

Abstract
Tumors override energy stress to grow. However, how nucleotide synthesis is regulated under energy stress is unclear. We demonstrate here that glucose deprivation or hypoxia results in the AMPK-mediated phosphorylation of phosphoribosyl pyrophosphate synthetase (PRPS) 1 S180 and PRPS2 S183, leading to conversion of PRPS hexamers to monomers and thereby inhibiting PRPS1/2 activity, nucleotide synthesis, and nicotinamide adenine dinucleotide (NAD) production. Knock-in of non-phosphorylatable PRPS1/2 mutants, which have uninhibited activity, in brain tumor cells under energy stress exhausts cellular ATP and NADPH and increases ROS levels, thereby promoting cell apoptosis. The expression of those mutants inhibits brain tumor formation and enhances the inhibitory effect of the glycolysis inhibitor 2-deoxy-D-glucose on tumor growth. Our findings highlight the significance of recalibrating tumor cell metabolism by fine tuning nucleotide and NAD synthesis in tumor growth.
Keywords: PRPS, AMPK, nucleotide synthesis, energy stress, tumorigenesis
INTRODUCTION
Cancer metabolism is programmed to facilitate the survival and proliferation of cancer cells in the non-native tumor microenvironment (1). A rapidly growing tumor encounters nutritional stresses and recalibrates cell metabolism to survive and grow. Many tumor cells exhibit enhanced Warburg effect, which is a high rate of glycolysis followed by lactic acid fermentation in the cytosol even in the presence of ample oxygen (2, 3). The enhanced production of glycolytic intermediates in the tumor can be used to synthesize cellular building blocks (nucleotides, amino acids, and lipids) to meet the demands of cell proliferation. The pentose phosphate pathway (PPP), which is derived from glycolytic intermediate glucose-6-phosphate and fructose-6-phosphate, produces ribose-5-phosphate (R5P) for the de novo synthesis of nucleotides and nucleic acids (4, 5). Growth signaling through the mechanistic target of rapamycin complex 1 (mTORC1) pathway stimulates de novo pyrimidine and purine synthesis (6–8). Phosphoribosyl pyrophosphate synthetase (PRPS) catalyzes the first and rate-limiting reaction for nucleotide synthesis, producing phosphoribosyl pyrophosphate (PRPP) from R5P by transferring the β, γ-diphosphoryl moiety of ATP to the C1-hydroxy group of R5P (9, 10). PRPP is then used for the synthesis of purine and pyrimidine nucleotides (Supplementary Fig. S1A), the pyridine nucleotide cofactors NAD and NADP, and the amino acids histidine and tryptophan (11).
Human PRPS family has three isoforms that share very high sequence identity: PRPS1 and PRPS2, which have 95% amino acid sequence identity, are expressed in a wide range of tissues, whereas PRPS3 is expressed specifically in the testis. PRPS1-3 are activated by Mg2+, sulfate (SO42−), and phosphate, while PRPS1 is inhibited by the nucleotide biosynthesis products ADP, AMP, and GDP (12, 13). PRPS1 forms a hexamer, which is facilitated by ATP (14). The catalytic active site, which consists of the ATP binding site and the R5P binding site, is located at the interface of two domains of one subunit; the allosteric site for phosphate and ADP is located at the interfaces between three subunits of the hexamer (13), indicating that a hexamer is required for PRPS1 activity. Ketohexokinase-A (KHK-A; also known as fructokinase-A) phosphorylates PRPS1 T225 and activates PRPS1 by blocking the binding of ADP, AMP, and GDP, which is required for hepatocellular carcinoma development (15, 16). Mutations of PRPS1, which reduced the feedback inhibition of purine biosynthesis, were identified in relapsed childhood B cell acute lymphoblastic leukemia (ALL) (17). In addition, PRPS2 was shown to be crucial for cancer cell survival (18–20). However, the mechanism through which PRPS and nucleotide synthesis are regulated under energy stress is unclear.
In this study, we showed that glucose deprivation results in the AMPK-mediated phosphorylation of PRPS1 S180 and PRPS2 S183, disruption of the PRPS1/2 hexamers, and inhibition of PRPS1/2 activity and nucleic acid synthesis. The expression of non-phosphorylatable PRPS1/2 mutants greatly decreased cellular ATP and NADPH levels, increased ROS levels and cell apoptosis, and inhibited brain tumorigenesis.
RESULTS
Energy stresses induce rapid inhibition of PRPS1/2 activity and nucleic acid synthesis
To determine the effects of energy stress on the regulation of nucleic acid synthesis, we removed glucose from the culture medium of U87 and U251 glioblastoma (GBM) cells for 3 h or treated the cells with the glucose metabolism inhibitor 2-deoxy-D-glucose (2-DG) for 4 h, followed by incubation of a limited amount of D-[6-14C] glucose (0.01 mM). We found that glucose deprivation (Fig. 1A) or 2-DG treatment (Supplementary Fig. S1B) largely decreased the production of glucose-derived 14C-RNA and 14C-DNA. In line with this finding, the levels of both purine (IMP, AMP, and GMP) and pyrimidine (UMP and CMP) intermediates were decreased in U87 (Fig. 1B) and U251 cells (Supplementary Fig. S1C) upon glucose deprivation. However, the amount of R5P was not affected by such a short period of glucose deprivation (Fig. 1C), strongly suggesting that the decrease in nucleotide production in response to acute glucose deprivation was not regulated through PPP-derived R5P production. PRPS-catalyzed conversion of R5P to PRPP is a rate-limiting reaction (11). Quantification of PRPS1 and PRPS2 mRNA levels by PCR amplification of their cDNA, in which the PRPS1 but not the PRPS2 fragment was cut by the HindIII restriction enzyme, showed that both PRPS1 and PRPS2 were comparably expressed in U87 and U251 cells (Supplementary Fig. S1D). We immunoprecipitated PRPS1/2 with an antibody recognizing both PRPS1 and PRPS2 from U87 and U251 cells and showed that their activities were inhibited by glucose deprivation (Fig. 1D) and 2-DG treatment (Supplementary Fig. S1E). In line with this finding, PRPP levels were decreased by glucose deprivation (Fig. 1E) and 2-DG treatment (Supplementary Fig. S1F).
Figure 1. Energy stresses induce rapid inhibition of PRPS1/2 and suppress nucleic acid synthesis.
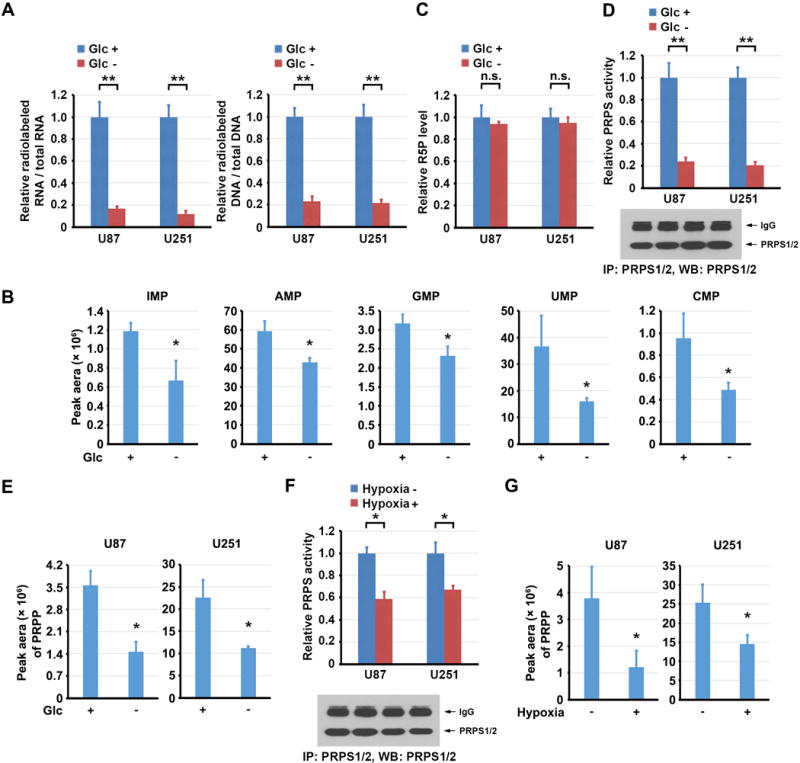
A, C, D and F, Data represent the mean ± SD from three independent experiments. B, E, and G, Data represent the mean ± SD from triplicate samples. *P < 0.05; **P < 0.001; n.s., no significance.
A, U87 or U251 cells were cultured in the presence or absence of glucose (Glc) for 3 h, followed by labeling with D-[6-14C] glucose (1 μCi, 0.01 mM) for 0.5 h. The amounts of 14C-RNA and 14C-DNA were measured.
B, U87 cells were cultured in the presence or absence of Glc for 3 h. Levels of nucleotide monophosphate were measured by LC-MS/MS.
C, U87 or U251 cells were cultured in the presence or absence of Glc for 3 h. R5P levels were measured.
D, E, U87 or U251 cells were cultured in the presence or absence of Glc for 3 h. The activity of immunoprecipitated PRPS1/2 was measured. Immunoblot analysis was performed with the indicated antibody (D). Levels of PRPP were measured by LC-MS/MS (E).
F, G, U87 or U251 cells were cultured with or without hypoxia for 6 h. The activity of immunoprecipitated PRPS1/2 was measured. Immunoblot analysis was performed with the indicated antibody (F). Levels of PRPP were measured by LC-MS/MS (G).
To further determine whether acute PRPS1/2 regulation is independent of glucose level and whether it is also regulated by other metabolic stress, we treated U87 cells with hypoxia for a short period (6 h) in the presence of an ample glucose supply. Hypoxia stimulation resulted in reduced PRPS1/2 activity (Fig. 1F) and PRPP level (Fig. 1G) without affecting R5P production (Supplementary Fig. S1G). These results indicate that PRPS1/2 inhibition and the subsequent decrease of nucleic acid synthesis are rapid responses of cells to energy stresses induced by glucose deprivation and hypoxia.
AMPK phosphorylates PRPS1 S180 and PRPS2 S183 in response to glucose deprivation and hypoxic stimulation
Posttranslational modifications of proteins often result in the alteration of their structures and activities (21). We immunoprecipitated Flag-tagged PRPS1 from glucose-deprived U87 cells and performed a liquid chromatography-coupled Orbitrap tandem mass spectrometric analysis of tryptic digests of immunoprecipitated PRPS1. We found that PRPS1 was phosphorylated at S180 (Supplementary Fig. S2A), which corresponds to S183 in PRPS2 (Supplementary Fig. S2B); both residues are conserved among species (Supplementary Fig. S2C). Of note, the Scansite analyses (http://scansite3.mit.edu/) of PRPS1/2 protein sequences revealed that PRPS1 S180 and PRPS2 S183 are potential phosphorylation residues of AMP-activated protein kinase (AMPK) with -4 (Lys), -3 (Arg), -2 (Val), and +3 (Asp) residues matched to the AMPK recognition motifs (Supplementary Fig. S2D) (22, 23). Glucose deprivation or 2-DG treatment, which activated AMPK, as reflected by phosphorylation of AMPKα T172 and its substrate acetyl-CoA carboxylase 1 (ACC1) S79, induced enhanced binding of endogenous AMPKα to endogenous PRPS1/2 (Fig. 2A) in U87 cells. These results were further validated by coimmunoprecipitation of HA-AMPKα with Flag-PRPS1 or Flag-PRPS2 (Supplementary Fig. S2E).
Figure 2. AMPK phosphorylates PRPS1 S180 and PRPS2 S183 in response to glucose deprivation and hypoxic stimulation.

Immunoprecipitation or immunoblot analyses were performed with the indicated antibodies.
A, U87 cells were cultured in the presence or absence of Glc for 3 h or treated with or without 2-DG (25 mM) for 4 h.
B, Bacterially purified WT His-PRPS1 or His-PRPS1 S180A mutant (upper panel) or WT His-PRPS2 or His-PRPS2 S183A mutant (lower panel) was incubated with or without bacterially purified active AMPK in the presence of [γ-32P]ATP. Autoradiography and immunoblot analyses were performed.
C, D, U87 cells expressing WT Flag-PRPS1 or Flag-PRPS1 S180A (upper panel) or WT Flag-PRPS2 or Flag-PRPS2 S183A (lower panel) were treated with or without 2-DG (25 mM) (C) or A769662 (0.5 mM) (D) for 4 h.
E, U87 cells were pretreated with or without compound C (5 μM) for 30 min and then treated with or without 2-DG (25 mM) for 4 h.
F, WT or AMPKα1/2 double-knockout (DKO) MEFs were cultured in the presence or absence of Glc for 3 h (left panel) or treated with or without A769662 (0.5 mM, right panel) for 4 h.
G, U87 cells were pretreated with or without compound C (5 μM) for 30 min and cultured in hypoxia for the indicated time periods.
To determine whether AMPK phosphorylates PRPS, we performed an in vitro phosphorylation analysis by incubating purified bacteria-expressed human His-PRPS1 or His-PRPS2 with active AMPK protein in the presence of [γ-32P]ATP, followed by autoradiography and immunoblot analysis by a specific anti-phospho-PRPS1/2 S180/S183 antibody, which recognizes both PRPS1 S180 and PRPS2 S183 phosphorylation (Supplementary Fig. S2F). We found that AMPK phosphorylated PRPS1 S180 and PRPS2 S183; those phosphorylations were abrogated by mutating the Ser residues into Ala (Fig. 2B). In addition, stoichiometry analyses of PRPS phosphorylation by AMPK showed incorporation of ~1 mol of phosphate per mol of PRPS1 or PRPS2 protein (Supplementary Fig. S2G). These results, which are consistent with the finding that S180A mutation in PRPS1 and S183A mutation in PRPS2 abolished AMPK-mediated phosphorylation (Fig. 2B), suggest that S180 and S183 are the only AMPK-phosphorylated residues in PRPS1 and PRPS2, respectively.
Consistent with the in vitro results, treatment with 2-DG (Fig. 2C) or A769662 AMPK activator (Fig. 2D) induced the phosphorylation of PRPS1 S180 and PRPS2 S183, but not PRPS1 S180A or PRPS2 S183A mutant, in U87 cells. Notably, PRPS1 S180 and PRPS2 S183 phosphorylation was rapidly induced by 2-DG treatment; this enhanced phosphorylation was detected within 15 min of treatment and was sustained for at least 8 h (Supplementary Fig. S2H). In contrast, treatment of U87 cells with compound C, a non-selective AMPK inhibitor (Fig. 2E), or deficiency of AMPKα1 and 2 in mouse embryonic fibroblasts (MEFs) (Fig. 2F) blocked 2-DG- (Fig. 2E), glucose-deprivation-, and A769662- (Fig. 2F) induced PRPS1/2 S180/S183 phosphorylation. Furthermore, the inhibitory effect of compound C on 2-DG-induced PRPS1/2 S180/S183 phosphorylation was also detected in U251, LKB1-overexpressed A549 (A549/LKB1) human non-small cell lung cancer cells, BxPC-3 human pancreatic cancer cells, and MDA-MB-231 human breast cancer cells (Supplementary Fig. S2I). As expected, hypoxia also induced PRPS1/2 S180/S183 phosphorylation, and compound C treatment inhibited this phosphorylation (Fig. 2G). These results indicate that AMPK activation, which is induced by glucose deprivation and hypoxic stimulation, is sufficient to phosphorylate PRPS1 S180 and PRPS2 S183.
AMPK-mediated PRPS1/2 phosphorylation converts PRPS1/2 hexamers to monomers
Structural analyses of PRPS1 hexamer revealed that PRPS1 S180 is located proximal to the interface of the subunits of the hexamer (Fig. 3A). We hypothesized that such phosphorylation regulates the hexameric structure of PRPS1/2. To test that, we immunoprecipitated endogenous PRPS1/2 and performed size-exclusion chromatography. According to the molecular weight calibration standard (Supplementary Fig. S3A), the fractions 17^#x02013;19 that co-eluted with a molecular weight around 200 kDa contained hexamers of PRPS1/2, which were shown to be non-phosphorylated (Fig. 3B, upper panel). In contrast, the fractions 42–44 that co-eluted with a molecular weight around 29 kDa contained monomers of PRPS1/2 (34 kDa), which were phosphorylated (Fig. 3B, lower panel). Incubation of wild-type (WT) or non-phosphorylatable mutants of PRPS1/2 in the presence or absence of active AMPK revealed that both WT and mutated PRPS1/2 existed as hexamers in the absence of AMPK (Fig. 3C). However, the presence of AMPK, which phosphorylated WT but not mutated PRPS1/2, converted WT but not mutated PRPS1/2 into a monomer.
Figure 3. AMPK-mediated PRPS1/2 phosphorylation converts PRPS1/2 hexamers to monomers.
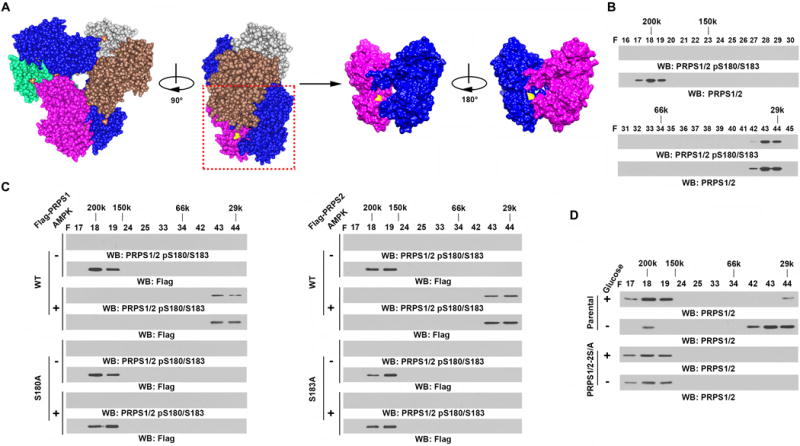
B–D, Immunoprecipitation and immunoblot analyses were performed using the indicated antibodies.
A, Structural view of PRPS1 (PDB: 2H06). Three homodimers formed a hexamer in a propeller shape, in which the N-terminal domains formed the inner circle and the C-terminal domains formed the “blades” towards the outside. Serine 180 (in yellow) was located close to the inter-surface between each homodimer.
B, U87 cell lysates were fractionated on a size-exclusion column. PRPS1/2 was eluted in fraction F17-19 (around 200 kDa) or in F42-44 (around 29 kDa) on the same column under identical conditions, as determined by chromatography molecular weight calibration.
C, Purified WT Flag-PRPS1 or Flag-PRPS1 S180A (left panel) or WT Flag-PRPS2 or Flag-PRPS2 S183A (right panel) from U87 cells was incubated with or without active AMPK in the presence of ATP (0.5 mM). Fractionation was performed on a size-exclusion column. The indicated fractions were used for the immunoblot analysis.
D, Parental U87 cells or U87 cells with knock-in of PRPS1/2 S180A/S183A (PRPS1/2-2S/A) were cultured in the presence or absence of Glc for 3 h. Cell lysates were fractionated on a size-exclusion column. The indicated fractions were used for the immunoblot analysis.
To further validate our findings in vivo, we used CRISPR/Cas9 genome editing knock-in technology to simultaneously replace endogenous PRPS1/2 with PRPS1 S180A and PRPS2 S183A (PRPS1/2-2S/A, Supplementary Fig. S3B–D) in U87 cells (24). Glucose deprivation (Fig. 3D) or A769662 treatment (Supplementary Fig. S3E) converted WT PRPS1/2 but not their mutants from hexamers into monomers. These results indicate that AMPK-mediated PRPS1/2 phosphorylation disrupts PRPS1/2 hexamers and converts them into monomers.
AMPK-mediated PRPS1/2 phosphorylation inhibits PRPS1/2 activity, nucleotide synthesis, and NAD production
PRPS1 may exist as a hexamer to maintain PRPS1 activity (13). To determine the effect of AMPK-dependent PRPS1/2 phosphorylation on the regulation of PRPS1/2 activity, we performed an in vitro phosphorylation assay by mixing WT or non-phosphorylatable mutants of PRPS1/2, with or without active AMPK. The presence of AMPK largely inhibited the activity of WT PRPS1 and WT PRPS2 but not of their mutants (Fig. 4A). Consistent with those results, glucose deprivation largely inhibited the activity of ectopically expressed WT Flag-PRPS1 and Flag-PRPS2 (Fig. 4B) and endogenously expressed PRPS1/2 (Fig. 4C). Similar inhibition was also observed by treating U87 cells with A769662 AMPK activator (Supplementary Fig. S4A and S4B). In contrast, glucose deprivation or A769662 treatment did not affect the activity of ectopically expressed Flag-PRPS1 S180A or Flag-PRPS2 S183A (Fig. 4B; Supplementary Fig. S4A) or knock-in-expressed PRPS1 S180A and PRPS2 S183A (Fig. 4C; Supplementary Fig. S4B). These results indicate that glucose deprivation or activation of AMPK inhibits PRPS1/2 activity by the AMPK-mediated phosphorylation of PRPS1/2 and subsequent disruption of PRPS1/2 hexamers.
Figure 4. AMPK-mediated PRPS1/2 phosphorylation inhibits PRPS1/2 activity and nucleotide synthesis.
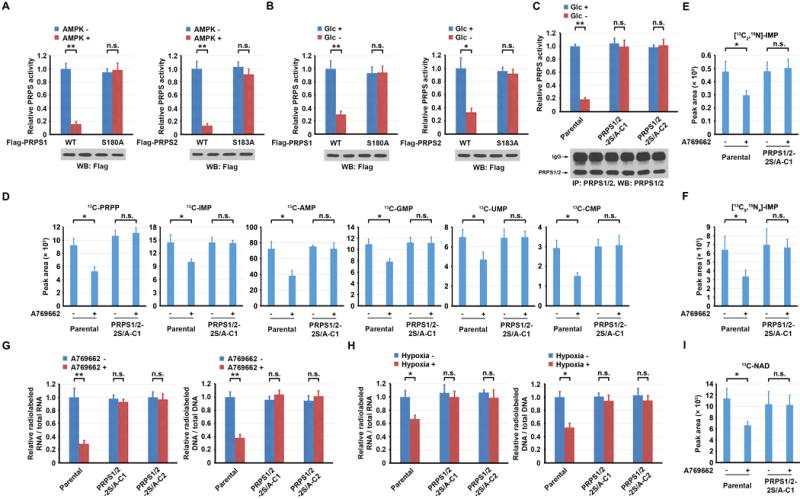
A–C, Immunoprecipitation and immunoblot analyses were performed using the indicated antibodies. A-C, G and H, Data represent the mean ± SD from three independent experiments. D–F and I, Data represent the mean ± SD from triplicate samples. *P < 0.05; **P < 0.001; n.s., no significance.
A, Purified WT Flag-PRPS1 or Flag-PRPS1 S180A (left panel) or WT Flag-PRPS2 or Flag-PRPS2 S183A (right panel) from U87 cells was incubated with or without active AMPK in the presence of ATP (0.5 mM). After the removal of AMPK by Ni-NTA resin, the activities of the remaining PRPS1/2 proteins were measured.
B, U87 cells expressing WT Flag-PRPS1, Flag-PRPS1 S180A (left panel), WT Flag-PRPS2, or Flag-PRPS2 S183A (right panel) were cultured in the presence or absence of Glc for 3 h. The activity of immunoprecipitated Flag-PRPS1/2 was measured.
C, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were cultured in the presence or absence of Glc for 3 h. The activity of immunoprecipitated PRPS1/2 was measured. PRPS1/2-2S/A-C1, clone 1; PRPS1/2-2S/A-C2, clone 2.
D, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were pretreated with or without A769662 (0.5 mM) for 3 h, followed by metabolically labeling with 13C6-glucose (10 mM) for 30 min. 13C-labeled metabolite intermediates of PRPP, IMP, AMP, GMP, UMP, and CMP were measured by LC-MS/MS.
E, F, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were pretreated with or without A769662 (0.5 mM) for 3 h, followed by metabolically labeling with [13C2,15N]-glycine (0.4 mM) (E) or [13C5,15N4]-hypoxanthine (0.1 mM) (F) for 2 h. 13C-labeled IMP was measured by LC-MS/MS.
G, H, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were pretreated with or without A769662 (0.5 mM) for 6 h (G) or hypoxia for 12 h (H), followed by labeling with D-[6-14C] glucose (1 μCi, 0.01 mM) for 30 min. The amounts of 14C-RNA (left panel) and 14C-DNA (right panel) were measured.
I, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were pretreated with or without A769662 (0.5 mM) for 3 h, followed by metabolic labeling with 13C6-glucose (10 mM) for 30 min. 13C-labeled NAD was measured by LC-MS/MS.
The allosteric site of PRPS1, bound phosphate for activation or ADP and AMP for inhibition, is located at the interfaces among three subunits of the hexamer (12, 13). We found that purified WT PRPS1 and PRPS1 S180A mutant were activated by phosphate or inhibited by ADP and AMP to a similar extent (Supplementary Fig. S4C), suggesting that mutation of this phosphorylation residue does not affect allosteric regulation of PRPS1. In contrast, the presence of AMPK, which phosphorylated and converted WT PRPS1 hexamers to monomers, made PRPS1 unresponsive to regulation by phosphate, ADP, or AMP (Supplementary Fig. S4C). These results suggest that rapid PRPS1 phosphorylation by AMPK disrupted the allosteric regulation of PRPS1 by converting PRPS1 hexamers to monomers and is an early response of tumor cells to energy stress.
PRPS1 S103T and A190T mutations, identified in ALL, reduce feedback inhibition by nucleotide biosynthesis products (17). S103 is in the allosteric site of PRPS1 hexamer, and A190 is in the dimer interface. It was previously shown that mutations of these two residues destabilize the dimer interface and allosteric sites for ADP or GDP (17). We treated U87 cells expressing Flag-PRPS1 S103T or Flag-PRPS1 A190T mutant with A769662 and found that AMPK activation induced S180 phosphorylation of both mutants (Supplementary Fig. S4D). Size-exclusion chromatography showed that the presence of AMPK converted both mutants from hexamers to monomers (Supplementary Fig. S4E). These data suggest that AMPK-mediated PRPS1 phosphorylation is able to inhibit PRPS1 S103T and PRPS1 A190T mutants identified in ALL.
To determine whether PRPS1/2 inhibition by activated AMPK decreases nucleotide synthesis, we used targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) to measure the incorporation of stable isotope-labeled glucose (13C6-glucose) into purine and pyrimidine intermediates. A769662 treatment of U87 cells significantly decreased the amount of 13C-labeled PRPP, and purine (IMP, AMP, and GMP) and pyrimidine (UMP and CMP) intermediates; that decrease was abrogated by knock-in expression of PRPS1 S180A and PRPS2 S183A (Fig. 4D). To determine whether AMPK-meditated PRPS1/2 inhibition decreases nucleotide synthesis through de novo or salvage pathways, we used [13C2,15N]-glycine and [13C5,15N4]-hypoxanthine to label U87 cells, respectively (Supplementary Fig. S4F). A769662 treatment reduced IMP incorporation of both [13C2,15N]-glycine (Fig. 4E) and [13C5,15N4]-hypoxanthine (Fig. 4F); this reduction was abolished by knock-in expression of PRPS1 S180A and PRPS2 S183A, suggesting that both de novo and salvage nucleotide synthesis pathways are regulated by AMPK-mediated PRPS1/2 inhibition.
A769662 treatment (Supplementary Fig. S4G) and the expression of active AMPKα11-312 mutant (25) but not AMPKα11-312 T172A kinase-inactive mutant (Supplementary Fig. S4H) in U87 or U251 cells inhibited the synthesis of RNA and DNA. A similar inhibitory effect was observed in A769662-treated A549/LKB1, BxPC-3, and MDA-MB-231 cells (Supplementary Fig. S4I). Those inhibitory effects induced by AMPK activation were abrogated by a deficiency of AMPKα1/2 (Supplementary Fig. S4J and S4K) and combined knock-in expression of PRPS1 S180A and PRPS2 S183A (Fig. 4G). In addition, knock-in of PRPS1 S180A and PRPS2 S183A blocked the hypoxia-induced inhibition of the synthesis of RNA and DNA (Fig. 4H). These results indicate that activation of AMPK, induced by glucose deprivation or hypoxic stimulation, inhibits nucleic acid synthesis by AMPK-mediated phosphorylation of PRPS1/2.
In addition to being used for nucleotide synthesis, PRPP is also an intermediate for NAD synthesis (26). Glucose deprivation reduced levels of NAD both in U87 and U251 cells (Supplementary Fig. S4L). In addition, A769662 treatment of U87 cells cultured with 13C6-glucose reduced the amount of 13C-labeled NAD, and this reduction was abrogated by knock-in expression of PRPS1 S180A and PRPS2 S183A (Fig. 4I). These results indicate that AMPK-mediated PRPS1/2 phosphorylation inhibits NAD biosynthesis.
AMPK-mediated PRPS1/2 phosphorylation copes with energy stress in tumorigenesis
Nucleotide synthesis is an energy-intensive process that consumes several sources of carbon and nitrogen and a large amount of ATP (27). A collapse of cellular ATP levels induces apoptosis and necrosis (27–30). To determine the effect of AMPK-inhibited PRPS1/2 on cellular levels of ATP, we cultured U87 cells with a low amount of glucose (0.5 mM) to maintain a low level of glycolysis, which resulted in partially inhibited RNA and DNA synthesis (Supplementary Fig. S5A), as evidenced by phosphorylation of PRPS1 S180 and PRPS2 S183 (Supplementary Fig. S5B). However, knock-in expression of PRPS1 S180A and PRPS2 S183A in U87 cells had no inhibitory effect on the nucleic acid synthesis (Supplementary Fig. S5A) and resulted in largely decreased cellular ATP levels (Fig. 5A). Similar results were obtained by treating U87 cells with hypoxia and revealed that knock-in of PRPS1 S180A and PRPS2 S183A decreased cellular ATP levels (Supplementary Fig. S5C).
Figure 5. AMPK-mediated PRPS1/2 phosphorylation maintains tumor cell homeostasis in energy stress.
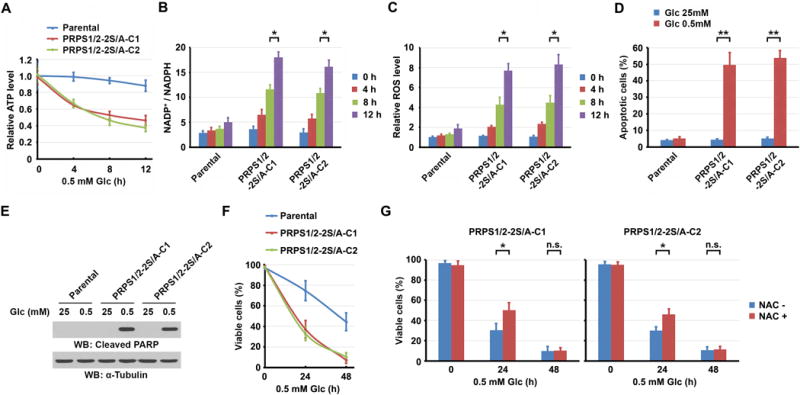
A–D and F, G, Data represent the mean ± SD from three independent experiments. *P < 0.05; **P < 0.001; n.s., no significance.
A, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were cultured in 0.5 mM Glc for the indicated periods of time. Intracellular ATP levels were measured.
B, C, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were cultured in 0.5 mM Glc for the indicated periods of time. Intracellular NADP+ and NADPH levels (B) or ROS levels (C) were measured.
D, E, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were cultured in 25 mM or 0.5 mM Glc for 8 h. An apoptotic assay was performed (D). Immunoblot analysis was performed with the indicated antibodies (E).
F, Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were cultured in 0.5 mM Glc for the indicated periods of time. Viable cells were measured.
G, U87 cells with knock-in of PRPS1/2-2S/A, pretreated with or without NAC (2 mM) for 30 min, were cultured in 0.5 mM Glc for the indicated periods of time. Viable cells were measured. Left panel, clone 1 (C1); right panel, clone 2 (C2).
NADPH eliminates reactive oxygen species (ROS). The cellular level of NADPH is the balance between NADPH synthesis, which occurs mainly through the PPP and mitochondrial metabolism, and NADPH consumption, which fuels fatty acid synthesis, redox reactions, and de novo DNA synthesis (31–33) (Supplementary Fig. S5D). Ribonucleotide reductase (RR), which is downstream from PRPS in the de novo DNA synthesis pathway, consumes a large amount of NADPH and catalyzes the conversion of ribonucleoside diphosphates to their corresponding deoxyribonucleotides (34, 35). Consistent with a previous report showing that AMPK maintains NADPH levels in the absence of glucose (31), we found that NADPH levels were not dramatically altered in U87 cells during the short period of limited glucose supply (Fig. 5B). However, significant decrease of NADPH levels (Fig. 5B) with large increase of ROS levels (Fig. 5C) were observed in U87 cells with knock-in of PRPS1 S180A and PRPS2 S183A. These results suggest that the unrestricted and activated PRPS-dependent nucleic acid synthesis mediated by PRPS1 S180A and PRPS2 S183A leads to consumption of a substantial amount of ATP and NADPH, leading to increased ROS levels under these metabolic stresses. As expected, knock-in of PRPS1 S180A and PRPS2 S183A enhanced apoptosis (Fig. 5D) and caspase 3-mediated poly (ADP-ribose) polymerase (PARP) cleavage (Fig. 5E).
To determine the effect of AMPK-mediated PRPS1/2 activity inhibition in cell survival, we cultured cells with a low amount of glucose (0.5 mM). Knock-in of PRPS1 S180A and PRPS2 S183A increased sensitivity of cells to glucose restriction and reduced the survival of these cells (Fig. 5F). Treating with N-acetyl-L-cysteine (NAC), a scavenger of free radicals, increased survival of U87 cells with knock-in of PRPS1 S180A and PRPS2 S183A in a short exposure time of low glucose (24 h), but failed to do so in a long exposure time (48 h) (Fig. 5G). Consistent with the finding that AMPK-mediated PRPS1 phosphorylation inhibits PRPS1 S103T and PRPS1 A190T activity, the cell viability of Hep3B or Huh7 hepatocellular carcinoma cells with depletion of primarily expressed endogenous PRPS1 (16) and reconstituted expression of WT Flag-rPRPS1, Flag-rPRPS1 S103T, or Flag-rPRPS1 A190T was comparable to each other in response to low glucose (0.5 mM) stimulation (Supplementary Fig. S5E and S5F). These results suggest that PRPS1 S103T or A190T mutation does not alter the sensitivity of cells to energy stress.
We next compared the viability of normal and tumor cells by culturing normal human astrocytes (NHA), U87 cells, U251 cells (Supplementary Fig. S5G), human pancreatic duct epithelial (HPDE) cells, and BxPC-3 pancreatic cancer cells (Supplementary Fig. S5H) in medium with a low glucose concentration. We found that tumor cells had more cell death than did normal cells. Combined treatment of compound C with a low glucose concentration caused more death of tumor cells than of normal cells. These results suggest that normal cells, which usually have limited and controlled proliferation, do not need high PRPS activity to produce nucleotides and that AMPK-dependent inhibition of PRPS activity is a primarily regulatory mechanism for tumor cells under energy stress that has been induced by their rapid proliferation.
To determine the role of PRPS1/2 regulation by AMPK in tumorigenesis, we intracranially injected athymic nude mice with U87 cells, with or without knock-in of PRPS1 S180A and PRPS2 S183A mutants. The expression of the PRPS1/2 mutants inhibited tumor growth (Fig. 6A and 6B) with abrogated PRPS1 S180 and PRPS2 S183 phosphorylation (Fig. 6C), as detected using a specificity-validated antibody (Supplementary Fig. S5I) and increased tumor cell apoptosis (Fig. 6D), supporting the critical role of AMPK-meditated phosphorylation and inhibition of PRPS1/2 in counteracting nutritional stress to maintain tumor growth. To further support this finding, we inhibited glucose use by tumor cells via the intraperitoneal injection of 2-DG. As expected, treatment with 2-DG decreased the growth of tumors expressing WT PRPS1/2 (Fig. 6A and 6B) and increased PRPS1 S180 and PRPS2 S183 phosphorylation (Fig. 6C). Of note, we observed that 2-DG treatment induced greater inhibition of the growth of tumors and apoptosis with knock-in of PRPS1 S180A and PRPS2 S183A mutants (Fig. 6A, 6B and 6D). These results indicate that inhibition of glycolysis results in PRPS1/2 phosphorylation and inhibition by AMPK, leading to recalibration of cellular homeostasis during tumor growth.
Figure 6. AMPK-mediated PRPS1/2 phosphorylation copes with energy stress in tumorigenesis.
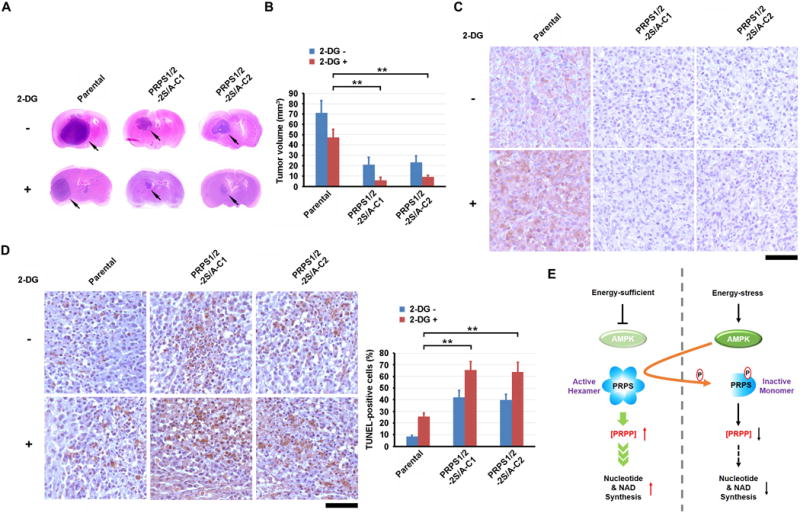
A–D, 1 × 106 of Parental U87 cells or U87 cells with knock-in of PRPS1/2-2S/A were intracranially injected into athymic nude mice (n = 7 per group). Two weeks after cell injection, 0.2 ml of 2-DG (500 mg/kg) was intraperitoneally administrated daily for 14 days. Mice were sacrificed and examined for tumor growth 28 days after injection. A, Representative H&E-stained coronal brain sections are shown. Arrows point to tumor. B, Tumor volumes were calculated. Data represent the mean ± SD of seven mice. **P < 0.001. C, Immunohistochemical analyses of xenograft brain tumor tissues were performed with an anti-PRPS1/2 pS180/S183 antibody. Bar, 100 μm. D, TUNEL analyses of the indicated tumor tissues were performed. Apoptotic cells were stained brown. Bar, 100 μm (left panel). Apoptotic cells were quantified, and the data represent the mean ± SD from 10 microscope fields (right panel). **P < 0.001.
E, Schematic model of AMPK-mediated nucleic acid and NAD synthesis under energy stress.
DISCUSSION
Tumor cells reprogram their metabolism to counteract energy stress (1). In this study, we demonstrated that glucose deprivation and hypoxia resulted in AMPK-mediated phosphorylation of PRPS1 S180 and PRPS2 S183. That phosphorylation converted PRPS1/2 hexamers to monomers and inhibited PRPS1/2 activity and subsequent synthesis of nucleotide and NAD (Fig. 6E).
Expression of non-phosphorylatable mutants, PRPS1 S180A and PRPS2 S183A, whose activity was unaffected under low glucose conditions or hypoxia conditions, greatly decreased cellular ATP levels and cell survival. Those results strongly suggest that tumor cells cope with energy stress by decreasing nucleotide synthesis, cell proliferation, and unnecessary consumption of ATP for maintaining cell homeostasis. Animal studies showed that the expression of those non-phosphorylatable mutants inhibited brain tumorigenesis. Importantly, 2-deoxy-D-glucose treatment, which inhibits tumor cell glycolysis, inhibited tumor growth, and that inhibition was enhanced by the expression of PRPS1 S180A and PRPS2 S183A. These findings highlight the significant role of reduced PRPS1/2 activity in counteracting energy stress.
The identified phosphorylation sequence of PRPS1 and PRPS2 is sub-optimal to the AMPK recognition motif. However, our in vitro and in vivo results supported the finding that AMPK directly phosphorylates PRPS1 S180 and PRPS2 S183. This finding, together with the reports that AMPK substrates such as histone H2B and eukaryotic elongation factor 2 kinase (eEF2K) containing a non-classic AMPK recognition motif (36–38), indicate that AMPK tolerates some flexibility in its recognition motif for phosphorylation.
AMPK is a central metabolic switch that is found in all eukaryotes; it governs glucose and lipid metabolism in response to alterations in nutrients and intracellular energy levels (39, 40). As a crucial cellular energy sensor, AMPK regulates energy balance at the cellular level by activating catabolic pathways, including glucose uptake, glycolysis, fatty acid uptake and oxidation, and autophagy, and by inhibiting anabolic pathways, including the transcription of lipogenic and gluconeogenic enzymes and syntheses of fatty acid, triglyceride, cholesterol, glycogen, proteins, and ribosomal RNA (39, 40). Our findings elucidate yet another function of AMPK—direct regulation of nucleic acid synthesis in response to metabolic stress. Ultimately, therapeutic targeting of AMPK in the context of metabolic stress on tumor cells may be an effective strategy for disrupting PRPS1/2-mediated tumor growth.
METHODS
Materials
Primary rabbit polyclonal antibody against PRPS1/2 pS180/S183 was custom produced by Signalway Biotechnology (Pearland, TX). Mouse monoclonal antibodies against PRPS1/2 (sc-376440), GST (sc-138), α-tubulin (sc-5286), normal rabbit IgG (sc-2027), and normal mouse IgG (sc-2025) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal antibodies against AMPKα (#5831), AMPKα pT172 (#2535), ACC1 (#3676), and ACC1 pS79 (#11818) were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal anti-Flag (F1804), 2-DG (D8375), gel filtration calibration markers kit (MWGF1000), ribose-5-phosphate, ribulose-5-phosphate, NADH, phosphoenolpyruvate, pyruvate kinase, lactic dehydrogenase, myokinase, thiamine pyrophosphate, α-glycerophosphate dehydrogenase, triosephosphate isomerase, transketolase, nicotinamide, thiazolyl blue, phenazine ethosulfate, glucose-6-phosphate dehydrogenase, glucose 6-phosphate, AMP, ADP, ATP, and N-acetyl-L-cysteine (NAC) were purchased from Sigma (St. Louis, MO). Anti-DYKDDDDK (Flag) tag affinity gel was purchased from BioLegend (San Diego, CA). [γ-32P]ATP was purchased from MP Biochemicals (Santa Ana, CA). D-[6-14C]-glucose was purchased from American Radiolabeled Chemicals (St. Louis, MO). D-[U-13C6]-glucose, [13C2,15N]-glycine, and [13C5,15N4]-hypoxanthine were purchased from Cambridge Isotope Laboratories (Tewksbury, MA). D-ribulose-5-phosphate 3-epimerase was obtained from Abcam (Cambridge, MA). Active AMPK proteins (#P47-10H) were obtained from SignalChem (Richmond, BC, Canada). A769662 was purchased from Tocris Bioscience (Avonmouth, Bristol, United Kingdom). Compound C was obtained from Millipore (Billerica, MA).
Cell culture and transfection
U87, A549, BxPC-3, MDA-MB-231, Hep3B, Huh7 and 293T cells were purchased from ATCC between 2003 and 2009. U251 cell was purchased from Sigma in 2010. Normal human astrocytes (NHA) cell was from Lonza in 2015. Cells were maintained in DMEM medium supplemented with 10% bovine calf serum (Hyclone, Logan, UT). AMPKα1 and AMPKα2 double-knockout mouse embryonic fibroblasts (obtained from B. Viollet, Institut Cochin, Paris, France, in June of 2015) were maintained in DMEM medium supplemented with 10% fetal bovine serum. Human pancreatic duct epithelial (HPDE) cells (obtained from Ming-Sound Tsao, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada, in August of 2017) were maintained in Keratinocyte-SFM medium (Thermo Fisher, #17005042). U87 and U251 cell lines were authenticated using short tandem repeat profiling at The University of Texas MD Anderson Cancer Center (Houston, Texas) in November of 2010. NHA, A549, BxPC-3, MDA-MB-231, Hep3B and Huh7 cell lines were authenticated in September of 2017. Mycoplasma was not detected in these cells.
Glucose deprivation was carried out by culturing cells in DMEM medium without glucose (Gibco, #11966), supplemented with dialyzed bovine calf serum or fetal bovine serum. Cells were plated at a density of 4 × 105 per 60-mm dish or 1 × 105 per well in a six-well plate 18 h before transfection. The transfection procedure was performed as previously described (41).
DNA construction and mutagenesis
Polymerase chain reaction (PCR)-amplified human PRPS1 and PRPS2 were cloned into pcDNA3.1/hygro(+)-Flag or pColdI (His) vector, respectively. Human AMPKα1 was cloned into pcDNA3-HA vector. GST-AMPKα11-312 was obtained from Addgene (#27632). Flag-PRPS1 S180A, S103T and A190T, Flag-PRPS2 S183A, His-PRPS1 S180A, His-PRPS2 S183A, and GST-AMPKα11-312 T172A were constructed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). shRNA targeting PRPS1 (5′-GATAATATGATCTGCACCT-3′) was constructed into the pGIPZ vector.
Purification of recombinant proteins
WT His-PRPS1, His-PRPS1 S180A, WT His-PRPS2, and His-PRPS2 S183A were expressed in bacteria and purified as described previously (41).
Immunoprecipitation and immunoblot analysis
The extraction of proteins using a modified buffer (50 mM Tris-HCl [pH 7.5], 0.01% SDS, 1% Triton X-100, 150 mM NaCl, 1 mM dithiothreitol, 0.5 mM EDTA, 100 μM PMSF, 100 μM leupeptin, 1 μM aprotinin, 100 μM sodium orthovanadate, 100 μM sodium pyrophosphate, and 1 mM sodium fluoride) from cultured cells was followed by immunoprecipitation and immunoblot analysis using corresponding antibodies, as described previously (42).
Mass spectrometry analysis
Immunoprecipitated Flag-PRPS1 protein from glucose-deprived U87 cells was digested and analyzed by LC-MS/MS on an Orbitrap-Ellite mass spectrometer (Thermo Fisher Scientific, Waltham, MA), as described previously (16).
Analysis of intermediate metabolites by LC-MS/MS
To determine the incorporation of glucose carbon (13C6-glucose) into intracellular nucleotide metabolites, extracts were prepared and analyzed by high-resolution mass spectrometry (HRMS). Approximately 80% confluent cells were seeded in 10 cm dishes in triplicate. For glucose labeling, cells were washed with glucose-free DMEM medium and incubated in fresh medium containing 13C6-glucose (10 mM) for 30 min. For glycine or hypoxanthine labeling, cells were incubated with DMEM medium supplemented with [13C2,15N]-glycine (0.4 mM) or [13C5,15N4]-hypoxanthine (0.1 mM) for 2 h. Nucleotide metabolites and NAD were extracted using 90/9/1 (v/v/v) acetonitrile/water/formic acid. Samples were centrifuged at 17,000 g for 5 min at 4°C, and supernatants were transferred to clean tubes, followed by evaporation to dryness using nitrogen. Samples were reconstituted in 0.2% ammonium hydroxide in ammonium acetate (10 mM), then 10 μl was injected into a Thermo Scientific Vanquish liquid chromatography (LC) system containing a Thermo Hypercarb 100×3 mm 3 μm HPLC column heated to 35°C with mobile phase A (MPA) consisting of 0.2% ammonium hydroxide in ammonium acetate (10 mM) and mobile phase B (MPB) consisting of 0.2% ammonium hydroxide in acetonitrile. Using a flowrate of 0.3 ml/min, the gradient elution program was: 0 min (0%MPB) – 2.0 min (0% MPB) – 15.0 min (30% MPB) – 15.1 min (95% MPB) – 20.0 min (95% MPB) – 20.1 min (0% MPB) – 25.0 min (STOP). Data were acquired using a Thermo Orbitrap Fusion Tribrid Mass Spectrometer via Selected Ion Mode (SIM) electrospray positive mode. For measurement of PRPP, metabolites were extracted by 75/25 (v/v) ethanol/10 mM HEPES buffer, and followed by similar protocol for measuring nucleotides. Data acquisition was performed via SIM electrospray negative mode. Peak integration and area calculation were performed by using Thermo TraceFinder software.
To determine the abundance of intracellular PRPP, nucleotide intermediates, and NAD, approximately 80% confluent cells were seeded in 10 cm dishes in triplicate. Extracts were prepared and analyzed by high-resolution mass spectrometry as described above.
In vitro kinase assay
The kinase reactions were performed as described previously (43). In brief, purified active AMPK proteins (including AMPKα1/β1/γ1) were incubated with purified WT His-PRPS1, His-PRPS1 S180A, WT His-PRPS2, or His-PRPS2 S183A (100 ng) in 25 μl of kinase buffer (50 mM Tris-HCl [pH 7.5], 100 mM KCl, 50 mM MgCl2, 1 mM Na3VO4, 1 mM DTT, 5% glycerol, 0.2 mM AMP, 0.5 mM ATP, and 10 μCi [γ-32P]ATP) at 25°C for 1 h. The reaction was terminated by adding SDS-PAGE loading buffer and heating at 100°C for 5 min. The reaction mixture was then subjected to an SDS-PAGE analysis.
For stoichiometry measurement of PRPS1 or PRPS2 phosphorylation by AMPK, a modified method was performed, as described previously (44). Active AMPK protein (25 ng) was incubated with purified His-PRPS1 or His-PRPS2 (200 ng) in 25 μl of kinase buffer (50 mM Tris-HCl [pH 7.5], 100 mM KCl, 50 mM MgCl2, 1 mM Na3VO4, 1 mM DTT, 5% glycerol, 0.2 mM AMP). The reaction was started by adding [γ-32P]ATP (0.1 mM, 1000 cpm/pmol) at 25°C. At various time points, samples were taken for SDS-PAGE analysis. Protein bands corresponding to PRPS1 or PRPS2 were cut directly from the gel and dissolved in vials containing 500 μl of 3% (w/w) H2O2 by heating for 2 h at 80°C. 32P incorporation was quantified by Cerenkov counting in a Beckman LS6500 scintillation counter and converted to moles of Pi incorporated per mole of PRPS1 or PRPS2.
CRISPR/Cas9-mediated genome editing
Genomic mutations were introduced into cells using the CRISPR/Cas9 system, as described previously (45). Single guided RNAs (sgRNAs) were designed to target the genomic area adjacent to the PRPS1/2 mutation sites using the CRISPR design tool (http://crispr.mit.edu/). The annealed guide RNA oligonucleotides were inserted into PX458 vector (Addgene, Cambridge, MA) digested with the BbsI restriction enzyme (46). Cells were seeded at 60% confluence, followed by co-transfection of sgRNAs (0.5 μg) and single-stranded donor oligonucleotide (10 pmol) as a template to introduce mutations. Twenty-four hours after transfection, cells were trypsinized and diluted for single cells and seeded into 96-well plates. Genomic DNA was extracted from GFP-positive cells, followed by sequencing of the PCR products spanning the mutation sites. The oligonucleotide and primers used for sgRNA cloning and genomic DNA sequencing are listed in Supplementary Table S1.
Size-exclusion chromatography
Size-exclusion chromatography was performed using a HiPrep 16/60 Sephacryl S-300 HR column (GE), as described previously (47). Cell lysates (1 ml of 20 mg) or purified proteins (20 μg) were loaded onto the column and fractionated at a flow rate of 0.5 ml/min. After passing through the void volume, the sample fractions were collected (1 ml for each) and analyzed by immunoblotting assay. The standard curve of the elution was plotted against LogMW by using size-exclusion chromatography calibration marker kit (Sigma) according to the vendor’s instructions.
Influx of glucose into nucleic acids
An assay of the influx of glucose to nucleic acids was performed as described previously, with modifications (16). In brief, cells were seeded in a six-well plate and incubated in 2 ml of fresh medium spiked with 1 μCi of D-[6-14C] glucose (0.01 mM) for 0.5 h, followed by washing with PBS. RNA or DNA was extracted, and 14C-RNA or 14C-DNA was assayed using a scintillation counter.
RT-PCR analysis of PRPS1/2 expression
The expression pattern of PRPS in cells was identified as described previously (16). In brief, total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. A cDNA library was generated by reverse-transcribing 1 μg of RNA using an iScript cDNA synthesis kit (Bio-Rad) and amplified with a pair of primers (listed in Supplementary Table S1) that recognized both PRPS1 and PRPS2 cDNA. For distinguishing the expression of PRPS1/2, 1 μg of PCR products was digested by HindIII, and the products of digestion were resolved on a 1.5% agarose gel.
Measurement of PRPS1/2 activity
PRPS1/2 activity was determined as described before (16). In brief, purified recombinant or immunoprecipitated WT or mutant PRPS1 or PRPS2 (10 ng) was incubated in 100 μl of reaction buffer (50 mM Tris-HCl [pH 7.5], 4.7 mM R5P, 0.4 mM NADH, 3.2 mM ATP, 1.8 mM phosphoenolpyruvate, 6 mM MgCl2, 31 mM NaHCO3, 7 U pyruvate kinase, 10 U lactic dehydrogenase, and 10 U myokinase) at 25°C in a 96-well plate. The absorbance was read at 340 nm in kinetic mode for 5 min using a multi-detection microplate reader (BMG Labtech).
Measurement of intracellular ATP levels
Cells (1 × 106) were washed and lysed in ATP assay buffer. Samples were deproteinized using 10 kDa spin columns (EMD Millipore). ATP levels were then determined using a BioVision ATP assay kit.
Measurement of intracellular R5P levels
The intracellular concentration of R5P was measured using previously described methods, with some modifications (48). In brief, 1 × 106 cells were washed, collected, and homogenized in 1 ml of hypotonic lysis buffer. The homogenates were centrifuged at 4°C for 10 min at maximum speed, and the supernatants were deproteinized using 10 kDa spin columns (EMD Millipore). The flow-through was mixed with 2× reaction buffer (100 mM Tris-HCl [pH 8.0], 3.4 mM ribulose-5-P, 0.004% w/v thiamine pyrophosphate, 30 mM MgCl2, 0.4 mM NADH, 1 U α-glycerophosphate dehydrogenase, 10 U triosephosphate isomerase, and 1 U transketolase). The reaction was initiated by adding D-ribulose-5-phosphate 3-epimerase (0.5 U) and incubated at 25°C for 30 min. The absorbance was read at 340 nm using a multi-detection microplate reader.
Measurement of intracellular NADPH and NADP+ levels
The intracellular levels of NADPH and total NADP (NADPH + NADP+) were measured according to previously described methods (31, 49). In brief, 2 × 106 cells were lysed in 400 μl of extraction buffer containing 20 mM nicotinamide, 20 mM NaHCO3, and 100 mM Na2CO3 and centrifuged for 10 min at maximum speed. The supernatant was kept on ice in the dark until use. For extracting NADPH, 150 μl of the supernatant was incubated at 60°C for 30 min because heating destroys the oxidized form (NADP+) while having no effect on the reduced form (NADPH). Next, 160 μl of NADP-cycling buffer (100 mM Tris-HCl [pH 8.0], 0.5 mM thiazolyl blue [MTT], 2 mM phenazine ethosulfate, and 5 mM EDTA) containing glucose-6-phosphate dehydrogenase (1.3 U) was added to a 96-well plate containing 20 μl of sample. After a 1 min incubation in the dark at 30°C, 20 μl of glucose 6-phosphate (10 mM) was added to the mixture, and the change in absorbance at 570 nm was measured every 30 s for 4 min at 30°C with a microplate reader. The concentration of NADP+ was calculated by subtracting NADPH from the total NADP.
Measurement of intracellular ROS
The fluorescence probe DCFDA (Abcam) was used to measure intracellular ROS following the manufacturer’s instructions. In brief, 2 × 104 cells were seeded in a clear-bottom 96-well plate. After treatment, cells were washed with HBSS, followed by incubation with DCFDA (25 μM)-containing HBSS for 30 min at 37°C in the dark. Cells were washed and green fluorescence was measured with a Synergy HT microplate reader (BioTek) at 485/535 nm.
Apoptosis analysis
Cells were stained with the Annexin V-Alexa Fluor 488 apoptosis detection kit (ThermoFisher), followed by cytometry (BD Biosciences).
TUNEL assay
Mouse tumor tissues were sectioned at 5 μm thickness. Apoptotic cells were counted using the DeadEnd Colorimetric TUNEL System (Promega) according to the manufacturer’s instruction.
Structural view of PRPS1
The structural view of PRPS1 hexamer (PDB: 2H06) was demonstrated by Cn3D macromolecular structure viewer (https://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml) (50), while the view of PRPS1 dimer was displayed by USFC Chimera (https://www.cgl.ucsf.edu/chimera/) (51).
Cell viability analysis
2 × 105 cells were plated in DMEM with 10% FBS. The viable cells were stained with trypan blue (0.5%) and counted using Beckman Coulter (Brea, CA).
Intracranial injection
Cells (1 × 106) suspended in 5 μl of Dulbecco’s modified Eagle’s medium were intracranially injected into 4-week-old female athymic nude mice, as described previously (52). Seven mice were included in each group. 2-DG treatment was initiated at the fourteenth day after the intracranial injection of tumor cells. 2-DG (0.2 ml, 500 mg/kg) was intraperitoneally administered daily for 14 days. Mice were sacrificed 28 days after injection. Their brains were harvested, fixed in formaldehyde (4%), and embedded in paraffin. Tumor formation was determined by H&E staining. Tumor volume was calculated by 0.5 × L × W2 (L, length; W, width). The use of animals was approved by the Institutional Review Board of MD Anderson Cancer Center.
Statistical analysis
The statistical analysis was conducted using the two-tailed unpaired Student’s t-test. All data represent the mean ± SD of three independent experiments/samples unless specifically indicated.
Supplementary Material
Significance.
Our findings elucidate an instrumental function of AMPK in direct regulation of nucleic acid and NAD synthesis in tumor cells in response to energy stress. AMPK phosphorylates PRPS1/2, converts PRPS1/2 hexamers to monomers, and inhibits PRPS1/2 activity and subsequent nucleotide and NAD synthesis to maintain tumor cell growth and survival.
Acknowledgments
We thank Dr. Li Li (Clinical and Translational Proteomics Service Center, Institute of Molecular Medicine, The University of Texas Health Center at Houston) for technical assistance and Ann Sutton (The University of Texas MD Anderson Cancer Center) for her critical reading of this manuscript.
Financial support
This work was supported by National Institute of Neurological Disorders and Stroke grant R01 NS089754 (Z.L.); National Cancer Institute grants 2R01 CA109035 (Z.L.), and R01 CA169603 (Z.L.); 2P50 CA127001 (Brain Cancer SPORE), MD Anderson’s Cancer Center Support Grant CA016672; and the National Natural Science Foundation of China (NSFC) 816228011 (H.W.), 81572499, and 81572700. Z.L. is a Ruby E. Rutherford Distinguished Professor.
Footnotes
Disclosure of Potential Conflicts of Interest
Zhimin Lu has owned shares in Signalway Biotechnology (Pearland, TX), which supplied rabbit antibodies recognizing PRPS1/2 pS180/S183. Dr. Lu’s interest in this company had no bearing on choosing it to supply these reagents.
References
- 1.Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell. 2014;159:1492–4. doi: 10.1016/j.cell.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature reviews Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyssiotis CA, Cantley LC. Metabolic syndrome: F stands for fructose and fat. Nature. 2013;502:181–2. doi: 10.1038/502181a. [DOI] [PubMed] [Google Scholar]
- 5.Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4320–5. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–8. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Sahra I, Hoxhaj G, Ricoult SJ, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–33. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–3. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 9.Kornberg A, Lieberman I, Simms ES. Enzymatic synthesis and properties of 5-phosphoribosylpyrophosphate. The Journal of biological chemistry. 1955;215:389–402. [PubMed] [Google Scholar]
- 10.Khorana HG, Fernandes JF, Kornberg A. Pyrophosphorylation of ribose 5-phosphate in the enzymatic synthesis of 5-phosphorylribose 1-pyrophosphate. The Journal of biological chemistry. 1958;230:941–8. [PubMed] [Google Scholar]
- 11.Hove-Jensen B. Mutation in the phosphoribosylpyrophosphate synthetase gene (prs) that results in simultaneous requirements for purine and pyrimidine nucleosides, nicotinamide nucleotide, histidine, and tryptophan in Escherichia coli. Journal of bacteriology. 1988;170:1148–52. doi: 10.1128/jb.170.3.1148-1152.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nosal JM, Switzer RL, Becker MA. Overexpression, purification, and characterization of recombinant human 5-phosphoribosyl-1-pyrophosphate synthetase isozymes I and II. The Journal of biological chemistry. 1993;268:10168–75. [PubMed] [Google Scholar]
- 13.Li S, Lu Y, Peng B, Ding J. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. The Biochemical journal. 2007;401:39–47. doi: 10.1042/BJ20061066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen P, Liu Z, Wang X, Peng J, Sun Q, Li J, et al. Crystal and EM structures of human phosphoribosyl pyrophosphate synthase I (PRS1) provide novel insights into the disease-associated mutations. PloS one. 2015;10:e0120304. doi: 10.1371/journal.pone.0120304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Qian X, Lu Z. Fructokinase A acts as a protein kinase to promote nucleotide synthesis. Cell Cycle. 2016;15:2689–90. doi: 10.1080/15384101.2016.1204861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Qian X, Peng LX, Jiang Y, Hawke DH, Zheng Y, et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nature cell biology. 2016;18:561–71. doi: 10.1038/ncb3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Li H, Bai Y, Kirschner-Schwabe R, Yang JJ, Chen Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21:563–71. doi: 10.1038/nm.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–103. doi: 10.1016/j.cell.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, et al. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle. 2008;7:2392–400. doi: 10.4161/cc.6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMahon SB. Control of nucleotide biosynthesis by the MYC oncoprotein. Cell Cycle. 2008;7:2275–6. [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitination. Annual review of biochemistry. 2009;78:435–75. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardie DG, Schaffer BE, Brunet A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends in cell biology. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingram-Smith C, Woods BI, Smith KS. Characterization of the acyl substrate binding pocket of acetyl-CoA synthetase. Biochemistry. 2006;45:11482–90. doi: 10.1021/bi061023e. [DOI] [PubMed] [Google Scholar]
- 25.Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. The Journal of biological chemistry. 1998;273:35347–54. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- 26.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell metabolism. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–85. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–12. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 29.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer research. 1997;57:1835–40. [PubMed] [Google Scholar]
- 30.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. The Journal of experimental medicine. 1997;185:1481–6. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nature reviews Cancer. 2016;16:650–62. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- 33.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–9. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 35.Jordan A, Reichard P. Ribonucleotide reductases. Annual review of biochemistry. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 36.Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–5. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. The Journal of biological chemistry. 2004;279:12220–31. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 38.Johanns M, Pyr Dit Ruys S, Houddane A, Vertommen D, Herinckx G, Hue L, et al. Direct and indirect activation of eukaryotic elongation factor 2 kinase by AMP-activated protein kinase. Cellular signalling. 2017;36:212–21. doi: 10.1016/j.cellsig.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews Molecular cell biology. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nature reviews Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia Y, Wang J, Liu TJ, Yung WK, Hunter T, Lu Z. c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Molecular cell. 2007;25:219–32. doi: 10.1016/j.molcel.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Activation of protein kinase C triggers its ubiquitination and degradation. Molecular and cellular biology. 1998;18:839–45. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. The Journal of biological chemistry. 2007;282:11221–9. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horman S, Morel N, Vertommen D, Hussain N, Neumann D, Beauloye C, et al. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. The Journal of biological chemistry. 2008;283:18505–12. doi: 10.1074/jbc.M802053200. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Molecular cell. 2016;61:705–19. doi: 10.1016/j.molcel.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang W, Lu Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle. 2013;12:3154–8. doi: 10.4161/cc.26182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin R, Elf S, Shan C, Kang HB, Ji Q, Zhou L, et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nature cell biology. 2015;17:1484–96. doi: 10.1038/ncb3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner TC, Scott MD. Single extraction method for the spectrophotometric quantification of oxidized and reduced pyridine nucleotides in erythrocytes. Analytical biochemistry. 1994;222:417–26. doi: 10.1006/abio.1994.1511. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Geer LY, Chappey C, Kans JA, Bryant SH. Cn3D: sequence and structure views for Entrez. Trends in biochemical sciences. 2000;25:300–2. doi: 10.1016/s0968-0004(00)01561-9. [DOI] [PubMed] [Google Scholar]
- 51.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–22. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.