

Abstract
Objective
Chronic sleep deficiency is associated with early mortality. In epileptic population, there is a higher prevalence of sleep disorders, and individuals with severe refractory epilepsy are at greater risk of premature mortality than the general population. Sudden unexpected death in epilepsy (SUDEP) affects 1:1000 people each year. Ketogenic diet (KD) treatment is one of the few effective options for refractory seizures. Despite the KD reducing seizures and increasing longevity in Kv1.1 knockout(KO) mice, they still succumb to sudden death. This study aims to determine whether (1) the rest profiles of KO and KOKD mice resemble each other as a function of either age or proximity to death; and (2) the timing of death correlates with acute or chronic changes in rest.
Methods
Non-invasive actimetry was used to monitor rest throughout the lives of KO and wild type (WT) littermates administered standard diet or KD.
Results
As KO mice age, rest is reduced (p<0.0001). Rest is significantly improved in KD-treated KO mice (p<0.0001), resembling WT values at several ages. When age is removed as a variable and data are re-aligned to the day of death, the rest profiles of KO and KOKD groups worsen to similar degrees as a function of proximity to death. The amount of rest acutely is not sensitive to the timing of death, whereas chronic rest deficiency profiles (10-15 days prior to death) of both groups were indistinguishable. Chronic accumulation of rest deficiency over the final 15 days was associated with 75% of deaths.
Significance
Our data suggest that the accumulated rest deficiency is associated with sudden death in Kv1.1 KO mice. These data (i) support the proposed clinical hypothesis that chronic sleep deficiency may be associated with early mortality in epileptic patients (ii) warrant future pre-clinical and clinical studies on sleep monitoring in epileptic patients.
Keywords: Sleep disorders, epilepsy, sudden unexpected death in epilepsy, Kv1.1 knockout, Ketogenic diet, SUDEP
Introduction
Epilepsy is a broad spectrum neurological disease characterized by recurrent and unprovoked seizures. It is the fourth most common neurological disorder affecting approximately 50 million people globally.1 It is estimated that about 1 in 26 people, will develop epilepsy during their lifetime.2 Within the epilepsy population, there is an increased risk for co-morbid sleep disorders, resulting in approximately 30% of epileptic patients experiencing inadequate sleep.3 Sleep deprivation can worsen seizures and the seizures themselves can further disrupt sleep, thus, promoting a negative-feedforward cycle.4,5
Individuals (with and without epilepsy) that suffer from chronic sleep deficiency are more likely to experience cognitive impairments, cardiac arrhythmias, apnea and have shorter lifespans.6–14,41 Indeed, there is a higher risk of premature mortality in epilepsy than the general population with the incidence rate of sudden unexpected death in epilepsy (SUDEP) approximating 1 in 1000 cases every year.15,16 Risk factors associated with SUDEP include severe and refractory seizures, lower IQ and cardiac arrhythmias.17 Despite the role of chronic sleep deficiency in seizure exacerbation, cognitive impairments, cardiac arrhythmias and mortality, whether sleep deficiency is associated with SUDEP remains unknown.
To begin to determine whether insufficient sleep is associated with SUDEP risk, we selected a model of epilepsy with comorbid sleep disorders and experience sudden death. Functional mutations in the Kcna1 gene, which encodes for the alpha subunit of the Kv1.1 channel (a delayed rectifier, voltage-gated potassium channel) in humans and mice is associated with episodic ataxia, partial epilepsy, sleep disorders and sudden death in epilepsy.5,18–24 Infra-red telemetric actimetry was implemented immediately after weaning throughout life until sudden death occurred in Kv1.1 knockout (KO) mice.
Here, we aimed to determine whether: (1) The rest profiles of KO worsened as a function of age; (2) A treatment known to improve longevity in KO mice, the ketogenic diet (KD), influenced the rest parameters. The KD is a high fat:low carbohydrate/protein diet effective in treating refractory seizures and improving sleep problems in epileptic patients and animal epilepsy models.20,24–31 We recently reported that longevity increased by approximately 47% in KO mice treated with the KD, however these mice still experienced sudden death.24 Here, we specifically determined whether the rest profiles of KO and KOKD mice resemble each other as a function of age or proximity to death; (3) The timing of death correlated with either acute or chronic changes in rest.
Materials and methods
Animals
C3HeB/FeJ KO mice and WT mice were reared in a quiet room on a 12-hr light/dark cycle (lights on at Zeitgeber time (ZT) 00:00 hr) in Creighton University's animal resource facility and provided food and water ad libitum. Genotype was determined from tail clips by Transnetyx, Inc (Cordova, TN, USA). KO mice and their wild-type (WT) littermates were weaned onto either a standard diet (SD) or a KD (6.3:1, fat to carbohydrates plus proteins; Bio-Serv F3666, Frenchtown, NJ, U.S.A.) at P21. Experiments were conducted in accordance with the guidelines of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee.
Actimetry
We utilized continuous infra-red telemetric actimetry and switch-closure activity monitoring (Vital View data acquisition system, Mini Mitter Company, Inc; Bend, OR, USA), a proven high-throughput non-invasive method to accurately assess rest and activity patterns.20,32 Actimetry was implemented immediately after weaning and recorded rest-activity in 3-min epochs throughout life.20 WT mice were euthanized to match the ages of the KO littermates. The data were analyzed using Actiview Biological Rhythm Analysis software (Mini Mitter Company, Inc. OR, USA). Parameters during the resting phase between ZT 00:00 and 10:30-hr were calculated for each day.
Statistics
We performed the statistical analysis using Prism 6 (GraphPad Software, Inc., La Jolla, CA, U.S.A.). Data were expressed as the mean ± SEM and analyzed using ANOVA with Dunnett's multiple comparison post hoc test unless otherwise noted.
Results
As KO mice aged, the number of rest epochs was reduced
The first aim of this study was to identify whether rest profiles worsen as a function of age. The representative actograms in Figure 1A depict the differences between the rest-active cycles of KO mice and WT littermates. The total number of daily rest epochs was determined and plotted for each animal (Figure 1B). When compared within genotype, WT rest did not change with age (Figure 1C). In contrast, there was a prominent reduction in the number of rest epochs as KO mice aged, beginning at P38. Post hoc analyses indicated that when compared to their youngest age, the number of KO rest epochs was reduced by 32% at P42 and by 47% at P50 (Figure 1C). Between genotypes, the rest values of KO and WT mice were similar at P26. However, with age KO mice rested for significantly fewer epochs when compared to their WT littermates beginning at P30 (Figure 1C).
Figure 1. The number of rest epochs decline as KO mice age.
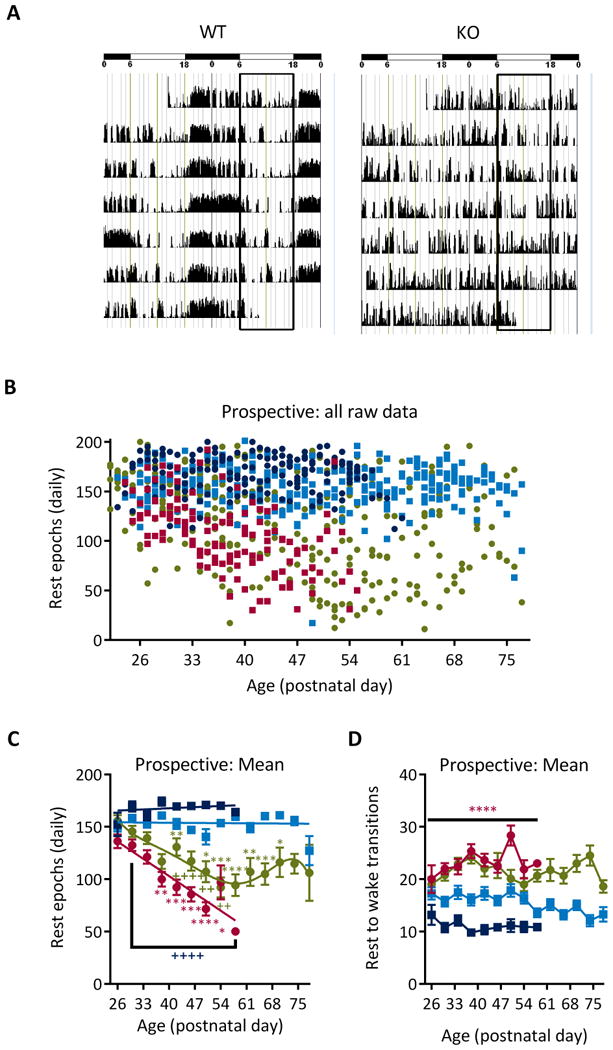
(A) Representative actograms from a WT and KO mouse over 7 days. The horizontal bars at the top indicate the light/resting phase (empty bars) and the dark/active phase (filled bars). The two vertical rectangles outline the resting phase. During each 3-min epoch, activity is scored in 0-50 units. Each epoch is plotted against time on the x-axis. (B) Scatterplot of all of the raw data. Each data point represents the total number of rest epochs for a single day for a single animal: WT (dark blue), WTKD (light blue), KO (red), KOKD (green). (C) Daily values were pooled into 4-day bins and averaged per group. Linear regression analyses indicated that both WT groups did not deviate from zero and had a minimal slope (WT: Y = -0.18*X + 161.4 and WTKD: Y = -0.03*X + 154.9), indicating that rest remained stable with age. Rest reduced with age in KO mice beginning at P38 (F(7,252)=4.99, p<0.0001) and when compared to WT (F(1,252)=365.9; p<0.0001). KD treatment of KO mice improved rest (F(3,533)=96.00; p<0.0001). Within group analyses indicates effects were not linear (F(12,536)=3.650; p<0.0001). Rest does not change among P50 and older ages in KOKD mice (F(7,140)=1.03, p=0.4). Data are expressed as the mean ± SEM; * indicates significantly differs within group from P26 value (red KO, green KOKD); + indicates statistical difference between KO (red KO × WT; green KO × KOKD); * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. (D) The daily number of rest-active transitions were pooled for each age bin. KO mice have significantly higher rest-to-wake transitions when compared to WT controls beginning on P30 (F (3,534) = 98.6, p < 0.0001). Data are expressed as the mean ± SEM; **** p < 0.0001 indicates statistical difference compared to WT. KO mice died on P50 ± 2 days; thus n=5 KO mice from P26-46; n=3 at P50; n = 2 at P54; n=1 at P57. At all ages, WT n=5, KOKD=8 and WTKD=6. KD, ketogenic diet; KO, Kv1.1 knockout mice; WT, wild type.
Ketogenic diet (KD) treatment increased rest of KO mice
The second aim of the study was to determine whether the rest profiles of KO and KOKD mice resemble each other as a function of age. Overall, KD treatment significantly improves rest in KO mice. However, the polynomial non-linear regression used to fit KOKD data indicates temporal phasic influence of KD treatment on rest in KO mice (Figure 1C). During the first two weeks of either KD or SD, KO groups did not differ. Compared with the youngest age, protective effects became apparent when KOKD mice reached P38. At P42, KOKD experience ∼25% less rest compared to WT and 31% less rest at P50. During this time, the trajectory of rest reduction for KO and KOKD groups are similar. Interestingly, once KOKD mice reach P50, the negative trajectory of the rest decline halts and the number of daily rest epochs stabilizes with no difference between P50 and older age groups. There was a significant difference among groups on the daily number of transitions from rest-active, however transitions remained stable across age (Figure 1D). Collectively, these data indicate that rest diminishes with age in KO. In addition, KD treatment provides some protection against the age-related severity of the rest decline in KO mice.
Rest diminished as KO and KOKD mice approach death
In our colony, 100% of KO mice die prematurely. KD treatment significantly increased longevity of KO mice from P49±2 days to P68±2 days (Figure 2A), however, KOKD mice still experienced premature mortality. Thus, we next determined whether the rest profiles of KO and KOKD mice resemble each other as a function of proximity to death. Age was removed as a variable from the subsequent analyses and the data were re-aligned starting at the day prior to sudden death (PTD). The rest epoch data were analyzed retrospectively to determine whether the severity of the rest decline that occurred immediately prior to death were similar between groups. Beginning at 1 day PTD and going backwards to younger ages, the daily rest epochs were re-pooled into 4-day bins. WT and WTKD rest were stable at all ages.
Figure 2. The decline in rest worsens as KO and KOKD mice approach death.
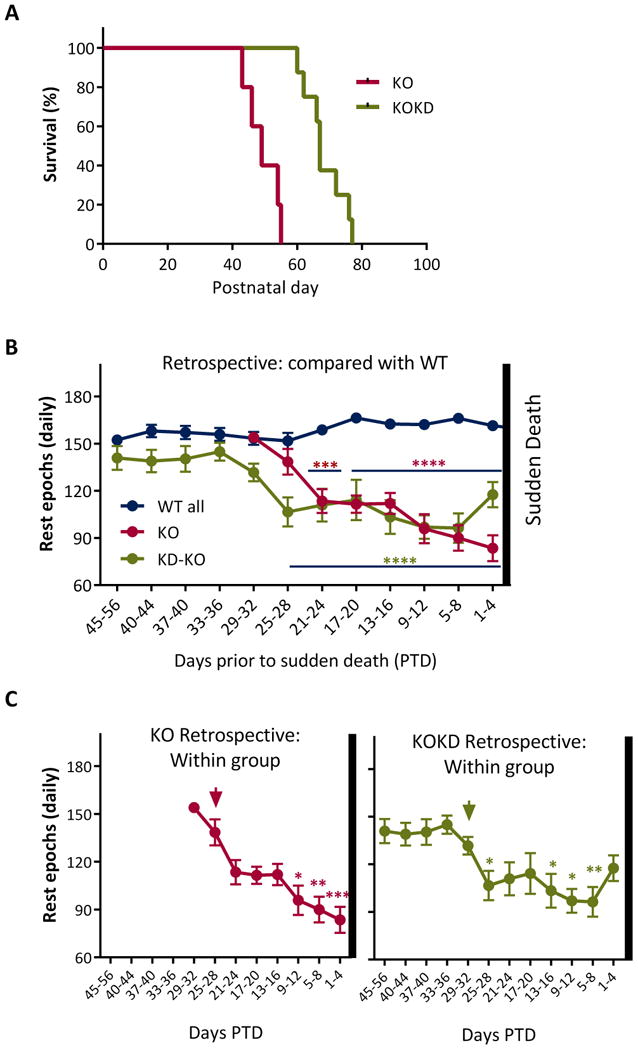
(A) Survival plot of KO (red line) and KOKD (green line) mice. KD treatment increases longevity (p<0.0001; Gehan-Breslow-Wilcoxon test; KO range: P43-55 days and KOKD range: is 60-77 days). (B) Retrospective analyses of data between groups. WT and WTKD rest epochs did not differ and were therefore combined (WT all, blue line). The vertical bar at the right indicates sudden death for KO and KOKD groups. The number of rest epochs significantly changed as both KO and KOKD groups approached sudden death (F (7,623) = 4.2, p < 0.0002). Red asterisks indicate KO group differs significantly from WT at all ages spanning the blue lines (in the middle of the graph). Green asterisks indicate KOKD group differs significantly from WT at all ages spanning the blue line (bottom on graph). (C) KO (left graph, red line) and KOKD (right graph, green line) data were separated from the graph in (B) and replotted to highlight the within group differences (ANOVA with Tukey's multiple comparisons post hoc test). All data were compared to the last bin when rest levels resembled WT levels (arrow). The rest deficiency occurred during the final 12days PTD and 16days PTD in KO and KO-KD mice, respectively (F(7,623)=4.16, p<0.005) and remained low until death. Data are expressed as the mean ± SEM; * p < 0.05, ** p< 0.01, *** p < 0.001, **** p < 0.0001.
Irrespective of age, the number of rest epochs significantly changed as both KO and KOKD groups approached sudden death. At the youngest ages (i.e. furthest from death), both KO and KOKD groups resembled WT values. After approximately 8 days of resting at WT levels (from 25-32 days PTD), KO mice began experiencing significant and chronic reduction of rest epochs (Figure 2B). In contrast, KD treatment allowed KO mice to rest at WT levels for approximately 30 days (from 29-56 days PTD). The onset of diminishing rest began at 25-28 days PTD and became chronic thereafter (Figure 2B). During the weeks prior to sudden death, this rest profile of KO and KOKD mice were indistinguishable. These data indicate that a subject that rests ∼30% less than WT will die within 24 days (KO: 29±5% and KOKD: 30±7%) and a subject that rests ∼40% less than WT will die within 12 days (KO: 41±5% and KOKD: 40±5%) in this model of epilepsy, sleep disorders and SUDEP.
Within group comparison was used to determine how severe and for how long the chronic reduction of rest occurred prior to death. When compared to 25-28 days PTD, KO mice were the most deficient in rest during the final 12 days PTD (Figure 2C). Following onset of rest reduction for KOKD mice, their rest profiles resembled the KO group prior to sudden death with the lowest levels occurring during the final 16 days PTD (Figure 2C).
Acute rest epochs from 1 or 2 days PTD is not unique to the time of death
Next, we determined whether the number of rest epochs that occurred acutely during 1 or 2 days PTD was unique within each group to the timing of death. As the number of rest epochs declined, mortality increased to similar degrees for KO and KOKD groups (Figure 3A). However, the mortality plot inaccurately suggests that KO mice will die if they rest for only 50 or fewer epochs. This is depicted by the range of the raw data (i.e. the number of rest epochs for each animal for each day) shown in the scatterplot superimposed on the lower portion of the percent mortality graph in Figure 3A. The scatterplot highlights the frequency of each daily rest epoch number in relation to mortality. The occurrence rate for the daily rest epoch value at 1 day PTD was calculated to determine how often that amount of rest or less occurred throughout the lifespan of that animal or any other animal within the same group. Resting for 50 or fewer epochs occurred 7% of the KO rest periods and death did not follow. As depicted in Figure 3B, the number of rest epochs at 1 day PTD occurred 10–39% of the time and death did not follow. Similarly, the rest experienced by KOKD mice at 1 day PTD occurred 7–98% of the time and death did not follow (Figure 3B). These data suggest that the amount of rest experienced 1 day PTD is not unique to the occurrence of death.
Figure 3. Acute rest epochs from 1 or 2 days PTD is not unique to the time of death.
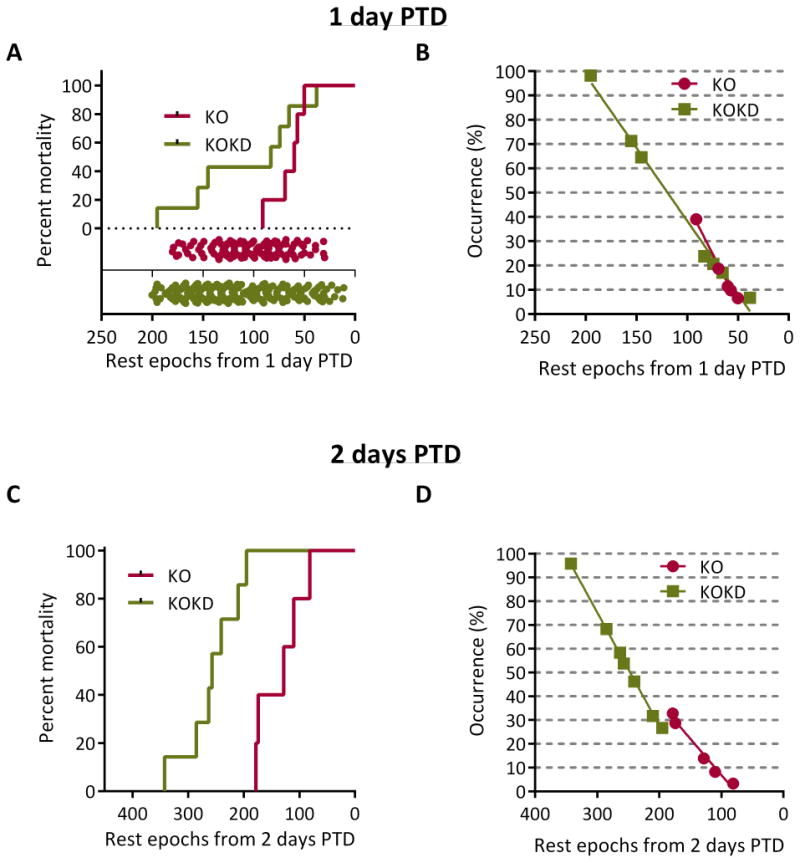
Data analyzed for (A,B) 1 day prior to death (PTD) or (C, D) 2 days PTD. (A) The number of rest epochs from 1 day PTD was plotted against the mortality and did not differ between groups (p=0.09; Log-rank Mantel-Cox text). For KO mice, the number of rest epochs 1 day PTD was 50, 57, 60, 69 and 91. For KOKD mice, the number of rest epochs 1 day PTD was 38, 65, 74, 83, 145, 155 and 195 rest epochs. The range of the raw data (i.e. the number of rest epochs for each animal for each day) shown in the scatterplot superimposed on the lower portion of the percent mortality graph (red for KO and green for KOKD). (B) The occurrence rate for the daily rest epoch value at 1 day PTD was calculated to determine how often that amount of rest or less occurred throughout the lifespan of that animal or any other animal within the same group. (C) The number of rest epochs that occurred during 2 days PTD were summed and plotted against the mortality (p<0.0005, Log-rank Mantel-Cox test). (D) The occurrence rate for the daily rest epoch value during 2 days PTD was calculated to determine how often that amount of rest or less occurred throughout the lifespan of that animal or any other animal within the same group (ranges for KO: 3–33% and KOKD: 27–96%).
Interestingly, when considering the total amount of rest experienced 2 days PTD, KOKD mice died with significantly more rest when compared to KO mice (Figure 3C). The range of occurrence rates for both KO and KOKD mice were common through life (Figure 3D). Collectively, these data suggest that the amount of rest during 1 or 2 days PTD is not unique to the occurrence of death.
Rest deficiency
In clinical sleep studies that focus on how much sleep an individual is deprived of each night, the terms (i) partial or full sleep deprivation distinguishes between a few hours vs. an entire night of deprivation and (ii) acute vs. chronic indicates the duration of the deprivation protocol (i.e. for a few nights vs. many weeks or months). To increase translational relevance, we assessed the deficiency of rest. Data were plotted in days PTD and analyzed prospectively to assess the accumulation of rest deficiency over time. This cumulative deficiency was analyzed during three time frames, (i) during their lifespan (Figure 4), (ii) during the final 10 days PTD (Figure 5) and (iii) during the final 15 days PTD (Figure 6), to determine whether one may be sensitive to and a more accurate predictor of the occurrence of death.
Figure 4. KOKD mice accumulate more rest deficiency prior to death (PTD).
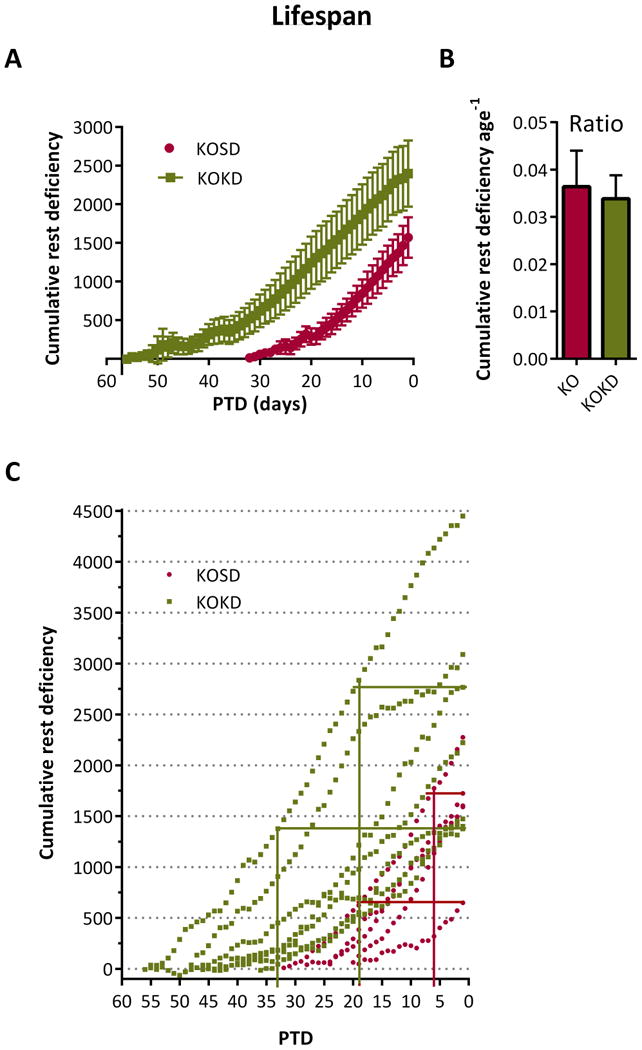
(A) Optimal rest was defined as the stable WT value throughout life (168± 6 rest epochs, n= 20-40 periods per mouse/10 mice). Therefore, we assessed the rest deficiency of KO mice, or the amount of rest KO mice were deprived of during each rest phase relative to age-matched WT controls. Data were aligned at the day of death. The number of rest epochs for each rest period of KO and KOKD mice was subtracted from the WT value (168 rest epochs) to yield the rest deficiency for each period PTD. Data were averaged within group and the cumulative rest deficiency was prospectively plotted for each day PTD over the animal's lifetime. Rest deficiency correlates with PTD for KO (R2 0.94, p<0.0001) and KOKD (R2 0.95, p<0.0001; Pearson's correlation). KOKD mice experienced more rest deficiency throughout their lives (area under the curve (AUC) KOKD 49,329±12,355 vs KO 16,797±3,793; Mann Whitney test: p=0.01). (B) Cumulative rest deficiency was normalized to lifespan. Data are expressed as the mean ± SEM. (C) Scatterplot of cumulative rest deficiency. The lower horizontal line indicate the least amount of deficiency accumulated prior to the first death for each group (KO is red and KOKD is green). The horizontal line is drawn towards the y axis until the last within group data point is reached. The corresponding vertical intersection indicates the number of days PTD in which all mice within that group died (i.e. once KOKD mice accumulate 1376 epochs of deficiency, mortality occurs within 33 days). The upper horizontal bar begins at 0 days PTD with the final three (KOKD) or two (KO) mice. At this accumulated deficiency, the percent mortality and the number of remaining days was determined (i.e. by 1724 epochs, 80% of KO mice have passed and the remaining will within 6 days).
Figure 5. Chronic accumulation of rest deficiency over the final 10 days PTD is sensitive to the timing of death for 58% of subjects.
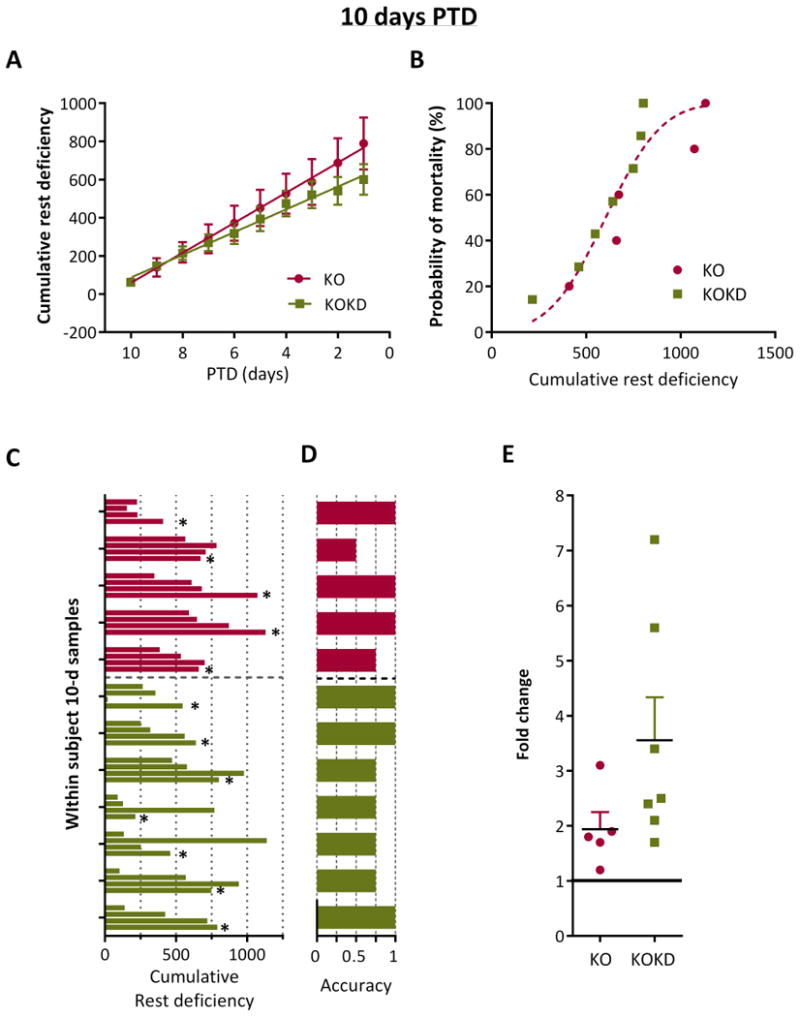
(A) Cumulative probability of rest deficiency (in epochs) significantly correlated with days prior to sudden death (PTD) for KO (R2 0.99, p<0.0001) and KOKD groups (R2 0.99, p<0.0001; Pearson's correlation). Linear regression indicates no statistical difference between slopes (F (1.116)=3.22, p=0.08) or y-intercept (F(1,117) = 3.6, p=0.06). Data are expressed as the mean ± SEM. (B) Histogram for the probability of death as a function of the cumulative rest deficiency for 10 days PTD was generated. A cumulative Gaussian nonlinear regression analyses fit both data set with a single curve (R2 = 0.92 for KO and 0.93 for KOKD). (C) Samples of accumulated deficiency in rest over 10 days were taken at three other time points throughout each animal's life in addition to the final 10 days PTD. Deficiency over the first 10 days is represent is represented as the first bar of the 4 bar clusters and the final 10 days is the last bar (denoted by the asterisks). KO are in red and KOKD are in green. (D) Fractional depiction of whether the highest amount of rest deficiency was accurately associated with death when considering the ranking of within subject samples from highest to lowest deficiency in rest. (E) The fold change in rest deficiency during the 10 final days PTD when compared to the first 10 days of data acquisition.
Figure 6. Chronic accumulation of rest deficiency over the final 15 days PTD is sensitive to the timing of death for 75% of subjects.
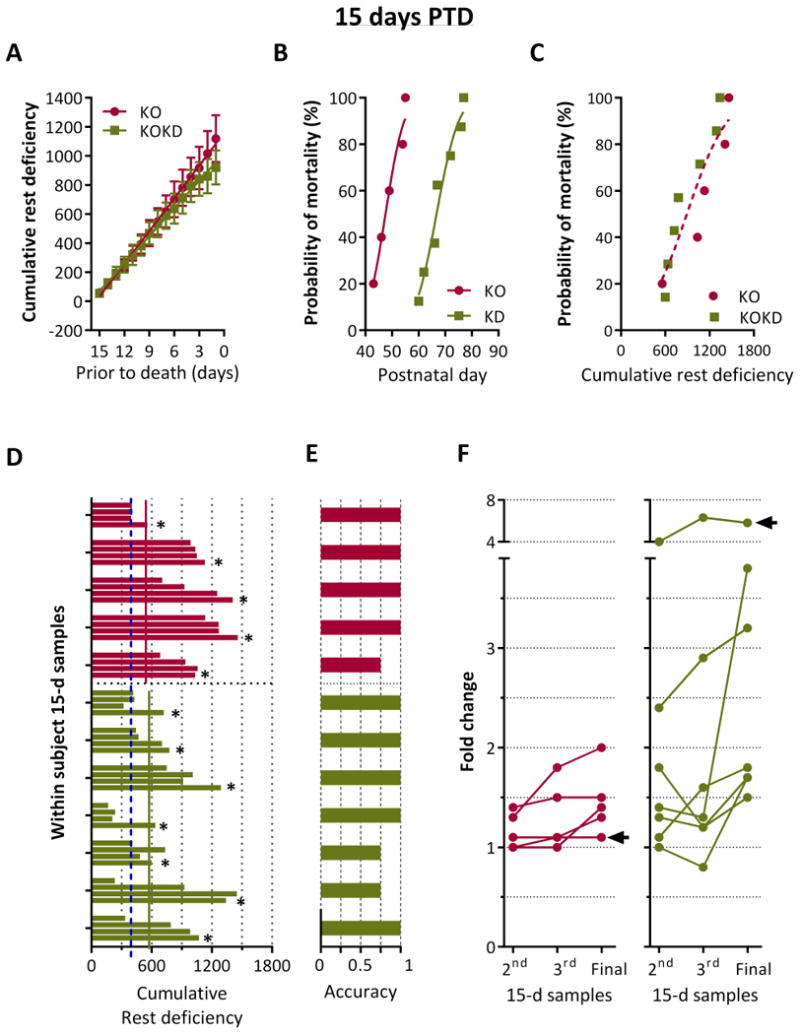
(A) Cumulative probability of rest deficiency (in epochs) significantly correlated with days prior to sudden death (PTD) for KO (R2 0.99, p < 0.0001) and KOKD groups (R2 0.99, p < 0.0001; Pearson's correlation). Linear regression indicates no statistical difference between slopes (F (1,176) = 2.5, p = 0.12) or y-intercept (F (1, 177) = 2.0, p = 0.16). Data are expressed as the mean ± SEM. (B) Cumulative probability of mortality as a function of age for KO and KOKD groups (the histogram of the frequency distribution was analyzed using a cumulative Gaussian nonlinear fit; p < 0.0001). (C) Cumulative probability of mortality as a function of cumulative rest deficiency. Gaussian nonlinear regression analysis fits both data set with a single curve (R2 = 0.91 for KO and 0.93 for KOKD). (D) Samples of accumulated deficiency in rest over 15 days were taken at four time points. Deficiency over the first 15 days is represented as the first bar of the 4 bar clusters and the final 15 days is the last bar (denoted by the asterisks). KO mice are in red and KOKD mice are in green. The blue dashed line depicts the lowest amount of rest deficiency experienced by KO mice. The horizontal lines indicate the least amount of deficiency at 15 days PTD for KO (red) and KOKD (green) groups. (E) Fractional depiction of whether the highest amount of rest deficiency was accurately associated with death when considering the ranking of within subject samples from highest to lowest deficiency in rest. (F) The fold change in rest deficiency compared the 2nd, 3rd and final 15-day samples to the first 15-day sample. The first sample began at data acquisition and was considered baseline. The final sample was 15 days PTD. Arrow indicates a subject in which the ratio of rest deficiency was not increased during the Final 15-days PTD.
KOKD mice accumulate more rest deficiency prior to death
The rest deficiency was determined for each rest phase over the lifespan of each subject (Figure 4A). Cumulative deficiency was plotted against days prior to death. At the youngest ages, KO and KOKD groups resembled WT, thus their deficiency was near 0. Cumulative rest deficiency was significantly correlated with PTD for KO and KOKD groups. Interestingly, KOKD mice experienced more rest deficiency throughout their lives when compared to KO mice. However, when the normalized to lifespan, the ratio of rest deficiency prior to death did not differ between KO and KOKD mice (Figure 4B). These data indicate that both groups experienced similar overall amounts of rest deficiency when normalized to longevity. These data also indicate that once KO mice accumulate 648 epoch of deficiency, mortality will occur within 19 days (Figure 4C, lower red horizontal line and vertical intersection). In addition, by 1724 epochs, 80% of mice have passed and the remaining will within 6 days (Figure 4C, upper red horizontal line and vertical intersection). Similarly, once KOKD mice accumulate 1376 epochs of deficiency, mortality occurs within 33 days; by 2767 epochs, 71% of mice have passed and the remaining will pass within 19 days (Figure 4C, green lines).
Monitoring the lifetime cumulative rest deficiency is only feasible in animal models and provides useful information. However, this type of monitoring is not feasible in the clinic. Thus, we analyzed the chronic accumulation of rest deficiency over manageable time frames of 10-day and 15-day increments.
Chronic accumulation of rest deficiency over the 10 days PTD is sensitive to the timing of death for 58% of subjects
Both KO and KOKD groups experienced similar degrees of rest deficiency during the final 10 days PTD (Figure 5A). This is apparent with similar AUC (KO 3,707±776 and KOKD 3,209±448; p=0.9) and by the single curve fit when assessing the cumulative probability of death by their cumulative rest deficiency (F(2,8)=3.0, p=0.1; Figure 5B). These data indicate that even though KOKD mice life significantly longer, their 10 d rest profile prior to death resembles KO values.
To control for whether these amounts of rest deficiency had occurred earlier in each subject's life, rest deficiency over 10 d was examined at three earlier time points: at the beginning of data acquisition and twice in middle of the animal's life (equidistant from the first and last time points). These within subject 10 d samples were compared to the final 10 days PTD for each subject (Figure 5C: the asterisks indicate the data from the final 10 days PTD time point). This graph depicts the high inter-subject variability within each group and thus highlights the importance of using a within subject design to analyze the data. The fractional accuracy of this metric was determined within subject by whether death followed the highest amount of cumulative rest deficiency (indicated by a value of 1; Figure 5D). Using this metric, 3/5 KO and 4/7 KOKD mice died following the greatest amount of accumulated rest deficiency. In addition, 4 of the remaining 5 mice died immediately following their second largest amount of rest deficiency. The data suggest that each subject reaches a critical mass of accumulated rest deficiency prior to death. When data from the final 10 days PTD was compared to the first 10 days of data acquisition (baseline), KO mice died when rest deficiency was 1.9±0.3 fold higher than baseline. Interestingly, KOKD died when deficiency was 3.6±0.8 fold greater than baseline (however, this does not differ from KO; p=0.08).
Chronic accumulation of rest deficiency over the 15 days PTD is sensitive to the timing of death for 75% of subjects
To determine whether data from 15 days PTD increased the fractional accuracy, we conducted the same analyses as described above. Similar degrees of rest deficiency occurred for both groups during the final 15 days PTD (similar AUC (KO 7,771±1182 and for KOKD 7,192±1092; p=0.8, Mann-Whitney test; Figure 6A). Even though KD treatment significantly increased longevity (Figure 6B), when age was removed as a variable, the amount of rest deficiency both groups accumulated over 15 days PTD was indistinguishable (F (2,8)=4.0, p=0.06; Figure 6C).
To control for whether the amounts of deficiency had occurred previously, three earlier time points were compared to the final 15 days PTD for each subject (Figure 6D). These data indicate that KO mice died with at least a 558-epoch deficiency (red line), however 85% of KO samples were greater than this. Interestingly, this value did not change much with KD treatment. KOKD mice died with at least a 601-epoch deficiency (green line) with 57% of KOKD samples being greater than this. Interestingly, of all the 15-day samples analyzed for both groups, note the lowest KO deficiency sample (blue dashed line at 396 epochs). KD treatment did allow 4/7 mice to experience less rest deficiency when compared to the lowest KO value. The fractional accuracy of this metric illustrates that 4/5 KO and 5/7 KOKD mice died following the greatest amount of accumulated rest deficiency (Figure 6E). In addition, the remaining 3 subjects died immediately following their second largest amount of rest deficiency.
Compared to baseline, there was a significant fold increase among the 2nd, 3rd and Final samples for both KO and KOKD groups (F(2,20)=4.18; p<0.05; two-factor ANOVA; Figure 6F). Arrows indicate that only 2/12 mice did not have an increase from the 3rd sample to the final sample. Overall, KO mice died when rest deficiency was increased by 1.4±0.2 fold. The final 15 days PTD for the KOKD group was the greatest fold increase with an average of 2.8 fold higher than baseline (p<0.01, Sidak's multiple comparisons test). Collectively, these data indicate 15 days of accumulated deficiency in rest is more sensitive to the timing of death for 75% of subjects.
Discussion
To our knowledge, this is the first study to assess whether deficiencies in rest are associated with sudden death in a pre-clinical epilepsy animal model in which mortality occurs during a predictable window. We report that (1) as KO mice age, rest is reduced when compared within group to younger ages and between groups to age-matched WT mice. (2) Rest is improved in KO mice treated with the KD. (3) When aligning the data points at the time of death, the rest profile of KOKD mice resemble those of KO mice. (4) Acute levels are rest (during the last day or two days PTD) are not sensitive to the timing of death. (5) The chronic rest deficiency profiles at 10 and 15 d prior to death were similar for KO and KOKD groups. (6) The chronic accumulation of rest deficiency over the final 15 d was associated with 75% of deaths (9/12 mice). On average, mice died when rest deficiency was 1.5 (KO) and 2.8 (KOKD) fold higher than baseline. These data highlight the importance of using a within subject design to analyze the data due to the high inter-subject variability within each group.
There is a dearth of knowledge regarding whether sleep problems precede/exacerbate promote SUDEP risk factors and/or SUDEP, and this is largely due to the fact that it is not feasible for clinical studies to be designed to monitor rest as SUDEP approaches and there is a lack of preclinical studies in the relatively few and imperfect animal models. Here, we provide pre-clinical data using the KO mouse model, which offer many advantages including: (i) The age of epilepsy onset is known as KO mice develop spontaneous behavioral seizures during the third postnatal week. (ii) The seizure phenotype is well characterized and generalizable (temporal lobe, idiopathic and juvenile epilepsies). (iii) Sleep disorders have been well characterized in KO mice. (iv) We and others have demonstrated that KO mice exhibit multiple risk factors for SUDEP including severe and frequent seizures (myoclonic, unilateral and bilateral clonic, generalized tonic-clonic (GTCs) and cardiac arrhythmias. (v) KO mice experience premature death during the sixth postnatal week on average.5,18–24,33,34 This known lifespan allows for lifelong monitoring of endpoints and subsequent retrospective analyses.
Here, we report that KO mice have increased fragmentation and rest deficiency, supporting our previous findings in adult KO mice.5,20 Interestingly, the fragmentation did not have a progressive ontogeny. At the youngest ages assessed, KO mice experienced more rest-to-wake transitions when compared to WT mice and this difference between genotypes remained constant throughout life. A previous report found that during the time period analyzed herein, KO mice experience minimal REM sleep, thus it is likely the transitions are predominantly between NREM rest and wake.5 Rest fragmentation may contribute to promoting SUDEP risk factors. Indeed, increased sleep fragmentation is associated with cognitive impairments, and apnea10,35,36, however, sleep fragmentation has not been associated with worsening a seizure phenotype and/or promoting cardiac arrhythmias. Due to the fact that the number of transitions did not change as a function of age, rest fragmentation does not provide temporal indication of SUDEP imminence in this model.
Alternatively, our data indicate that progressive reduction in rest does precede sudden death. Seizures and sleep deprivation can exacerbate the presence of one another thus creating a negative feed forward cycle.4,5 Moreover, studies report that more severe sleep deficiency is associated with the greater degree of cognitive impairment, arrhythmias and seizure severity.5–14,35,36 Here, we found that after epilepsy onset, young KO mice rest for similar amounts of time as WT mice. However, as KO mice age, the deficiency in rest begins to progressively worsen with maximal rest deficiency occurring immediately prior to death. When rest was ∼30% less than WT, both KO and KOKD died within 24 days; when rest was ∼40% less than WT, death occurred within 12 days. When considering the various timeframes of analyses, data from 1-2 days PTD was not reliable and the lifespan data, while informative, is not clinically translational. However, monitoring rest deficiency for 15 day periods and comparing it to a within subject baseline provided the most accurate temporal indicator of SUDEP proximity for 75% of mice.
In rodent studies, prolonged sleep deprivation causes death.42,43 Chronic partial sleep deprivation and sleep problems can promote cognitive impairment, cardiac arrhythmias, apnea and premature death, all risk factors for SUDEP.6–14,17,41 However, whether insufficient sleep is associated with SUDEP and/or promotes SUDEP risk factors remains unknown. Previous studies have reported that in another SUDEP animal model, the genetic mouse model of Dravet syndrome (DS), circadian rhythm and sleep architecture abnormalities occur,44,45 supporting our previously reported findings in Kcna1-null mice.5,20Indeed, a limitation of the published literature is the analyses of one temporal measurement. Whether these changes worsen as DS mice approach death has yet to be determined. Interestingly, the number of animals that survived pentylenetetrazole (PTZ)-injections and subsequent seizures positively correlated with previous sleep; sleep deprivation increased mortality and restored sleep improved survivability.46 These data suggest that restored sleep may improve ability to recover from a severe seizure .
Our current experimental design was not able to decouple whether the seizures caused the rest deficiency or whether the rest deficiency occurred independent of the seizures. Video-EEG monitoring of mice with SCN1A mutation R1648H, a genetic model for spontaneous seizures with susceptibility to febrile seizures, also indicated sleep architecture disruptions (i.e. more wakefulness and less NREM and REM sleep) during time periods in which seizures were absent.47 These data suggest that seizures were not the direct cause of sleep architecture deficiencies. In contrast, clinical and animal model studies have found that the more severe seizures can trigger wake from sleep.7,48-49 We have previously reported that seizures increase in severity and frequency with age, and herein we found the rest deficiency follows a similar timeline.24 In Knca1-null mice, severe seizures can propagate to the lateral hypothalamus, a region involved in promoting wake from sleep and increased seizure burden (which accounts for frequency, severity and duration) positively correlates with the severity of sleep architecture disruption.5 Specifically, the higher the seizure burden, the longer it took for REM sleep onset to occur. Collectively, data suggest that seizures may promote wake and may be at least partially responsible for sleep disruptions.
Recently, Haden et al. (2017) performed a detailed review of clinical studies concerning SUDEP and concluded that the major risk factor for SUDEP is the occurrence of GTCs and that risk increases with increased frequency of GTCs.37 Therefore, the authors concluded that a clinical strategy to reduce SUDEP risk is to achieve freedom from seizures, in particular GTCs. The KD is one of the only effective, non-surgical clinical treatments for refractory epilepsy.31 In humans, KD treatment has been known to abolish seizures by about 13% of the patients with more than 50% reduction in seizures in two-thirds of patients with refractory epilepsy.25 In PTZ and pilocarpine seizure rodent models, KD increases seizure threshold and attenuates epileptogenesis.28,29 In KO mice KD treatment decreases hippocampal excitability, reduces seizures by ∼75%, delays GTC seizure progression and postpones SUDEP.20,24,30
Previously, we demonstrated that GTC occurrence does increase with age in KO mice paralleling the progressive worsening of rest deficiency found here.24 Currently, it is unknown whether the epilepsy and sleep phenotypes of KO mice have dependent (causative or negative feedback) or independent relationships. However, we have found that somnogenic drugs used to treat insomnia and other sleep disorders, do indeed improve sleep and reduce seizures in KO mice.5,20 In addition, the KD improves REM sleep, reduces excessive daytime sleepiness and improves the overall quality of sleep in pediatric patients of refractory epilepsy.25,26 In KO mice, KD improves the diurnal rhythms of adult KO mice.5,20 In the current study, the KD similarly delays the worsening of rest deficiency and SUDEP. But again, whether the KD-mediated SUDEP delay is due to effects on GTCs or rest is unclear. Before KD-treated KO mice experience SUDEP, both GTCs and rest deficiency reach levels comparable to untreated KO mice. These data suggest that the KD is not necessarily a cure in this model of severe, genetic epilepsy. Nevertheless, this study does indicate that long-term monitoring of rest/activity may enable real-time assessment of SUDEP risk.
Specific sub-populations of people with epilepsy have specific identified cardiac abnormalities and genomic variants (in genes including KCNA1, SCN1A, SCN8A, HCN2, KCNQ1, KCNH2, RYR3, and HTR2C), which are postulated to be potential risk factors of SUDEP.38–40,50 Here, we present pre-clinical evidence that the onset and progressive decline in rest efficiency within subject may provide a temporal biomarker for SUDEP imminence. Future pre-clinical and clinical studies are required to identify the combination of genomic and physiological risk factors that may classify not only SUDEP risk, but provide temporal information regarding SUDEP imminence, and thus hopefully provide a window of opportunity for aggressive proactive and preventative treatment strategies in high risk individuals.
Supplementary Material
Supplemental Figure 1. Operational definition of rest using actimetry. (A) Each infra-red beam break caused by activity is scored as an actimetry unit. The amount of activity per epoch ranges from 0 to 50 units (a score of 0 units indicates an absence of beam breaks for the entire 3 min epoch). The actimetry score of each epoch was recorded. All of the behaviors that occurred during each epoch were recorded using time-linked video-actimetry monitoring: active behaviors, included grooming, eating, drinking, moving, immobile (still with eyes open); rest was defined as a subject being still with its eyes closed. The epochs with low actimetry scores were analyzed (0-11 units, n=442 epochs). In epochs with scores of 0-3 units, resting was the dominant behavior and thus defined as rest. Epochs with the lowest scoring actimetry value in which active behaviors were dominant (by at least 50%), and all subsequent values were defined as active. Thus, epochs with actimetry values of 4 units or greater were defined as active. (B) In our colony, seizures of KO mice are scored with a modified Racine Scale Type: 1= brief myoclonic jerks; 2=automatisms and side-to-side head movement; 3=forelimb/hindlimb clonus, tail extension, and/or a single rearing event; 4=seizures are continuous rearing and falling; 5=tonic-clonic seizures. Examples of representative spectral density plots using fast Fourier transformation of typical Racine Scale Type 1 (spike and wave discharge), 3 and 5 seizures (increased spectral density from 1-40 Hz) and their corresponding EEG traces commonly recorded in KO mice. (C) Behavioral identification of seizures during each epoch were verified via video recordings (n=116 epochs). Seizure distribution across actimetry scores. Data are expressed as the percentage of epochs during which a seizure occurred divided the total number of epochs for each actimetry score.
Key point box.
Rest deficiency progressively worsens with age in the Kv1.1 Knockout (KO) mice, an animal model of temporal lobe epilepsy and sudden death.
Ketogenic diet (KD) treatment improves rest in the KO mice.
However viewed retrospectively, from time of death, the rest profiles of both KD treated and untreated KO mice are similar.
Acute deficiency in rest (during last 1-2 days prior to death) do not correlate accurately to the timing of death in the KO mice.
Chronic rest deficiency profiles at 10 and 15 days prior to death were similar for both KD treated and untreated KO mice.
The chronic accumulation of rest deficiency over the final 15 days was associated with 75% of deaths in the KO mice.
Acknowledgments
The authors thank Chaz Johnson for his technical assistance during the conception of this study and Sarah Budney for her mathematical assistance. This work was supported by the National Institutes of Health R01 NS072179 (KAS) and the Citizens United for Research in Epilepsy Foundation grant (KAS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Disclosure: None of the authors have any conflict of interest to disclose. We confirm that our work is consistent with the Journal's guidelines for ethical publication.
References
- 1.Leonardi M, Ustun TB. The Global Burden of Epilepsy. Epilepsia. 2002;43:21–25. doi: 10.1046/j.1528-1157.43.s.6.11.x. [DOI] [PubMed] [Google Scholar]
- 2.Hesdorffer DC, Logroscino G, Benn EKT, et al. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology. 2011;76:23–7. doi: 10.1212/WNL.0b013e318204a36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manni R, Terzaghi M. Comorbidity between epilepsy and sleep disorders. Epilepsy Res. 2010;90:171–177. doi: 10.1016/j.eplepsyres.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Matos G, Andersen ML, do Valle AC, et al. The relationship between sleep and epilepsy: Evidence from clinical trials and animal models. J Neurol Sci. 2010;295:1–7. doi: 10.1016/j.jns.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Roundtree HM, Simeone TA, Johnson C, et al. Orexin Receptor Antagonism Improves Sleep and Reduces Seizures in Kcna1-null Mice. Sleep. 2016;39:357–68. doi: 10.5665/sleep.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maganti R, Sheth RD, Hermann BP, et al. Sleep Architecture in Children with Idiopathic Generalized Epilepsy. Epilepsia. 2005;46:104–109. doi: 10.1111/j.0013-9580.2005.06804.x. [DOI] [PubMed] [Google Scholar]
- 7.Malow BA. Sleep and Epilepsy. Neurol Clin. 2005;23:1127–1147. doi: 10.1016/j.ncl.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Maganti R, Hausman N, Koehn M, et al. Excessive daytime sleepiness and sleep complaints among children with epilepsy. Epilepsy Behav. 2006;8:272–277. doi: 10.1016/j.yebeh.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Kotagal P, Yardi N. The Relationship Between Sleep and Epilepsy. Semin Pediatr Neurol. 2008;15:42–49. doi: 10.1016/j.spen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Mullington JM, Haack M, Toth M, et al. Cardiovascular, Inflammatory, and Metabolic Consequences of Sleep Deprivation. Prog Cardiovasc Dis. 2009;51:294–302. doi: 10.1016/j.pcad.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludka O, Konecny T, Somers V. Sleep apnea, cardiac arrhythmias, and sudden death. Texas Hear Inst J. 2011;38:340–3. [PMC free article] [PubMed] [Google Scholar]
- 12.Simeone KA, Johnson CJ, Samson KK, et al. Sleep. In: Masino, Boison, editors. Homeostatic Control of Brain Function. Oxford Press; 2015. pp. 314–332. [Google Scholar]
- 13.Steinsbekk S, Wichstrøm L. Stability of Sleep Disorders From Preschool to First Grade and Their Bidirectional Relationship with Psychiatric Symptoms. J Dev Behav Pediatr. 2015;36:243–251. doi: 10.1097/DBP.0000000000000134. [DOI] [PubMed] [Google Scholar]
- 14.Boly M, Jones B, Findlay G, et al. Altered sleep homeostasis correlates with cognitive impairment in patients with focal epilepsy. Brain. 2017;140:1026–1040. doi: 10.1093/brain/awx017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forsgren L, Hauser WA, Olafsson E, et al. Mortality of Epilepsy in Developed Countries: A Review. Epilepsia. 2005;46:18–27. doi: 10.1111/j.1528-1167.2005.00403.x. [DOI] [PubMed] [Google Scholar]
- 16.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: Assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- 17.Walczak TS, Leppik IE, D'Amelio M, et al. Incidence and risk factors in sudden unexpected death in epilepsy: a prospective cohort study. Neurology. 2001;56:519–25. doi: 10.1212/wnl.56.4.519. [DOI] [PubMed] [Google Scholar]
- 18.Smart SL, Lopantsev V, Zhang CL, et al. Deletion of the KV1.1 Potassium Channel Causes Epilepsy in Mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 19.Zuberi SM, Eunson LH, Spauschus A, et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122:817–825. doi: 10.1093/brain/122.5.817. [DOI] [PubMed] [Google Scholar]
- 20.Fenoglio-Simeone KA, Wilke JC, Milligan HL, et al. Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia. 2009;50:2027–34. doi: 10.1111/j.1528-1167.2009.02163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glasscock E, Yoo JW, Chen TT, et al. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–75. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbins CA, Tempel BL. Kv1.1 and Kv1.2: Similar channels, different seizure models. Epilepsia. 2012;53:134–141. doi: 10.1111/j.1528-1167.2012.03484.x. [DOI] [PubMed] [Google Scholar]
- 23.Moore BM, Jou CJ, Tatalovic M, et al. The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP) Epilepsia. 2014;55:1808–1816. doi: 10.1111/epi.12793. [DOI] [PubMed] [Google Scholar]
- 24.Simeone KA, Matthews SA, Rho JM, et al. Ketogenic diet treatment increases longevity in Kcna1 -null mice, a model of sudden unexpected death in epilepsy. Epilepsia. doi: 10.1111/epi.13444. Epub ahead of print June 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallböök T, Köhler S, Rosén I, et al. Effects of ketogenic diet on epileptiform activity in children with therapy resistant epilepsy. Epilepsy Res. 2007;77:134–40. doi: 10.1016/j.eplepsyres.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 26.Hallböök T, Lundgren J, Rosén I. Ketogenic Diet Improves Sleep Quality in Children with Therapy-resistant Epilepsy. Epilepsia. 2007;48:59–65. doi: 10.1111/j.1528-1167.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- 27.Acharya MM, Hattiangady B, Shetty AK. Progress in neuroprotective strategies for preventing epilepsy. Prog Neurobiol. 2008;84:363–404. doi: 10.1016/j.pneurobio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen SL, Nielsen AH, Knudsen KE, et al. Ketogenic diet is antiepileptogenic in pentylenetetrazole kindled mice and decrease levels of N-acylethanolamines in hippocampus. Neurochem Int. 2009;54:199–204. doi: 10.1016/j.neuint.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Hori A, Tandon P, Holmes GL, et al. Ketogenic Diet: Effects on Expression of Kindled Seizures and Behavior in Adult Rats. Epilepsia. 1997;38:750–758. doi: 10.1111/j.1528-1157.1997.tb01461.x. [DOI] [PubMed] [Google Scholar]
- 30.Simeone TA, Samson KK, Matthews SA, et al. In vivo ketogenic diet treatment attenuates pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices from epileptic K v 1.1α knockout mice. Epilepsia. 2014;55:e44–e49. doi: 10.1111/epi.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felton EA, Cervenka MC. Dietary therapy is the best option for refractory nonsurgical epilepsy. Epilepsia. 2015;56 doi: 10.1111/epi.13075. n/a–n/a. [DOI] [PubMed] [Google Scholar]
- 32.Pack AI, Galante RJ, Maislin G, et al. Novel method for high-throughput phenotyping of sleep in mice. Physiol Genomics. 2007;28:232–238. doi: 10.1152/physiolgenomics.00139.2006. [DOI] [PubMed] [Google Scholar]
- 33.Wenzel HJ, Vacher H, Clark E, et al. Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia. 2007;48:2023–2046. doi: 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim DY, Simeone KA, Simeone TA, et al. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann Neurol. 2015;78:77–87. doi: 10.1002/ana.24424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drummond S, Brown GG. The Effects of Total Sleep Deprivation on Cerebral Responses to Cognitive Performance. Neuropsychopharmacology. 2001;25:S68–S73. doi: 10.1016/S0893-133X(01)00325-6. [DOI] [PubMed] [Google Scholar]
- 36.Vgontzas AN, Chrousos GP. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: Multiple interactions and disturbances in sleep disorders. Endocrinology and Metabolism Clinics of North America. 2002;31:15–36. doi: 10.1016/s0889-8529(01)00005-6. [DOI] [PubMed] [Google Scholar]
- 37.Harden C, Tomson T, Gloss D, et al. Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2017;88:1674–1680. doi: 10.1212/WNL.0000000000003685. [DOI] [PubMed] [Google Scholar]
- 38.DeGiorgio CM, Miller P, Meymandi S, et al. RMSSD, a Measure of Heart Rate Variability, Is Associated With Risk Factors For SUDEP: The SUDEP-7 Inventory. Epilepsy Behav. 2011;19:78–81. doi: 10.1016/j.yebeh.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klassen TL, Bomben VC, Patel A, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–e12. doi: 10.1111/epi.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glasscock E. Genomic biomarkers of SUDEP in brain and heart. Epilepsy Behav. 2014;38:172–179. doi: 10.1016/j.yebeh.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colten HR, Altevogt BM. Sleep Disorders and Sleep Deprivation: An Unmet Public Health Problem. Washington (DC): National Academies Press (US); 2006. pp. 0–425. [PubMed] [Google Scholar]
- 42.Rechtschaffen A, Gilliland MA, Bergmann BM, et al. Physiological correlates of prolonged sleep deprivation in rats. Science. 1983;221:182–4. doi: 10.1126/science.6857280. [DOI] [PubMed] [Google Scholar]
- 43.Kushida CA, Bergmann BM, Rechtschaffen A. Sleep Deprivation in the Rat: IV. Paradoxical Sleep Deprivation. Sleep. 1989;12:22–30. doi: 10.1093/sleep/12.1.22. [DOI] [PubMed] [Google Scholar]
- 44.Han S, Yu FH, Schwartz MD, et al. Na(V)1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci U S A. 2012;109:E368–77. doi: 10.1073/pnas.1115729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalume F, Oakley JC, Westenbroek RE, et al. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol Dis. 2015;77:141–54. doi: 10.1016/j.nbd.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ni LY, Zhu MJ, Song Y, et al. Pentylenetetrazol-induced seizures are exacerbated by sleep deprivation through orexin receptor-mediated hippocampal cell proliferation. Neurol Sci. 2014;35:245–252. doi: 10.1007/s10072-013-1495-5. [DOI] [PubMed] [Google Scholar]
- 47.Papale LA, Makinson CD, Christopher Ehlen J, et al. Altered sleep regulation in a mouse model of SCN1A - derived genetic epilepsy with febrile seizures plus (GEFS+) Epilepsia. 2013;54:625–634. doi: 10.1111/epi.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bazil CW, Walczak TS. Effects of Sleep and Sleep Stage on Epileptic and Nonepileptic Seizures. Epilepsia. 1997;38:56–62. doi: 10.1111/j.1528-1157.1997.tb01077.x. [DOI] [PubMed] [Google Scholar]
- 49.Hellier JL, Dudek FE. Spontaneous motor seizures of rats with kainate-induced epilepsy: effect of time of day and activity state. Epilepsy Res. 1999;35:47–57. doi: 10.1016/s0920-1211(98)00127-2. [DOI] [PubMed] [Google Scholar]
- 50.Bagnall RD, Crompton DE, Semsarian C. Genetic Basis of Sudden Unexpected Death in Epilepsy. Front Neurol. 2017;8:348. doi: 10.3389/fneur.2017.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Operational definition of rest using actimetry. (A) Each infra-red beam break caused by activity is scored as an actimetry unit. The amount of activity per epoch ranges from 0 to 50 units (a score of 0 units indicates an absence of beam breaks for the entire 3 min epoch). The actimetry score of each epoch was recorded. All of the behaviors that occurred during each epoch were recorded using time-linked video-actimetry monitoring: active behaviors, included grooming, eating, drinking, moving, immobile (still with eyes open); rest was defined as a subject being still with its eyes closed. The epochs with low actimetry scores were analyzed (0-11 units, n=442 epochs). In epochs with scores of 0-3 units, resting was the dominant behavior and thus defined as rest. Epochs with the lowest scoring actimetry value in which active behaviors were dominant (by at least 50%), and all subsequent values were defined as active. Thus, epochs with actimetry values of 4 units or greater were defined as active. (B) In our colony, seizures of KO mice are scored with a modified Racine Scale Type: 1= brief myoclonic jerks; 2=automatisms and side-to-side head movement; 3=forelimb/hindlimb clonus, tail extension, and/or a single rearing event; 4=seizures are continuous rearing and falling; 5=tonic-clonic seizures. Examples of representative spectral density plots using fast Fourier transformation of typical Racine Scale Type 1 (spike and wave discharge), 3 and 5 seizures (increased spectral density from 1-40 Hz) and their corresponding EEG traces commonly recorded in KO mice. (C) Behavioral identification of seizures during each epoch were verified via video recordings (n=116 epochs). Seizure distribution across actimetry scores. Data are expressed as the percentage of epochs during which a seizure occurred divided the total number of epochs for each actimetry score.