

Abstract
CLIP170 is a CAP-Gly domain containing protein that is associated with the plus end of growing microtubules and implicated in various cellular processes including the regulation of microtubule dynamics, cell migration and intracellular transport. Our studies revealed a previously unrecognized property and role of CLIP170. We identified CLIP170 as one of the interacting partners of Brucella effector protein TcpB that negatively regulates TLR2 and TLR4 signaling. In this study, we demonstrate that CLIP170 interacts with the TLR2 and TLR4 adaptor protein TIRAP. Further, our studies revealed that CLIP170 induces ubiquitination and subsequent degradation of TIRAP to negatively regulate TLR4-mediated pro-inflammatory responses. Overexpression of CLIP170 in mouse macrophages suppressed the LPS induced expression of IL-6 and TNF-α whereas silencing of endogenous CLIP170 potentiated the levels of pro-inflammatory cytokines. In vivo silencing of CLIP170 in C57BL6 mice by CLIP170-specific siRNA enhanced LPS-induced IL-6 and TNF-α expression. Furthermore, we found that LPS modulates the expression of CLIP170 in mouse macrophages. Overall, our experimental data suggest that CLIP170 serves as an intrinsic negative regulator of TLR4 signaling that targets TIRAP.
Introduction
Toll-like receptors (TLRs) are essential components of innate immune response, which is at the first level of host defense against invading microorganisms. TLRs carry numerous leucine rich repeats containing an extracellular domain that binds specific conserved pathogen associated molecular pattern (PAMPs) and an intracellular Toll/IL-1R1 intracellular (TIR) domain that interacts with specific adaptor proteins such as MyD88, TIRAP, TRIF, TRAM and SARM to initiate the signaling (1, 2), (3). TIRAP is essential for TLR2 and TLR4 mediated signaling where it recruits MyD88 to the TIR domain of plasma membrane localized TLR2 and TLR4 (4). Activation of TLR signaling by PAMPs leads to the activation of transcription factors including NF-κB that in turn triggers expression of many pro-inflammatory cytokine genes (5). Subsequent secretion of pro-inflammatory cytokines activates innate immune cells that leads to various anti-microbial responses and promotes adaptive immunity.
TLR activities are tightly regulated to maintain cellular homeostasis following activation by microbial components. Ubiquitination and deubiquitination play essential roles in the regulation of TLR signaling pathways (6, 7). Ubiquitination comprises covalent attachment of ubiquitin to the lysine residues of target proteins, which involves activation of ubiquitin residues by ubiquitin-activating enzyme (E1) followed by transfer of activated ubiquitin to the catalytic cysteine of the ubiquitin-conjugating enzyme (E2) (8). Subsequently, the ubiquitin-conjugating enzyme transfers the ubiquitin moiety to the lysine residues of target proteins, which are catalyzed by ubiquitin ligases (E3). Ubiquitination can be monoubiquitination where a single ubiquitin moiety is attached to the substrate or polyubiquitination where polymerized ubiquitin chains are linked through the lysine residues of ubiquitin to the target protein. Ubiquitination and proteosomal degradation of TLRs or adaptor proteins have been implicated in negative regulation of TLR signaling to restore cellular homeostasis. Triad3A is an E3 ubiquitin ligase that negatively regulates TLR4 and TLR9 signaling by promoting their K48-linked ubiquitination and proteolytic degradation (9). Triad3A was also reported to induce proteolytic degradation of TIRAP (10). Suppressor of Cytokine Signaling-1 (SOCS-1) inhibits TLR2 and TLR4 receptor signaling by inducing polyubiquitination and degradation of the adaptor protein TIRAP (11).
Brucella melitensis, an infectious intracellular bacterial pathogen, encodes the TIR domain containing protein, TcpB that suppresses TLR2 and TLR4 signaling to subvert host innate immune responses (12-16). TcpB targets the TLR2 and TLR4 adaptor protein TIRAP and induces ubiquitination and subsequent degradation of TIRAP (12, 17). However, the mechanism of TcpB- mediated ubiquitination of TIRAP remains obscure. We performed a high-throughput yeast two-hybrid screening that identified the microtubule plus end tracking protein, Cytoplasmic Linker Protein 170 kDa (CLIP170), as the interacting partner of TcpB. CLIP170 is a microtubule associated protein that specifically binds to plus ends of growing microtubules (18). It is characterized by two conserved Cytoskeleton Associated Protein Glycine-rich (CAP-Gly) domains present at the N-terminus that function as the microtubule binding module (19). CLIP-170 harbors two tandem repeated zinc knuckle motifs in the C-terminus that have been implicated in the interaction with endocytic vesicles and other microtubule plus-end-tracking proteins (+TIP) (20-22). CLIP170 is a multifunctional protein that plays essential roles in regulating microtubule dynamics, dynein localization to the microtubule tips, microtubule interaction with the cell cortex and linking of endosomes to microtubules (18, 23-28). Here we report that CLIP170 enhances the ubiquitination of TIRAP, which promotes the proteasome-mediated degradation of TIRAP. Further, we demonstrate that CLIP170 serves as a negative regulator of TLR4 signaling, and CLIP170 expression is modulated by the TLR4 ligand, lipopolysaccharide (LPS).
Methods
Cell Culture and Transfections
Human Embryonic Kidney (HEK) 293T (American Type Culture Collection, ATCC) or HEK293FT cells (Thermo Fisher Scientific) were cultured in Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10% fetal bovine serum (FBS) (Sigma), 1X penicillin-streptomycin solution (Gibco) and 100 μg/ml normocin (Invivogen). RPMI (Sigma) supplemented with 10% FBS and 1X penicillin-streptomycin solution (Gibco) was used to culture J774 mouse macrophages (ATCC) and mouse embryonic fibroblast cells (ATCC). Cells were grown in a 37° C humidified atmosphere of 5% CO2. All the transfections were performed using Lipofectamine 2000 (Invitrogen) or JetPEI Macrophage (Polyplus Transfections) according to the manufacturer’s instructions.
Co-immunoprecipitation assays
To analyze interaction between TIRAP and CLIP170, HEK293T cells (1×106) were co-transfected with FLAG-TIRAP (gifted by Dr. Douglas Golenbock) and HA-CLIP170 or empty vector using Lipofectamine 2000 reagent in 6-well plates. Forty-eight hours after transfections, cells were lysed and clarified by centrifugation. The cell lysates were pre-cleared with Protein G plus agarose beads followed by addition of 5 μg of anti-FLAG antibody (Sigma) and incubation overnight at 4° C on a rotator. Next, Protein G plus agarose beads were added into the samples and incubated further for 3 h at 4° C on a rotator. Subsequently, agarose beads were washed three times with TNT buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1% Triton X100) and re-suspended in 30 μl of SDS sample buffer (Bio-Rad) and boiled for 10 minutes followed by SDS PAGE and immunoblotting. The membrane was probed with horseradish peroxidase (HRP)-conjugated anti-HA antibody (Sigma) in 5% milk in TBST overnight at 4° C. Subsequently, the membrane was washed 3 times with TBST for 5 min each and incubated with SuperSignal West Pico Chemiluminescent Substrate (Pierce) for 5 min followed by acquiring the luminescent signal using a Chemi-documentation system (Syngene). After that, the membrane was treated with Restore Western Blot Stripping buffer (Pierce) and re-probed with anti-FLAG antibody to detect FLAG-TIRAP. The whole cell lysates were also subjected to immunoblotting and probed with HRP-conjugated anti-HA and anti-FLAG antibodies to detect HA-CLIP170 and FLAG-TIRAP, respectively.
Co-transfections to analyze protein degradation
HEK293T cells (0.5 × 106) were seeded into 12-well plates and co-transfected with 300 ng of FLAG-tagged TIRAP/MyD88 (gifted by Dr. Douglas Golenbock)/hTLR2 (gift from Ruslan Medzhitov; Addgene plasmid # 13082)/TLR4/mTLR9 (gift from Ruslan Medzhitov; Addgene plasmid # 13091)) plasmids and increasing concentrations (300 ng, 600 ng, 900 ng and 1.2 μg) of HA-CLIP170 plasmid. Twenty-four hours after the transfections, cells were lysed in RIPA buffer and the protein concentration was estimated using Bradford reagent. Equal amounts of protein samples were loaded on 4-20% Tris-Glycine SDS PAGE gel and subjected to immunoblotting. The membrane was probed with HRP-conjugated anti-FLAG antibody (Sigma, #A8592) to detect FLAG-tagged proteins and HRP conjugated anti-HA antibody (Sigma, #H6533) for detecting HA-CLIP170. Actin was detected using monoclonal anti-β-actin peroxidase-conjugate antibody (Sigma, #A3854).
To analyze the degradation of endogenous TIRAP or MyD88, RAW264 cells (0.5×106) were seeded into 12-well plates and transfected with 3 μg of HA-CLIP170 plasmid or empty vector using Fugene HD transfection reagent (Promega) according to the manufacturer’s protocols. Twenty-four hours post-transfections, cells were lyzed in RIPA buffer and subjected to immunoblotting. Endogenous TIRAP was detected using anti-TIRAP antibodies (Cell Signaling Technology, # 13077S); (Santa Cruz Biotechnology, #sc-166149) or (Abcam, #ab17218). Anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, #7074) was used as the secondary antibody for the blots probed with anti-TIRAP antibody from Cell Signaling Technology and Abcam. Anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology, #7076) was used as the secondary antibody for the blot probed with anti-TIRAP antibody from Santa Cruz Biotechnology. Endogenous MyD88 was detected using anti-MyD88 antibody (Santa Cruz Biotechnology, #sc-74532).
Reverse transcript and quantitative PCR analysis
Total RNA was isolated from HEK293T cells transfected with FLAG-TIRAP + HA-CLIP170 or FLAG-MyD88 + HA-CLIP170 plasmids followed by cDNA synthesis using PrimeScript first strand cDNA synthesis kit (Takara). Primers used for amplification are CLIP170 (CLIP170.F: GTGGCGTGGAGTTAGATGAG; CLIP170.R: TTTGGCTGGTGTAGTGGAAG); TIRAP (TIRAP.F: CTCCTACTTGGAAGGCAGCAC; TIRAP.R: ACGAAAGCCACCATCAGGG); MyD88 (MyD88.F: TGCTGGAGCTGGGACCCAGCATTGAGGA; MyD88.R: TCAGACACACACACAACTTCAGTCGATA) and GAPDH (GAPDH.F: AATCCCATCACCATCTTCCA; GAPDH.R: TGGACTCCACGACGTACTCA). Relative gene expression of CLIP170, TIRAP and MyD88 was analyzed by Comparative 2-ΔΔCt method using ABI 7500 software (Applied Biosystems). Data was normalized with an endogenous control, GAPDH.
Ubiquitination assays
HEK293FT cells (1×106 in 6-well plates) were co-transected with 1.5 μg of MYC-CLIP170, 1.5 μg of FLAG-TIRAP or FLAG-MyD88 and 1 μg of wild-type or mutant versions of HA-ubiquitin (gifted by Dr. Shigeki Miyamoto) plasmids in various combinations, identified in the results. Total DNA concentration was maintained at 4 μg using the empty vector. Twenty hours after the transfection, the cells were treated with proteosome inhibitor MG132 (Sigma) at a concentration of 20 μm for 4 h. Cells were then washed once with PBS and lysed in 300 μl of lysis buffer containing 20 mM Tris-HCI [pH 7.4] and 1% SDS (17). Cell lysates were transferred into Eppendorf tubes and boiled for 10 min. Lysates were then clarified by centrifugation at 13,000 rpm for 15 min and diluted with buffer containing 20mM Tris-HCl [pH 7.5]; 150 mM NaCl, 2% Triton X100 and 0.5% NP40 (17). Five micrograms of anti-FLAG antibody was added into the lysates and incubated overnight at 4° C on a rotator. Immunoprecipitation and western analysis were performed with samples as described before. The membrane was probed with HRP-conjugated anti-HA antibody to detect HA-Ubiquitin.
Immunocytochemistry
Mouse embryonic fibroblast cells (1×106) were seeded into 30 mm glass-bottom Petri plates (Eppendorf) and allowed to adhere overnight. Cells were then transfected with 3 μg of MYC-CLIP170 or HA-TcpB plasmid using Lipofectamine 3000 reagent. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde in PBS for 10 min, followed by 3 washes for 5 min each with PBS and incubated with 50 mM NH4Cl for 10 min. Cells were then permeabilized with 0.1 % Triton X100 for 5 min. Subsequently, cells were washed 3 times with PBS and incubated with FITC-conjugated anti-MYC antibody (Sigma, # F2047) to stain MYC-tagged proteins, FITC-conjugated anti-HA (Sigma, #H7411) to stain HA-TcpB and Cy3-labelled anti-β tubulin antibody (Sigma, # C4585) to stain microtubules for 1 h. Cells were washed and mounted in ProLong Gold anti-fading reagent (Molecular Probes). Cells were analyzed using a confocal microscope (Leica SP8). Experiments were repeated thrice and 15-20 cells were analyzed per experiment.
NF-κB reporter assays
For TLR4 activation assay, 12 h prior to the transfections, HEK293T cells expressing humanTLR4, CD14 and MD2 (293/hTLR4A-MD2-CD14; Invivogen) were seeded into 12-well plates at a density of 0.5×106/well. Cells were then co-transfected with various amounts of HA-CLIP170 (50, 100, and 250 ng), pNF-κB-Luc (100 ng; Stratagene), and pRL-TK (50 ng; Promega) plasmids. The total amount of DNA was made constant by adding empty vector (pCMV-HA). Twenty-four hours post-transfection, cells were challenged with 300 ng of LPS for 12 h. Cells were then lysed and luciferase activity was assayed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocols. Firefly luciferase activity was normalized to the Renilla luciferase activity and the reporter assays were expressed as mean fold induction over un-induced controls. For TLR9 activation assay, HEK293T cells were co-transfected with mTLR9 (100 ng) expression plasmid and other reporter plasmids mentioned above followed by induction with 1μg of Oligodeoxynucleotides (ODN) and measurement of luciferase activity. Assays were performed in triplicate, and the experiments were repeated thrice.
Over expression and silencing of CLIP170 in mouse macrophages
To overexpress CLIP170 in mouse macrophages, full-length CLIP170 cds segment was ligated to the lentivirus expression plasmid, pMSCV-PIG (a gift from David Bartel; Addgene plasmid # 21654. To prepare lentiviral particles harboring pMSCV-PIG-CLIP170, HEK293FT cells (1x106 in 6-well plates) were co-transfected with packaging vector psPAX2 (a gift from Didier Trono, Addgene plasmid # 12260) and the envelop vector and pMD2.G (a gift from Didier Trono, Addgene plasmid # 12259) and pMSCV-PIG-CLIP170 or empty vector. Seventy-two hours after transfection, culture supernatant that contains lentiviral particles were collected and clarified by centrifugation and passed through a 0.45 μm syringe filter (Millipore). The lentiviral titer in the clarified cell supernatant was assessed using Lenti-X GoStix (Clontech) followed by transduction of J774 cells (1×106) with lentiviral particles. Forty-eight hours after transduction, cells were lysed in RIPA buffer and analyzed by immunoblotting to confirm the over expression of CLIP170 using anti-CLIP170 antibody (Santa Cruz Biotechnology, #sc-28325). To analyze the overexpression of CLIP170 by qPCR, total RNA was extracted from lentivirus transduced cells followed by cDNA synthesis and qPCR analysis. Primers used for CLIP170 amplification are as follows: CLIP. F: CCTCAAGATGAAGGTGGAGATG; CLIP. R: AGGTCAAAGCAGTCACAGATG. CLIP170 qPCR primers were derived from a region that spans the exon-intron boundary of pre-mRNA of CLIP170 transcript.
To study effect of CLIP170 on cytokine secretion by macrophages, J774 cells were transduced with CLIP170 or empty vector for 48 hours, followed by treatment with LPS (100 ng/ml), Pam3CSK4 (100 ng/ml), ODN (1μg/ml), or TNF-α (20 ng/ml). Subsequently, culture supernatants and cells were collected at 0, 1, and 2 h after treatment. Total RNA was extracted from the LPS challenged cells followed by cDNA synthesis and qPCR analysis. Primers for qPCR analysis were TNF-α.F: GACGTGGAAGTGGCAGAAGAG; TNF-α.R: TGCCACAAGCAGGAATGAGA, IL-6.F: TCCAGTTGCCTTCTTGGGAC; IL-6.R: GTACTCCAGAAGACCAGAGG; IFN-β.F: CTGGCTTCCATCATGAACAA; IFN-β.R: CATTTCCGAATGTTCGTCCT; mGAPDH.F: AACGACCCCTTCATTGAC; mGAPDH.R: TCCACGACATACTCAGCAC. IL-6, TNF-α, IFN-β and CLIP170 expression data were normalized to the expression levels of GAPDH. Quantification of IL-6 and TNF-α in the cell culture supernatant was performed by ELISA using Quantikine ELISA kit (R & D Systems) according to the manufacturer’s protocols. All the assays were performed at least three times.
To silence the endogenous CLIP170 in J774 cells, we synthesized CLIP170 specific shRNA, derived from CLIP170 siRNA reported previously (23). The non-targeting control shRNA sequence used was CCGGTTCTCCGAACGTGTCACGTTTCTGCAGAAACGTGAC ACGTTCGGAGAATTTTTG. The shRNA oligos were annealed and cloned into pLKO-1 vector (a gift from David Root, Addgene plasmid # 10878). Lentiviral particles harboring the CLIP170 shRNA or control shRNA were prepared as described above. J774 cells were transduced with lentivirus and silencing of endogenous CLIP170 was assessed by immunoblotting using anti-CLIP170 antibody and qPCR. To analyze the levels of IL-6 and TNF-α in CLIP170 silenced cells, J774 cells were transduced with lentivirus harboring CLIP170 specific shRNA or control shRNA. Forty-eight hours after the transduction, cells were challenged with various ligands followed by quantification of IL-6, TNF-α and IFN-β in the culture supernatants and cells by ELISA and qPCR, respectively as described above.
Silencing of CLIP170 in mice by delivery of siRNA in vivo
Mice experiments were performed after obtaining Institutional Animal Ethics Committee approvals. The in vivo siRNA targeting the CLIP170 (5-GGAGAAGCAGCAGCACAUUTT-3) or scrambled siRNA (5-UUCUCCGAACGUGUCACGUTT-3) were synthesized from Ambion. In vivo siRNA was complexed with the in vivo siRNA delivery reagent, Invivofectamine 2.0 (Invitrogen) according to the manufacturer’s protocols. One hundred microliters of siRNA complex that contained 50 μg of siRNA, was injected into 6 weeks old female C57BL/6 mice through tail vein. To examine the silencing of endogenous CLIP170 in the liver, 48 h after the siRNA delivery, Kupffer cells were harvested from the mice employing a gradient centrifugation protocol as described by Li et al., 2014 (29). Adhered Kupffer cells were lysed and subjected to immunoblotting using anti-CLIP170 antibody. Spleens were also harvested and dispersed in RPMI1640 in 30 mm culture dishes. Spleen homogenate was then filtered through a cell strainer and seeded into 6-well plates. Splenocytes were allowed to adhere for 2 hours followed by cell lysis and immunoblotting.
Quantification of TNF-α and IL-6 in CLIP170 silenced mice
To analyze levels of IL-6 and TNF-α in splenocytes, cells were prepared as described before. Cells were then challenged with LPS (100 ng/ml) followed by quantification of IL-6 and TNF-α levels in the culture supernatants and mRNA in cells by ELISA and qPCR, respectively. To analyze the levels of pro-inflammatory cytokines in the liver of siRNA treated mice, mice were injected intraperitoneally with sub-lethal dose of LPS (25 mg/kg) after 72 hours of siRNA delivery. Two hours after the LPS injection, mice were sacrificed followed by collection of liver. Total RNA was isolated from the liver and cDNA was synthesized. The levels of IL-6 and TNF-α mRNA were measured in the liver by qPCR analysis.
Analysis of CLIP170 expression in macrophages
J774 cells (0.5×106) were seeded into 12-well plates and allowed to adhere overnight. Cells were then challenged with LPS (100 ng/ml) for various times (0, 15, 30, 60 and 120 minutes) followed by lysis in RIPA buffer for immunoblotting or isolation of total RNA for qPCR analysis. For immunoblot analysis, membranes were probed with anti-CLIP170 antibody followed by anti-β-actin antibody where β-actin served as the loading control. For qPCR analysis, cDNA was prepared from total RNA followed by quantification of endogenous levels of CLIP170.
Results
CLIP170 promotes ubiquitination and degradation of TIRAP
We analyzed the interaction between CLIP170 and the TLR2/4 adaptor protein TIRAP using co-immunoprecipitation assays. HEK293T cells were co-transfected with FLAG-TIRAP and HA-CLIP170 followed by immunoprecipitation with anti-FLAG antibody. HA-CLIP170 could be co-immunoprecipitated with FLAG-TIRAP indicating potential interaction between CLIP170 and TIRAP (Fig. 1a). SOCS-1 promoted polyubiquitination and subsequent degradation of TIRAP to negatively regulate TLR4 signaling (11). The Brucella effector protein, TcpB that interacted with CLIP170 also induced enhanced ubiquitination and degradation of TIRAP (17). Therefore, we examined whether CLIP170 promotes degradation of TIRAP. We co-transfected HEK293T cells with a constant amount of FLAG-TIRAP and increasing concentrations of HA-CLIP170 followed by immunoblot analysis using anti-FLAG antibody to detect the FLAG-TIRAP. We observed enhanced degradation of FLAG-TIRAP with increasing concentrations of HA-CLIP170 (Fig. 1b). To rule out the possibility that CLIP170 may affect the expression of TIRAP, we analyzed the TIRAP mRNA levels in the presence or absence of CLIP170. TIRAP mRNA levels were not altered by overexpression of CLIP170 (Supplementary Figure 1).
Figure 1. CLIP170 induces ubiquitination and degradation of TIRAP.
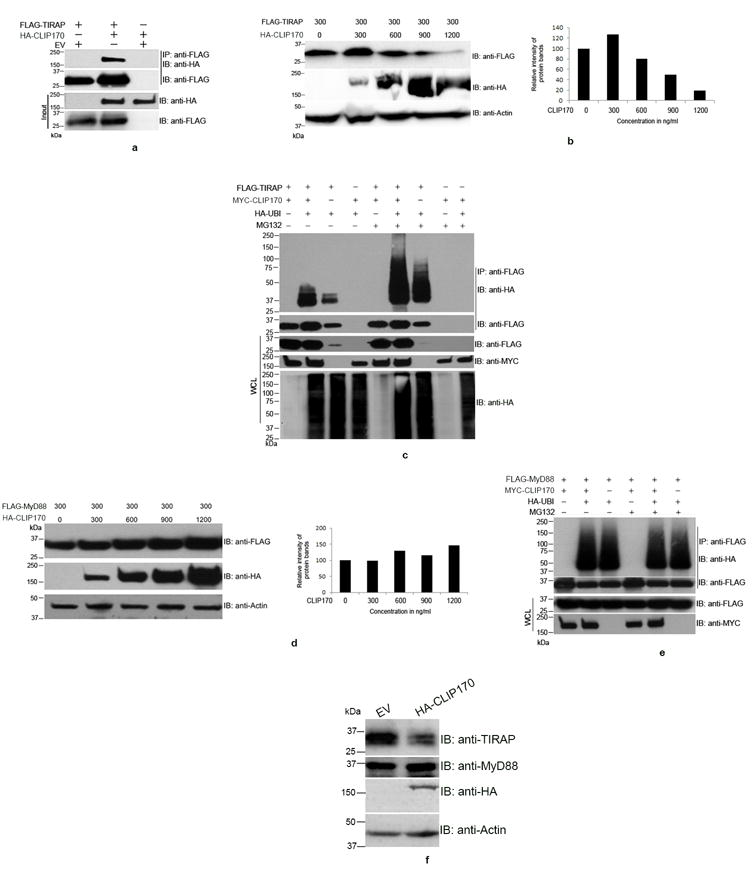
(a) CLIP170 interacts with TIRAP. HEK293T cells were co-transfected with equal amounts of HA-CLIP170 and FLAG-TIRAP plasmids. Twenty four hours post-transfection, cells were lysed and FLAG-TIRAP was immunoprecipitated using anti-FLAG antibody followed by immunoblotting. The blot was probed with anti-HA antibody to detect the co-immunoprecipitated HA-CLIP170 followed by detection of FLAG-TIRAP by anti-FLAG antibody. The whole cell lysates were also subjected to immunoblotting followed by immuno-detection of HA-CLIP170 and FLAG-TIRAP. (b) CLIP170 promotes degradation of TIRAP. HEK293T cells were co-transfected with FLAG-TIRAP and increasing concentrations of HA-CLIP170 plasmids. Twenty four hours post-transfection, cells were lysed and subjected to immunoblotting. The blot was probed with anti-FLAG and anti-HA antibodies to detect FLAG-TIRAP and HA-CLIP170, respectively. Actin served as the loading control. Right panel of the immunoblot shows the densitometry analysis of FLAG-TIRAP bands normalized to actin; (c) CLIP170 induces ubiquitination of TIRAP. HEK293T cells were co-transfected with various combinations of FLAG-TIRAP, MYC-CLIP170 and HA-Ubiquitin as indicated. Twenty four hours post-transfection, cells were treated with 20 μm MG132 for 4 h as indicated in the figure. Cells were then lysed and FLAG-TIRAP was immunoprecipitated followed by immunoblotting. The blot was probed with anti-HA antibody to detect the HA-Ubiquitin-conjugated FLAG-TIRAP. CLIP170 enhanced the ubiquitination of CLIP170 in MG132 treated or untreated cells. Similar concentrations of FLAG-TIRAP and MYC-CLIP170 resulted in accumulation of ubiquitinated FLAG-TIRAP in the immunoprecipitated samples. (d) CLIP170 did not induce degradation of MyD88. HEK293T cells were co-transfected with 300 ng of FLAG-MyD88 and increasing concentrations of HA-CLIP170. Twenty four hours post-transfection, cells were lysed and subjected to immunoblotting. The blot was probed with anti-FLAG and anti-HA antibodies to detect FLAG-MyD88 and HA-CLIP170, respectively. Degradation of MyD88 was not observed with increasing concentrations of HA-CLIP170. Right panel of the immunoblot shows the densitometry analysis of FLAG-MyD88 bands normalized to actin; (e) CLIP170 did not promote the ubiquitination of FLAG-MyD88. Ubiquitination assay was performed as described before with the FLAG-MyD88 as the substrate. Ubiquitination status of MyD88 was not affected by CLIP170; (f) CLIP170 induces degradation of endogenous TIRAP. RAW264 cells were transfected with EV or HA-CLIP170 followed by detection of endogenous TIRAP and MyD88 using anti-TIRAP and anti-MyD88 antibodies, respectively. Actin served as the loading control. Immunoblots are representative of two independent experiments. EV: Empty vector; IP: immunoprecipitation; IB: immunoblotting.
Selective protein degradation in eukaryotic cells is achieved by ubiquitination of target proteins and their degradation by the 20S proteasome complex. Therefore, we sought to examine whether CLIP170 could promote the ubiquitination of TIRAP. To analyze this, we performed an ubiquitination assay by co-transfecting HEK293T cells with various combinations of FLAG-TIRAP, MYC-CLIP170 and HA-Ubiquitin constructs as described in the Fig. 1c. Twenty-four hours after the transfections, cells were treated with the irreversible proteasome inhibitor, MG132 for 4 h to inhibit the degradation of ubiquitinated FLAG-TIRAP. Next, FLAG-TIRAP was immunoprecipitated, and conjugation of HA-Ubiquitin to the FLAG-TIRAP was analyzed by immunoblotting using anti-HA antibody. Ubiquitinated TIRAP could be detected in MG132 treated or untreated cells and the levels of TIRAP ubiquitination was dramatically increased in the presence of CLIP170 (Fig. 1c). This suggests that CLIP170 promotes ubiquitination of TIRAP.
TIRAP serves as the bridging adaptor for recruiting the signaling adaptor, MyD88 to the plasma membrane bound TLR2 and TLR4 (4). Therefore, we wanted to analyze whether CLIP170 promotes degradation of MyD88. To examine this, we co-transfected HEK293T cells with a constant amount of FLAG-MyD88 expression plasmid and increasing concentrations of HA-CLIP170 plasmid and they were sampled for immunoblot analysis. In contrast to a decrease in TIRAP overexpression upon increasing co-overexpression of CLIP170, the levels of Myd88 expression did not decrease but slightly increased following increasing CLIP170 co-overexpression (Fig. 1d). However, reverse transcriptase or quantitative PCR analyses did not indicate altered mRNA levels of MyD88 with CLIP170 (Supplementary Fig. 1). To confirm the substrate specificity for the ubiquitin ligase-like property of CLIP170, we performed an ubiquitination assay with FLAG-MyD88. CLIP170 did not induce ubiquitination of MyD88 as an equal amount of ubiquitinated MyD88 was observed in the presence or absence of CLIP170 (Fig. 1e). The levels of MyD88 did not change in the MG132 treated or untreated cells suggesting that MyD88 did not undergo proteasome mediated degradation (Fig. 1e). Next, we analyzed the levels of endogenous TIRAP and MyD88 in the presence of CLIP170 in macrophages. RAW264 mouse macrophages were transfected with HA-CLIP170 or empty vector and analyzed for the levels of TIRAP and MyD88 by immunoblotting using specific antibodies. We observed an enhanced degradation of endogenous TIRAP in the RAW cells transfected with HA-CLIP170 whereas MyD88 was not degraded (Fig. 1f). The immunoblot analysis of RAW cell lysates using anti-TIRAP antibodies from various suppliers detected a band of ~35 kDa, which was degraded upon overexpression of CLIP170 (Supplementary Fig. 2a, b and c). The experimental data suggest that CLIP170 targets TIRAP for degradation.
Next, we analyzed whether CLIP170 induces degradation of TLR2, TLR4 and TLR9. HEK293 cells were co-transfected with constant amounts of FLAG tagged TLRs and increasing concentrations of MYC-CLIP170. We observed slight degradation of FLAG-TLR2 with increasing concentrations of MYC-CLIP170 (Supplementary Fig. 2d) whereas FLAG-TLR4 underwent slightly enhanced ubiquitination (Supplementary Fig. 2e). Levels of FLAG-TLR9 remained unchanged with increasing concentrations of MYC-CLIP170 (Supplementary Fig. 2f).
CLIP170 promotes multiple mono- and poly-ubiquitination of TIRAP
Ubiquitination involves attachment of ubiquitin to the lysine residues of the substrate protein that can result in monoubiquitination or polyubiquitination. In monoubiquitination, a single ubiquitin moiety is attached to the lysine residues of the substrate where as in polyubiquitination K48 or K63 linked homotypic ubiquitin chains are attached to the substrate (30). We analyzed the nature of CLIP170-induced ubiquitination of TIRAP by ubiquitination assay using mutant versions of ubiquitin. HEK293T cells were co-transfected with FLAG-TIRAP, MYC-CLIP170 and various mutants of HA-tagged Ubiquitin, viz UBI-K48R, UBI-K48 only, UBI-K63R, UBI-K63 only, UBI-7KR. The UBI-K48R cannot form K48-linked ubiquitin chains whereas UBI-K48 only mutant can form only K48-linked ubiquitin chains. Similarly, the UBI-K63R cannot form K63-linked ubiquitin chains whereas UBI-K63only can form only K63-linked ubiquitin chains. In the UBI-7KR, all the seven lysine residues in ubiquitin were mutated to arginine which cannot form homotypic chains. All the mutants are capable of mono or multiple-mono ubiquitination of target proteins. Interestingly, CLIP170 promoted conjugation of all the mutant versions of ubiquitin including UBI-7KR to TIRAP (Fig. 2a and 2b). Conjugation of UBI-7KR to TIRAP suggests that CLIP170 promotes the monoubiquitination of TIRAP. The smeared appearance of the TIRAP band with UBI-7KR suggests that TIRAP undergoes multiple mono-ubiquitinations in the presence of CLIP170. Similarly, presence of high-molecular weight smears of TIRAP with other ubiquitin mutants, which could form homotypic chains, suggest that CLIP170 also promotes the polyubiquitination of TIRAP (Fig. 2a and 2b).
Figure 2. TIRAP undergoes poly-ubiquitination and multiple mono-ubiquitinations in the presence of CLIP170.
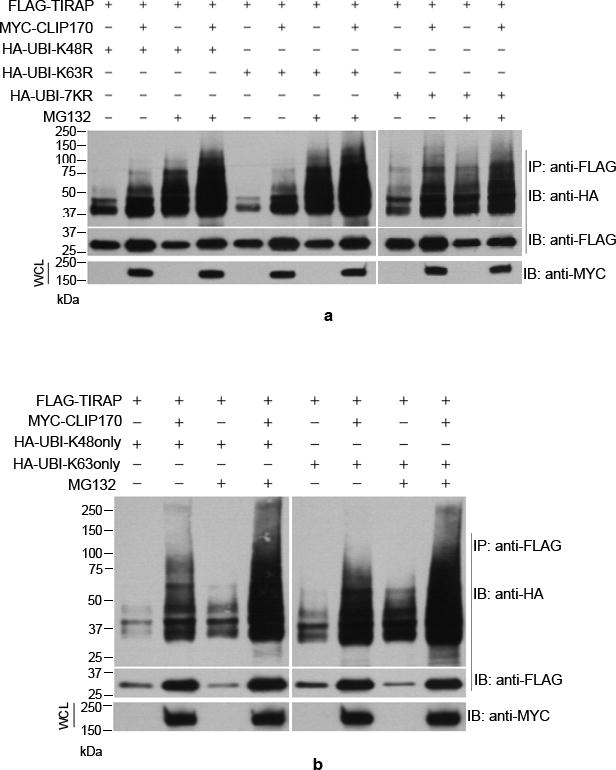
HEK293T cells were co-transfected with FLAG-TIRAP, CLIP170 and various Lysine to Arginine mutants of HA-Ubiquitin followed by immunoprecipitation of FLAG-TIRAP and immunoblotting. CLIP170 mediated conjugation of all the mutant versions of Ubiquitin including UBI7KR where all the Lysine residues are mutated to Arginine. The vertical white lines indicate where the figure panels were joined.
CLIP170 regulates microtubule dynamics and has been implicated in stabilization of microtubules (31). We analyzed the subcellular localization of CLIP170 in mouse embryonic fibroblast (MEF) cells. MYC-CLIP170 was overexpressed in MEF cells followed by immunocytochemistry using anti-MYC-FITC to visualize CLIP170 (Supplementary Fig. 3; Panels a-c). CLIP170 induced dramatic microtubule bundling in MEF cells. As we demonstrated before, Brucella effector protein TcpB that interacted with CLIP170 also induced severe microtubule bundling and strongly co-localized with microtubules (Supplementary Fig. 3; Panels d-f).
Overexpression of CLIP170 attenuates LPS-induced NF-κB activation and pro-inflammatory cytokine release
TIRAP is indispensable for MyD88-dependent pathway of TLR4 signaling and, therefore, the negative regulation of this pathway can be achieved by selected elimination of TIRAP by proteins like SOCS-1. Given that CLIP170 promotes ubiquitination and degradation of TIRAP, we wished to examine whether CLIP170 affects the LPS-induced signaling mediated by TLR4. We performed luciferase reporter assays to analyze the effect of CLIP170 in TLR4 mediated NF-κB activation. HEK293 cells, stably expressing TLR4, CD14 and MD2, were co-transfected with reporter plasmids and increasing concentrations of CLIP170 followed by challenge with LPS and NF-κB reporter assay. The assay indicated that CLIP170 could efficiently suppress TLR4-induced NF-κB activation in a dose dependent manner (Fig. 3a). To examine whether the action of CLIP170 is specific for TLR4, we analyzed the effect of CLIP170 on TLR9 signaling, which does not require TIRAP for transduction. HEK293T cells were co-transfected with mTLR9 plasmid, reporter plasmids and increasing concentrations of CLIP170 followed by induction with ODN and NF-κB reporter assay. CLIP170 did not affect the activation of NF-κB induced by TLR9 signaling (Fig. 3b). This suggests that CLIP170 targets the TLR4 signaling pathway where the role of TIRAP is indispensable.
Figure 3. CLIP170 attenuates TLR4- but not TLR9-induced NF-κB activation.
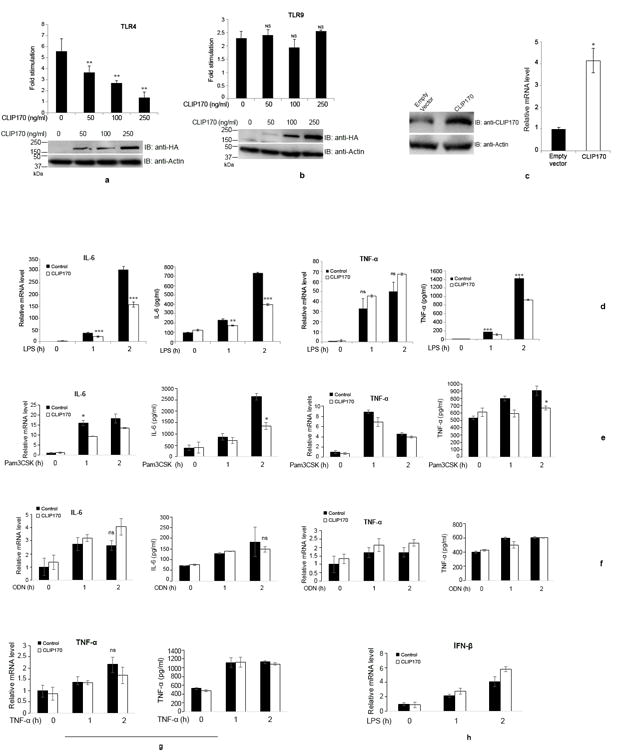
(a) HEK293 cells expressing human TLR4/CD14/MD2 were transfected with increasing concentrations of HA-CLIP170 (50, 100, and 250 ng), pNF-κB-Luc reporter plasmid (100 ng), and pRL-TK (50 ng). (b) For TLR9 assay, HEK293T cells were co-transfected with mTLR9 (200 ng) plasmid along with other plasmids mentioned above. The total amount of DNA was made constant by adding empty vector. Twenty four hours post-transfection, cells were challenged overnight with LPS (300 ng/ml) and ODN (1 μg/ml), respectively. Luciferase activity was measured using the Dual-Luciferase Reporter assay system; the lower panels are immunoblots demonstrating the overexpression of HA-CLIP170. Actin was used as the loading control; (c) Overexpression of CLIP170 in mouse macrophages. J774 cells were transduced with lentiviral particles harboring CLIP170 or empty vector followed by immunoblotting and qPCR. The blot was probed with anti-CLIP170 antibody to examine the overexpression of CLIP170. Actin served as the loading control; (d) CLIP170 suppresses LPS-induced activation of IL-6 and TNF-α in mouse macrophages. J774 cells transduced with CLIP170 expression plasmid or empty vector were challenged with LPS (100 ng/ml) for indicated time points, followed by ELISA and qPCR to quantify the levels of IL-6 and TNF-α. (e - h) Effect of CLIP170 on Pam3CSK4-, ODN-, TNF-α-induced pro-inflammatory cytokines and LPS-induced IFN-β secretion. CLIP170 weakly suppressed Pam3CSK4-induced IL-6 and TNF-α levels (e) whereas it did not affect ODN- (f), TNF-α (g)-induced pro-inflammatory cytokines or LPS-induced IFN-β (h) levels. Data are presented as mean ± SD from at least three independent experiments (*P < 0.05, **P < 0.01 and ***P < 0.001); ns: not significant.
Next, we examined whether the overexpression of CLIP170 attenuates pro-inflammatory cytokine secretion by macrophages. J774 cells were transduced with lentivirus harboring CLIP170 expression plasmid or empty vector and the overexpression of CLIP170 was confirmed by immunoblotting using anti-CLIP170 antibody and qPCR (Fig. 3c). Next, lentivirus transduced J774 cells were challenged with LPS and the levels of IL-6 and TNF-α were analyzed by ELISA and qPCR. J774 macrophages overexpressing CLIP170 secreted diminished levels of IL-6 and TNF-α compared to cells transduced with the empty vector (Fig. 3d). We did not observe a significant difference in TNF-α mRNA levels between CLIP170 overexpressed and control J774 cells (Fig. 3d). Next, we examined whether CLIP170 affects signaling from other TLR or non-TLR receptors. J774 cells that were transduced with CLIP170 expression plasmid were stimulated with Pam3CSK4 (Fig. 3e), ODN (Fig. 3f) and TNF-α (Fig. 3g). CLIP170 could weakly inhibit Pam3CSK4-mediated signaling (Fig. 3e) whereas ODN- or TNF-α-mediated signaling was not affected (Fig. 3f and 3g). LPS induces production of type I interferons, which is mediated by the MyD88-independent pathway of TLR4 signaling (32). We did not observe suppression of LPS-induced IFN-β in J774 cells overexpressing CLIP170 (Fig. 3h). The experimental data suggest that CLIP170 mainly targets the MyD88-dependent pathway of TLR4 signaling.
Silencing of CLIP170 potentiates the pro-inflammatory cytokine levels in macrophages and mice
As overexpression of CLIP170 attenuated TLR4-induced NF-κB activation and pro-inflammatory cytokine secretion, we wanted to examine whether silencing of endogenous CLIP170 leads to potentiation of pro-inflammatory cytokines. J774 cells were transduced with lentivirus harboring CLIP170 specific shRNA expression plasmid or control shRNA and the downregulation of CLIP170 was confirmed by immunoblotting and qPCR (Fig. 4a). Next, CLIP170 silenced cells were challenged with LPS and levels of IL-6 and TNF-α were analyzed by qPCR and ELISA. CLIP170 silenced J774 cells expressed elevated levels of IL-6 compared to the cells transduced with scrambled shRNA (Fig. 4b). We observed significantly elevated levels of TNF-α only at the one hour time point in CLIP170-silenced J774 cells (Fig. 4b). As noticed before, mRNA levels of TNF-α did not change significantly in CLIP170-silenced J774 cells in comparison to the control (Fig. 4b). Stimulation of CLIP170-silenced cells with Pam3CSK4 resulted in suppression of IL-6 and TNF-α at the early time points whereas no suppression was observed in ODN or TNF-α induced cells (Fig. 4c, d & e). As observed previously, IFN-β levels were not significantly altered in CLIP170-silenced cells compared to the control (Fig. 4f).
Figure 4. Silencing of endogenous CLIP170 potentiates LPS-induced pro-inflammatory cytokines in macrophages.
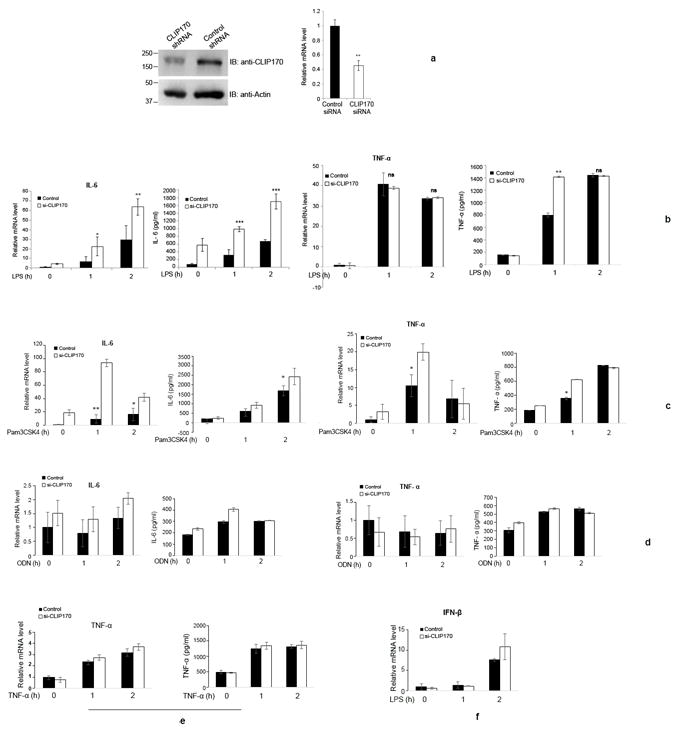
(a) shRNA-mediated knockdown of endogenous CLIP170 in mouse macrophages. J774 cells were transduced with lentiviral particles harboring CLIP170 shRNA or control shRNA expression plasmid followed by immunoblotting and qPCR analysis to examine the levels of endogenous CLIP170. The blot was probed with anti-CLIP170 antibody to detect the endogenous CLIP170. Actin served as the loading control; (b) Knockdown of endogenous CLIP170 potentiates LPS-induced expression of IL-6 and TNF-α in mouse macrophages. J774 cells transduced with CLIP170 shRNA expression plasmid or control shRNA plasmid were challenged with LPS (100 ng/ml) for indicated time points, followed by ELISA and qPCR to quantify the levels of IL-6 and TNF-α. (c - e) Expression of IL-6 and TNF-α in CLIP170 silenced J774 cells challenged with Pam3CSK4, ODN and TNF-α. CLIP170 weakly suppressed Pam3CSK4-induced IL-6 and TNF-α levels (c) whereas it did not affect ODN- (d), TNF-α- (e) induced pro-inflammatory cytokines. (f) Expression levels of IFN-β in CLIP170 silenced cells challenged with LPS. IFN-β levels were not significantly altered in CLIP170 silenced cells compared to control. Data are presented as mean ± SD from at least three independent experiments (*P < 0.05, **P < 0.01 and ***P < 0.001); ns: not significant.
Next, we examined whether the silencing of endogenous CLIP170 in mice potentiated the LPS-induced inflammatory responses by employing an in vivo siRNA delivery technique. In vivo siRNA targeting CLIP170 or scrambled siRNA was complexed with the siRNA delivery agent, invivofectamine, followed by intravenous injection to 6 weeks old C57BL/6 female mice (50 μg of siRNA/mice). To analyze the silencing of endogenous CLIP170, mice were sacrificed after 48 h of siRNA delivery followed by isolation of splenocytes and Kupffer cells. Subsequently, the endogenous levels of CLIP170 were analyzed in splenocytes and Kupffer cells by immunoblotting and qPCR. Both cell types displayed diminished levels of CLIP170 expression compared to the control indicating the silencing of endogenous CLIP170 (Fig. 5a & b). We obtained a robust silencing of CLIP170 in the splenocytes (Fig. 5a). Next, we examined the levels of IL-6 and TNF-α in the splenocytes and liver of CLIP170-silenced or control mice. To determine the cytokine levels in splenocytes, isolated cells were challenged with LPS for various time points, followed by quantification of IL-6 and TNF-α by ELISA and qPCR. We observed elevated levels of both IL-6 and TNF-α in splenocytes from CLIP170-silenced mice (Fig. 5c). Next, we analyzed the levels of pro-inflammatory cytokines in the liver of CLIP170-silenced or control mice. Seventy-two hours after the siRNA delivery, mice were given sub-lethal injection of LPS (25 mg/kg) intraperitoneally and harvested the liver after two hours post-injection. Total RNA was isolated from a portion of the liver followed by cDNA synthesis and qPCR analysis to quantify the levels of IL-6 and TNF-α. We observed potentiated levels of IL-6 and TNF-α expression in the CLIP170-silenced mice compared to the control (Fig. 5d). The experimental data suggest that CLIP170 acts as the negative regulator of LPS-induced TLR4 signaling in mouse.
Figure 5. Silencing of CLIP170 enhances LPS-induced pro-inflammatory cytokines in mice.
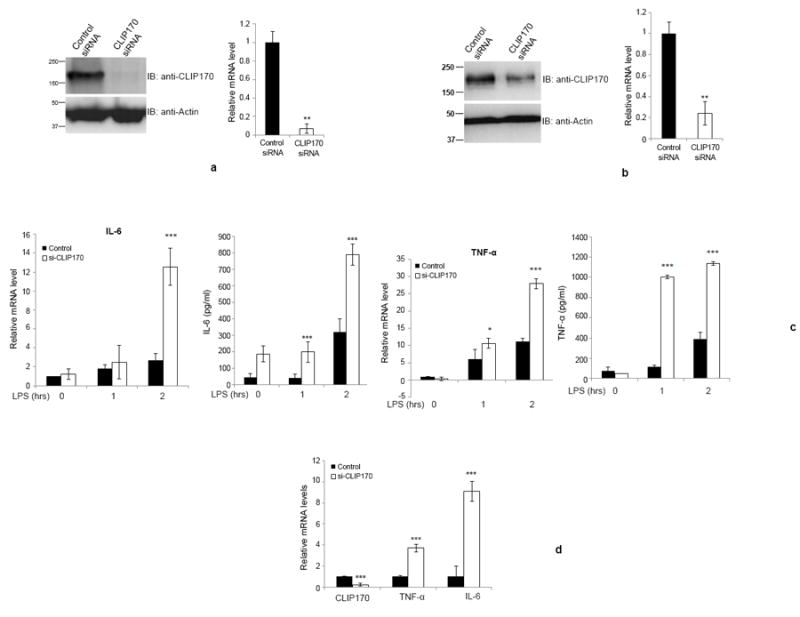
(a & b) Depletion of CLIP170 in splenocytes or Kupffer cells derived from CLIP170 siRNA treated mice. Mice were treated with CLIP170 siRNA or control siRNA for 48 h, followed by collection of splenocytes (a) or Kupffer cells (b). The cells were analyzed by immunoblotting and qPCR to examine the levels of endogenous CLIP170. The blot was probed with anti-CLIP170 to detect the endogenous CLIP170. Actin served as the loading control. (c) Depletion of CLIP170 potentiates the expression of IL-6 and TNF-α in splenocytes. The splenocytes isolated from mice injected with CLIP170-specific or control siRNA were challenged with LPS (100 ng/ml) for the indicated time points followed by quantification of IL-6 and TNF-α by ELISA and qPCR. (d) Potentiated levels of IL-6 and TNF-α in the liver of CLIP170-silenced mice. Seventy-two hours after the siRNA delivery, mice were injected with LPS (25 mg/kg) intraperitoneally and the levels of IL-6 and TNF-α were quantified by qPCR. Data are presented as mean ± SD from two independent experiments of six mice per group; *P < 0.05, **P < 0.01 and ***P < 0.001.
Negative regulators of TLR signaling are induced by the agonists of respective TLRs (33, 34). Since CLIP170 negatively regulates the TLR4 signaling, we examined if CLIP170 expression is induced by the TLR4 ligand, LPS. To analyze this, J774 cells were challenged with LPS followed by analysis of endogenous levels of CLIP170 mRNA and protein by qPCR and immunoblotting, respectively. We observed diminished levels of CLIP170 transcript and protein expression during the first 15 min of LPS induction compared to the 0-h time point (Supplementary Fig. 4a & b). This is followed by a gradual increase of CLIP170 expression up to 2 to 3 hours. It appears that CLIP170 expression was down-regulated at the initial stages of TLR4 signaling followed by its upregulation at later stages. TLR4 signaling activates the transcription factor NF-κB which in turn activates many genes that harbor NF-κB responsive elements. Interestingly, experimentally validated NF-κB1 binding sites were reported in the promoter region of both mouse and human CLIP170 (Qiagen).
Discussion
CLIP170 is multifunctional protein that is involved in important cellular processes including intracellular transport and signaling. CLIP170 interacts with the Brucella effector protein, TcpB that promotes ubiquitination and degradation of TIRAP to subvert the TLR2 and TLR4 signaling (17). TcpB is unlikely to harbor a ubiquitin ligase property, and it may recruit host protein(s) for ubiquitination and degradation of TIRAP (17). Here we report that CLIP170 interacts with TIRAP and induces its ubiquitination and degradation. Human CLIP170 maps on the reverse strand of chromosome 12 at 12q24.3 position, which spans 152.26 kb and the locus is reported to be complex (35). The CLIP170 gene is predicted to transcribe 29 different mRNAs, 21 alternatively spliced variants and 8 unspliced forms (35). The gene is highly expressed, 4.3 times the average gene, and the RNA-Seq expression analysis indicated its expression in various tissues including brain, skeletal muscle, liver, spleen, colon, lymph nodes, testes and thymus (35, 36). Four isoforms of CLIP170 viz, Restin, CLIP170, CLIP170 (11) and CLIP170(11+35) have been cloned and analyzed from human and chicken (37-40). All four CLIP170 isoforms appear to carry the identical CAP-Gly domains at the N-terminus and zinc knuckles at the C-terminus. Structural analysis indicated that CLIP170 forms parallel dimers through its central α-helical coiled-coil rod-domain (23). Rod domain of CLIP170 has two kinks, which may contribute to the folded back state of CLIP170 that regulates the activity of the protein (41). The changes in the rod domain of CLIP170 by alternative splicing may regulate its activity and recruitment for diverse cellular functions.
Our analysis revealed a previously unrecognized ubiquitin ligase-like property of CLIP170. Interestingly, CLIP170 does not possess classical ubiquitin ligase domains such as HECT, RING or U box (42). Therefore, CLIP170 may act as a scaffold to facilitate the ubiquitination of target proteins. Familial cylindromatosis tumor suppressor (CYLD) contains three CAP-Gly domains and inhibits NF-ĸB activation by cleaving the K-63 linked poly-ubiquitin chains from the ubiquitin ligases, TNF receptor-associated factor (TRAF)-2, TRAF-6 and NEMO (43-45). In contrast, CLIP170 promoted ubiquitination of target proteins. SOCS1 suppresses TLR4 signaling through proteasome-mediated degradation of TIRAP (11). We assume that CLIP170 also employs a similar mechanism to attenuate TLR4 signaling. Our studies suggest that CLIP170 mediates conjugation of mono- and poly-ubiquitin to TIRAP. Polyubiquitination through K48 results in proteasome-mediated degradation of the target proteins (46, 47). Monoubiquitination has also been shown to direct the proteins for degradation (48-50).
Signaling through TLR4 is heavily regulated owing to the extreme toxicity of TLR4 signaling (51). The regulators target multiple points of the complex TLR4 signaling pathway. We observed that the expression of CLIP170 in mouse macrophages was modulated by the TLR4 agonist, LPS. A transient downregulation of CLIP170 was observed in the first 15 min of LPS induction followed by a steady increase of CLIP170 expression. TLR4 signaling occurs in two phases, an early phase mediated by TLR4-MyD88-TIRAP complex and a late phase mediated by TLR4-TIRAP-4-1BBL complex (52). Early phase may last for the first 15 minutes of LPS induction as TIRAP degradation is initiated during this period (11). Subsequently, TIRAP may be resynthesized for the late phase signaling of TLR4 (52). Hence, in the early phase of TLR4 signaling, TIRAP degradation should be prevented to facilitate the unhindered signaling from TLR4. Therefore, it is conceivable that a transient downregulation of CLIP170 at the early stage of TLR4 signaling may ensure the availability of signal competent TIRAP. Early TLR4 signaling may upregulate CLIP170 expression through NF-κB that in turn ubiquitinate and degrade TIRAP resulting in the negative regulation of TLR4 signaling.
Over expression of CLIP170 or TcpB induced severe microtubule bundling in various cell types. Taxol-stabilized microtubules have been reported to relieve the auto-inhibition of CLIP170 (53). We previously demonstrated that TcpB mimics the properties of taxol by acting as a microtubule stabilization agent (14). Therefore, it is possible that the stabilization of microtubules by TcpB may activate the auto-inhibited CLIP170 molecules. Besides this, the perturbation of microtubule dynamics by taxol or nocodazole was reported to cause dissociation of CLIP170 from the microtubule ends (54, 55). Hence, it can be speculated that TcpB-induced microtubule modulation may cause displacement of CLIP170 from the microtubule tips, which may facilitate the recruitment of CLIP170 for the suppression of TLR4 signaling.
In summary, we report an unexpected finding that CLIP170 negatively regulates TLR4 signaling by the targeted ubiquitination and degradation of TIRAP. Furthermore, we observed that CLIP170 expression is modulated by LPS to maintain the cellular homeostasis. The role of CLIP170 in TLR signaling and inflammation has not been recognized previously. However, CLIP170 has been implicated in disease conditions like Hodgkin’s disease, anaplastic large cell lymphoma and autosomal recessive intellectual disability (40, 56, 57). The role of CLIP170 has recently been reported in improving the sensitivity of paclitaxel in breast cancer cells (58). Our findings help to understand the underlying mechanisms of CLIP170-mediated disease pathogenesis, which may ultimately help to develop novel therapeutic interventions.
Supplementary Material
Acknowledgments
We thank G. Ramadevi and Shashikant Gawai for their help in mice experiments and confocal microscopy, respectively. We thank Dr. Sathya Velmurugan for proof-reading of the manuscript.
Funding:
Work in the laboratory of GR is supported by funding from the Department of Biotechnology, Ministry of Science and Technology, Government of India, through the National Institute of Animal Biotechnology, Hyderabad, India. Work in the laboratory of GS is funded by National Institutes of Health (NIH) grant R01-AI-073558. GR was awarded NIH grant R03AI101611 to initiate this study. PJ acknowledges research fellowship for doctoral studies from INSPIRE, Department of Science and Technology, Government of India. SM acknowledges post-doctoral fellowship from University Grant Commission, Government of India.
Abbreviations
- CLIP170
Cytoplasmic linker protein 170
- +TIP
Microtubule plus-end-tracking proteins
- TIRAP
Toll/IL-1 receptor domain-containing adapter proteins
- MyD88
myeloid differentiation primary response 88
- TLR2
Toll-Like Receptor-2
- TLR4
Toll-Like Receptor-4
- TLR9
Toll-Like Receptor-9
- PAMP
Pathogen Associated Molecular Pattern
- LPS
Lipopolysaccharide
- ODN
Oligodeoxynucleotide
- SOCS-1
Suppressor of cytokine signalling-1
- TIR domain
Toll/IL-1 receptor domain
- TNF-α
Tumour Necrosis Factor-Alpha
- IL-6
Interleukins 6
- IFN-β
Interferon-beta
References
- 1.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 2.Miggin SM, O’Neill LA. New insights into the regulation of TLR signaling. J Leukoc Biol. 2006;80:220–226. doi: 10.1189/jlb.1105672. [DOI] [PubMed] [Google Scholar]
- 3.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 4.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 6.Lowe EL, Doherty TM, Karahashi H, Arditi M. Ubiquitination and de-ubiquitination: role in regulation of signaling by Toll-like receptors. J Endotoxin Res. 2006;12:337–345. doi: 10.1179/096805106X118915. [DOI] [PubMed] [Google Scholar]
- 7.O’Neill LA. Regulation of signaling by non-degradative ubiquitination. J Biol Chem. 2009;284:8209. doi: 10.1074/jbc.R800070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 9.Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5:495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- 10.Fearns C, Pan Q, Mathison JC, Chuang TH. Triad3A regulates ubiquitination and proteasomal degradation of RIP1 following disruption of Hsp90 binding. J Biol Chem. 2006;281:34592–34600. doi: 10.1074/jbc.M604019200. [DOI] [PubMed] [Google Scholar]
- 11.Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O’Neill LA, Hertzog PJ. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 12.Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR Domain-containing Protein Mimics Properties of the Toll-like Receptor Adaptor Protein TIRAP. J Biol Chem. 2009;284:9892–9898. doi: 10.1074/jbc.M805458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radhakrishnan GK, Splitter GA. Biochemical and functional analysis of TIR domain containing protein from Brucella melitensis. Biochem Biophys Res Commun. 2010;397:59–63. doi: 10.1016/j.bbrc.2010.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radhakrishnan GK, Harms JS, Splitter GA. Modulation of microtubule dynamics by a TIR domain protein from the intracellular pathogen Brucella melitensis. Biochem J. 2011;439:79–83. doi: 10.1042/BJ20110577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirl C, Wieser A, Yadav M, Duerr S, Schubert S, Fischer H, Stappert D, Wantia N, Rodriguez N, Wagner H, Svanborg C, Miethke T. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat Med. 2008;14:399–406. doi: 10.1038/nm1734. [DOI] [PubMed] [Google Scholar]
- 16.Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, Muller A, Lapaque N, Demaria O, Alexopoulou L, Comerci DJ, Ugalde RA, Pierre P, Gorvel JP. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008;4:e21. doi: 10.1371/journal.ppat.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sengupta D, Koblansky A, Gaines J, Brown T, West AP, Zhang D, Nishikawa T, Park SG, Roop RM, 2nd, Ghosh S. Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter, MAL. J Immunol. 2010;184:956–964. doi: 10.4049/jimmunol.0902008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierre P, Scheel J, Rickard JE, Kreis TE. CLIP-170 links endocytic vesicles to microtubules. Cell. 1992;70:887–900. doi: 10.1016/0092-8674(92)90240-d. [DOI] [PubMed] [Google Scholar]
- 19.Slep KC, Vale RD. Structural basis of microtubule plus end tracking by XMAP215, CLIP-170, and EB1. Mol Cell. 2007;27:976–991. doi: 10.1016/j.molcel.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 2001;104:923–935. doi: 10.1016/s0092-8674(01)00288-4. [DOI] [PubMed] [Google Scholar]
- 21.Coquelle FM, Caspi M, Cordelieres FP, Dompierre JP, Dujardin DL, Koifman C, Martin P, Hoogenraad CC, Akhmanova A, Galjart N, De Mey JR, Reiner O. LIS1, CLIP-170’s key to the dynein/dynactin pathway. Mol Cell Biol. 2002;22:3089–3102. doi: 10.1128/MCB.22.9.3089-3102.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holzbaur EL, Hammarback JA, Paschal BM, Kravit NG, Pfister KK, Vallee RB. Homology of a 150K cytoplasmic dynein-associated polypeptide with the Drosophila gene Glued. Nature. 1991;351:579–583. doi: 10.1038/351579a0. [DOI] [PubMed] [Google Scholar]
- 23.Lansbergen G, Komarova Y, Modesti M, Wyman C, Hoogenraad CC, Goodson HV, Lemaitre RP, Drechsel DN, van Munster E, Gadella TW, Jr, Grosveld F, Galjart N, Borisy GG, Akhmanova A. Conformational changes in CLIP-170 regulate its binding to microtubules and dynactin localization. J Cell Biol. 2004;166:1003–1014. doi: 10.1083/jcb.200402082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunner D, Nurse P. CLIP170-like tip1p spatially organizes microtubular dynamics in fission yeast. Cell. 2000;102:695–704. doi: 10.1016/s0092-8674(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 25.Dujardin D, Wacker UI, Moreau A, Schroer TA, Rickard JE, De Mey JR. Evidence for a role of CLIP-170 in the establishment of metaphase chromosome alignment. J Cell Biol. 1998;141:849–862. doi: 10.1083/jcb.141.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komarova YA, Akhmanova AS, Kojima S, Galjart N, Borisy GG. Cytoplasmic linker proteins promote microtubule rescue in vivo. J Cell Biol. 2002;159:589–599. doi: 10.1083/jcb.200208058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanenbaum ME, Galjart N, van Vugt MA, Medema RH. CLIP-170 facilitates the formation of kinetochore-microtubule attachments. Embo j. 2006;25:45–57. doi: 10.1038/sj.emboj.7600916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson P, Stephens DJ. Microtubule plus-end loading of p150(Glued) is mediated by EB1 and CLIP-170 but is not required for intracellular membrane traffic in mammalian cells. J Cell Sci. 2006;119:2758–2767. doi: 10.1242/jcs.02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li PZ, Li JZ, Li M, Gong JP, He K. An efficient method to isolate and culture mouse Kupffer cells. Immunology Letters. 158:52–56. doi: 10.1016/j.imlet.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 31.Pierre P, Pepperkok R, Kreis TE. Molecular characterization of two functional domains of CLIP-170 in vivo. J Cell Sci. 1994;107(Pt 7):1909–1920. doi: 10.1242/jcs.107.7.1909. [DOI] [PubMed] [Google Scholar]
- 32.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 33.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 35.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7(Suppl 1: S12):11–14. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pipes L, Li S, Bozinoski M, Palermo R, Peng X, Blood P, Kelly S, Weiss JM, Thierry-Mieg J, Thierry-Mieg D, Zumbo P, Chen R, Schroth GP, Mason CE, Katze MG. The non-human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Res. 2013;41:D906–914. doi: 10.1093/nar/gks1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griparic L, Keller TC., 3rd Differential usage of two 5’ splice sites in a complex exon generates additional protein sequence complexity in chicken CLIP-170 isoforms. Biochim Biophys Acta. 1999;1449:119–124. doi: 10.1016/s0167-4889(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 38.Griparic L, Keller TC. Identification and expression of two novel CLIP-170/Restin isoforms expressed predominantly in muscle. Biochim Biophys Acta. 1998;1405:35–46. doi: 10.1016/s0167-4889(98)00096-2. [DOI] [PubMed] [Google Scholar]
- 39.Griparic L, Volosky JM, Keller TC., 3rd Cloning and expression of chicken CLIP-170 and restin isoforms. Gene. 1998;206:195–208. doi: 10.1016/s0378-1119(97)00585-4. [DOI] [PubMed] [Google Scholar]
- 40.Bilbe G, Delabie J, Bruggen J, Richener H, Asselbergs FA, Cerletti N, Sorg C, Odink K, Tarcsay L, Wiesendanger W, et al. Restin: a novel intermediate filament-associated protein highly expressed in the Reed-Sternberg cells of Hodgkin’s disease. Embo j. 1992;11:2103–2113. doi: 10.1002/j.1460-2075.1992.tb05269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheel J, Pierre P, Rickard JE, Diamantopoulos GS, Valetti C, van der Goot FG, Haner M, Aebi U, Kreis TE. Purification and analysis of authentic CLIP-170 and recombinant fragments. J Biol Chem. 1999;274:25883–25891. doi: 10.1074/jbc.274.36.25883. [DOI] [PubMed] [Google Scholar]
- 42.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 43.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 44.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 45.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 46.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 47.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 48.Boutet SC, Biressi S, Iori K, Natu V, Rando TA. Taf1 regulates Pax3 protein by monoubiquitination in skeletal muscle progenitors. Mol Cell. 2010;40:749–761. doi: 10.1016/j.molcel.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, Engelender S. alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A. 2011;108:18666–18671. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin H, Gui Y, Du G, Frohman MA, Zheng XL. Dependence of phospholipase D1 multi-monoubiquitination on its enzymatic activity and palmitoylation. J Biol Chem. 2010;285:13580–13588. doi: 10.1074/jbc.M109.046359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 52.Ma J, Bang BR, Lu J, Eun SY, Otsuka M, Croft M, Tobias P, Han J, Takeuchi O, Akira S, Karin M, Yagita H, Kang YJ. The TNF family member 4-1BBL sustains inflammation by interacting with TLR signaling components during late-phase activation. Sci Signal. 2013;6:ra87. doi: 10.1126/scisignal.2004431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weisbrich A, Honnappa S, Jaussi R, Okhrimenko O, Frey D, Jelesarov I, Akhmanova A, Steinmetz MO. Structure-function relationship of CAP-Gly domains. Nat Struct Mol Biol. 2007;14:959–967. doi: 10.1038/nsmb1291. [DOI] [PubMed] [Google Scholar]
- 54.Perez F, Diamantopoulos GS, Stalder R, Kreis TE. CLIP-170 highlights growing microtubule ends in vivo. Cell. 1999;96:517–527. doi: 10.1016/s0092-8674(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 55.Dragestein KA, van Cappellen WA, van Haren J, Tsibidis GD, Akhmanova A, Knoch TA, Grosveld F, Galjart N. Dynamic behavior of GFP-CLIP-170 reveals fast protein turnover on microtubule plus ends. J Cell Biol. 2008;180:729–737. doi: 10.1083/jcb.200707203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larti F, Kahrizi K, Musante L, Hu H, Papari E, Fattahi Z, Bazazzadegan N, Liu Z, Banan M, Garshasbi M, Wienker TF, Ropers HH, Galjart N, Najmabadi H. A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delabie J, Shipman R, Bruggen J, De Strooper B, van Leuven F, Tarcsay L, Cerletti N, Odink K, Diehl V, Bilbe G, et al. Expression of the novel intermediate filament-associated protein restin in Hodgkin’s disease and anaplastic large-cell lymphoma. Blood. 1992;80:2891–2896. [PubMed] [Google Scholar]
- 58.Sun X, Li D, Yang Y, Ren Y, Li J, Wang Z, Dong B, Liu M, Zhou J. Microtubule-binding protein CLIP-170 is a mediator of paclitaxel sensitivity. J Pathol. 2012;226:666–673. doi: 10.1002/path.3026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.