

Abstract
Voltage-gated potassium (KV) channels are key regulators of vascular smooth muscle contractility and vascular tone, and thus have major influence on the microcirculation. KV channels are important determinants of vascular smooth muscle membrane potential (Em). A number of KV subunits are expressed in the plasma membrane of smooth muscle cells. Each subunit confers distinct kinetics and regulatory properties that allow for fine control of Em to orchestrate vascular tone. Modifications in KV subunit expression and/or channel activity can contribute to changes in vascular smooth muscle contractility in response to different stimuli and in diverse pathological conditions. Consistent with this, a number of studies suggest alterations in KV subunit expression and/or function as underlying contributing mechanisms for small resistance artery dysfunction in pathologies such as hypertension and metabolic disorders, including diabetes. Here, we review our current knowledge on the effects of these pathologies on KV channel expression and function in vascular smooth muscle cells, and the repercussions on (micro)vascular function.
Introduction
The microcirculation is greatly influenced by vascular smooth muscle cells lining the arterial wall of small resistance arteries and arterioles. The contractile state of vascular smooth muscle determines the diameter of these vessels and helps establish the level of vascular tone. This contributes to proper regulation of blood pressure and blood flow to meet metabolic demands of surrounding tissue. The association of microvascular dysfunction in humans with a number of pathological conditions, including hypertension and diabetes, underscores the significance of this mechanism (40, 47, 67).
Vascular tone is largely determined by a dynamic interplay between different ionic conductances in vascular smooth muscle that help control its membrane potential (Em) and the level of intracellular calcium (Ca2+) (75). Accordingly, the activity of potassium (K+) channels, including a number of voltage-gated K+ (KV) channels, is a major regulator of vascular smooth muscle Em (38). KV channels, through their regulation of Em, have a major influence on intracellular Ca2+ by modulating the open probability of L-type Ca2+ channels (and perhaps T-type channels (25)). This is important as Ca2+ influx through L-type Ca2+ channel CaV1.2 (e.g. Ca2+ sparklets) is essential for vascular smooth muscle contraction (3, 37, 52). Physiological activation of KV channels hyperpolarizes and relaxes vascular smooth muscle by decreasing the activity of voltage-gated Ca2+ channels and therefore Ca2+ influx, whereas their inhibition promotes contraction (56). Thus, KV channels are important regulators vascular tone.
KV channels represent a diverse group of membrane proteins, with 12 distinct families identified to date (KV1-KV12) (24). Each KV channel comprises a KV α subunit that forms the ion conducting pore and an ancillary KV β subunit that modulates activity of KV α subunits. The KV α subunit consists of six transmembrane helices (S1-S6) with the S4 transmembrane domain containing the voltage sensor. A tetramer of KV α subunits forms the ion-conducting pore, through interactions of the S6 domain and the P-loop between the S5 and S6 domains. Vascular smooth muscle cells express a variety of KV channels in different vascular beds, including KV1 (KV1.1, KV1.2, KV1.3, KV1.5, KV1.6), KV2 (KV2.1) as well as members of the KV7 (KV 7.1-5) and silent KV subunits (KV9.3) (Table 1) (2, 5, 15, 75, 81). KV channels can exist as homo- or heterotetramers with distinct biophysical and pharmacological properties. In vascular smooth muscle, for example, KV1.2 and KV1.5, as well as KV7.4 and KV7.5 can form heteromeric channels with profound implications for cell excitability (36, 75). Likewise, co-assembly with auxiliary KV β subunits and silent KV subunits confers further functional diversity (36).
Table 1. Expression of KV subunits in blood vessels from different species.
| vascular bed | species | KV subunits | references |
|---|---|---|---|
| cerebral arteries | mouse | 1.2, 1.5, 1.6, 2.1 | (15, 58) |
| rabbit | 1.5, 1.6 | (14) | |
| rat | 1.2, 1.5, 2.1, 7.4, 7.5, 9.3 | (2, 4, 5, 80, 81) | |
| mesenteric arteries | mouse | 1.2, 1.5, 2.1, 6.3, 7.4 | (48, 58, 78) |
| rat | 1.2, 1.5, 2.1 | (18, 44, 76) | |
| coronary arteries | mouse | 1.2, 1.5, 2.1, 7.4, 7.5 | (63, 78) |
| rat | 1.2, 1.5, 7.1, 7.4, 7.5 | (30, 35) | |
| human | 1.5 | (59) |
KV channels represent key substrates underlying vascular smooth muscle excitability in response to not only pressure-induced depolarization (e.g. vascular tone), but also vasoactive substances. For example, KV channels have been shown to participate in the response to vasodilators such as adenosine and β-adrenergic agonists. These vasodilators promote cAMP production, activation of protein kinase A (PKA) and phosphorylation of KV channels to positively modulate KV function to promote vasodilation (1). KV inhibitors also blunt nitric oxide (NO)-induced arterial dilation, suggesting that KV channels, at least partially mediate NO-dependent vasorelaxant effects in some vascular beds (21, 68, 70). Conversely, molecules that activate protein kinase C (PKC) are generally associated with KV channel inhibition and membrane potential depolarization. For example, the potent vasoactive peptide angiotensin II and elevations in extracellular glucose have been shown to induce vasoconstriction, at least in part by inhibiting KV channels through a PKC-mediated pathway (16, 65, 71). Therefore, changes in the expression of one or more KV subunits or signaling pathways regulating KV function may impact vascular smooth muscle excitability and (micro)vascular function during physiological and pathological conditions. KV channel remodeling could represent an underlying mechanism for microvascular dysfunction affecting vascular tone and blood flow leading to organ damage. In support of this, studies using animal models have shown that modifications in KV channel expression and/or function can contribute to changes in vascular smooth muscle contractility during different pathological conditions such as hypertension and metabolic disorders, including diabetes. Here, we review our current knowledge related to the changes in KV expression and function in vascular smooth muscle cells associated with hypertension and metabolic disorders, and its repercussions on (micro)vascular function. The role of KV channels in pulmonary artery hypertension and in the renal vasculature will not be addressed here, but extensive, recent reviews on the subject can be found elsewhere (26, 46, 50, 66).
Vascular voltage-gated KV channels in hypertension
In hypertension, the function of the microcirculation is altered. Enhanced vascular tone and reduced vasodilator response have been suggested to contribute, at least in part, to impaired tissue perfusion and end-organ damage during this pathological condition (40). Changes in KV channel expression and/or function may contribute to this outcome. A reduction in KV channel activity during hypertension has been reported in vascular smooth muscle from rat mesenteric arteries (9, 19, 32, 79), rat thoracic aorta (17, 32) mouse aortic arteries (48), rat renal arteries (12, 45), rat pial arteries (4, 74), mouse mesenteric arteries (32, 48) and mouse pial arteries (4). These changes are independent of the animal model employed. An early study however, reported augmentation of KV channel activity in hypertensive vascular smooth muscle (18). The difference between this study and the rest has been attributed to methodological differences associated with intracellular Ca2+ levels that mediate KV channel inhibition. This was not apparent in subsequent work examining the influence of intracellular Ca2+ on KV channel function in hypertensive vascular smooth muscle from the same arterial bed (9). Thus, additional research is still required to further elucidate this conundrum.
The mechanisms for changes in KV channel activity in hypertensive vascular smooth muscle have been associated, at least in part, with altered expression in the mRNA and/or protein levels of one or more of the KV subunits underlying the KV current in these cells. A reduction in the expression of mRNA and/or protein levels for KV1.2, KV1.5 or both subunits has been found in different vascular beds from genetic animal models of hypertension (8, 74, 79). A number of other studies have reported downregulation of KV2.1 subunit mRNA/protein expression with no apparent change in KV1.X levels in cerebral and mesenteric arteries from an angiotensin II-induced hypertension model and a genetic mouse model of hypertension, respectively (4, 5, 48). More recently, downregulation in the expression of KV7.4 was suggested to contribute to increased vascular smooth muscle contractility in spontaneous hypertensive rats (12, 32, 35). These results suggest a key role for several KV subunits in the regulation of KV currents, vascular smooth muscle contraction, and vascular reactivity during hypertension. Furthermore, they underscore the importance of the methodological conditions, species, animal models of hypertension and vascular beds in the development of experimental design and interpretation of results.
Detailed mechanistic information for changes in KV2.1 expression in vascular smooth muscle from pial and mesenteric arteries during angiotensin II-induced hypertension involving the activation of the Ca2+-dependent calcineurin - nuclear factor of activated T cells isoform c3 (NFATc3) signaling pathway (Figure 1) (4, 5). Accordingly, chronic activation of angiotensin II signaling mediated by targeted PKC stimulates L-type Ca2+ channel activity leading to enhanced Ca2+ influx and calcineurin activity (51, 53). Calcineurin-mediated dephosphorylation of the transcription factor NFATc3 promotes its nuclear translocation (57). Once in the nucleus, NFATc3 suppresses the expression of KV2.1 mRNA and protein levels, which results in decreased voltage-dependent K+ currents. Decreased KV2.1 function leads to membrane depolarization, further activation of L-type Ca2+ channels and Ca2+ influx (4, 57). This may create a positive feedback loop that could potentially perpetuate the pathological signal. This loop may be interrupted by inhibition of L-type Ca2+ channels, calcineurin or NFATc3 (4). Conversely, the molecular mechanisms underlying alterations in KV1.X and KV7.X subunit expression in vascular smooth muscle during hypertension are unclear, and will certainly need further scrutiny. Moreover, given that most studies use rodent male tissues/cells, it will be important to extend studies to samples using female tissue, and when possible, the human vasculature.
Figure 1. Proposed mechanism for suppression of KV channel expression and function during angiotensin II-induced hypertension.
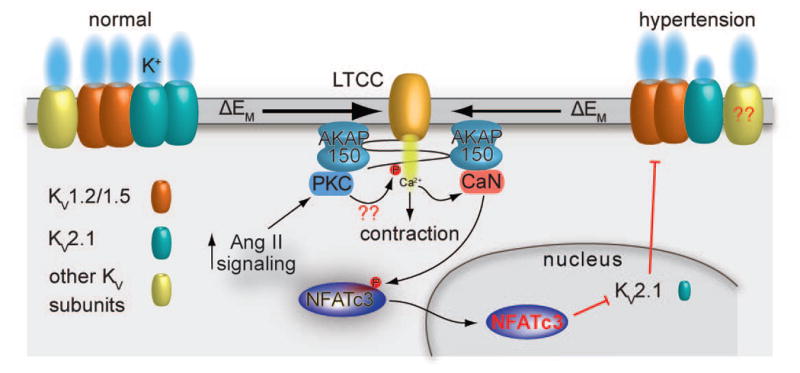
Under physiological conditions, expression and function of several KV subunits, including KV1.X, KV2.1 and KV7.X, oppose pressure-induced depolarization to limit LTCC activity and vascular smooth muscle contraction. During chronic angiotensin II signaling activation, as in hypertension, activation of PKC stimulates LTCC activity, thus increasing global Ca2+ influx and promoting contraction. This increase in Ca2+ influx can also stimulate the activation of the Ca2+/calmodulin-dependent, AKAP150-targeted phosphatase calcineurin. This phosphatase dephosphorylates the transcription factor NFATc3, allowing its translocation to the nucleus. Once in the nucleus, NFATc3 can regulate gene expression, including suppression of the expression of KV2.1 (but not KV1.2 and KV1.5) subunits. This reduces feedback membrane potential hyperpolarization leading to increased activity of LTCC and Ca2+ influx, vascular smooth muscle contraction and enhanced vascular tone during angiotensin II-induced hypertension. This feedback loop may be interrupted by LTCC blockers, inhibition of calcineurin or NFATc3, or disruption of the interaction between AKAP150 and calcineurin. Whether direct phosphorylation of LTCCs by PKC, as well as alterations in KV7.X expression in response to angiotensin II-induced hypertension occurs is unclear (indicated by ??). Illustration of the interaction between AKAP150 and LTCC does not necessarily reflect the native interaction between these proteins. The cartoon was drawn as such for simplicity.
Vascular voltage-gated KV channels in metabolic disorders
As with hypertension, changes in the function of the microcirculation are also apparent in metabolic disorders. Diet-mediated changes in plasma membrane lipid composition may impact cellular function by influencing the activity of ion channels, including KV channels (39, 69). Initial studies suggested a reduction in K+ channel function in rabbit portal vein smooth muscle during dietary hypercholesterolemia (20) and in the mouse aorta from a genetic mouse model of atherosclerosis (34). Subsequently, a reduction in vascular KV channel function was confirmed in arteries and arterioles from animal models of diet-induced hypercholesterolemia, obesity and metabolic syndrome (7, 22, 28, 29, 33, 60, 77). This reduction in vascular KV channel function resulted in impaired coronary vasodilation and blood flow during diet-induced metabolic syndrome (7, 60), aberrant adenosine-mediated dilation of porcine coronary arteries and arterioles in animal models of hypercholesterolemia (22, 29) that is not corrected with exercise (28, 77), and reduced sildenafil and sodium nitroprusside-induced penile artery relaxation in a genetic model of metabolic syndrome (33). Interestingly, KV channel function in coronary smooth muscle varies under basal conditions, in response to diet and vasoactive molecules, and during exercise in a sex-dependent manner (27, 77).
For the most part, the mechanisms underlying aberrant KV channel function in smooth muscle cells during diet-induced hypercholesterolemia, obesity and metabolic syndrome are unclear. No change in the mRNA expression of KV subunits in coronary arterioles has been observed in a Yucatan swine model of hypercholesterolemia (28). Rather, it was suggested that changes in the signaling pathway by which adenylyl cyclase regulates KV channel activity seemed to contribute to impaired adenosine-mediated vasodilation of coronary arterioles in this animal model (28). In contrast, a reduction in protein levels of KV1.5 in coronary arteries was associated with decreased KV channel function, leading to impaired coronary blood flow in an Ossabaw swine model of metabolic syndrome (7). The differences between these two studies may be accounted for by differences in the extent of metabolic abnormalities in response to diet between animal models (55). More recently, altered function of KV7 channels, but not changes in mRNA or protein expression levels, was suggested to contribute to impaired dilation in different arterial beds in several animal models of metabolic syndrome (33, 60). These studies raise important questions about the specific mechanisms underlying altered KV channel activity in smooth muscle that contribute to impair vasodilation during metabolic disorders, which may be the basis for future experiments. Therefore, a concerted effort should be undertaken to rigorously assess the effects of changes in vascular smooth muscle membrane lipid composition as well as cholesterol content, as a potential mechanism impacting its fluidity and ion channel function, including that of KV channels.
Vascular voltage-gated KV channels in hyperglycemia
High blood glucose (e.g. hyperglycemia) is a major metabolic abnormality that contributes to vascular complications in diabetes. Changes in extracellular glucose content may have a major impact on KV channel function and vascular reactivity. Accordingly, studies have demonstrated distinctive regulation of KV channel function in vascular smooth muscle cells from several vascular beds in response to acute and chronic elevations in extracellular glucose concentration (31, 41-43, 58, 65, 71, 72). During acute increases in extracellular glucose, KV channel activity is inhibited in vascular smooth muscle cells from small diameter mesenteric arteries and cerebral parenchymal arterioles (31, 65, 71). This glucose-mediated inhibition of KV channels is concentration dependent and resulted in vascular smooth muscle Em depolarization (31, 65), leading to enhanced vasoconstriction (31, 71) (also see Figure 2) and impaired neurovascular coupling (71). The mechanism underlying the reduction in KV channel activity in response to acute increases in extracellular glucose did not appear to depend on changes in KV subunit expression, but was attributed to a PKC-dependent pathway (31, 65, 71). More recently, a study revealed that differential engagement of PKCβ and PKCα isoforms may contribute to inhibition of KV channels in a glucose concentration-dependent manner (31). However, whether glucose-mediated inhibition of KV channel function is due to direct KV subunit phosphorylation or activation of a different PKC-dependent pathway is unclear. Furthermore, and in contrast to the proposed PKC-mediated inhibition of KV channels in response to elevated glucose, we recently found that PKA activity was necessary for glucose-induced vasoconstriction of cerebral parenchymal arterioles (Figure 2). These results are consistent with recent work in pial arteries (62), and suggest an unexpected role for PKA in vasoconstriction of these small resistance arteries that requires further investigation.
Figure 2. PKA is required for vasoconstriction of cerebral parenchymal arteries in response to elevated glucose.
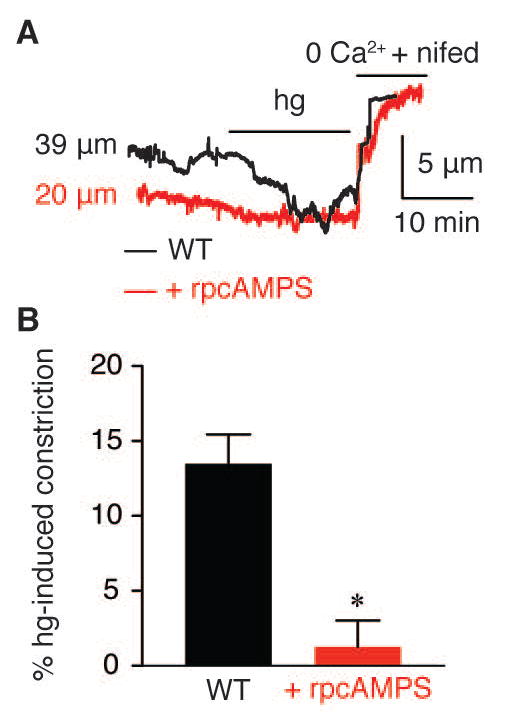
A) Representative diameter recordings and B) summary hg-induced constriction in the absence or presence of the PKA inhibitor rpcAMPs (10 μM). A solution containing 0 mM extracellular Ca2+ and the LTCC blocker nifedipine (1 μM) was used to obtain the passive diameter. *P < 0.05.
A reduction in the function of KV channels in response to chronic elevations in extracellular glucose has also been reported for vascular smooth muscle from small coronary and cerebral arteries (41-43, 58, 72). Interestingly, multiple pathways have been described to account for reduced KV channel activity in response to chronic hyperglycemia. Earlier studies correlated a reduction in KV currents in vascular smooth muscle of small coronary arteries incubated in elevated glucose for 24 hours to glucose-mediated production of superoxide and peroxynitrite. This was associated with specific nitration of the KV1.2 subunit (with no change in KV1.2 or KV1.5 protein expression), impairment of KV channel function and loss of cAMP-mediated dilation of small coronary arteries (41-43). A recent study using primary coronary vascular smooth muscle cells incubated in elevated glucose for 48 hours, linked the reduction in KV channel function to a decrease in KV1.2 and KV1.5 mRNA and protein levels (72). This process was mediated by advanced glycation end products (AGE) and required AGE interaction with its surface receptor RAGE (72). A reduction in KV2.1 mRNA expression was observed in mouse cerebral arteries incubated in elevated glucose for 48 hours through a mechanism that requires targeting of the phosphatase calcineurin by the scaffold protein A kinase anchoring protein 150 (AKAP150) (Figure 3) (58). On the other hand, the effects of acute and chronic elevations in extracellular glucose on KV7 function are poorly understood and require additional studies. Nevertheless, the differences in mechanisms between the aforementioned studies (even in cells/arteries from the same vascular bed) may be due to experimental conditions and use of distinct animal models. Furthermore, these observations also raise the possibility that different mechanisms may synergize to impair KV channel function and vascular reactivity during chronic elevations in extracellular glucose with major implications for vascular complications in diabetes.
Figure 3. Model for suppression of KV channel expression and function in diabetes.
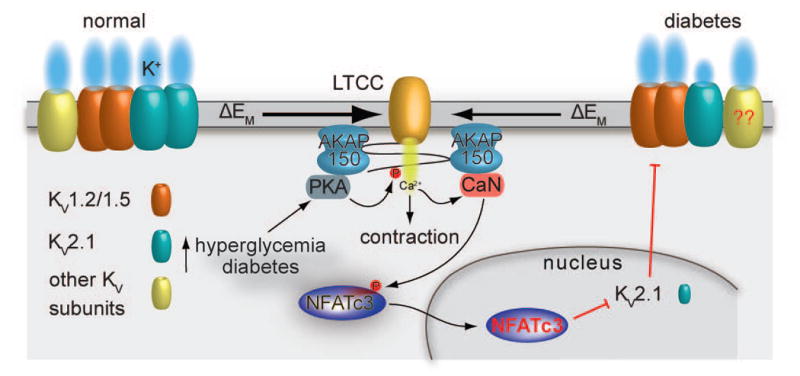
PKA-mediated phosphorylation of activation of serine 1928 induces potentiation of LTCCs in response to hyperglycemia and during diabetes (62). This leads to increased global Ca2+ influx and contraction. The increase in Ca2+ influx promotes activation of the AKAP150-targeted calcineurin, which dephosphorylates NFATc3 and allows its nuclear translocation where the transcription factor can suppress the expression of KV2.1 (but not KV1.2 and KV1.5) subunits. This reduction in KV2.1 expression and function decreases voltage-gated K+ currents and the negative feedback membrane potential hyperpolarization, thus leading to membrane potential depolarization, further Ca2+ influx through LTCCs, vascular smooth muscle contraction and enhanced vascular tone. Whether changes in KV7.X subunits during diabetes proceed through the calcineurin/NFATc3 signaling pathway and similar changes occur in the human vasculature require further investigation. Illustration of the interaction between AKAP150 and LTCC does not necessarily reflect the native interaction between these proteins. The cartoon was drawn as such for simplicity.
Vascular voltage-gated KV channels in diabetes
The function of vascular smooth muscle cells in the microcirculation during diabetes is impaired (47), and changes in KV channel expression and/or function may contribute to this outcome. Indeed, the bulk of the published data suggest a reduction in KV channel function in smooth muscle from different vascular beds and in several models of diabetes (10, 11, 13, 58, 72). Not surprisingly and similar to chronic hyperglycemia, multiple mechanisms have been described to account for altered KV channel activity in vascular smooth muscle during diabetes. In a rat model of type 1 diabetes (e.g. streptozotocin-induced diabetes), a reduction in KV channel function in vascular smooth muscle from small coronary arteries was associated with downregulation of KV1.2 subunit expression and increased KV1.2 nitration (but not KV1.5) due to enhanced superoxide production (10, 11). This was shown to contribute to impaired cAMP-mediated dilation (10, 11, 13). Interestingly, treatment with the anti-oxidant compound Ebselen decreased KV1.2 nitration, and improved KV1.2 expression, KV channel activity as well as cAMP-mediated coronary dilation, in type 1 diabetic rats (11). These results suggest a potential beneficial effect of Ebselen in treating vascular complications during diabetes.
In a high fat diet (HFD) rat model of type 2 diabetes however, aberrant KV channel activity in coronary vascular smooth muscle and impaired small coronary artery dilation were correlated with reduced mRNA and protein levels of KV1.2 and KV1.5 subunits (72). In this study, altered KV subunit expression, KV channel function and forskolin-mediated coronary artery dilation during diabetes were ameliorated in diabetic rats treated with the AGE inhibitor aminoguanidine (72). Interestingly, aminoguanidine treatment did not improve blood pressure in diabetic rats when compared to non-diabetic rats, perhaps suggesting a distinct role for AGEs in regulation of KV channels or any other target in different vascular beds. No changes in KV subunit expression, KV channel function and forskolin-mediated coronary artery dilation were observed in arteries/cells from non-diabetic rats treated with aminoguanidine. These results suggest that excessive production of AGEs may be an upstream pathological signal leading to impaired KV channel function and coronary artery reactivity during diabetes.
Alterations in KV channel function and impaired vascular reactivity were also described for cerebral and mesenteric arteries in a HFD mouse model of type 2 diabetes (58). In this study, selective transcriptional suppression and reduced protein levels of the KV2.1 subunit (but not KV1.2 and KV1.5) were associated with impaired KV currents and enhanced vascular tone during diabetes. This result is important as changes in KV2.1 expression and function may have prominent effects on intracellular Ca2+ and Em regulation, as revealed by mathematical simulation experiments (49, 61). The mechanisms underlying this selective suppression of KV2.1 expression and function are dependent on L-type Ca2+ channel activity and targeting of PKA and calcineurin by AKAP150 (Figure 3) (54, 58, 62). Accordingly, a PKA-mediated increase in Ca2+ influx through L-type Ca2+ channels activates a subpopulation of AKAP150-targeted calcineurin (54, 62). Calcineurin then dephosphorylates NFATc3 and promotes its nuclear translocation (61). Once in the nucleus, NFATc3 can downregulates KV2.1 mRNA expression, leading to a reduction in KV2.1 protein levels and impaired KV channel function (58). As is the case in hypertension, blocking L-type Ca2+ channels, preventing the interaction between calcineurin and AKAP150 or inhibiting NFATc3 could disrupt this pervasive pathway. Taken together, all of these studies on KV function leading to vascular complications during diabetes reveal fundamental differences and divergent mechanisms that vary between vessels and animal models. Thus, the relative contributions of all of the aforementioned pathways to altered KV channel expression and function during diabetes should be carefully evaluated and integrated.
Conclusions
KV channels represent major substrates underlying vascular smooth muscle excitability in small resistance arteries and arterioles. Under physiological conditions, the ability of KV channels to respond to pressure-induced depolarization, as well as to vasoactive molecules, such as vasodilators and/or vasoconstrictors, helps maintain a delicate balance between constriction and relaxation of small resistance arteries and arterioles that is necessary for appropriate myogenic response and tissue perfusion. Not surprisingly, alterations in KV channel function have been associated with impaired vascular reactivity in a variety of diseases affecting the vasculature. KV channel function is impaired in hypertension and in several metabolic disorders, including diabetes. The mechanisms underlying impaired KV channel function in different pathological conditions (i.e. hypertension vs. diabetes) are variable. This most likely reflects the activation of unique signaling pathways that distinctively impact KV expression and/or function in vascular smooth muscle cells for a specific disease. Interestingly, different mechanisms have also been reported to account for changes in KV expression and/or function within the same pathological condition. The differences, in this case, may be related to different experimental conditions, use of cells from different vascular beds, and /or the disease state at which the experiments were performed. Regardless of the cause, these observations indicate that a single mechanism does not account for all the remodeling in KV function during a pathological condition and suggest that multiple pathways could synergize to alter KV channel activity with major implications for vascular smooth muscle membrane potential and vascular reactivity, particularly in the microcirculation. Therefore, further research is required to completely appreciate mechanisms underlying alteration in KV channel activity in disease states.
Given the diverse population of homo- and heteromeric KV channels in vascular smooth muscle from small resistance arteries, additional approaches are necessary to tease out and integrate the specific contribution of each subunit to the regulation of membrane potential and vascular tone in health and disease. Computational approaches, as well as variations of the membrane-clamp sequential dissection technique, which have been extensively employed in the cardiac field (6, 23, 49), can be implemented to quantify the relative contribution of each KV subunit that may interact non-linearly to regulate vascular smooth muscle excitability and vascular reactivity. In addition, the development of optical sensors that can selectively report the activity of different KV subunits in response to changes in membrane potential in live cells may aid in this task (73). Future studies should also address in more detail differences in KV function between sexes and vascular beds. Improved understanding on how inflammation and oxidative stress, which are processes common to many disease states, impact KV channel activity in native cells is also needed. The modulation of KV channels in vascular smooth muscle by differences in lipid membrane composition and its regulatory mechanisms, which may ensue in certain pathological conditions, has to be further explored. A recent study in HEK cells revealed a complex, yet functionally relevant interaction between phosphatidylinositol 4,5-bisphosphate (PIP2) and the G protein βγ subunit in control of KV7.4 channel activity (64). Whether this interaction between PIP2, Gβγ and KV7 or any other KV channel also occurs in native vascular smooth muscle to play a role during pathological conditions is not clear and should be addressed in future research. Finally, the expression, function, and regulation of KV channels in human vascular smooth muscle from small resistance arteries and arterioles should be comprehensively examined. This may reveal novel mechanisms regulating KV channel function in health and disease. A recent study using vascular smooth muscle from coronary arterioles of patients with coronary artery disease suggested a key role for KV1.5 in vascular reactivity and revealed that altered KV channel function in these cells was not due to changes in KV1.5 expression, but to reduced surface localization of this subunit (59). Thus, a better understanding of the mechanisms underlying alterations in KV function in human small resistance arteries and arterioles may lead to the identification of potential novel therapeutic targets to “correct” KV dysfunction and treat (micro)vascular complications during pathological conditions.
Acknowledgments
We thank Miss Maria Paz Prada for reading the manuscript. This work was supported by NIH grants R01-HL098200 and R01-HL121059 and AHA grant 14GRNT18730054 (to MFN), NIH grant T32HL086350 (to AUS and MAN), AHA 16SDG27260070 (to MAN) and a UC Davis Academic Federation Innovative Development Award (to MN-C).
References
- 1.Aiello EA, Walsh MP, Cole WC. Phosphorylation by protein kinase A enhances delayed rectifier K+ current in rabbit vascular smooth muscle cells. Am J Physiol. 1995;268:H926–934. doi: 10.1152/ajpheart.1995.268.2.H926. [DOI] [PubMed] [Google Scholar]
- 2.Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, Rusch NJ. Voltage-gated K+ Channels in Rat Small Cerebral Arteries: Molecular Identity of the Functional Channels. J Physiol. 2003 doi: 10.1113/jphysiol.2003.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amberg GC, Navedo MF, Nieves-Cintrón M, Molkentin JD, Santana LF. Calcium Sparklets Regulate Local and Global Calcium in Murine Arterial Smooth Muscle. J Physiol. 2007;579:187–201. doi: 10.1113/jphysiol.2006.124420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amberg GC, Rossow CF, Navedo MF, Santana LF. NFATc3 Regulates Kv2.1 Expression in Arterial Smooth Muscle. J Biol Chem. 2004;279:47326–47334. doi: 10.1074/jbc.M408789200. [DOI] [PubMed] [Google Scholar]
- 5.Amberg GC, Santana LF. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol. 2006;291:C348–356. doi: 10.1152/ajpcell.00086.2006. [DOI] [PubMed] [Google Scholar]
- 6.Banyasz T, Horvath B, Jian Z, Izu LT, Chen-Izu Y. Sequential dissection of multiple ionic currents in single cardiac myocytes under action potential-clamp. J Mol Cell Cardiol. 2011;50:578–581. doi: 10.1016/j.yjmcc.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berwick ZC, Dick GM, Moberly SP, Kohr MC, Sturek M, Tune JD. Contribution of voltage-dependent K(+) channels to metabolic control of coronary blood flow. J Mol Cell Cardiol. 2012;52:912–919. doi: 10.1016/j.yjmcc.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bratz IN, Dick GM, Partridge LD, Kanagy NL. Reduced molecular expression of K(+) channel proteins in vascular smooth muscle from rats made hypertensive with N{omega}-nitro-L-arginine. Am J Physiol Heart Circ Physiol. 2005;289:H1277–1283. doi: 10.1152/ajpheart.01052.2004. [DOI] [PubMed] [Google Scholar]
- 9.Bratz IN, Swafford AN, Jr, Kanagy NL, Dick GM. Reduced functional expression of K(+) channels in vascular smooth muscle cells from rats made hypertensive with N{omega}-nitro-L-arginine. Am J Physiol Heart Circ Physiol. 2005;289:H1284–1290. doi: 10.1152/ajpheart.01053.2004. [DOI] [PubMed] [Google Scholar]
- 10.Bubolz AH, Li H, Wu Q, Liu Y. Enhanced oxidative stress impairs cAMP-mediated dilation by reducing Kv channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol. 2005;289:H1873–1880. doi: 10.1152/ajpheart.00357.2005. [DOI] [PubMed] [Google Scholar]
- 11.Bubolz AH, Wu Q, Larsen BT, Gutterman DD, Liu Y. Ebselen reduces nitration and restores voltage-gated potassium channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol. 2007;293:H2231–2237. doi: 10.1152/ajpheart.00717.2007. [DOI] [PubMed] [Google Scholar]
- 12.Chadha PS, Zunke F, Zhu HL, Davis AJ, Jepps TA, Olesen SP, Cole WC, Moffatt JD, Greenwood IA. Reduced KCNQ4-encoded voltage-dependent potassium channel activity underlies impaired beta-adrenoceptor-mediated relaxation of renal arteries in hypertension. Hypertension. 2012;59:877–884. doi: 10.1161/HYPERTENSIONAHA.111.187427. [DOI] [PubMed] [Google Scholar]
- 13.Chai Q, Liu Z, Chen L. Effects of streptozotocin-induced diabetes on Kv channels in rat small coronary smooth muscle cells. The Chinese journal of physiology. 2005;48:57–63. [PubMed] [Google Scholar]
- 14.Cheong A, Dedman AM, Beech DJ. Expression and function of native potassium channel [K(V)alpha1] subunits in terminal arterioles of rabbit. J Physiol. 2001;534:691–700. doi: 10.1111/j.1469-7793.2001.00691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheong A, Dedman AM, Xu SZ, Beech DJ. K(V)alpha1 channels in murine arterioles: differential cellular expression and regulation of diameter. Am J Physiol Heart Circ Physiol. 2001;281:H1057–1065. doi: 10.1152/ajpheart.2001.281.3.H1057. [DOI] [PubMed] [Google Scholar]
- 16.Clement-Chomienne O, Walsh MP, Cole WC. Angiotensin II activation of protein kinase C decreases delayed rectifier K+ current in rabbit vascular myocytes. J Physiol. 1996;495(Pt 3):689–700. doi: 10.1113/jphysiol.1996.sp021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox RH. Comparison of K+ channel properties in freshly isolated myocytes from thoracic aorta of WKY and SHR. Am J Hypertens. 1996;9:884–894. doi: 10.1016/s0895-7061(96)00179-3. [DOI] [PubMed] [Google Scholar]
- 18.Cox RH, Folander K, Swanson R. Differential expression of voltage-gated K+ channel genes in arteries from spontaneously hypertensive and Wistar-Kyoto rats. Hypertension. 2001;37:1315–1322. doi: 10.1161/01.hyp.37.5.1315. [DOI] [PubMed] [Google Scholar]
- 19.Cox RH, Lozinskaya I, Dietz NJ. Differences in K+ current components in mesenteric artery myocytes from WKY and SHR. Am J Hypertens. 2001;14:897–907. doi: 10.1016/s0895-7061(01)02145-8. [DOI] [PubMed] [Google Scholar]
- 20.Cox RH, Tulenko TN. Altered contractile and ion channel function in rabbit portal vein with dietary atherosclerosis. Am J Physiol. 1995;268:H2522–2530. doi: 10.1152/ajpheart.1995.268.6.H2522. [DOI] [PubMed] [Google Scholar]
- 21.Favaloro JL, Kemp-Harper BK. Redox variants of NO (NO{middle dot} and HNO) elicit vasorelaxation of resistance arteries via distinct mechanisms. Am J Physiol Heart Circ Physiol. 2009;296:H1274–1280. doi: 10.1152/ajpheart.00008.2009. [DOI] [PubMed] [Google Scholar]
- 22.Franke R, Yang Y, Rubin LJ, Magliola L, Jones AW. High-fat diet alters K+-currents in porcine coronary arteries and adenosine sensitivity during metabolic inhibition. Journal of cardiovascular pharmacology. 2004;43:495–503. doi: 10.1097/00005344-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Grandi E, Sanguinetti MC, Bartos DC, Bers DM, Chen-Izu Y, Chiamvimonvat N, Colecraft HM, Delisle BP, Heijman J, Navedo MF, Noskov S, Proenza C, Vandenberg JI, Yarov-Yarovoy V. Potassium channels in the heart: structure, function and regulation. J Physiol. 2017;595:2209–2228. doi: 10.1113/JP272864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacological reviews. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 25.Harraz OF, Welsh DG. T-type Ca(2)(+) channels in cerebral arteries: approaches, hypotheses, and speculation. Microcirculation. 2013;20:299–306. doi: 10.1111/micc.12038. [DOI] [PubMed] [Google Scholar]
- 26.Hayabuchi Y. The Action of Smooth Muscle Cell Potassium Channels in the Pathology of Pulmonary Arterial Hypertension. Pediatric cardiology. 2017;38:1–14. doi: 10.1007/s00246-016-1491-7. [DOI] [PubMed] [Google Scholar]
- 27.Heaps CL, Bowles DK. Gender-specific K(+)-channel contribution to adenosine-induced relaxation in coronary arterioles. Journal of applied physiology. 2002;92:550–558. doi: 10.1152/japplphysiol.00566.2001. [DOI] [PubMed] [Google Scholar]
- 28.Heaps CL, Jeffery EC, Laine GA, Price EM, Bowles DK. Effects of exercise training and hypercholesterolemia on adenosine activation of voltage-dependent K+ channels in coronary arterioles. Journal of applied physiology. 2008;105:1761–1771. doi: 10.1152/japplphysiol.90958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heaps CL, Tharp DL, Bowles DK. Hypercholesterolemia abolishes voltage-dependent K+ channel contribution to adenosine-mediated relaxation in porcine coronary arterioles. Am J Physiol Heart Circ Physiol. 2005;288:H568–576. doi: 10.1152/ajpheart.00157.2004. [DOI] [PubMed] [Google Scholar]
- 30.Hu Z, Ma A, Zhang Y, Xi Y, Fan L, Wang T, Zhang T. Voltage-gated potassium+ channel expression in coronary artery smooth muscle cells of SHR and WKY. Cell Biochem Biophys. 2014;70:1725–1731. doi: 10.1007/s12013-014-0120-4. [DOI] [PubMed] [Google Scholar]
- 31.Jackson R, Brennan S, Fielding P, Sims MW, Challiss RA, Adlam D, Squire IB, Rainbow RD. Distinct and complementary roles for alpha and beta isoenzymes of PKC in mediating vasoconstrictor responses to acutely elevated glucose. Br J Pharmacol. 2016;173:870–887. doi: 10.1111/bph.13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jepps TA, Chadha PS, Davis AJ, Harhun MI, Cockerill GW, Olesen SP, Hansen RS, Greenwood IA. Downregulation of Kv7.4 channel activity in primary and secondary hypertension. Circulation. 2011;124:602–611. doi: 10.1161/CIRCULATIONAHA.111.032136. [DOI] [PubMed] [Google Scholar]
- 33.Jepps TA, Olesen SP, Greenwood IA, Dalsgaard T. Molecular and functional characterization of Kv 7 channels in penile arteries and corpus cavernosum of healthy and metabolic syndrome rats. Br J Pharmacol. 2016;173:1478–1490. doi: 10.1111/bph.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang J, Thoren P, Caligiuri G, Hansson GK, Pernow J. Enhanced phenylephrine-induced rhythmic activity in the atherosclerotic mouse aorta via an increase in opening of KCa channels: relation to Kv channels and nitric oxide. Br J Pharmacol. 1999;128:637–646. doi: 10.1038/sj.bjp.0702855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khanamiri S, Soltysinska E, Jepps TA, Bentzen BH, Chadha PS, Schmitt N, Greenwood IA, Olesen SP. Contribution of Kv7 channels to basal coronary flow and active response to ischemia. Hypertension. 2013;62:1090–1097. doi: 10.1161/HYPERTENSIONAHA.113.01244. [DOI] [PubMed] [Google Scholar]
- 36.Kilfoil PJ, Tipparaju SM, Barski OA, Bhatnagar A. Regulation of ion channels by pyridine nucleotides. Circ Res. 2013;112:721–741. doi: 10.1161/CIRCRESAHA.111.247940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol. 1995;269:H348–355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 39.Levitan I, Fang Y, Rosenhouse-Dantsker A, Romanenko V. Cholesterol and ion channels. Sub-cellular biochemistry. 2010;51:509–549. doi: 10.1007/978-90-481-8622-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy BI, Ambrosio G, Pries AR, Struijker-Boudier HA. Microcirculation in hypertension: a new target for treatment? Circulation. 2001;104:735–740. doi: 10.1161/hc3101.091158. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Chai Q, Gutterman DD, Liu Y. Elevated glucose impairs cAMP-mediated dilation by reducing Kv channel activity in rat small coronary smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H1213–1219. doi: 10.1152/ajpheart.00226.2003. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Gutterman DD, Rusch NJ, Bubolz A, Liu Y. Nitration and functional loss of voltage-gated K+ channels in rat coronary microvessels exposed to high glucose. Diabetes. 2004;53:2436–2442. doi: 10.2337/diabetes.53.9.2436. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Terata K, Rusch NJ, Gutterman DD. High glucose impairs voltage-gated K+ channel current in rat small coronary arteries. Circ Res. 2001;89:146–152. doi: 10.1161/hh1401.093294. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y, Hanna ST, Tang G, Wang R. Contributions of Kv1.2, Kv1.5 and Kv2.1 subunits to the native delayed rectifier K+ current in rat mesenteric artery smooth muscle cells. Life sciences. 2002;71:1465–1473. doi: 10.1016/s0024-3205(02)01922-7. [DOI] [PubMed] [Google Scholar]
- 45.Martens JR, Gelband CH. Alterations in rat interlobar artery membrane potential and K+ channels in genetic and nongenetic hypertension. Circ Res. 1996;79:295–301. doi: 10.1161/01.res.79.2.295. [DOI] [PubMed] [Google Scholar]
- 46.Montani D, Chaumais MC, Guignabert C, Gunther S, Girerd B, Jais X, Algalarrondo V, Price LC, Savale L, Sitbon O, Simonneau G, Humbert M. Targeted therapies in pulmonary arterial hypertension. Pharmacology & therapeutics. 2014;141:172–191. doi: 10.1016/j.pharmthera.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Montero D, Walther G, Perez-Martin A, Vicente-Salar N, Roche E, Vinet A. Vascular smooth muscle function in type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia. 2013;56:2122–2133. doi: 10.1007/s00125-013-2974-1. [DOI] [PubMed] [Google Scholar]
- 48.Moreno-Dominguez A, Cidad P, Miguel-Velado E, Lopez-Lopez JR, Perez-Garcia MT. De novo expression of Kv6.3 contributes to changes in vascular smooth muscle cell excitability in a hypertensive mice strain. J Physiol. 2009;587:625–640. doi: 10.1113/jphysiol.2008.165217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morotti S, Nieves-Cintrón M, Nystoriak MA, Navedo MF, Grandi E. Predominant contribution of L-type Cav1.2 channel stimulation to impaired intracellular calcium and cerebral artery vasoconstriction in diabetic hyperglycemia. Channels. 2017:1–7. doi: 10.1080/19336950.2017.1293220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moudgil R, Michelakis ED, Archer SL. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation. 2006;13:615–632. doi: 10.1080/10739680600930222. [DOI] [PubMed] [Google Scholar]
- 51.Navedo MF, Amberg GC. Local regulation of L-type ca(2+) channel sparklets in arterial smooth muscle. Microcirculation. 2013;20:290–298. doi: 10.1111/micc.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Navedo MF, Amberg GC, Westenbroek RE, Sinnegger-Brauns MJ, Catterall WA, Striessnig J, Santana LF. Cav1.3 channels produce persistent calcium sparklets, but Cav1.2 channels are responsible for sparklets in mouse arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2007;293:H1359–1370. doi: 10.1152/ajpheart.00450.2007. [DOI] [PubMed] [Google Scholar]
- 53.Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. 2008;102:e1–e11. doi: 10.1161/CIRCRESAHA.107.167809. [DOI] [PubMed] [Google Scholar]
- 54.Navedo MF, Takeda Y, Nieves-Cintron M, Molkentin JD, Santana LF. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. American journal of physiology Cell physiology. 2010;298:C211–220. doi: 10.1152/ajpcell.00267.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neeb ZP, Edwards JM, Alloosh M, Long X, Mokelke EA, Sturek M. Metabolic syndrome and coronary artery disease in Ossabaw compared with Yucatan swine. Comparative medicine. 2010;60:300–315. [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 57.Nieves-Cintron M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci U S A. 2008;105:15623–15628. doi: 10.1073/pnas.0808759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nieves-Cintron M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell'Acqua ML, Scott JD, Navedo MF. Selective downregulation of Kv2.1 function contributes to enhanced arterial tone during diabetes. Journal of Biological Chemistry. 2015;290:7918–7929. doi: 10.1074/jbc.M114.622811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishijima Y, Cao S, Chabowski DS, Korishettar A, Ge A, Zheng X, Sparapani R, Gutterman DD, Zhang DX. Contribution of KV1.5 Channel to Hydrogen Peroxide-Induced Human Arteriolar Dilation and Its Modulation by Coronary Artery Disease. Circ Res. 2017;120:658–669. doi: 10.1161/CIRCRESAHA.116.309491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noblet JN, Owen MK, Goodwill AG, Sassoon DJ, Tune JD. Lean and Obese Coronary Perivascular Adipose Tissue Impairs Vasodilation via Differential Inhibition of Vascular Smooth Muscle K+ Channels. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:1393–1400. doi: 10.1161/ATVBAHA.115.305500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nystoriak MA, Nieves-Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell'acqua ML, Scott JD, Santana LF, Navedo MF. AKAP150 Contributes to Enhanced Vascular Tone by Facilitating Large-Conductance Ca2+-Activated K+ Channel Remodeling in Hyperglycemia and Diabetes Mellitus. Circ Res. 2014;114:607–615. doi: 10.1161/CIRCRESAHA.114.302168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Dos Santos Fernandes J, Forbush K, Hofmann F, Sasse KC, Scott JD, Ward SM, Hell JW, Navedo MF. Ser1928 phosphorylation by PKA stimulates L-type Ca2+ channel Cav1.2 and vasoconstriction during acute hyperglycemia and diabetes. Science signaling. 2017;10 doi: 10.1126/scisignal.aaf9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nystoriak MA, Zhang D, Jagatheesan G, Bhatnagar A. Heteromeric complexes of aldo-keto reductase auxiliary KVbeta subunits (AKR6A) regulate sarcolemmal localization of KV1.5 in coronary arterial myocytes. Chemico-biological interactions. 2017 doi: 10.1016/j.cbi.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Povstyan OV, Barrese V, Stott JB, Greenwood IA. Synergistic interplay of Gbetagamma and phosphatidylinositol 4,5-bisphosphate dictates Kv7.4 channel activity. Pflugers Arch. 2017;469:213–223. doi: 10.1007/s00424-016-1916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rainbow RD, Hardy ME, Standen NB, Davies NW. Glucose reduces endothelin inhibition of voltage-gated potassium channels in rat arterial smooth muscle cells. J Physiol. 2006;575:833–844. doi: 10.1113/jphysiol.2006.114009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salomonsson M, Brasen JC, Sorensen CM. Role of renal vascular potassium channels in physiology and pathophysiology. Acta physiologica. 2017 doi: 10.1111/apha.12882. [DOI] [PubMed] [Google Scholar]
- 67.Serne EH, Stehouwer CD, ter Maaten JC, ter Wee PM, Rauwerda JA, Donker AJ, Gans RO. Microvascular function relates to insulin sensitivity and blood pressure in normal subjects. Circulation. 1999;99:896–902. doi: 10.1161/01.cir.99.7.896. [DOI] [PubMed] [Google Scholar]
- 68.Sobey CG, Faraci FM. Inhibitory effect of 4-aminopyridine on responses of the basilar artery to nitric oxide. Br J Pharmacol. 1999;126:1437–1443. doi: 10.1038/sj.bjp.0702439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spector AA, Yorek MA. Membrane lipid composition and cellular function. Journal of lipid research. 1985;26:1015–1035. [PubMed] [Google Scholar]
- 70.Stott JB, Barrese V, Jepps TA, Leighton EV, Greenwood IA. Contribution of Kv7 channels to natriuretic peptide mediated vasodilation in normal and hypertensive rats. Hypertension. 2015;65:676–682. doi: 10.1161/HYPERTENSIONAHA.114.04373. [DOI] [PubMed] [Google Scholar]
- 71.Straub SV, Girouard H, Doetsch PE, Hannah RM, Wilkerson MK, Nelson MT. Regulation of intracerebral arteriolar tone by K(v) channels: effects of glucose and PKC. Am J Physiol Cell Physiol. 2009;297:C788–796. doi: 10.1152/ajpcell.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Su W, Li W, Chen H, Liu H, Huang H, Li H. Advanced Glycation End Products Impair Voltage-Gated K+ Channels-Mediated Coronary Vasodilation in Diabetic Rats. PLoS One. 2015;10:e0142865. doi: 10.1371/journal.pone.0142865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tilley DC, Eum KS, Fletcher-Taylor S, Austin DC, Dupre C, Patron LA, Garcia RL, Lam K, Yarov-Yarovoy V, Cohen BE, Sack JT. Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc Natl Acad Sci U S A. 2014;111:E4789–4796. doi: 10.1073/pnas.1406876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tobin AA, Joseph BK, Al-Kindi HN, Albarwani S, Madden JA, Nemetz LT, Rusch NJ, Rhee SW. Loss of cerebrovascular Shaker-type K(+) channels: a shared vasodilator defect of genetic and renal hypertensive rats. Am J Physiol Heart Circ Physiol. 2009;297:H293–303. doi: 10.1152/ajpheart.00991.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tykocki NR, Boerman EM, Jackson WF. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Comprehensive Physiology. 2017;7:485–581. doi: 10.1002/cphy.c160011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu C, Lu Y, Tang G, Wang R. Expression of voltage-dependent K(+) channel genes in mesenteric artery smooth muscle cells. Am J Physiol. 1999;277:G1055–1063. doi: 10.1152/ajpgi.1999.277.5.G1055. [DOI] [PubMed] [Google Scholar]
- 77.Yang Y, Jones AW, Thomas TR, Rubin LJ. Influence of sex, high-fat diet, and exercise training on potassium currents of swine coronary smooth muscle. Am J Physiol Heart Circ Physiol. 2007;293:H1553–1563. doi: 10.1152/ajpheart.00151.2007. [DOI] [PubMed] [Google Scholar]
- 78.Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, Greenwood IA. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y, Gao YJ, Zuo J, Lee RM, Janssen LJ. Alteration of arterial smooth muscle potassium channel composition and BKCa current modulation in hypertension. European journal of pharmacology. 2005;514:111–119. doi: 10.1016/j.ejphar.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 80.Zhong XZ, Abd-Elrahman KS, Liao CH, El-Yazbi AF, Walsh EJ, Walsh MP, Cole WC. Stromatoxin-sensitive, heteromultimeric Kv2.1/Kv9.3 channels contribute to myogenic control of cerebral arterial diameter. J Physiol. 2010;588:4519–4537. doi: 10.1113/jphysiol.2010.196618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, Greenwood IA. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]