

Abstract
Heterozygous de novo mutations in the autophagy gene, WDR45, are found in beta-propeller protein-associated neurodegeneration (BPAN). BPAN is characterized by adolescent-onset dementia and dystonia; 66% patients have seizures. We asked whether WDR45 was associated with developmental and epileptic encephalopathy (DEE). We performed next generation sequencing of WDR45 in 655 patients with developmental and epileptic encephalopathies. We identified 3/655 patients with DEE plus four additional patients with de novo WDR45 mutations (6 truncations, one missense), all were female. Six presented with DEE and one with early-onset focal seizures and profound regression. Median seizure onset was 12 months, 6 had multiple seizure types and 5/7 had focal seizures. Three patients had MRI Susceptibility Weighted Imaging; blooming was noted in the globus pallidi and substantia nigra in the two older children aged 4 and 9 years, consistent with iron accumulation. We show that de novo mutations are associated with a range of developmental and epileptic encephalopathies with profound developmental consequences.
Introduction
De novo mutations in the X-linked gene WDR45 are associated with a large spectrum of neurological diseases ranging from a specific form of neurodegeneration with brain iron accumulation (NBIA), called beta-propeller protein-associated neurodegeneration (BPAN)1 (or static encephalopathy of childhood with neurodegeneration (SENDA))2 to isolated mild developmental delay (see supplementary references). BPAN comprises the largest group, beginning with childhood developmental delay and evolving to progressive dystonia, dementia and parkinsonism in late adolescence or early adulthood. Brain MRI shows iron deposition in the globus pallidus and substantia nigra, cerebral and cerebellar atrophy3. Two-thirds (37/56) of reported patients present with seizures, and at least ten also had Rett-like features (see supplementary references). Also, three males have been reported with West Syndrome4.
The developmental and epileptic encephalopathies (DEEs) are characterized by refractory seizures and frequent epileptiform activity that contribute to developmental slowing and often regression. Given that seizures occur frequently in patients with WDR45 diseases, we hypothesized that WDR45 mutations cause DEEs and sought to characterize the epilepsy phenotypes in WDR45 encephalopathy.
Methods
We recruited 655 patients with DEEs from our Australian epilepsy genetics research program and University of Washington. All patients had DEE according to ILAE classification criteria and did not have pathogenic variants in known DEE genes (5, unpublished data). An additional 59 Chinese patients with epilepsy were screened who had a range of phenotypes. Written informed consent was obtained for all patients from their parents or legal guardians. The Human Research Ethics Committees of Austin Health, Peking University First Hospital and University of Washington approved the study.
We performed targeted resequencing of WDR45 using molecular inversion probes (n= 655) or a custom-designed gene panel used in the Chinese cohort (n=59) to capture all exons and five base pairs of flanking intronic sequence, using established protocols5; 6. We priortized only variants that were non-synonymous, altered the acceptor/donor splice sites, or indels that disrupted the coding frame, and were not present in 65,000 individuals in ExAC (see URLs). We performed segregation analysis in parental DNA samples for all variants that met these criteria to determine if they arose de novo. Two additional patients were ascertained by clinical testing; one with DEE had whole-exome analysis (GeneDx) and one with ID and seizures underwent gene-panel testing6.
X-inactivation studies were performed in five patients to quantify the X-inactivation ratios; we assessed the variable number of tandem repeats (VNTR), CAG repeats of the MAOA region and human androgen receptor (HUMARA) locus as described previously (with modifications)7; 8. Briefly 200ng of DNA was digested with methylation-sensitive HhaI restriction enzyme; digested and un-digested DNA were PCR amplified and products sized and quantified by capillary electrophoresis.
Results
We identified seven de novo mutations, all in girls (Table 1, Figure 1A). 3/655 (0.5%) individuals with DEE were identified by targeted resequencing, while four additional patients were found through collaborations.
Table 1.
Phenotypic features of patients with WDR45 mutations
| Case ID Age, Gender | Epilepsy syndrome | Mutation* (skewed X-inactivation) |
Development prior to seizure onset | Age and seizure type at onset | Development after seizure onset/Regression | Other seizure types | Seizure offset | EEG | Neuroimaging MRI/FDG PET | Other features | Medications (current medication underlined) |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
#1 8y, F |
IS evolving to LGS | c.629delG p.Ser210X (mildly 84:16) | Delayed: fixed and followed by 6w, never rolled over or sat | 7m: IS (ceased by 18m, recurred at 5y) | Profound ID: non-verbal, non-ambulatory,
cannot sit Regression with seizure onset: stopped smiling, reduced eye contact |
Myo (onset uncertain, by
15m) Abs Tonic FIAS (onset 5y) |
6y 10m - following bilateral femoral osteotomy | Posterior quadrant epileptiform discharges
evolving to abundant posterior spike-wave discharges, PSW and low
voltage PFA, L>R Multifocal epileptiform activity Diffuse background slowing Spasms associated with bilateral slow & fast paroxysms (10m and 15m) Tonic seizures lasting 2–40 seconds associated with low voltage fast activity, R>L Myo jerks in sleep associated with PSW |
1y 4m: large ventricles
especially frontal horns, small incompletely rotated hippocampi, thin
CC, decreased white matter volume, delayed myelination (approx 9m),
large extra axial spaces 4y 1m: larger ventricles, round hippocampi, no internal architecture in hippocampi, bright on T2 and improved rotation, very thin CC, decreased white matter volume and very delayed myelination, blooming in cerebral peduncles and both globus pallidi FDG-PET 2y 4m: extensive bilateral frontal cortical hypometabolism, L > R |
Profound myopia Cortical visual impairment Asymmetric spastic quadriparesis Dislocated left hip Kyphosis Scoliosis No dysmorphic or behavioral features |
TPM, prednisolone (after 2 weeks stopped spasms for 4m), VPA, LTG, CZP (increased tonic seizures), VGB, CLB, ETX, LCM, ZNS, KD, VNS (more alert, fewer tonic seizures), GBP (5 weeks seizure free post-surgery) |
|
#2 7y, F |
LGS | c.1007_1008del p.Tyr336CysfsX5
recurrent (ND) |
Delayed: sat with support 6–8m, “babbling” and sitting independently at 16m | 16m: Tonic seizures | Profound ID: non-verbal, non-ambulatory,
cannot sit Regression with seizure onset: lost ability to sit, roll over, “babble”, use a fork |
Myo (onset 18m) FIAS (onset 3.5y) Abs (onset 4.5y) FIAS evolving to bilateral TCS (3y 10m) |
Ongoing | Occipital slowing and sharp waves evolving to
GSW in sleep followed by decrements, fast activity in wakefulness. Then
posterior predominant SSW, PSW and PFA in sleep. Tonic seizures associated with diffuse fast activity and bilateral PFA |
1y 2m: large ventricles
especially frontal horns, very thin CC, decreased white matter volume
and delayed myelination 1y 11m: large ventricles especially frontal horns, round hippocampi but no internal architecture, thin CC, decreased white matter volume and severe myelination delay, large extra axial CSF spaces |
Peripheral spasticity No behavioral features Brachycephaly |
ZNS, VPA (worse), CLB (increased duration tonic seizures, respiratory difficulties), LTG, TPM, LEV, prednisolone, 6m of no medication with no increase in seizures at 3.5y, Baclofen (seizures ceased), nil |
|
#3 11y, F |
DEE | c.700C>T p.Arg234X recurrent (No 49:50) | Delayed speech acquisition | 12m: Myo | Severe ID: single words 18m, rare word
combinations 9y, currently has 20 single words and follows simple
commands Regression with frequent seizures: loss of speech, less response to painful stimuli |
Febrile NCSE (14m) FIAS with clonic component |
10.5y | Frequent irregular GSW. Background slowing
with occipital predominance Myo jerks associated with irregular GSW. Staring episodes with irregular GSW with variable lead from central and posterior regions. Focal clonic seizures emanating from L or R central region. |
9y 4m: mild ventriculomegaly,
thin CC in posterior body and splenium, subtle white matter volume
reduction, normal myelination, SWI blooming in cerebral peduncles and
globus pallidi 10y 5m: mild subtle white matter volume reduction, normal myelination |
Sleep disturbance Dental issues Oro-motor apraxia Moderate pes planus Peripheral hypotonia Intoeing with wide-based gait Poor coordination High pain threshold Seizure trigger: fever, head flexion Aggression Broad nasal bridge Hypertelorism Mild facial asymmetry |
VPA (irritable), VGB, TPM, LEV, CLB (seizures ceased when added to VGB + TPM + LEV, VGB then weaned) |
|
#4 2y, F |
IS | c.627del p.Ser210GlnfsX78 (Yes 92:8) |
Delayed: rolled over 5m, sat unsupported 10m | 8m: IS | Delayed: nonverbal, pulls to stand, cruising,
eats with spoon No regression |
FIAS (9m) IS with head deviation to L (16m) |
Ongoing |
8m: modified
hypsarrhythmia 9m: bi-temporal epileptiform activity during sleep, hypsarrhythmia resolved From 10m: multifocal epileptiform activity, GSW |
7m: prominence of ventricles and extra axial CSF spaces, incomplete rotation of L hippocampi, generally thin CC, normal white matter volume and myelination | Hypertension (frusemide 1mg/kg
daily) Diarrhoea No behavioral features Cushingoid features |
Prednisolone (stopped spasms and hypsarrhythmia), VGB, TPM, pyridoxine, ACTH, medical cannabis, KD (metabolic acidosis requiring potassium bicarbonate supplementation) |
|
#5 7y, F |
MAE | c.454delT p.Cys152AlafsX9 (ND) |
Delayed smiling | 17m: Febrile seizure | Severe ID: few single words, walks
independently Regression with seizure onset: loss of speech |
Atonic (onset 24m) Myo (onset uncertain) NCSE (onset 2y 10m) Atypical Abs (onset 27m) |
5y Rare febrile TCS from 4y 2m |
GSW, PSW Biposterior quadrant epileptiform activity, L > R Slow background Atypical absence seizure with 1.5–2.5 Hz GSW |
23m:
normal 5y: mild cerebellar atrophy, mild reduction in white matter volume |
Genua valgum (knock knees) No dysmorphic or behavioral features |
LEV, LTG, VPA (stopped seizures) |
|
#6 4y, F |
Focal seizures with fever | c.726C>G p.Tyr242X (ND) |
Delayed speech acquisition: no spontaneous speech, could repeat and imitate intonation and speech sounds at 12m | 12m: Focal seizure with fever | Severe ID: few single words, walks
independently Regression with seizure onset: loss of “babble” |
Febrile FIAS (onset 1y) | 3y | No definite epileptiform
discharges Background slowing |
1y 6m: thin CC, normal white
matter volume and myelination 3y 6m: mild cerebellar atrophy of superior vermis, prominent ventricles and extra axial CSF spaces, thin CC, mild reduction in white matter, myelination normal |
Sleep disturbance No dysmorphic or behavioral features |
VPA (no change in seizure frequency),TPM (stopped seizures but withdrawn due to side effects), LEV (no seizures for 11m) |
| #7 3y, F |
DEE | c.614G>A p.Gly205Asp (no 34:66) |
Delayed: sat 2.5y, non-verbal, standing with support, not walking | 36m: Drop attacks | Severe ID: 2 single words, walks with assistance, reaches for spoon | FIAS | Ongoing | Diffuse moderate background slowing Multifocal discharges, GSW, GPFA | Normal | Episodes of hyperventilation Severe autistic behavior No dysmorphic features |
LEV, LTG, VPA, CLB, OCBZ, TPM, ZNS, RFM, LCM, no change to seizure frequency), KD, nil |
Mutation co-ordinates based on WDR45: NM_007075.3and protein NP_009006.2 # effect of frameshift mutation predicted by mutalyzer (see URLs).
All mutations arose de novo
Abs, Absence; ACTH, acetylcholinesterase; CC, corpus callosum; CLB, clobazam; CSE, status epilepticus; CSF, cerebrospinal fluid; CZP, clonazepam; ETX, ethosuxamide; F, female; FDG-PET, fluoro-deoxyglucose positron emission tomography; FIAS, focal impaired awareness seizure; FS, febrile seizures; GBP, gabapentin; GPFA, generalised paroxysmal fast activity; GPS, generalised polyspike; GSW, generalized spike-wave; ID, intellectual disability; IS, infantile spasms; KD, ketogenic diet; L, left; LCM, lacosamide; LGS, Lennox-Gastaut syndrome; LEV, levetiracetam; LTG, lamotrigine; m, months; MRI, magnetic resonsnace imaging; Myo, myoclonic; NCSE, non-convulsive status epilepticus; ND, not determined; OCBZ, oxcarbazepine; PET, positon emission tomography; PFA, paroxysmal fast activity; PPR, photoparoxysmal response; PSW, polyspike wave; R, right; sec, seconds; RFM, rufinamide; SE, status epilepticus; sec, seconds; SSW, slow spike and wave; SW, slow-wave; SWI, susceptibility weighted imaging; TCS, tonic-clonic seizures; TPM, topiramate; VGB, vigabatrin; VNS, vagal nerve stimulator; VPA, valproate; w, weeks; y, years; ZNS, zonisamide
Current medications underlined
Figure 1.
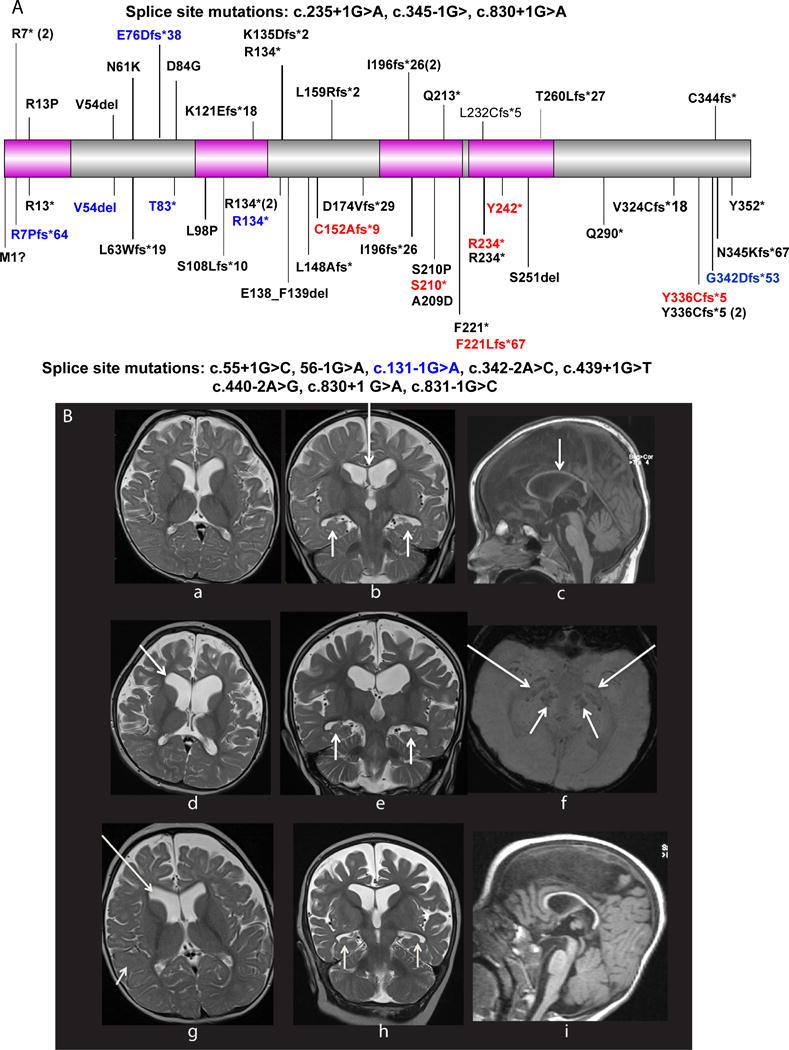
A Distribution of 63 all novel and published WDR45 mutations reported previously and in this study (in red). The majority of pathogenic mutations occur in females, with only eight males (in blue) described, the WD40 domains are shown in pink. Upper panel of the schematic shows all pathogenic mutations in patients in whom seizures are not present, while those mutations in the lower panel show those individuals who had seizures. Gray circles within the protein sketch represent missense mutations that are present >once in the ExAC dataset containing whole-exome data from ~65,000 individuals. 84% (52/63) mutations are either known or thought to lead to truncation of WDR45, suggesting a loss-of-function mechanism. Only 7 missense mutations and 3 single amino acid in-frame deletions have been reported, including the G205D mutation reported here. 5/7 missense mutations fall within the WD40 domains, and these regions tend to be devoid of missense variation in the general population, suggesting these missense mutations likely lead to a loss-of-function of the WD40 domains.
B (a–f) MRI scans of patient T20993 with WDR45 mutation at 16 months (a–b) and 4 years (c–f). a; axial T2 shows mild enlargement of the frontal horns of the lateral ventricles, prominent extra-axial CSF spaces and delayed myelination. b; coronal T2 shows small bilateral hippocampi (short arrows) and thin corpus callosum (long arrow). c; globally thinned corpus callosum (arrow) on sagittal T1. d; axial T2 shows marked atrophy, especially frontal with enlargement of the frontal horns, note that myelination has barely progressed and is commensurate with that of a 9 month old child. e; coronal T2 confirms atrophy, hippocampi remain tiny and rounded with no internal architecture (arrows), left hippocampus is more severe than the right. f; SWI demonstrates blooming in the basal ganglia (long arrows) and cerebral peduncles (short arrows) in keeping with iron deposition. MRI scans of patient T24729 with WDR45 mutation (g–i) at 9 years (g) and 12 years (h–i). g; axial T2 weighted scan shows enlargement of the frontal horns of the lateral ventricles (long arrow) and large frontal extra-axial CSF spaces, note very delayed myelination with lack of normal T2 hypointensity of the subcortical white matter. h; coronal T2 scan shows further volume loss of the frontotemporal regions, no improvement in myelination which remains that of a 9 month old infant, the hippocampi are small and rounded with loss of normal internal architecture (short arrows). i; sagittal T1 scan shows generally thinned corpus callosum.
Six de novo mutations resulted in a truncation (n=1) or an indel (n=5) predicted to cause a frameshift alteration. In one individual with intractable seizures and severe ID, we identified a de novo missense mutation (Gly205Asp) in the third WD40 domain, at a highly conserved nucleotide (GERP 3.76, CADD 21) predicted to be probably damaging by PolyPhen2(0.994) and damaging by SIFT(0). X-inactivation studies revealed inactivation was skewed in one female, mildly skewed in another and random in two. A fifth case was uninformative as she was homozygous for both polymorphisms (Table 1).
Six patients had a range of DEEs including one each with infantile spasms (IS), Lennox-Gastaut syndrome (LGS), IS evolving to LGS, Epilepsy with Myoclonic-Atonic seizures (MAE), and two with unclassified DEE. One patient did not have an DEE: she had a history of eight focal seizures with fever and severe regression with seizure onset at 1 year, following isolated speech delay.
Median seizure onset was 12 months (range 7–36). Focal impaired awareness seizures occurred in 5/7 patients. Six developed multiple seizure types including tonic, spasms (Supplementary video), focal, absence and myoclonic seizures. Seizures were refractory to virtually all anti-epileptic drugs; two individuals were on no drugs.
All patients showed developmental delay prior to seizure onset (Table 1). Five had regression with reduced speech in all 5 patients, loss of smiling and reduced eye contact in 1 patient, loss of sitting and rolling over in 1 patient and less response to painful stimuli in 1 patient. Regression coincided with seizure onset in 4 patients. Information gathered from examinations by neurologists and paediatricians and from direct questioning of parents identified two patients with profound impairment who are non-verbal and cannot sit, four patients with severe impairment who have few single words but can walk and the youngest patient at age 2 years with global developmental delay; she is non-verbal but can eat with a spoon and pull herself to stand but not walk independently. The two Chinese patients underwent formal developmental assessments; patient 5 was assessed at chronological age 7 years to be at a developmental age of 3 years for fine and gross motor skills, 16 months for language skills and 3 years for social skills. Patient 6 was assessed at chronological age 4 years to be at a developmental age of 3 years for fine motor skills and 19.5 months for language skills. Neurological examination revealed generalized spasticity in two (Supplementary video). Typical hand stereotypies of Rett syndrome seen in patients with BPAN were not observed in any of our patients.
Six patients had 12 brain MRI studies. Median age at first MRI was 17 months (range 7 months to 9 years). 5/6 children had a structurally normal brain with thin corpus callosum, ventriculomegaly (3 mild, 2 moderately severe with frontal predominance) and prominent frontal extra-axial CSF spaces. White matter volume loss with normal myelination was present in 2/6, and severe progressive atrophy associated with severely delayed myelination occurred in 2/6 (Figure 1B). Two patients developed mild cerebellar atrophy and one increased T2 signal. Hippocampi could be evaluated in 5/6 cases as one had no coronal imaging. 4/5 had dysplastic, small hippocampi with loss of normal internal architecture. Of the three children with susceptibility sequences (SWI), two had marked blooming in the globus pallidi and substantia nigra. The youngest patient had normal SWI at 7 months. Patient 5, whose scan was normal at 2 and 3 years, showed mild cerebellar atrophy at 5 years.
Discussion
We expand the phenotypic spectrum of WDR45 encephalopathy to include DEEs characterized by regression with seizure onset resulting in severe to profound impairment. Our cohort showed infantile onset of regression in five patients; associated with seizure onset at age 7 to 17 months in four and with escalating seizures in the remaining child. Patient 4, aged 2 years, has shown a plateau in development and patient 7 has severe intellectual disability at 3 years. The importance of this early onset regression is further highlighted by patient 6 who regressed from isolated speech delay to severe retardation at age one year with seizure onset. Individuals with BPAN/SENDA show mild delay in childhood and regress later in adolescence or adult life3, contrasting significantly with the regression and severe disability early in life in WDR45 DEE.
There is no obvious genotype-phenotype correlation with respect to mutation type or location to explain our patients’ early regression (Figure 1A). Two patients with West syndrome and refractory seizures had a premature truncation at position 210 yet three patients with isolated mild delay had truncating mutations just several amino acids away at positions 196 (n=2) and 2132; 9. Our only patient with a missense mutation had seizure onset at 36 months, much later than the six individuals with truncating mutations (median onset 12 months). Further analysis of seizure onset in patients with missense mutations is required to establish if this correlation is significant.
Only eight males have been reported with WDR45 mutations, including three with West syndrome and truncation mutations4. A boy with an Xp11.23 microdeletion, including WDR45, has a more severe early onset DEE with onset of spasms and focal seizures at 3 months and profound impairment from birth5. Males seem to have a more severe phenotype, or are proposed to have somatic mosaic mutations1; 4, suggesting that mutations in WDR45 are lethal, or more severe in hemizygous males. However, we also describe girls with a severe phenotype. Skewing of X-inactivation towards the mutated allele has been proposed to explain this phenotypic variability2. To date, 17/19 females with BPAN and pathogenic WDR45 mutations have skewed X-inactivation. Only one of our patients with West syndrome and a truncation mutation had skewed X-inactivation (Table 1). Given these figures, X-inactivation cannot be the sole explanation for the phenotypic spectrum associated with WDR45 mutations therefore additional genetic or environmental factors may underlie this phenotypic variability. However, whether skewing patterns in the blood reflect those in the brain is unknown; different cerebral patterns of X-inactivation could explain the WDR45 phenotypic spectrum.
Two patients had severe spasticity with truncal hypotonia, associated with relatively little movement (Supplementary video). The lack of a movement disorder separates their phenotype from genetic DEEs associated with SCN2A, SCN8A and STXBP1 mutations10, and other encephalopathies such as Rett syndrome.
Although adults with mutations in WDR45 have a distinct imaging phenotype, this was not seen in our patients. Adults with BPAN have T1 hyperintensity of the peduncles, substantia nigra and to a lesser extent, the globus pallidi3; 11. These regions may show T2/FLAIR hypointensity corresponding to iron deposition. None of our cohort had T2 hypointensity in the globus pallidi although two, aged 4 and 9 years, had blooming on SWI which likely represents iron deposition. The youngest reported case with T2 hypointensity in the globus pallidi was an 11 year old girl12, and our oldest patient last had imaging performed at 10 years and 5 months, so this may yet evolve.
Our MRI findings are similar to those reported in adults including atrophy, thin corpus callosum, delayed myelination and large ventricles consistent with white matter volume loss14; 15. Two had severe progressive atrophy, with prominent frontal lobe involvement, and delayed myelination. Hippocampal appearances have not been described previously: 4/5 (67%) patients had hippocampal malformations, including our youngest patient. Small, dysplastic and malrotated hippocampi with loss of normal internal architecture are developmental abnormalities; quite distinct from hippocampal sclerosis.
We expand the phenotypic spectrum of WDR45 disorders to include infantile presentations with DEEs with abnormal early development, early seizure onset, multiple seizure types, regression and severe to profound impairment. Importantly, the MRI findings of brain iron accumulation associated with BPAN were not visible on FLAIR/T2 sequences in the first decade; however, the more sensitive SWI sequence showed blooming at 4 and 9 years of age but not in early life. Thus, a normal MRI should not preclude consideration of WDR45 testing in patients with DEEs.
Supplementary Material
Supplementary video of patient T20993 showing general paucity of movement and a series of epileptic spasms.
Acknowledgments
We thank the patients and their families for participating in our research. This work was supported by funding from the NIH (NINDS 5R01NS069605) to H.C.M. G.L.C is supported by a postdoctoral fellowship from the American Epilepsy Society and the Lennox and Lombroso Fund, the Citizens United for Research in Epilepsy (CURE) Taking Flight Award and NIH (NINDS) Pathway to Independence Award (K99/R00) (1K99NS089858-01). I.E.S is supported by a National Health and Medical Research Council of Australia (NHMRC) Program Grant and Practitioner Fellowship.
GLC is a member of the scientific advisory board for Ambry Genetics; IES serves on the editorial boards of Neurology and Epileptic Disorders; may accrue future revenue on patent WO61/010176 and WO2006/133508; has received speaker honoraria or travel funding from Athena Diagnostics, UCB, GSK and Transgenomics; the remaining authors have no conflicts of interest to declare.
Footnotes
MISS MICHAELA WAAK (Orcid ID: 0000-0002-8317-3331)
Accession numbers
WDR45 mRNA NM_007075.3and protein NP_009006.2
Web Resources
ExAC: http://exac.broadinstitute.org/
CADD: http://cadd.gs.washington.edu/
Polyphen: http://genetics.bwh.harvard.edu/pph2/
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines
References
- 1.Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–449. doi: 10.1038/ng.2562. 449e441. [DOI] [PubMed] [Google Scholar]
- 3.Hayflick SJ, Kruer MC, Gregory A, et al. beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013;136:1708–1717. doi: 10.1093/brain/awt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakashima M, Takano K, Tsuyusaki Y, et al. WDR45 mutations in three male patients with West syndrome. J Hum Genet. 2016 doi: 10.1038/jhg.2016.27. [DOI] [PubMed] [Google Scholar]
- 5.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong W, Zhang Y, Gao Y, et al. SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia. 2015;56:431–438. doi: 10.1111/epi.12925. [DOI] [PubMed] [Google Scholar]
- 7.Hendriks RW, Chen ZY, Hinds H, et al. An X chromosome inactivation assay based on differential methylation of a CpG island coupled to a VNTR polymorphism at the 5′ end of the monoamine oxidase A gene. Human molecular genetics. 1992;1:662. doi: 10.1093/hmg/1.8.662. [DOI] [PubMed] [Google Scholar]
- 8.Allen RC, Zoghbi HY, Moseley AB, et al. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 9.Nishioka K, Oyama G, Yoshino H, et al. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging. 2015;36:2004.e2009–2004.e2015. doi: 10.1016/j.neurobiolaging.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 10.McTague A, Howell KB, Cross JH, et al. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2015;15:304–316. doi: 10.1016/S1474-4422(15)00250-1. [DOI] [PubMed] [Google Scholar]
- 11.Doorn JM, Kruer MC. Newly characterized forms of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2013;13:413. doi: 10.1007/s11910-013-0413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohba C, Nabatame S, Iijima Y, et al. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J Hum Genet. 2014;59:292–295. doi: 10.1038/jhg.2014.18. [DOI] [PubMed] [Google Scholar]
- 13.Abidi A, Mignon-Ravix C, Cacciagli P, et al. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur J Hum Genet. 2015 doi: 10.1038/ejhg.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffjan S, Ibisler A, Tschentscher A, et al. WDR45 mutations in Rett (-like) syndrome and developmental delay: Case report and an appraisal of the literature. Mol Cell Probes. 2016;30:44–49. doi: 10.1016/j.mcp.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Yoganathan S, Arunachal G, Sudhakar SV, et al. Beta Propellar Protein-Associated Neurodegeneration: A Rare Cause of Infantile Autistic Regression and Intracranial Calcification. Neuropediatrics. 2016 doi: 10.1055/s-0035-1571189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary video of patient T20993 showing general paucity of movement and a series of epileptic spasms.