

Abstract
Targeting the transcription factor NRF2 has been recognized as a feasible strategy for cancer prevention and treatment, but many of the mechanistic details underlying its role in cancer development and progression are lacking. Therefore, careful mechanistic studies of the NRF2 pathway in cancer initiation and progression are needed to identify which therapeutic avenue—activation or inhibition—is appropriate in a given context. Moreover, while numerous reports confirm the protective effect of NRF2 activation against chemical carcinogenesis little is known of its role in cancer arising from spontaneous mutations. Here, we tested the effects of NRF2 modulation (activation by sulforaphane or inhibition by brusatol) in lung carcinogenesis using a chemical (vinyl carbamate) model in A/J mice and a genetic (conditional KrasG12D oncogene expression, to simulate spontaneous oncogene mutation) model in C57BL/6J mice. Mice were treated with NRF2 modulators before carcinogen exposure or KrasG12D expression to test the role of NRF2 in cancer initiation, or treated after tumor development to test the role of NRF2 in cancer progression. Lung tissues were analyzed to determine tumor burden, as well as status of NRF2 and KRAS pathways. Additionally, proliferation, apoptosis, and oxidative DNA damage were assessed. Overall, NRF2 activation prevents initiation of chemically induced cancer, but promotes progression of pre-existing tumors regardless of chemical or genetic etiology. Once tumors are initiated, NRF2 inhibition is effective against the progression of chemically and spontaneously induced tumors. These results have important implications for NRF2-targeted cancer prevention and intervention strategies.
Keywords: chemoprevention, sulforaphane, brusatol, carcinogenesis, vinyl carbamate
INTRODUCTION
Cancer is the second leading cause of death in the United States (US) and worldwide1. Lung cancer is the most common cancer in the United States (excluding melanoma) with an estimated 158,000 deaths in 2016, which accounts for 27% of all cancer deaths, and makes it the leading cause of cancer deaths2. Non-small cell lung cancer (NSCLC) makes up ~85% of all diagnosed cases of lung cancer, and has a predicted 5-year survival rate of 15.9%3. Recent studies have also identified a high prevalence of mutations in genes of the nuclear factor erythroid 2-related factor 2 (NRF2) pathway that result in uncontrolled, constitutive activation of NRF2 in NSCLC4,5. NRF2 is the primary regulator of cellular redox homeostasis and xenobiotic metabolism6. Through its role as a transcription factor, NRF2 controls the expression of antioxidant response element (ARE) regulated genes7. NRF2 target genes can be classified into: drug metabolizing enzymes (phase I-III), redox proteins, transcription factors, anti-apoptotic proteins, carbohydrate and lipid metabolizing enzymes, proliferation and cell cycle regulators, proteostasis machinery (autophagy and proteasomal), and heme and iron metabolizing proteins8,9. NRF2 is expressed in all human organs, but its basal levels under homeostatic conditions are low due to tight regulatory mechanisms that continuously target it for degradation in the cytosol9. This is achieved by association of NRF2 with Kelch-like ECH-associated protein 1 (KEAP1), the main regulator of NRF2, which serves as an E3 ubiquitin ligase substrate adaptor protein10. KEAP1 dimers interact with the N-terminal Neh2 domain of NRF2, thus bringing it in close proximity to the Cullin 3-RING Box 1 (CUL3-RBX1) E3 ubiquitin ligase11,12. As a result, NRF2 is ubiquitylated and subsequently degraded by the 26S proteasome. However, under oxidative stress, critical cysteine residues in KEAP1 get oxidized, which affects its conformation, thereby leading to an NRF2-KEAP1 complex that does not favor ubiquitylation13. As a consequence, this protein complex is stabilized and newly synthesized NRF2 accumulates and translocates to the nucleus, where it dimerizes with a small MAF protein and performs its transcriptional function 7. Upon restoration of redox balance, KEAP1 translocates into the nucleus to escort NRF2 back into the cytosol and restore it to basal levels14. This mechanism of controlled NRF2 regulation is operative in the canonical NRF2 pathway15.
Numerous studies have demonstrated that Nrf2−/− mice are more susceptible than wild type mice to chemically induced carcinogenesis16,17, in agreement with the observation that canonical NRF2 activation by chemopreventive compounds, such as sulforaphane (SF), prevent carcinogenesis18. However, NRF2 has a dark side in cancer, as uncontrolled activation of NRF2 aids in the promotion, progression, and metastasis of cancer; contributes to chemo and radiotherapy resistance; and confers a poor prognosis15,19-22. Cancer cells constitutively activate NRF2 signaling by diverse mechanisms, including: somatic mutations, like gain-of-function NRF2 or loss-of-function KEAP1 or CUL3 mutations that prevent KEAP1-mediated degradation of NRF223-26; KEAP1 adduction by fumarate, an electrophilic oncometabolite that accumulates due to mutations in the metabolic enzymes fumarate hydratase (FH) or fumarylacetoacetate (FAH)27; KEAP1 promoter hypermethylation that reduces its expression28; and activation of KRAS, BRAF, and MYC oncogenes that increases NRF2 mRNA expression29,30. The resulting uncontrolled upregulation of NRF2 contributes to malignancy by increasing chemotherapeutic detoxification, maintaining reducing conditions, promoting metabolic reprogramming, increasing proliferation, suppressing apoptosis, inducing autophagy, and upregulating the proteasome15,31. Additionally, NRF2 maintains low ROS levels in cancer stem cells, which promotes quiescence, self-renewal, anchorage-independent growth, and protects them from chemotherapy32,33. Consistent with the dual roles of NRF2 in cancer, previous studies of lung carcinogenesis in mice found that at early time points (8-12 weeks) after urethane (ethyl carbamate, which is metabolized into VC) exposure, Nrf2−/− mice had more tumors than Nrf2+/+ mice, indicating the protective role of NRF2 against cancer initiation34,35. However, at later time points (24 weeks), Nrf2−/− mice had less and smaller tumors with clear borders, unlike their wild type counterparts, demonstrating that NRF2 promotes tumor growth34. Therefore, targeting NRF2 in cancer has been identified as a means to confer sensitivity to cancer therapies and improve prognosis36. To overcome the dark side of NRF2, our group determined that brusatol was able to inhibit NRF2 signaling and sensitize the human NSCLC cell line A549 (KEAP1 mutant, high NRF2) to cisplatin, carboplatin, 5-fluorouracil, etoposide, and paclitaxel37. In a xenograft model, brusatol was effective in combating both intrinsic and acquired resistance to cisplatin37. Furthermore, in an oncogenic KRAS-driven NSCLC mouse model, we identified that KRAS-driven NRF2 upregulation mediated chemoresistance to cisplatin, and that this could be overcome with brusatol co-treatment30. Importantly, oncogenic KRAS mutations are found in ~30% of NSCLC originating both from carcinogen exposures and spontaneous oncogene activation38,39.
Tomasetti and Vogelstein40 conjectured that replication errors (“bad luck”) that occur in stem cells determine cancer risk. They consider that “deterministic tumors” are those associated with specific environmental or hereditary risk factors, and can be prevented by primary prevention strategies (such as altered lifestyles or vaccinations); whereas “replicative tumors” are mostly affected by stochastic factors (replication errors leading to spontaneous mutations in proto-oncogenes or tumor suppressors), so they cannot be prevented. Here we investigated the role of NRF2 in carcinogenesis induced by (i) environmental risk factors (a chemical carcinogenesis model using VC), or (ii) due to spontaneous mutations in oncogenes (a genetic model, using Cre-mediated activation of the KrasG12D oncogene). Using these two models we investigated the effects of NRF2 on (a) cancer initiation: NRF2 was activated or inhibited before carcinogen exposure or before expression of oncogenic KrasG12D, or (b) cancer progression: NRF2 was modulated after tumor development. Our results support this overall conclusion: NRF2 prevents the initiation of environmental factor-induced cancer but has no effect on spontaneous cancer arising from oncogene activation. Furthermore, NRF2 promotes the progression of pre-existing tumors, while NRF2 inhibition reduces pre-existing tumor growth in both models.
MATERIALS AND METHODS
Antibodies and chemicals
Sulforaphane (SF) was purchased from LKT laboratories, vinyl carbamate was purchased from Toronto Research Chemicals. Brusatol (Bru) was purified in our laboratory as described previously37. Corn oil was purchased from Sigma. The primary antibodies against NRF2, KEAP1, GCLM, NQO1, AKR1C1, AKR1B10, p-ERK, ERK, KRAS, GAPDH, as well as horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnologies.
Mouse lung cancer models
All mice were housed in a specific pathogen-free room with food and water ad libitum at the University of Arizona Animal Care facility. Protocols were approved by the UA Institutional Animal Care and Use Committee and all experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals. Six-week-old male mice were acclimated to laboratory conditions for one week and then were randomly allocated (n=5/group) to the treatment groups: (i) non-treatment (vehicle control: corn oil), (ii) SF (12.5 mg/kg i.p.) pre-treatment, (iii) SF (12.5 mg/kg i.p.) post-treatment, (iv) Bru (2 mg/kg i.p.) pre-treatment, (v) Bru (2 mg/kg i.p.) post-treatment. For the chemical carcinogenesis model with vinyl carbamate, A/J mice were obtained from The Jackson Laboratory. Mice received two i.p. injections of VC (total dose 32 mg/kg), one week apart, and were sacrificed at 15 weeks, as described previously41. For the genetic model, LSL-K-rasG12D/+ mice were obtained from The Jackson Laboratory42. Mice were intratracheally instilled with Cre adenovirus to activate expression of the KrasG12D oncogene. These mice were sacrificed 6 weeks after the start of the experiment. See Figure 1 for a detailed description of treatment schedules. Mice were weighed once a week for the duration of the experiments. At the time of euthanasia, lungs were collected for gross analysis of total surface tumors and weight. Then, the lungs were divided and were either fixed in neutral buffered formalin for histological analyses or snap frozen for protein and RNA analyses.
Figure 1. NRF2 modulation in chemical and genetic lung cancer models.
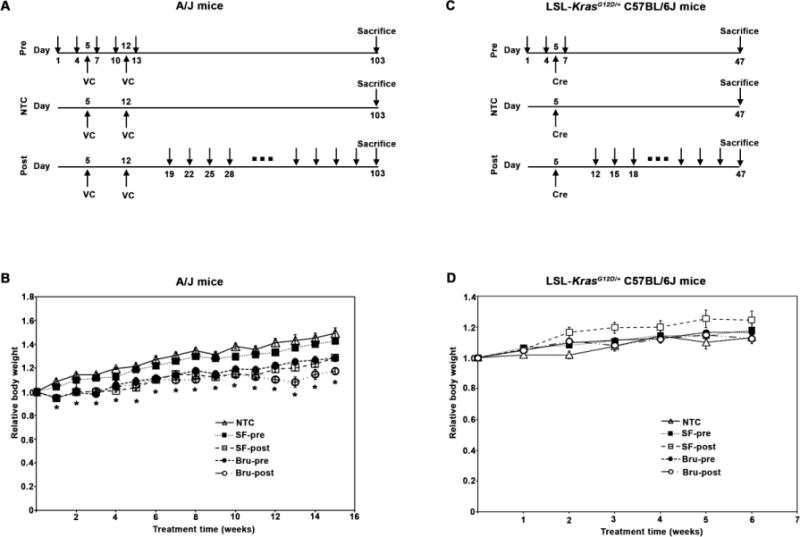
(A) Chemical carcinogenesis model of lung cancer. Seven-week-old A/J mice (n=5/group) were randomly allocated to non-treatment control (NTC), pre-treatment (SF-pre or Bru-pre), or post-treatment (SF-post or Bru-post) groups. Mice in all the groups were injected intraperitoneally (i.p.) twice with the chemical carcinogen vinyl carbamate (VC, 16 mg/kg each injection) at days 5 and 12 to induce lung tumors as indicated. Mice in the pre-treatment (SF-pre or Bru-pre) groups received either sulforaphane (SF, 12.5 mg/kg) or Brusatol (Bru, 2 mg/kg) every three days, as indicated with arrows, for five times. Mice in the post-treatment (SF-post or Bru-post) groups were injected with SF or Bru every three days, starting one week after the second VC injection for a total of 13 weeks (Fig. 1A). Mice were sacrificed the next day after the last injection. (B) A/J mice were weighed weekly and relative body weights (changes in weight relative to weight at the start of the experiment) are reported. Results are presented as means ± SD. *, P<0.05 compared to NTC. (C) Genetic model of lung cancer. Seven-week-old LSL-KrasG12D/+ C57BL/6J mice (n=5/group) were randomly allocated to the NTC, SF-pre, Bru-pre, SF-post, or Bru-post groups. Mice were intratracheally (i.t.) instilled with Cre adenovirus at day 5 to activate the KrasG12D allele to induce lung tumors. Mice in the SF-pre or Bru-pre group received i.p. injections of SF or Bru every three days as indicated with arrows for three times. Mice in the SF-post or Bru-post group were injected with SF or Bru every three days, starting one week after i.t. for a total of 5 weeks. Mice were sacrificed the next day after the last injection. (D) LSL-KrasG12D/+ C57BL/6J mice were weighed weekly and relative body weights are reported. Results are presented as means ± SD. No significant differences in relative body weights among the groups were observed.
Immunoblot analyses
Lung tissues were homogenized in sample buffer containing 50 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS), 10% glycerol, and 100 mM dithiothreitol (DTT). Lysates were boiled for 10 min and sonicated. Lysates were resolved by SDS-PAGE, transferred onto a nitrocellulose membrane, and subjected to immunoblot analyses with the indicated antibodies. The relative intensity of the bands was quantified using the ChemiDoc CRS gel documentation system and Quantity One software (Biorad).
Histological analyses
Staining was performed using the EnVision+System-HRP kit (Dako) according to the manufacturer’s instructions and nuclei were counterstained with hematoxylin. Images were acquired using a Nikon Eclipse 50i microscope with the NIS Elements F software. H&E images were acquired using a 2X objective and images were superimposed to compose a whole lung image using Photoshop 7.0. Apoptotic cells in tissue sections were detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using the In Situ Cell Death Detection Kit (Roche) according to the manufacturer’s instructions. Briefly, tissue sections were treatment with proteinase K (20 μg/mL) in 10 mM Tris-HCl (pH 7.8) at 37 °C for 30 min. Tissue sections were incubated with TUNEL reaction mixture for 1 h at 37 °C in the dark and nuclei were counterstained with Hoechst. Slides were analyzed using a Zeiss Observer.Z1 microscope with the Slidebook 5.0 software (Intelligent Imaging Innovations).
Statistical analyses
The results are presented as fold changes to the NTC group. Data are all shown as mean ± SEM (n=5). Statistical tests were performed using SPSS 20.0. Unpaired student’s t-tests were used to compare the means of two groups. One-way analysis of variance was applied to compare the means of three or more groups. P<0.05 was considered to be significant.
RESULTS
NRF2 modulation in chemical and genetic lung cancer models
To test the roles of NRF2 in cancer initiation and progression, two different models of lung adenocarcinoma were used: chemical carcinogenesis (the VC model) and genetic (simulating spontaneous mutation, KrasG12D) models. The chemical carcinogenesis model for lung cancer was established in A/J mice, known for their sensitivity to chemically-induced lung tumors. A/J mice were injected twice with VC (16 mg/kg i.p. for each injection) to induce lung cancer as shown in Fig. 1A. NRF2 modulation was achieved by injecting the mice with either the activator sulforaphane (SF, 12.5 mg/kg i.p.) or the inhibitor brusatol (Bru, 2 mg/kg i.p.) before (Pre) or after (Post) VC injections (Fig. 1A). All the mice survived during the course of the experiment. Animal body weights were recorded weekly. There was a significant reduction in body weight of mice in the SF-post, Bru-pre, and Bru-post group, compared to the non-treated control (NTC) or SF-pre group (Fig. 1B). The genetic model of lung cancer was established using LSL-KrasG12D/+ C57BL/6J mice, in which addition of Cre recombinase promotes the expression of the oncogenic KrasG12D mutant that promotes tumor development (Fig. 1C). NRF2 was activated or suppressed by injecting SF or Bru either before (SF-pre or Bru-pre) or after (SF-post or Bru-post) administration of Cre (Fig. 1C). No significant reduction in body weight was noticed in any group, indicating neither the treatment nor the tumor burden affected the body weight of C57BL/6J mice (Fig. 1D).
NRF2 modulation had different effects on chemical vs. genetic lung cancer models
In the VC model, mice in the NTC group had an average of 27 surface tumors per mouse (Fig. 2A and B). Pre-treatment with SF significantly reduced the number of surface tumors (20 in the SF-pre group), consistent with the protective role of NRF2 in chemically induced carcinogenesis, whereas post-treatment with SF marginally increased the number of tumors (30 in the SF-post group). Pre-treatment with Bru significantly increased the number of tumors (32.5 in the Bru-pre group), while Bru post-treatment reduced the number of tumors (15 in the Bru-post group). The relative weight of the lungs of the treatment groups did not differ from the NTC group (Fig. 2C). Consistent with the results from surface tumors, histological analysis of the lungs also revealed differences in the number, and particularly in the size, of internal tumors (Fig. 2D). Quantification of tumor area showed that the mice in the SF-pre or Bru-post groups had smaller tumors than the NTC mice, but the mice in the SF-post or Bru-pre group had larger tumors (Fig. 2E). These results indicate that pre-activation of NRF2 by SF prevents tumor initiation in response to chemical carcinogen exposure as both tumor number and size were decreased (Fig. 2B and 2E). However, after tumor initiation, activation of NRF2 by SF promotes the growth of pre-existing tumors, as indicated by larger tumors, but only a marginal increase in tumor number (Fig. 2B and 2E). Bru showed opposite results, i.e. pre-inhibition of NRF2 increased tumor number and size, while post-inhibition reduced both tumor burden (Fig. 2B and 2E).
Figure 2. Nrf2 modulation had different effects on chemical vs. genetic lung cancer models.
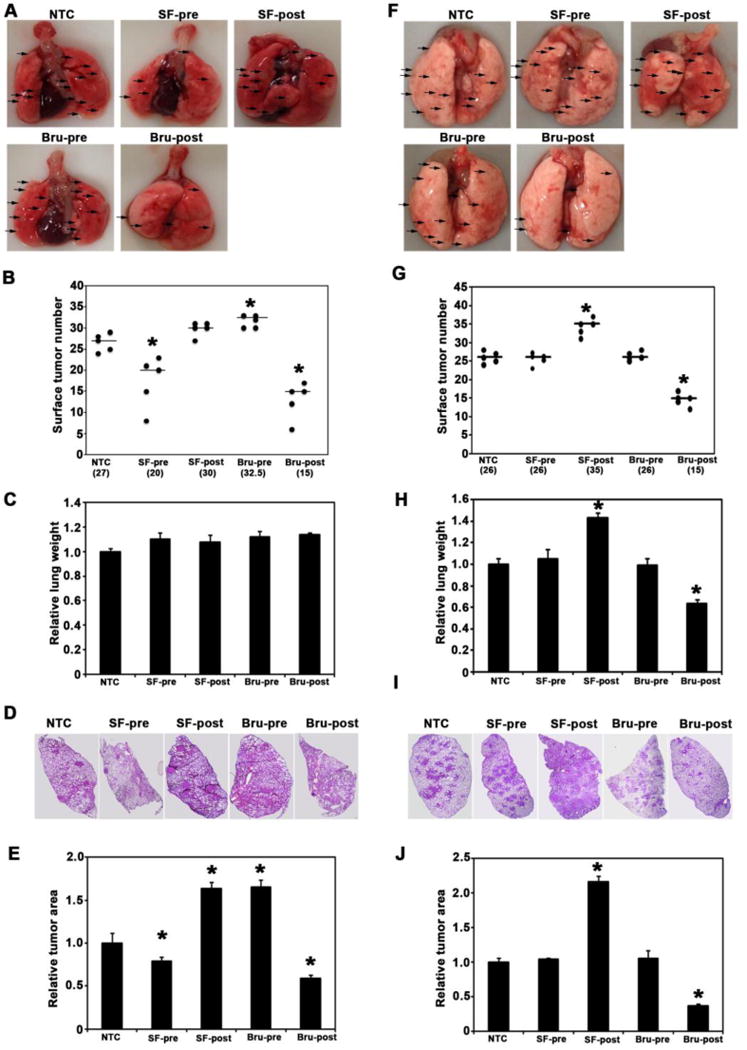
(A) Representative image of whole lungs from each group of A/J mice in the VC model (arrows point to surface tumors). (B) Average number of total surface tumors in the whole lungs from each group of A/J mice. *, P<0.05 compared to NTC. (C) Wet lungs were weighed for each A/J mouse and relative lung weights (normalized to NTC group) were obtained. Results are presented as means ± SD. No significant differences were observed. (D) Representative H&E staining of lung tissue sections of A/J mice. (E) The ratio of tumor area to total area of lung tissue sections in A/J mice was quantified and the relative tumor area (normalized to the NTC group) is presented. Results are presented as means ± SD. *, P<0.05 compared to NTC. (F) Representative image of whole lungs from each group of LSL-KrasG12D/+ C57BL/6J mice in the KrasG12D model (arrows are pointing at surface tumors. (G) Average number of total surface tumors in the whole lungs from each group of KrasG12D mice was counted. *, P<0.05 compared to NTC. (H) Wet lungs were weighed for each KrasG12D mouse and relative lung weights (normalized to NTC group) were obtained. Results are presented as means ± SD. *, P<0.05 compared to NTC. (I) Representative H&E staining of lung tissue sections of C57BL/6J mice. (J) The ratio of tumor area to total area of lung tissue sections in C57BL/6J mice was quantified and the relative tumor area (normalized to the NTC group) is presented. Results are presented as means ± SD. *, P<0.05 compared to NTC.
In the KrasG12D model, the number of surface tumors in the NTC group was 26. Pre-treatment with SF or Bru had no effects on tumor number (Fig. 2F and G) or tumor size (Fig. 2I and J). However, after tumor initiation, activation of NRF2 by SF post-treatment, or suppression of NRF2 by Bru post-treatment increased or decreased tumor number and size, respectively (Fig. 2F, G, I, and J). Interestingly, these trends were also observed for relative lung weight in KrasG12D C57BL/6J mice (Fig. 2H). These results indicate that upon the acquisition of high-risk spontaneous mutation of oncogenes, such as KrasG12D, chemoprevention strategies are ineffective at preventing tumor initiation. Once tumors are initiated, NRF2 inhibition slows down tumor progression, while NRF2 activation enhances tumor growth and multiplicity.
SF and Bru modulate NRF2 and KRAS signaling in lung tissues
Based on our previous findings that VC- or KrasG12D-induced lung tumor tissues have higher levels of NRF2 and its target genes, as well as activation of the KRAS-ERK pathway (as measured by an increase in the phosphorylation of ERK)30,41, modulation of the NRF2 and KRAS pathways was examined in lung tissues of mice at the end of the experiments by analyzing protein and mRNA expression of relevant genes. In the VC model, SF-pretreatment slightly lowered NRF2 protein levels compared to non-treatment, although no significant changes in the expression of NRF2 target genes (GCLM, NQO1, AKR1C1, AKR1B10, and HMOX1) at mRNA or protein levels were observed (Fig. 3A, B, and C). As expected, SF post-treatment greatly enhanced the protein levels of NRF2 and the expression of its target genes at both mRNA and protein levels, while the expression of KEAP1 (mRNA or protein levels) was unchanged (Fig. 3A, B, and C). Consistent with the results in Figures 2B and E, a significant increase in the phosphorylation of ERK (p-ERK) was observed in the SF-post and Bru-pre groups, indicating higher tumor burden in these groups (Fig. 3A, B, and C). As expected, NRF2 signaling and ERK phosphorylation were decreased in the Bru-post group (Fig. 3A, B, and C). Overall, total ERK and KRAS protein levels remained unchanged across all treatment groups.
Figure 3. SF and Bru modulate NRF2 and KRAS signaling in lung tissues.
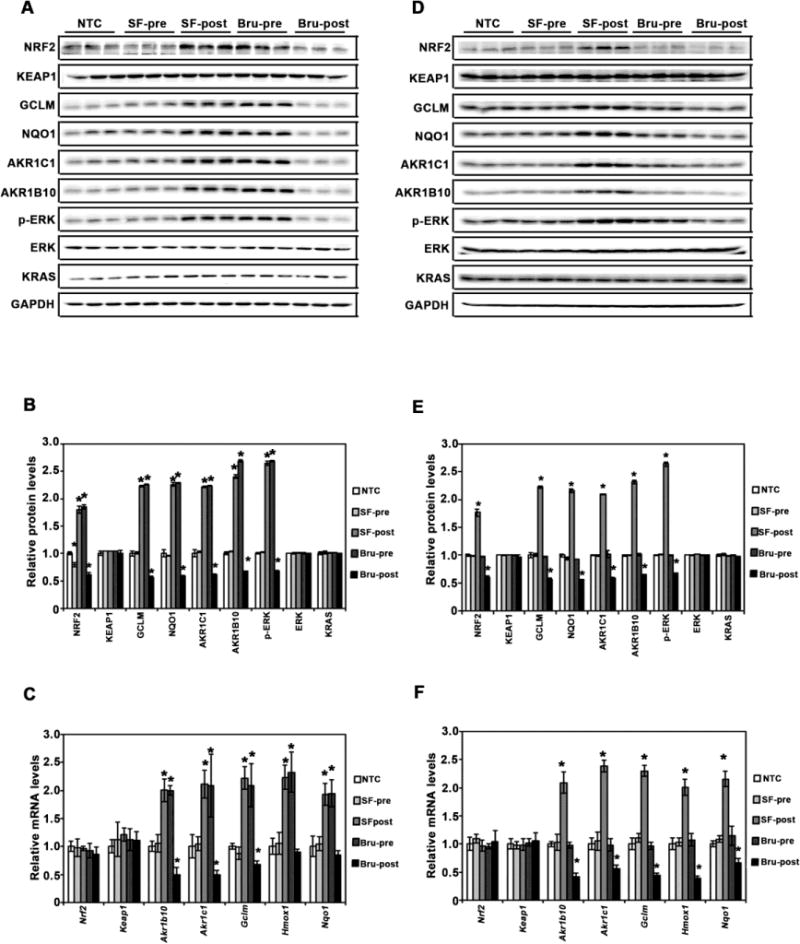
(A and D) Protein expression of the NRF2 and KRAS pathways. Total lung tissue lysates of mice (n=3/group) from the VC model (A) and the Kras model (D) were subjected to immunoblot analyses with the indicated antibodies. (B and E) Band intensities were quantified and normalized to the loading control (GAPDH) to obtain relative protein levels for the VC model (B) and the Kras model (E). The relative protein level in the NTC group was set to 1, and the relative protein levels in other groups were normalized to the NTC group. Results are presented as means ± SD. *, P<0.05 compared to NTC. (C and F) mRNA expression of the indicated genes in the NRF2 and KRAS pathways. Total RNA extracted from lung tissue (n=3/group) from the VC model (C) and the Kras model (F) was subjected to qRT-qPCR analysis. Results are presented as means ± SD. *, P<0.05 compared to NTC.
In the KrasG12D model, SF or Bru pre-treatment did not affect NRF2 or KRAS signaling, compared to the NTC group (Fig. 3D, E, and F), which is consistent with the result showing that pre-treatment had no effects on tumor burden (Fig. 2G and J). SF post-treatment increased NRF2 signaling and ERK phosphorylation, while Bru post-treatment reduced NRF2 signaling and p-ERK (Fig. 3D, E, and F). As expected, none of the treatments affected KEAP1 (mRNA or protein levels were measured, Fig. 3D, E, and F), as well as the total protein levels of ERK or KRAS (Fig. 3D and E).
NRF2 modulation by SF and Bru in lung tumor tissues correlates with proliferation, oxidative DNA damage, and apoptosis
Histological examination of lung tissue sections revealed a correlation in the levels of NRF2, GCLM, NQO1, and AKR1C1 (Fig. 4 and 5, top four panels) with proliferation, oxidative DNA damage, and apoptosis of tumor tissues in both the VC and KrasG12D models (Fig. 4 and 5, bottom four panels). In the VC model, IHC staining clearly showed an increase in NRF2 signaling in lung tumor tissues from the SF-post or Bru-pre groups, and a decrease in the Bru-post group (Fig 4, top four panels). These results are consistent with the NRF2 and target genes protein levels measured by immunoblot analyses in total lung tissue lysates (Fig. 3B). The proliferative marker Ki67 was most strongly expressed in the SF-post or Bru-pre group (Fig. 4, Ki67 panel). These results are consistent with previous observations that NRF2 promotes proliferation31. Furthermore, high NRF2 expression also correlates with lower phosphorylated histone H2AX (γ-H2AX) and 8-oxo-dG staining (Fig. 4, γ-H2AX and 8-oxo-dG panels), consistent with the reported potential of NRF2 signaling to reduce endogenous ROS generation, which directly affect DNA integrity by causing oxidative modifications (8-oxo-dG) and double strand breaks (γ-H2AX)43. In contrast, lower NRF2 expression, which correlates with high γ-H2AX and 8-oxo-dG, also correlates with a higher degree of cells undergoing apoptosis, as measured by TUNEL (Fig. 4, bottom panel).
Figure 4. NRF2 modulation by SF and Bru in lung tumor tissues correlates with proliferation, oxidative DNA damage, and apoptosis in the VC model.
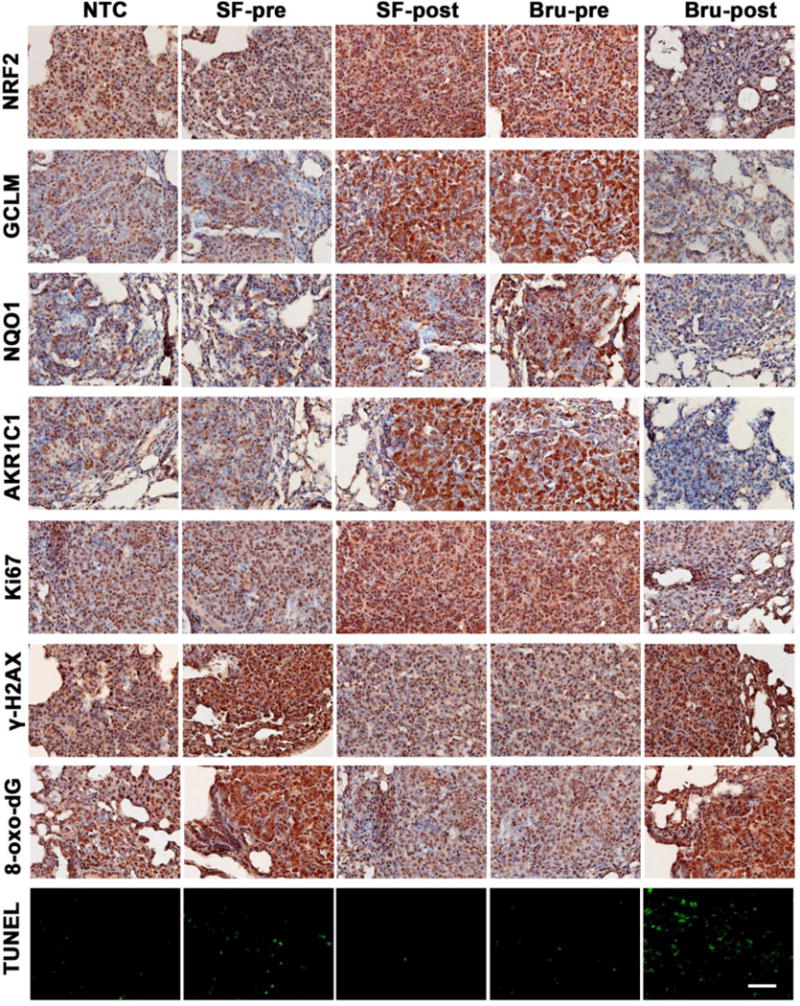
A representative image of IHC staining of lung tissue sections containing tumors from each group (n=3) in the VC model. Expression levels of the following proteins in tissues were measured: NRF2 pathway proteins (NRF2, GCLM, NQO1, and AKR1C1 in the top four panels), the proliferation marker Ki67 (Ki67 panel), and phosphorylated histone H2AX (γ-H2AX panel). The oxidative DNA marker 8-oxo-deoxyguanosine (8-oxo-dG panel) was also detected. The bottom panel shows in situ detection of apoptotic cells by TUNEL. Scale bar: 100 μm.
Figure 5. NRF2 modulation by SF and Bru in lung tumor tissues correlates with proliferation, oxidative DNA damage, and apoptosis in the Kras model.
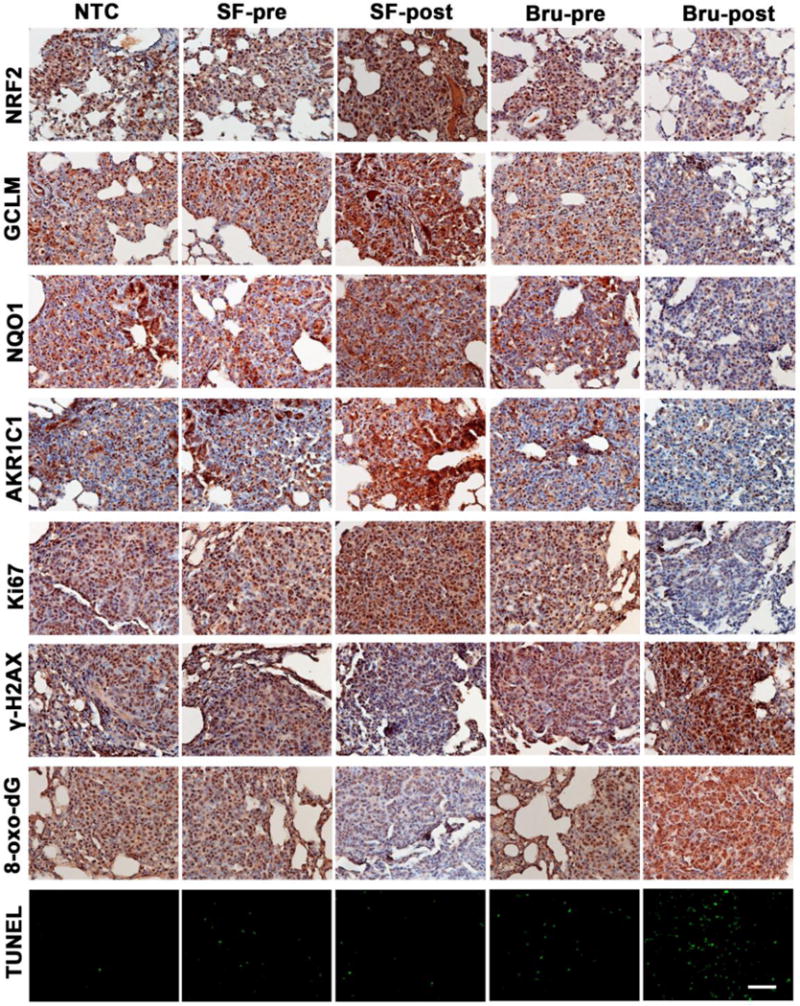
A representative image of IHC staining of lung tissue sections containing tumors from each group (n=3) in the Kras model. Expression levels of the following proteins in tissues were measured: NRF2 pathway proteins (NRF2, GCLM, NQO1, and AKR1C1 in the top four panels), the proliferation marker Ki67 (Ki67 panel), and phosphorylated histone H2AX (γ-H2AX panel). The oxidative DNA marker 8-oxo-deoxyguanosine (8-oxo-dG panel) was also detected. The bottom panel shows in situ detection of apoptotic cells by TUNEL. Scale bar: 100 μm.
In the KrasG12D model, IHC of lung tumor sections clearly showed enhanced or reduced expression of NRF2 and its target genes (GCLM, NQO1, AKR1C1) in the SF-post or Bru-pre group (Fig. 5, top four panels), respectively. These results were consistent with the immunoblot analyses of lung tissue lysates (Fig 3E). Interestingly, a positive Ki67, but a negative 8-oxo-dG and γ-H2AX correlation with enhanced NRF2 signaling was observed (Fig 5). These correlations are consistent in both the VC and KrasG12D models, highlighting the universal role of NRF2 in controlling oxidative damage to determine the fate of cells: survival, proliferation and death.
DISCUSSION
In the present study, we tested the roles of NRF2 in chemically and genetically induced lung cancer. In the chemical model, NRF2 pathway activation by pre-treatment with the chemopreventive inducer SF before carcinogen (VC) exposure was protective, as indicated by fewer and smaller tumors. Molecular analyses revealed that SF-pre tumors had lower levels of NRF2 and Ki67, which could account for less aggressive growth of these tumors. Additionally, these SF-pre tumors had higher oxidative DNA damage than their control counterparts. Interestingly, SF-post and Bru-pre mice displayed similar phenotypes: higher tumor multiplicity, greater tumor area, high Ki67, and activation of the NRF2 pathway and ERK phosphorylation. These results suggest that ablation of NRF2 before initiation (Bru-pre) increases susceptibility to carcinogens, and activation of NRF2 during tumor progression (SF-post) accelerates tumor growth. Bru-post greatly reduced tumor area and multiplicity, reduced Ki67 expression, and enhanced oxidative DNA damage and tumor cell apoptosis. Our results are consistent with numerous studies regarding the role of NRF2 in chemical carcinogenesis: activation of NRF2 prevents or reduces carcinogenesis, while inhibition of NRF2 is an effective therapeutic strategy15. Overall, this implies that chemoprevention strategies that activate NRF2 could be effective to decrease cancer incidence of populations with a high risk attributable to carcinogenic exposures. However, for those individuals who have developed tumors, therapies that include NRF2 inhibitors could be beneficial.
Interestingly, in the genetic KrasG12D model we observed different results. Pre-activation or suppression of NRF2 by SF or Bru had no effects on tumor burden. This supports the fact that oncogenic Kras is a strong driver of tumorigenesis and the modulation of NRF2 has no consequences on tumor initiation that is driven by the activation of an oncogene. However, activation of NRF2 signaling after tumors have developed by SF post-treatment increased proliferation, and thus tumor burden, but decreased oxidative damage. Though oncogenic Kras has been reported to activate NRF2 signaling through upregulation of NRF2 mRNA29,30, these results demonstrate that this activation is not maximized and can be further enhanced by SF, thus increasing proliferation and the growth rate of these tumors. Bru-post was again proven effective as an anti-cancer intervention. Our current study showed that Bru alone was also an effective anticancer agent, but we and others have shown that Bru is most effective in combination with chemotherapy30,31,37. Although Bru was recently found to be a global protein translation inhibitor, it has a very strong inhibitory effect on NRF2 due to NRF2’s short protein half-life44. Even though it is unlikely that Bru will become a drug for cancer treatment, it still serves as an important research tool and provides proof-of-concept demonstrating the effectiveness of NRF2 inhibition for cancer treatment while new specific NRF2 inhibitors are being developed.
Our results rekindle the debate over NRF2 activation for cancer prevention. While pre-activation of NRF2 is a very powerful means to prevent cancer induced by environmental carcinogen exposure, presumably by blocking cancer initiation, activation of NRF2 once cancer has developed was detrimental in both the chemical and genetic models. It has been widely speculated that the failure of antioxidants in human clinical trials might be due to the promotion of preexisting malignancies45, which may be true for compounds that induce NRF2, as seen in our results for SF post-treatment in both models. Therefore, while increases in the consumption of natural products containing NRF2 inducers in healthy populations could greatly reduce cancer incidence, it might be unadvisable in cancer patients. To further support the importance of NRF2 activation for chemoprevention, key studies by Yamamoto’s group found that systemic NRF2 activation enhances antitumor immunity and prevents metastasis in Keap1−/− mice46,47. Antitumor immunity is largely dependent on CD8+ T cells and macrophages48, and a study has shown that NRF2 activation in macrophages is necessary for CD8+ T cell function49.
Regardless of the cause of tumor initiation (environmental exposure, replication-associated mutations, inheritance of susceptibility genes), inhibition of NRF2 seems to be a very promising strategy to slow down tumor growth or to sensitize tumors to chemo or radiation therapies. NRF2 is very frequently upregulated in cancer and NRF2 target genes are involved in processes related to the hallmarks of cancer50 (sustained proliferation, insensitivity to apoptosis, metabolic reprogramming, angiogenesis, etc.), as well as chemoresistance. Therefore, careful NRF2 modulation before cancer development and during the different stages of tumorigenesis would maximize the pleiotropic anticancer effects of this critical regulator of cancer. As such, the development of safe, specific and targeted NRF2 activators and inhibitors should be a priority. In addition, systemic delivery of NRF2 activators, but tumor localized delivery of NRF2 inhibitors may prove to be the best approach to reduce unwanted side effects.
Acknowledgments
This work was supported by the National Institutes of Health grants R01 CA154377 and R01 ES026845 to D.D.Z.; R01 ES023758 to E.C. and D.D.Z.
Grant support: This work was supported by the National Institutes of Health grants R01 CA154377 and R01 ES026845 to D.D.Z., and R01 ES023758 to E.C. and D.D.Z.
Abbreviations
- γ-H2AX
gamma-histone 2A family member X (phosphorylated)
- 8-oxo-dG
8-oxo-deoxyguanosine
- AKR1B10
aldo-ketoreductase family 1 member B10
- AKR1C1
aldo-ketoreductase family 1 member C1
- ARE
antioxidant response element
- BRU
brusatol
- CUL3
Cullin 3
- DTT
dithiothreitol
- ERK
extracellular regulated kinase
- FAH
fumarylacetoacetate
- FH
fumarate hydratase
- GAPDH
glyceraldehyde phosphate dehydrogenase
- GCLM
glutamate-cysteine ligase, modifier subunit
- H&E
hematoxylin and eosin
- HMOX1
heme oxygenase 1
- HRP
horseradish peroxidase
- IHC
immunohistochemistry
- KEAP1
Kelch ECH-associated protein 1
- Ki67
marker of proliferation Ki-67
- KRAS
Kirsten rat sarcoma proto-oncogene
- mRNA
messenger ribonucleic acid
- NRF2
nuclear factor erythroid 2-related factor 2
- NQO1
NAD(P)H quinone dehydrogenase 1
- NSCLC
non-small cell lung cancer
- NTC
non-treated control
- PAGE
polyacrylamide gel electrophoresis
- RBX1
ring-box 1
- SEM
standard error mean
- SDS
sodium dodecyl sulfate
- SF
sulforaphane
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- VC
vinyl carbamate
References
- 1.Global Burden of Disease Cancer C. Fitzmaurice C, Dicker D, et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1(4):505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88(Pt B):93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 8.Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88(Pt B):108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24(24):10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23(22):8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27(18):6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramos-Gomez M, Kwak MK, Dolan PM, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98(6):3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khor TO, Huang MT, Prawan A, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila) 2008;1(3):187–191. doi: 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu C, Huang MT, Shen G, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006;66(16):8293–8296. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 19.Grossman R, Ram Z. The dark side of Nrf2. World Neurosurg. 2013;80(3-4):284–286. doi: 10.1016/j.wneu.2011.09.055. [DOI] [PubMed] [Google Scholar]
- 20.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58(5-6):262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeong Y, Hoang NT, Lovejoy A, et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017;7(1):86–101. doi: 10.1158/2159-8290.CD-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Liu X, Long M, et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med. 2016;8(334):334ra351. doi: 10.1126/scitranslmed.aad6095. [DOI] [PubMed] [Google Scholar]
- 23.Shibata T, Ohta T, Tong KI, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105(36):13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata T, Kokubu A, Gotoh M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135(4):1358–1368. 1368 e1351–1354. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 25.Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68(5):1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 26.Ooi A, Dykema K, Ansari A, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73(7):2044–2051. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 27.Ooi A, Wong JC, Petillo D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Wang R, An J, Ji F, Jiao H, Sun H, Zhou D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun. 2008;373(1):151–154. doi: 10.1016/j.bbrc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 29.DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tao S, Wang S, Moghaddam SJ, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74(24):7430–7441. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami S, Motohashi H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med. 2015;88(Pt B):168–178. doi: 10.1016/j.freeradbiomed.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 32.Ryoo IG, Choi BH, Kwak MK. Activation of NRF2 by p62 and proteasome reduction in sphere-forming breast carcinoma cells. Oncotarget. 2015;6(10):8167–8184. doi: 10.18632/oncotarget.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu T, Harder BG, Wong PK, Lang JE, Zhang DD. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol Carcinog. 2015;54(11):1494–1502. doi: 10.1002/mc.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73(13):4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 35.Bauer AK, Cho HY, Miller-Degraff L, et al. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One. 2011;6(10):e26590. doi: 10.1371/journal.pone.0026590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang XJ, Sun Z, Villeneuve NF, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108(4):1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 39.Massey TE, Devereux TR, Maronpot RR, Foley JF, Anderson MW. High frequency of K-ras mutations in spontaneous and vinyl carbamate-induced lung tumors of relatively resistant B6CF1 (C57BL/6J x BALB/cJ) mice. Carcinogenesis. 1995;16(5):1065–1069. doi: 10.1093/carcin/16.5.1065. [DOI] [PubMed] [Google Scholar]
- 40.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen T, Jiang T, Long M, et al. A curcumin derivative that inhibits vinyl carbamate-induced lung carcinogenesis via activation of the Nrf2 protective response. Antioxid Redox Signal. 2015;23(8):651–64. doi: 10.1089/ars.2014.6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeggo PA, Lobrich M. DNA double-strand breaks: their cellular and clinical impact? Oncogene. 2007;26(56):7717–7719. doi: 10.1038/sj.onc.1210868. [DOI] [PubMed] [Google Scholar]
- 44.Harder B, Tian W, La Clair JJ, et al. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol Carcinog. 2017;56(5):1493–1500. doi: 10.1002/mc.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371(2):177–178. doi: 10.1056/NEJMcibr1405701. [DOI] [PubMed] [Google Scholar]
- 46.Satoh H, Moriguchi T, Saigusa D, et al. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016;76(10):3088–3096. doi: 10.1158/0008-5472.CAN-15-1584. [DOI] [PubMed] [Google Scholar]
- 47.Hiramoto K, Satoh H, Suzuki T, et al. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev Res (Phila) 2014;7(8):835–844. doi: 10.1158/1940-6207.CAPR-14-0094. [DOI] [PubMed] [Google Scholar]
- 48.Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer. 2015;1(1):66–75. doi: 10.1016/j.trecan.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sha LK, Sha W, Kuchler L, et al. Loss of Nrf2 in bone marrow-derived macrophages impairs antigen-driven CD8(+) T cell function by limiting GSH and Cys availability. Free Radic Biol Med. 2015;83:77–88. doi: 10.1016/j.freeradbiomed.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]