7,9,14,16,17. The detection of intracellular pools of KV1.5 and KV2.1 raised the possibility that the surface abundance of these channels may be dynamically altered by physiological and/or pathological stimuli that modulate KV currents. 9,14 This possibility was particularly high for KV1.5, for which a large proportion of total protein was intracellular. 9,14 As mechanisms that regulate the surface abundance of KV1 and KV2 channels in arterial smooth muscle cells are best described in the literature, this review article will primarily discuss these proteins.
Regulation of KV1 channel trafficking and surface protein in arterial smooth muscle cells
To control smooth muscle cell membrane potential, KV channels must traffic from the ER-golgi complex to the plasma membrane. A recent study reported the regulation of KV1.5 trafficking by physiological intravascular pressure and membrane potential in smooth muscle cells of rat mesenteric arteries. 9 Using a combination of Western blotting, biotinylation and immunofluorescence, the authors showed that arterial depressurization reduced surface KV1.5 protein by ~60%. Internalized KV1.5 did not accumulate in an intracellular compartment, suggesting that the protein was degraded. 9 In line with this finding, KV1.5 loss was prevented by both lysosomal and proteasomal degradation inhibitors. Physiological intravascular pressure and membrane depolarization, through the activation of CaV1.2 channels, prevented the loss of both surface and total KV1.5 protein in arteries. Concanavalin A, an internalization inhibitor, increased the plasma membrane abundance of KV1.5 protein, consistent with the concept that KV1.5 channels continuously recycle between an intracellular compartment and the plasma membrane and that membrane potential controls this process in smooth muscle cells. 9 The loss of surface KV1.5 protein reduced KV1.5 currents in isolated arterial smooth muscle cells through a process that was prevented by bafilomycin, a lysosomal degradation inhibitor. 9 To examine the functional impact of intravascular pressure on KV1.5 trafficking, mesenteric artery segments were pressurized to pre-experimental pressures of either 10 or 80 mmHg prior to measurement of contractile responses at 80 mmHg. Arteries maintained at a pre-experimental pressure of 80 mmHg constricted more to Psora-4, an inhibitor of KV1.5 channels, than those that had been maintained at 10 mmHg. These data indicate that intravascular pressure stimulates CaV1.2 channels, leading to an increase in [Ca2+]i that inhibits proteasomal and lysosomal degradation of KV1.5 channels in arterial smooth muscle cells. As KV1.5 channels continuously recycle between the plasma membrane and an intracellular compartment, Ca2+-dependent inhibition of degradation allows these proteins to return to the surface increasing their abundance and KV1.5 current density (Fig. 1). 9 Thus, membrane depolarization increases KV1.5 currents (I) in arterial smooth muscle cells through two distinct mechanisms: 1) through elevating the number (N) of channel proteins at the plasma membrane and 2) by directly increasing the open probability (P) of surface-localized channels. These mechanisms combine to oppose pressure-induced vasoconstriction.
Figure 1. Mechanisms that regulate KV1.5 channel trafficking in arterial smooth muscle cells.

KV1.5 continuously recycles between the cytosol and plasma membrane. Intravascular pressure stimulates arterial smooth muscle cell membrane depolarization, leading to CaV1.2 channel activation. The increase in [Ca2+]i inhibits KV1.5 channel degradation, allowing KV1.5 to recycle to the plasma membrane. A reduction in intravascular pressure leads to membrane hyperpolarization which promotes the degradation of KV1.5 protein through a process that involves both proteasomes and lysosomes. Angiotensin II binding to a surface receptor (ATIIR) activates protein kinase C, which stimulates the degradation of internalized KV1.5 channels, leading to a reduction in the amount of protein that is returned to the surface. Through this mechanism angiotensin II reduces surface KV1.5 protein, which decreases KV1.5 current density, leading to vasoconstriction. Green and red arrows indicate activation and inhibition, respectively.
The regulation of KV1.5 trafficking by angiotensin II (Ang II), a potent vasoconstrictor, has also been investigated in arterial smooth muscle cells. 14 Ang II reduced both total and surface KV1.5 protein without altering the relative cellular distribution of channels. Data suggested that Ang II binding to a Gq/11-coupled receptor activated protein kinase C (PKC), which reduced both surface and total KV1.5. 14,32 Ang II did not promote KV1.5 internalization, but stimulated lysosomal degradation of constitutively internalized KV1.5 protein, thereby reducing the number of channels that could recycle back to the plasma membrane, decreasing surface abundance. 14 Patch-clamp electrophysiology experiments indicated that Ang II-induced PKC activation decreased KV1.5 current density by reducing the number of functional proteins at the plasma membrane. 14 Similarly, experiments in pressurized mesenteric arteries showed that the Ang II-induced reduction in surface KV1.5 protein in arterial smooth muscle cells reduced the ability of these channels to regulate contractility (Fig. 1). 14 Thus, Ang II stimulates the degradation of KV1.5 channels, leading to a reduction in KV1.5 current density in arterial smooth muscle cells. As KV1.5 channel activation leads to vasodilation, a reduction in surface channel number promotes vasoconstriction.
These studies described how different physiological stimuli can act through distinct mechanisms to regulate KV1.5 trafficking and surface expression. Membrane depolarization stimulates CaV1.2 channels, leading to an increase in nanomolar [Ca2+]i in arterial smooth muscle cells. 9 Thus, KV1.5 channel degradation may be inhibited by proteins that are activated by [Ca2+]i within the nanomolar range, such as calmodulin. 9 Ang II has been shown to selectively stimulate protein kinase Cε (PKCε), a DAG-dependent PKC. 33 Ang II-mediated PKCε activation reduced whole-cell KV currents in smooth muscle cells and PKCε inhibition reduced Ang II-induced vasoconstriction in mesenteric artery rings. 34 Thus, Ang II may stimulate Ca2+-independent PKCε, leading to KV1.5 degradation. Collectively, these studies not only show that different physiological stimuli regulate the abundance of surface KV1.5 channels, but support the concept that KV1.5 degradation is regulated by both Ca2+-dependent and Ca2+-independent processes. Future studies will be required to understand the mechanisms by which an increase in [Ca2+]i inhibits KV1.5 degradation and protein kinase C activation stimulates KV1.5 degradation to differentially control arterial contractility.
Serotonin (5-hydroxytryptamine, 5-HT) internalized KV1.5 channels in rat pulmonary artery smooth muscle cells, as measured using immunofluorescence. 12 Consistent with this finding, 5-HT reduced KV currents in isolated smooth muscle cells and contracted pulmonary arteries. 12 Data pointed to 5-HT signaling through a Gq/11-coupled 5-HT2A receptor to stimulate a signaling cascade involving PLC, which activates PKC or tyrosine kinases to modify vascular tone. 12 KV1.5 protein coimmunoprecipitated with 5-HT2A receptors and caveolin-1, a caveolar protein, in pulmonary artery homogenate. The authors suggested that an interaction between KV1.5, 5-HT2A and caveolin-1 may underlie 5-HT-induced KV1.5 internalization, leading to a decrease in surface KV1.5 protein and pulmonary vasoconstriction. 12
KV1.2 channels coimmunoprecipitated with Postsynaptic density-95 (PSD95), a scaffolding protein, in cerebral arteries. 13 KV1.2 also co-localized with PSD95 in immunofluorescence experiments performed on cerebral artery smooth muscle cells. Antisense knockdown of PSD95 reduced KV1.2 channel protein and KV current density in cerebral artery smooth muscle cells. A peptide sequence identical to the KV1.2 PSD95 binding sequence decreased smooth muscle cell KV currents and stimulated depolarization and vasoconstriction in cerebral arteries, suggesting that an interaction with PSD95 is essential for function. 15 This study suggested that PSD95 acts as a molecular scaffold that increases the surface abundance of KV1.2 channels to control cerebral artery contractility.
KV1.5 channel activation underlies H2O2-induced vasodilation in adipose arterioles from healthy human subjects. 17,18,35,36 In contrast, in adipose arterioles from patients with coronary artery disease (CAD), H2O2-induced vasodilation is preferentially mediated through the activation of large-conductance Ca2+-activated K+ channels (BKCa). 35–40 PCR, Western blotting and immunofluorescence data identified the expression of KV1.3, KV1.4, Kv1.5 and KV1.6 proteins, with KV1.5 abundance being the highest in human adipose arteries. 17 Immunofluorescence suggested a lower abundance of surface KV1.5 protein in smooth muscle cells of CAD arteries compared to those of non-CAD subjects. KV currents sensitive to DPO-1, a KV1.5 channel specific blocker, were also smaller in smooth muscle cells of CAD patients. Accordingly, H2O2-elicited vasodilation was attenuated in CAD arteries due to the loss of KV1.5 channels. 17 A chronic elevation in reactive oxygen species (ROS), reported to reduce KV1.5 expression in hypoxia, hyperglycemia and hypertension, was proposed to underlie reduced KV1.5 surface expression and KV1.5 function in arteries from CAD subjects. 17,41–44 This study demonstrated that a reduction in KV1.5 surface expression in arterial smooth muscle cells leads to a loss of KV1.5-mediated vasodilation to H2O2 in CAD patients.
Pial arteriole smooth muscle cells from (Tg)Notch3R169C mice, a genetic model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), had larger whole-cell KV current density than control cells. 45 Isolated pial artery segments from (Tg)Notch3R169C mice also produced greater vasoconstriction in response to KV channel inhibitors than control arteries. Stimulation of surface KV1 endocytosis with HB-EGF, an epidermal growth factor receptor agonist, decreased KV current density and restored myogenic responses in pial arteries from (Tg) Notch3R169C mice. 45 Thus, an increase in surface KV1 channels that blunts pressure-induced depolarization and vasoconstriction in cerebral arteries contributes to the cerebrovascular manifestations of CADASIL. 45
Following subarachnoid hemorrhage, an increase in extracellular oxyhemoglobin stimulates cerebral artery vasospasm. 46 Immunofluorescence data showed that oxyhemoglobin stimulated KV1.5 endocytosis. 47 Consistent with this finding, oxyhemoglobin decreased 4-AP-sensitive KV currents in rabbit cerebral artery smooth muscle cells. 47 Inhibition of tyrosine kinases abolished both oxyhemoglobin-induced suppression of KV currents in isolated smooth muscle cells and oxyhemoglobin-induced constriction of isolated cerebral arteries. 47 Data suggested a model in which oxyhemoglobin reduces KV1.5 channels via a mechanism involving an increase in tyrosine kinase activity and channel endocytosis, leading to depolarization and vasoconstriction. 48–50
Mechanisms that regulate KV2 channel surface abundance
KV2 channels are important contributors to KV currents in smooth muscle cells of cerebral and mesenteric arteries. 7 KV2.1 and KV2.2 transcripts each represented the second highest level of message when several different KV1 and KV2 isoforms were quantified in fresh dissociated rat mesenteric artery smooth muscle cells. 9 Arterial biotinylation experiments revealed that KV2.1 is primarily plasma membrane-localized, with ~80% of total protein located at the surface. 9 In contrast to KV1.5, the surface abundance of KV2.1 protein was not regulated by intraluminal pressure, membrane potential or Ang II, at least during acute exposures in rat mesenteric arteries. 9,14 Thus, distinct processes control the surface abundance of KV1.5 and KV2.1 channels in arterial smooth muscle cells. Additional studies will be required to dissect out the differential mechanisms involved, but possibilities include that KV1.5 channels continuously recycle, whereas KV2.1 channels do not and that the regulatory mechanisms controlling surface abundance occur intracellularly, such as degradation. If this is the case, only once channels are internalized can their abundance be reduced to decrease surface levels of these proteins.
KV2.1 transcription was investigated in cerebral artery smooth muscle cells of Ang II-infused hypertensive rats. 7 Chronic Ang II exposure stimulated arterial depolarization and Ca2+ influx, leading to an increase in [Ca2+]i and the activation of calcineurin (CaN), a Ca2+/calmodulin-dependent protein phosphatase. Activated CaN stimulated the transcription factor NFATc3 to promote its nuclear translocation, leading to transcriptional downregulation of KV2.1. Studies suggested that while short-term Ang II exposure does not alter KV2.1 trafficking, chronic Ang II treatment reduces KV2.1 transcription and the amount of protein available for surface trafficking, leading to a decrease in current density and vasoconstriction (Fig. 2). 7,14
Figure 2. KV2.1 expression in diabetes and hypertension.
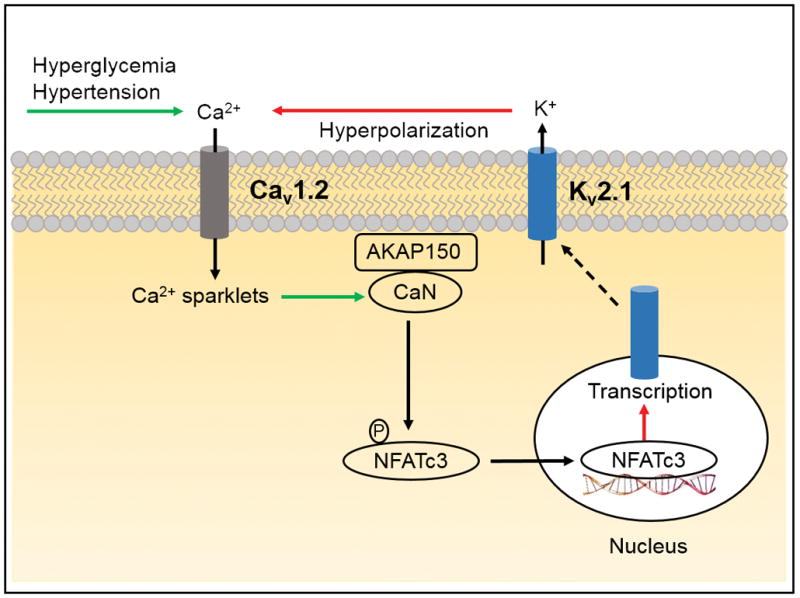
Chronic hyperglycemia and in-vivo Ang II treatment, a model of hypertension, activate CaV1.2 channel-mediated Ca2+ sparklets and calcineurin (CaN). AKAP150 anchors activated CaN to dephosphorylate and activate NFATc3. Subsequently, nuclear translocation of activated NFATc3 leads to reduced transcription of KV2.1, which decreases KV current density. Green and red arrows indicate activation and inhibition, respectively.
Western blotting and immunofluorescence experiments demonstrated that KV2.1 protein was reduced in cerebral and mesenteric artery smooth muscle cell from mice fed a high-fat diet, a model of type-2 diabetes. 16 Whole-cell currents sensitive to stromatoxin, a KV2 channel blocker, were smaller in smooth muscle cells and KV2 function was lost in pressurized arteries of diabetic mice, leading to an increase in arterial tone. 16 This reduction in KV2.1 channels was attributed to hyperglycemia-induced suppression of KV2.1 transcription. Downregulation of KV2.1 transcription was mediated via AKAP150, which anchored CaN to dephosphorylate and activate the transcription factor NFATc3. Nuclear translocation of activated NFATc3 reduced KV2.1 transcription and surface expression in smooth muscle cells. These studies provided a functional link between AKAP150-CaN/NFATc3-mediated nuclear signaling, reducing the pool of KV2.1 protein available for trafficking in arterial smooth muscle cells and enhanced arterial tone during diabetes (Fig. 2). 7,16
Conclusions
Plasma membrane ion channels regulate smooth muscle cell membrane potential and [Ca2+]i to alter arterial contractility. Understanding mechanisms that control the surface abundance of ion channels in arterial smooth muscle cells is therefore, important to determine. Although ion channel trafficking in smooth muscle cells is poorly understood, some recent studies have identified mechanisms that control KV channel surface abundance and their functional significance. Future studies should identify mechanisms that regulate the trafficking of these and other KV channel isoforms, their functional significance, pathological modification and potential for therapeutic intervention in arterial smooth muscle cells.
Acknowledgments
Funding information
NHLBI Grants HL67061, HL133256 and HL137745 to J.H.J. and an American Heart Association Fellowship Award (16POST30960010) to R. H.
This work is supported by NHLBI grants HL67061, HL133256 and HL137745 to J.H.J. and an American Heart Association Fellowship to R. H.
Footnotes
Conflict of Interest
The authors declare that they have no competing interests.
References
- 1.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 2.Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 3.Earley S, Brayden JE. Transient Receptor Potential Channels in the Vasculature. Physiol Rev. 2015;95:645–690. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 5.Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res. 1995;77:370–378. doi: 10.1161/01.res.77.2.370. [DOI] [PubMed] [Google Scholar]
- 6.Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, Rusch NJ. Voltage-gated K+ channels in rat small cerebral arteries: molecular identity of the functional channels. J Physiol. 2003;551:751–763. doi: 10.1113/jphysiol.2003.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amberg GC, Santana LF. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol. 2006;291:C348–356. doi: 10.1152/ajpcell.00086.2006. [DOI] [PubMed] [Google Scholar]
- 8.Cheong A, Dedman AM, Beech DJ. Expression and function of native potassium channel (KVα1) subunits in terminal arterioles of rabbit. J Physiol. 2001;534:691–700. doi: 10.1111/j.1469-7793.2001.00691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kidd MW, Leo MD, Bannister JP, Jaggar JH. Intravascular pressure enhances the abundance of functional Kv1.5 channels at the surface of arterial smooth muscle cells. Sci Signal. 2015;8:ra83. doi: 10.1126/scisignal.aac5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Hanna ST, Tang G, Wang R. Contributions of Kv1.2, Kv1.5 and Kv2.1 subunits to the native delayed rectifier K+ current in rat mesenteric artery smooth muscle cells. Life Sci. 2002;71:1465–1473. doi: 10.1016/s0024-3205(02)01922-7. [DOI] [PubMed] [Google Scholar]
- 11.Plane F, Johnson R, Kerr P, Wiehler W, Thorneloe K, Ishii K, Chen T, Cole W. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res. 2005;96:216–224. doi: 10.1161/01.RES.0000154070.06421.25. [DOI] [PubMed] [Google Scholar]
- 12.Cogolludo A, Moreno L, Lodi F, Frazziano G, Cobeño L, Tamargo J, Perez-Vizcaino F. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: role of 5-HT 2A receptors, caveolin-1, and KV1.5 channel internalization. Circ Res. 2006;98:931–938. doi: 10.1161/01.RES.0000216858.04599.e1. [DOI] [PubMed] [Google Scholar]
- 13.Joseph BK, Thakali KM, Pathan AR, Kang E, Rusch NJ, Rhee SW. Postsynaptic density-95 scaffolding of Shaker-type K+ channels in smooth muscle cells regulates the diameter of cerebral arteries. J Physiol. 2011;589:5143–5152. doi: 10.1113/jphysiol.2011.213843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kidd MW, Bulley S, Jaggar JH. Angiotensin II reduces the surface abundance of KV1.5 channels in arterial myocytes to stimulate vasoconstriction. J Physiol. 2017;595:1607–1618. doi: 10.1113/JP272893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore CL, Nelson PL, Parelkar NK, Rusch NJ, Rhee SW. Protein kinase A-phosphorylated KV1 channels in PSD95 signaling complex contribute to the resting membrane potential and diameter of cerebral arteries. Circ Res. 2014;114:1258–1267. doi: 10.1161/CIRCRESAHA.114.303167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nieves-Cintrón M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell’Acqua ML, Scott JD, Navedo MF. Selective Down-regulation of KV2.1 Function Contributes to Enhanced Arterial Tone during Diabetes. J Biol Chem. 2015;290:7918–7929. doi: 10.1074/jbc.M114.622811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishijima Y, Cao S, Chabowski DS, Korishettar A, Ge A, Zheng X, Sparapani R, Gutterman DD, Zhang DX. Contribution of KV1.5 Channel to Hydrogen Peroxide-Induced Human Arteriolar Dilation and Its Modulation by Coronary Artery Disease. Circ Res. 2017;120:658–669. doi: 10.1161/CIRCRESAHA.116.309491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 20.Aiello EA, Clement-Chomienne O, Sontag DP, Walsh MP, Cole WC. Protein kinase C inhibits delayed rectifier K+ current in rabbit vascular smooth muscle cells. Am J Physiol. 1996;271:H109–H119. doi: 10.1152/ajpheart.1996.271.1.H109. [DOI] [PubMed] [Google Scholar]
- 21.Garcia ML, Garcia-Calvo M, Hidalgo P, Lee A, MacKinnon R. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 1994;33:6834–6839. doi: 10.1021/bi00188a012. [DOI] [PubMed] [Google Scholar]
- 22.Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- 23.Immke D, Wood M, Kiss L, Korn SJ. Potassium-dependent changes in the conformation of the Kv2.1 potassium channel pore. J Gen Physiol. 1999;113:819–836. doi: 10.1085/jgp.113.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu C, Lu Y, Tang G, Wang R. Expression of voltage-dependent K+ channel genes in mesenteric artery smooth muscle cells. Am J Physiol. 1999;277:G1055–1063. doi: 10.1152/ajpgi.1999.277.5.G1055. [DOI] [PubMed] [Google Scholar]
- 25.Yuan XJ, Wang J, Juhaszova M, Golovina VA, Rubin LJ. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol. 1998;274:L621–635. doi: 10.1152/ajplung.1998.274.4.L621. [DOI] [PubMed] [Google Scholar]
- 26.Miguel-Velado E, Moreno-Domínguez A, Colinas O, Cidad P, Heras M, Pérez-García MT, López-López JR. Contribution of Kv Channels to Phenotypic Remodeling of Human Uterine Artery Smooth Muscle Cells. Cir Res. 2005;97:1280–1287. doi: 10.1161/01.RES.0000194322.91255.13. [DOI] [PubMed] [Google Scholar]
- 27.Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ channels: structure, function, and clinical significance. Physiol Rev. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- 28.Xu C, Tang G, Lu Y, Wang R. Molecular basis of voltage-dependent delayed rectifier K+ channels in smooth muscle cells from rat tail artery. Life Sci. 2000;66:2023–2033. doi: 10.1016/s0024-3205(00)00529-4. [DOI] [PubMed] [Google Scholar]
- 29.Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, Greenwood IA. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, Greenwood IA. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong XZ, Abd-Elrahman KS, Liao C-H, El-Yazbi AF, Walsh EJ, Walsh MP, Cole WC. Stromatoxin-sensitive, heteromultimeric KV2.1/KV9.3 channels contribute to myogenic control of cerebral arterial diameter. J Physiol. 2010;588:4519–4537. doi: 10.1113/jphysiol.2010.196618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berk BC, Corson MA. Angiotensin II signal transduction in vascular smooth muscle: role of tyrosine kinases. Circ Res. 1997;80:607–616. doi: 10.1161/01.res.80.5.607. [DOI] [PubMed] [Google Scholar]
- 33.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 34.Rainbow RD, Norman RI, Everitt DE, Brignell JL, Davies NW, Standen NB. Endothelin-I and angiotensin II inhibit arterial voltage-gated K+ channels through different protein kinase C isoenzymes. Cardiovasc Res. 2009;83:493–500. doi: 10.1093/cvr/cvp143. [DOI] [PubMed] [Google Scholar]
- 35.Park SW, Noh HJ, Sung DJ, Kim JG, Kim JM, Ryu S-Y, Kang K, Kim B, Bae YM, Cho H. Hydrogen peroxide induces vasorelaxation by enhancing 4-aminopyridine-sensitive Kv currents through S-glutathionylation. Pflügers Arch. 2015;467:285–297. doi: 10.1007/s00424-014-1513-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogers PA, Dick GM, Knudson JD, Focardi M, Bratz IN, Swafford AN, Saitoh S-i, Tune JD, Chilian WM. H2O2-induced redox-sensitive coronary vasodilation is mediated by 4-aminopyridine-sensitive K+ channels. Am J Physiol. 2006;291:H2473–H2482. doi: 10.1152/ajpheart.00172.2006. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H(2)O(2) is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for Hydrogen Peroxide in Flow-Induced Dilation of Human Coronary Arterioles. Circ Res. 2003;92:e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 39.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol. 2007;292:H93–H100. doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- 40.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res. 2012;110:471–480. doi: 10.1161/CIRCRESAHA.111.258871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berwick ZC, Dick GM, Moberly SP, Kohr MC, Sturek M, Tune JD. Contribution of voltage-dependent K+ channels to metabolic control of coronary blood flow. J Mol Cell Cardiol. 2012;52:912–919. doi: 10.1016/j.yjmcc.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Chai Q, Gutterman DD, Liu Y. Elevated glucose impairs cAMP-mediated dilation by reducing Kv channel activity in rat small coronary smooth muscle cells. Am J Physiol. 2003;285:H1213–H1219. doi: 10.1152/ajpheart.00226.2003. [DOI] [PubMed] [Google Scholar]
- 43.Tobin AA, Joseph BK, Al-Kindi HN, Albarwani S, Madden JA, Nemetz LT, Rusch NJ, Rhee SW. Loss of cerebrovascular Shaker-type K+ channels: a shared vasodilator defect of genetic and renal hypertensive rats. Am J Physiol. 2009;297:H293–H303. doi: 10.1152/ajpheart.00991.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Juhaszova M, Rubin LJ, Yuan XJ. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest. 1997;100:2347–2353. doi: 10.1172/JCI119774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dabertrand F, Krøigaard C, Bonev AD, Cognat E, Dalsgaard T, Domenga-Denier V, Hill-Eubanks DC, Brayden JE, Joutel A, Nelson MT. Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc Natl Acad Sci. 2015;112:E796–E805. doi: 10.1073/pnas.1420765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dietrich HH, Dacey RG., Jr Molecular keys to the problems of cerebral vasospasm. Neurosurgery. 2000;46:517–530. doi: 10.1097/00006123-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Ishiguro M, Morielli AD, Zvarova K, Tranmer BI, Penar PL, Wellman GC. Oxyhemoglobin-induced suppression of voltage-dependent K+ channels in cerebral arteries by enhanced tyrosine kinase activity. Cir Res. 2006;99:1252–1260. doi: 10.1161/01.RES.0000250821.32324.e1. [DOI] [PubMed] [Google Scholar]
- 48.Hayabuchi Y, Standen NB, Davies NW. Angiotensin II inhibits and alters kinetics of voltage-gated K+ channels of rat arterial smooth muscle. Am J Physiol. 2001;281:H2480–H2489. doi: 10.1152/ajpheart.2001.281.6.H2480. [DOI] [PubMed] [Google Scholar]
- 49.Holmes T, Fadool D, Levitan I. Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci. 1966;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koide M, Penar PL, Tranmer BI, Wellman GC. Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am J Physiol. 2007;293:H1750–H1759. doi: 10.1152/ajpheart.00443.2007. [DOI] [PubMed] [Google Scholar]