

Summary
Bacterial signal transduction systems commonly use receiver (REC) domains, which regulate adaptive responses to the environment as a function of their phosphorylation state. REC domains control cell physiology through diverse mechanisms, many of which remain understudied. We have defined structural features that underlie activation of the multi-domain REC protein, PhyR, which functions as an anti-anti-σ factor and regulates transcription of genes required for stress adaptation and host-microbe interactions in Alphaproteobacteria. Though REC phosphorylation is necessary for PhyR function in vivo, we did not detect expected changes in inter-domain interactions upon phosphorylation by solution X-ray scattering. We sought to understand this result by defining additional molecular requirements for PhyR activation. We uncovered specific interactions between unphosphorylated PhyR and an intrinsically disordered region (IDR) of the anti-σ factor, NepR, by solution NMR spectroscopy. Our data support a model whereby nascent NepR(IDR)-PhyR interactions and REC phosphorylation coordinately impart the free energy to shift PhyR to an open, active conformation that binds and inhibits NepR. This mechanism ensures PhyR is activated only when NepR and an activating phosphoryl signal are present. Our study provides new structural understanding of the molecular regulatory logic underlying a conserved environmental response system.
Keywords: stress response, Caulobacter, Brucella, signal transduction, intrinsically disordered protein, Alphaproteobacteria
Graphical Abstract
Receiver (REC) proteins are a diverse class of regulators that control physiological responses in microbes and plants as a function of their phosphorylation state. The conserved REC protein, PhyR, is an anti-anti-σ factor that regulates stress-dependent transcription in Alphaproteobacteria. We have applied a combination of experimental and computational approaches to uncover structural determinants of PhyR activation.
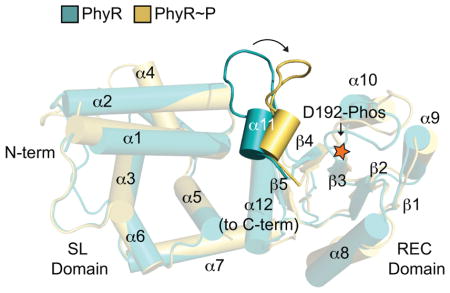
Introduction
Bacteria use numerous molecular response mechanisms to adapt to shifts in the physical or chemical state of the environment. In Alphaproteobacteria, environmental adaptation is largely controlled by the general stress response (GSR) system. The GSR system activates transcription of dozens of genes and mediates stress resistance and survival in species that inhabit niches ranging from the interior of mammalian cells (Kim et al., 2014, Sycz et al., 2015), to plant leaves (Kaczmarczyk et al., 2011) and roots (Flechard et al., 2010, Gourion et al., 2009), to freshwater and oceans (Alvarez-Martinez et al., 2007, Corrêa et al., 2013, Herrou et al., 2010). The environmental cues and regulatory responses regulated by this system vary across species, but the core proteins that control GSR-dependent transcription are conserved (Fiebig et al., 2015, Francez-Charlot et al., 2015).
The GSR combines two classic gene regulatory paradigms: two-component signaling and alternative σ factor regulation. At the center of this pathway is the PhyR protein, comprising a C-terminal receiver (REC) domain and an N-terminal σ-like (SL) domain (Francez-Charlot et al., 2009, Herrou et al., 2010). In the current structural model, aspartyl phosphorylation in the PhyR REC domain by a sensor kinase (Herrou et al., 2017, Kaczmarczyk et al., 2015, Kim et al., 2014, Lourenco et al., 2011), induces a conformational change that enables binding of the anti-σEcfG factor NepR to the PhyR SL domain. This process releases the extracytoplasmic function (ECF) σ-factor, σEcfG, to bind RNA polymerase and control gene expression (Figure 1A) (Campagne et al., 2012, Francez-Charlot et al., 2009). There is evidence from multiple systems that REC phosphorylation is allosterically regulated by interaction with protein or nucleic acid substrates, including PhyR by its binding partner NepR (Ames et al., 1999, Barbieri et al., 2010, Herrou et al., 2015, Kaczmarczyk et al., 2014, Schuster et al., 2001). The PhyR protein and the GSR system of Alphaproteobacteria thus provide an excellent model to investigate the structural basis by which substrate interaction and phosphorylation modulate REC protein conformation and function.
Figure 1. PhyR α11-β5 loop rearrangement decreases the energetic barrier to opening.

(A) A model of general stress response (GSR) activation. Upstream sensory kinases phosphorylate PhyR, leading to PhyR opening and NepR binding. This releases σEcfG to direct GSR transcription. (B) Alignment of PhyR (green) and PhyR~P (gold) MD trajectories. Rearrangement can occur on short MD timescales in the α11-β5 loop, which retracts toward the REC domain (shown by arrow). Structures shown are the average backbone positions over the final 10 ns of simulation. (C) NepR (pink) modelled into the PhyR-SL domain (green) binding site using a hypothetical open state model. Hydrophobic (red) and hydrophilic (gray) regions at the domain interface (brown) are highlighted. (D) Calculated solvent accessible surface area (SASA) values for PhyR (green) and PhyR~P (gold) at 50 ns for NepR-binding residues. (E) Umbrella sampling of PhyR and PhyR~P at increasing inter-domain distances. Arrows indicate points corresponding to the initial equilibrium state of the protein, collected from previous simulations, as well as the point in the PhyR~P (50 ns) sampling where solvation of the total interdomain interface began. The energetic barrier to open PhyR is lowered after phosphorylation (see 50 ns simulation). Energy to open PhyR~P prior to solvation is equivalent to unphosphorylated PhyR. Error bars represent standard deviation.
Recent nuclear magnetic resonance (NMR) spectroscopy studies of full-length Erythrobacter litoralis PhyR and its isolated REC domain demonstrate that (1) aspartyl modification of the PhyR REC domain by the phosphomimetic compound BeF3− shifts REC toward an active conformation in full-length protein, and (2) the isolated PhyR REC domain adopts an active conformation even in the absence of BeF3− modification (Corrêa & Gardner, 2016). Thus, the PhyR REC domain is held in an inactive conformation by its interaction with the SL domain on the same polypeptide. A model that emerges from these and other published data (reviewed in (Fiebig et al., 2015, Francez-Charlot et al., 2015)) is that the energy of aspartyl phosphorylation overcomes an inhibitory interaction between the PhyR REC and SL domains. Once free from SL, REC is predicted to adopt an active conformation that stabilizes an “open” state in which SL is undocked from activated REC and the NepR-binding site is fully exposed (Figure 1A) (Campagne et al., 2012, Herrou et al., 2012). While mutagenesis data provide support for this PhyR open state model (Campagne et al., 2012), a structure of this PhyR~P open state has not, to our knowledge, been reported in the literature. A challenge in studying PhyR~P is that addition of the anti-σ factor NepR is necessary to generate a large fraction of phosphorylated PhyR (PhyR~P) in vitro (Herrou et al., 2015, Kaczmarczyk et al., 2014), which confounds structural investigations of isolated PhyR~P. Enhancement of PhyR REC domain phosphorylation by NepR requires an intrinsically disordered region (IDR) at the NepR amino terminus (Herrou et al., 2015) via a mechanism that remains undefined.
In this study, we aimed to define the molecular requirements of PhyR activation by investigating the roles of both phosphorylation and nascent interactions with the IDR of NepR in promoting a fully-active PhyR open state. Experimental protein–protein interaction studies contribute to an emerging model in which restructuring of the α11-β5 loop (α4-β5 in standard REC domain nomenclature) plays a role in PhyR activation as an anti-anti-σ factor. Molecular dynamics (MD) umbrella sampling calculations provide evidence that aspartyl phosphorylation of the PhyR REC domain lowers the energy barrier to an active, open state conformation, though small-angle X-ray scattering (SAXS) experiments indicate that aspartyl phosphorylation is insufficient to shift a large fraction of PhyR to an open state in vitro. Thus, additional free energy may be required to disengage REC and SL domains and fully expose the NepR-binding site on SL. Two-dimensional biomolecular NMR experiments revealed a stable interaction between unphosphorylated PhyR and NepR that requires the IDR at the NepR N-terminus. We propose this nascent molecular interaction between the IDR of NepR and unphosphorylated PhyR is specific and serves an important role in priming PhyR for activation by phosphorylation, and providing free energy to form the open, active state that binds NepR with high affinity.
Results
Umbrella sampling calculations provide evidence that PhyR phosphorylation lowers the energetic barrier to an open state conformation
The NepR-binding site on the PhyR SL domain is partially occluded by the REC domain in the full-length, unphosphorylated protein (Campagne et al., 2012, Herrou et al., 2012). Thus, PhyR must undergo a large conformational change to expose the NepR-binding site that has been defined in both X-ray and NMR structures. The current model of PhyR activation as an anti-anti-σ factor entails a phosphorylation-dependent transition from a “closed” to “open” state, which enables NepR binding (Figure 1A) (Campagne et al., 2012, Corrêa & Gardner, 2016, Herrou et al., 2010, Herrou et al., 2012). While biomolecular NMR experiments have shown that modification of PhyR with the phosphomimetic compound BeF3− induces widespread chemical shift changes in the heteronuclear single quantum coherence (HSQC) spectrum of E. litoralis PhyR (Corrêa & Gardner, 2016), the hypothesis that REC domain phosphorylation is sufficient to shift a large fraction of PhyR molecules to an open conformation remains unproven.
To further interrogate the structural and thermodynamic basis of PhyR activation by phosphorylation, we performed a series of atomistic MD simulations and umbrella sampling calculations on high-resolution structural models of Caulobacter crescentus PhyR and PhyR~P. Ten fifty ns equilibration simulations of PhyR~P were performed to probe early structural transitions induced by phosphorylation. We also performed a 50 ns control simulation of unphosphorylated PhyR. As previously observed in longer timescale simulations (Herrou et al., 2012), there is no evidence that SL and REC domains of PhyR~P undock to expose the NepR-binding site in an open state conformation, though these timescales may be too short to observe such a large-scale structural change. In all simulations of PhyR~P, we observed restructuring of certain regions (particularly the α4 flanking loop) and, in some cases, of the REC domain α11-β5 loop region surrounding the phosphorylation site (residues 224–231) (Figures 1B and S1A).
To assess the degree to which these simulated changes may prime PhyR to adopt an open conformation that favors NepR binding, we conducted solvent accessible surface area (SASA) calculations on NepR-binding residues at the SL-REC domain interface in PhyR and one of the PhyR~P simulations (trajectory 1) (Figure 1C). Increased solvation was largely limited to charged residues on the α1 helix of the SL domain (Figure 1D). Prior to phosphorylation, this region of structure is directly associated with the REC domain α11-β5 loop. We calculated the root mean square fluctuation (RMSF) of each residue in our simulations by averaging the squared distance of polypeptide alpha carbons from their initial positions at 40 ns (after all simulations were well-relaxed). RMSF reports on the variance in the position of each residue, and was particularly elevated in the α4 loop after restructuring (Figure S1B–C); we also observed some modest fluctuation in the region surrounding the phosphorylation site that is consistent with increased dynamics as a function of phosphorylation (Campagne et al., 2012, Corrêa & Gardner, 2016, Herrou et al., 2012).
We next assessed the energetics of the transition from a closed to open state conformation following phosphorylation using an umbrella sampling approach. Specifically, we simulated the 10 ns (i.e. before structural rearrangement) and 50 ns (i.e. after structural rearrangement) states of PhyR~P (trajectory 1) and PhyR using the inter-domain distance between SL and REC as a proxy reaction coordinate for formation of a PhyR open state. Inter-domain distance was defined as the length between the center of mass of the SL (residues 1–120) and REC domains (residues 140–266). Simulations sampled inter-domain distances from their free equilibrium values of ~28.5 Å and ~29.5 Å for PhyR and PhyR~P, respectively, to a final distance of 34 Å. At this distance, most charged residues at the domain interface are fully solvated. The MD simulations and umbrella sampling provide evidence that PhyR does not undergo a spontaneous transition to an open state upon phosphorylation at this short timescale, though phosphorylation does lower the energy barrier to opening (Figures 1B–E and S1).
Phosphorylation of PhyR is not sufficient to induce a measureable open state conformation in vitro
MD and umbrella sampling approaches provide evidence that phosphorylation of the PhyR REC domain does not result in spontaneous formation of an open conformation on short timescales (Figure 1). NMR studies of E. litoralis PhyR (56% identical to C. crescentus PhyR; BLAST e < 3e−96) show widespread changes in chemical shifts as a function of modification with the phosphomimetic compound, beryllium fluoride (BeF3−) (Corrêa & Gardner, 2016). Thus, PhyR phosphorylation clearly alters PhyR structure. How these changes in chemical shift correspond to changes in PhyR tertiary structure is not well defined.
We used SAXS to directly test whether PhyR phosphorylation results in a measurable structural change that is consistent with an open state where the phosphorylated REC domain dissociates from the SL domain. We selected Brucella abortus PhyR (55% identical to C. crescentus PhyR; BLAST e < 8e−91) as our model for scattering studies because we have established robust protocols to produce stably phosphorylated PhyR (PhyR~P) using its cognate histidine kinase as a phosphoryl donor (Kim et al., 2014). We optimized conditions of the phosphoryl-transfer reaction to generate quantities of PhyR~P suitable for SAXS analysis; greater than 90% of PhyR was phosphorylated as estimated using a combination of size-exclusion chromatography (SEC) and the Phos-tag (Kinoshita et al., 2006) acrylamide mobility shift detection system (Figures 2A,B and S2A).
Figure 2. PhyR phosphorylation does not induce changes in overall global structure.

(A) Gel filtration elution profiles of B. abortus PhyR (blue), PhyR~P (red), and the PhyR~P:NepR complex (black). (B) Quantification of PhyR phosphorylation. Top: LovHK-dependent [γ-32P]-ATP phosphoryl transfer to PhyR as a function of time in minutes. Middle: Phos-tag acrylamide gels of PhyR (blue arrow), PhyR~P (red arrow), and PhyR~P:NepR (black arrow) samples. Bottom: SDS-PAGE of PhyR, PhyR~P, and PhyR~P:NepR. (C) Calculated (sim) SAXS scattering curves from PhyR (blue) and PhyR~P structures (red) taken from the 50 ns timepoint of MD simulations; scattering for the PhyR~P open conformation was calculated from umbrella sampling model at a SL-REC interdomain distance of 34 Å (green). SL-REC interdomain distance for the PhyR closed state is ≈28.5 Å. Structural models from which scattering was calculated are presented in panel G. (D) Pair-distance distribution functions (PDDF) calculated from simulated scattering curves in Panel C. (E) Experimental background-subtracted SAXS scattering data (dots) and fits for PhyR (blue, solid) and PhyR~P (red, dashed). Inset: calculated scattering for a PhyR~P open state (green) compared to experimental scattering from PhyR (blue) and PhyR~P (red) between q=0.13 and 0.22. (F) Pair-distance distribution function (PDDF) and Guinier analysis (inset) of PhyR and PhyR~P experimental SAXS data. Guinier plots were offset from each other for clarity. (G) Structural models derived from MD simulations for three states of PhyR (PhyR closed, PhyR~P closed, and PhyR~P open); models were used to backcalculate SAXS scattering curves and corresponding PDDF plots in panels C and D.
The SAXS data for PhyR and PhyR~P are nearly identical (Figures 2C–E and S3). The similarity in scattering is particularly apparent when comparing the pair-distance distribution function (PDDF) plots (Figure 2F). Radii of gyration (Rg), Dmax, and Porod volumes are also the same, within error (Table S1). The experimental scattering data do not have features of an open state, based on calculation of a theoretical scattering curve (Mertens & Svergun, 2010) from structural models in which the SL and REC domain of PhyR are partially hinged apart (Figure 2C–G). These data provide evidence that PhyR phosphorylation, though required for NepR binding and GSR transcriptional activation, is insufficient to drive a measurable fraction of protein into an open conformation. Neither PhyR phosphorylation nor complexation with NepR significantly affect protein stability as measured by thermal denaturation (Figure S2C and D). This is consistent with our MD simulations (Figure 1) in which there are significant but localized structural transitions. Based on these data and published biochemical data showing NepR-dependent stimulation of PhyR phosphorylation (Herrou et al., 2015, Kaczmarczyk et al., 2014), we support a model in which a specific interaction between NepR and unphosphorylated PhyR is an important step in generating an active PhyR~P open state that binds NepR with high affinity. This stated, we cannot rule out the possibility that a minority fraction of PhyR~P accesses an open state, which is subsequently bound by NepR.
The role of phosphorylation-induced structural changes in PhyR-NepR binding
The primary structural changes we observed in the C. crescentus PhyR~P MD trajectories were modest restructuring of the region near the α11-β5 loop and fluctuations in the conformation of the flexible loop N-terminal to α4. While these changes did not correlate significantly (Figure S1C), they accompanied a significant decrease in the cross-correlation between α4 loop residues and the residues of α6 and α7 (Figure S1A) indicating that the structural state of these regions may be coupled. As the structural changes near α4 were difficult to interpret, we focused our analysis on the α11-β5 loop. The α11-β5 loop corresponds to α4-β5 of typical receiver proteins, one of several regions where structural rearrangements occur upon phosphorylation that control REC domain function (Bourret, 2010). In contrast to canonical receiver proteins, PhyR contains an extension of the loop between α11 and β5 that directly interacts with and is stabilized by the SL domain (Campagne et al., 2012, Corrêa & Gardner, 2016, Herrou et al., 2012). A putative hydrogen bonding network in the α11-β5 loop is evident between the backbone nitrogen residues of T228 and G229, the side chain hydroxyl of T228, and the carboxyl oxygen of the E233 side chain (Figure 3A). These interactions appear to define the α11-β5 loop structure and stabilize a closed PhyR conformation. Many of these interactions are disrupted upon phosphorylation in some simulations.
Figure 3. Hydrogen bonding in the α11-β5 loop stabilizes the inactive state of PhyR.

(A) Phosphorylation–induced rearrangement of α11-β5 loop hydrogen bonds of PhyR (top) and PhyR~P following loop rearrangement (bottom) observed in select MD simulations. Sidechain–sidechain and sidechain–backbone hydrogen bonds are colored red and blue, respectively. (B) Bacteria two-hybrid (BTH) interaction assays with PhyR α11-β5 loop alanine mutants. Inset: Cartoon outlining BTH interaction assay. (C) BTH interaction assays for L227A, T228A or E233A combined with the D192A (non-phosphorylatable) PhyR mutation. (D) Activity of a GSR transcriptional reporter (PsigU) in the absence (blue) or presence (red) of hyperosmotic stress. Inset: Cartoon outlining transcriptional reporter scheme. Controls for BTH and PsigU assays are summarized in Figure S7. Error bars represent standard deviation.
To develop the existing model (Campagne et al., 2012) that destabilization of the α11-β5 loop plays a role in phosphorylation-dependent activation of C. crescentus PhyR, we prepared a series of alanine substitutions (i.e., an alanine scan) of residues 220–233 (excluding P223, G229, and P232) and assessed NepR binding using a bacterial two-hybrid (BTH) assay. PhyR mutations L227A, T228A, or E233A in the α11-β5 loop increased apparent NepR binding to a level similar to that of the isolated SL domain, which binds NepR constitutively (Figures 3B and S4A) (Campagne et al., 2012, Herrou et al., 2012, Herrou et al., 2015). These results are consistent with published data (Campagne et al., 2012) showing that the E235A mutation in Sphingomonas (corresponding to E233 in C. crescentus) derepresses binding to NepR even when the phosphorylation site (D194) is substituted to alanine. In C. crescentus, T228 and E233 form extensive hydrogen bonds in the α11-β5 loop and to the SL domain (Figure 3A) while the hydrophobic side chain of L227 is packed against SL-α1 at the domain interface. In SASA calculations, L227 is solvated upon phosphorylation (Figure 1C). These data support a model in which mutations that destabilize the α11-β5 loop result in a structural shift toward a PhyR~P-like state. Indeed, these mutant variants of PhyR retain their capacity to bind NepR in this BTH assay even when the D192 phosphorylation site is mutated to alanine (Figure 3C), and thus decouple PhyR phosphorylation from NepR interaction.
Two other α11-β5 loop mutants, T220A and R225A, did not interact with NepR in the BTH assay (Figure 3B). T220 is highly conserved and stabilizes the phosphoryl group in REC domains that utilize either Y-T coupling or FATGY signaling mechanisms (Cho et al., 2000, Campagne et al., 2016, Sheftic et al., 2014). This residue is required for conformational changes induced by BeF3− modification in E. litoralis PhyR (Corrêa & Gardner, 2016). The structural role of R225 is unclear, though we note that R225 stacks with F222 and may provide stability to the α11 helix. In our PhyR~P simulations, R225 interacts with the phosphoryl group on D192 and may function to stabilize aspartyl phosphate or assist in pulling α11 away from the REC-SL domain interface.
We next assessed the functional effect of the PhyR L227A, T228A, and E233A mutations (i.e. mutants with increased NepR interaction in the BTH assay) on GSR transcriptional activation in C. crescentus. To measure GSR transcription we used a PsigU-lacZ transcriptional fusion plasmid (Herrou et al., 2010) as a reporter, and quantified β-galactosidase activity in the presence and absence of osmotic stress. We expressed these mutant phyR alleles in a ΔphyR background from a vanillate-inducible promoter (Pvan). For each of these mutants, we observed a small increase in baseline GSR transcription compared with wild type, which is consistent with bias toward a derepressed state (i.e. a NepR-binding state). Transcription was activated under an osmotic stress condition, indicating that these mutant PhyR proteins remain stress responsive. Expression of PhyR L227A, T228A, or E233A alleles harboring a non-phosphorylatable D192A mutation did not support GSR transcription; D192 was required under all backgrounds to activate GSR transcription (Figure 3D). We conclude that although the L227A, T228A, or E233A mutants of PhyR have derepressed NepR binding in a BTH assay, REC phosphorylation is still required for these alleles to function as positive regulators of GSR transcription in vivo.
NepR interacts in a structured manner with unphosphorylated PhyR
An intrinsically disordered region of polypeptide (IDR) at the N-terminal end of NepR, termed FR1, is required for high-affinity C. crescentus PhyR~P-NepR interaction in vitro and in vivo, and for stimulation of PhyR autophosphorylation in vitro (Herrou et al., 2015). These results suggest a model whereby PhyR interacts with NepR in the absence of phosphorylation. This interaction is predicted to prime PhyR for activation as an anti-anti-σ factor during the GSR. However, previous studies (Herrou et al., 2012) and size exclusion chromatography data (Figure S2B) provide no evidence for a stable interaction between C. crescentus NepR and unphosphorylated PhyR.
We tested whether unphosphorylated PhyR and NepR form a weak stable interaction using a spectroscopic approach. Specifically, we purified 15N-labeled C. crescentus PhyR (15N-PhyR) and measured 2D 1H-15N transverse relaxation-optimized spectroscopy (TROSY) spectra in the presence and absence of a 1.25-fold molar excess of unlabeled NepR. We observed small but distinct changes in 15N-PhyR chemical shifts, which provide evidence that PhyR and NepR interact in a stable manner in the absence of PhyR phosphorylation (Figure 4). We note that unphosphorylated B. abortus PhyR is reported to form a complex with NepR that is sufficiently stable to purify by size-exclusion chromatography (SEC) (Figure S2A) (Kim et al., 2013). Thus the affinity of interaction between unphosphorylated PhyR and NepR varies across species, which may reflect differences in steady-state PhyR and NepR–σEcfG levels in the cell or other differences in GSR regulation among Alphaproteobacteria.
Figure 4. NepR induces structural shifts in unphosphorylated PhyR.

(A) 1H-15N TROSY spectra of 15N-labeled C. crescentus PhyR (blue) overlaid with 15N-PhyR in the presence of 1.25-fold molar excess of unlabelled C. crescentus NepR (red). Boxes 1 and 2 are magnified in panel B. (B) Selected PhyR peak shifts observed in the presence of NepR are marked with black arrowheads.
Given the spectroscopic evidence for interaction between 15N-PhyR and NepR, we performed a reciprocal set of experiments with 15N-NepR and unlabeled PhyR. We first measured a 2D 1H-15N heteronuclear single quantum coherence (HSQC) spectrum of 15N-NepR to assess its structure in isolation. This spectrum had poor chemical shift dispersion (Figure 5A) characteristic of unstructured polypeptide, which is consistent with spectra of S. melonis NepR (Campagne et al., 2012). Phase-modulated CLEAN chemical exchange (CLEANEX-PM) revealed fast exchange of nearly all amide hydrogens in this sample with water. These data indicate that C. crescentus NepR is dynamic and largely unstructured in the absence of a binding partner (Figures 5A and S5A). We next conducted a 1H-15N HSQC experiment on 15N-NepR in the presence of a 1.25-fold molar excess of unlabeled and unphosphorylated PhyR. We observed the gain, loss, or shift of multiple 15N-NepR peaks upon the addition of PhyR (Figure 5B). The majority of new peaks are restricted to a narrow region in the 1H dimension in these spectra (7.6–8.1 ppm) with greater dispersion in the 15N dimension, which is consistent with an intrinsically disordered protein (IDP) experiencing multiple stable conformational states on a millisecond timescale (Kragelj et al., 2013, Kosol et al., 2013, Konrat, 2014). Nearly all peaks that arise in this region of the spectrum are exchange protected as assessed by CLEANEX-PM, which provides evidence that NepR adopts an ordered conformation (or set of conformations) in the presence of unphosphorylated PhyR. PhyR, NepR and σEcfG are abundant proteins in the cell. Calculated concentrations inside C. crescentus approach 41, 90, and 12 μM, respectively, in the absence of stress based on published intracellular protein levels and ribosome profiling data (and assuming a cell volume of 0.3 femtoliters) (Li et al., 2014, Schrader et al., 2016, Schrader et al., 2014). Interactions detected in our NMR experiments may thus reflect interactions present in the cell.
Figure 5. NepR interaction with unphosphorylated PhyR affects NepR structure.
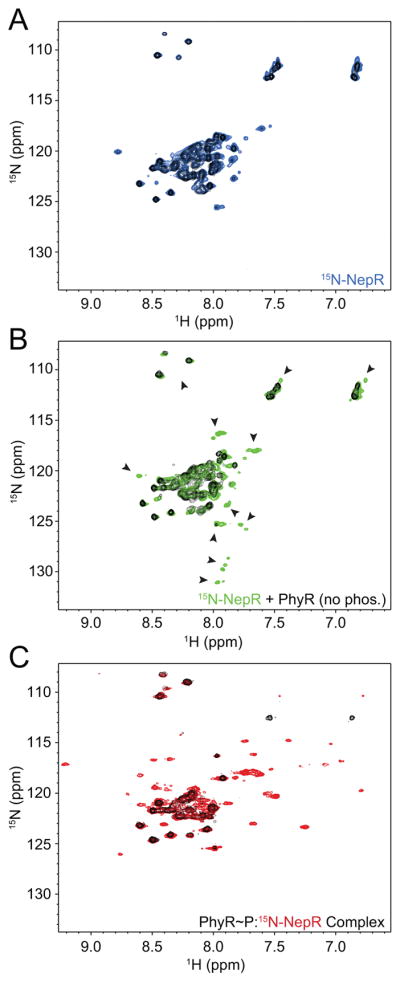
1H-15N HSQC spectra of uniformly 15N-labeled C. crescentus NepR; CLEANX-PM spectra for all samples are shown in black. (A) NepR alone (blue); (B) NepR in the presence of excess unlabelled, unphosphorylated C. crescentus PhyR (green, arrows indicate new, shifting, or lost peaks); (C) NepR in a complex with unlabeled PhyR~P, denoted PhyR~P:15N-NepR (red).
As controls, we collected HSQC spectra of 15N-NepR after 24 h incubation in NMR conditions or in the presence of a 3.5-fold molar excess of bovine serum albumin (BSA). We did not observe any significant chemical shifts in these spectra (Figure S5A). We conclude that the gain of structure in NepR requires the specific presence of PhyR and is not due to degradation or the simple presence of another protein at high concentration. We also collected an HSQC spectrum of 15N-NepR with a 1.25-fold excess of D192A PhyR and obtained an identical result to WT PhyR (Figure S5B) indicating changes in the spectra are not due to a small amount of phosphorylated PhyR in the sample tube.
15N-NepR peaks that appear in the presence of unphosphorylated PhyR are distinct from those measured in 1H-15N HSQC spectra of a PhyR~P:15N-NepR complex, which are characterized by improved peak dispersion, particularly in the hydrogen dimension, and protection of amide protons (Figure 5C). The peaks observed at 7.6–8.1 and 127–132 ppm in the hydrogen and nitrogen dimensions, respectively, in the PhyR:15N-NepR spectrum are not present in PhyR~P:15N-NepR (Figure 5B and C). Notably, when we mixed 15N-NepR from C. crescentus with a 1.25-fold molar excess of unphosphorylated B. abortus PhyR, we did not observe a similar appearance of peaks even after 24 h incubation. Only minor peak shifts were evident in this spectrum and the majority of peaks that appear between 7.6–8.1 and 127–132 ppm in the conspecific C. crescentus PhyR–NepR complex are absent (Figure S5C). This provides evidence that interaction between NepR and unphosphorylated PhyR requires a conspecific protein pairing.
The NMR spectroscopic studies reported here provide evidence for a structurally defined interaction between unphosphorylated PhyR and NepR, which may underlie NepR-dependent activation of PhyR phosphorylation (Herrou et al., 2015, Kaczmarczyk et al., 2014). Such a model is in line with other models of response regulator activation, in which the presence of substrates, additional domains, or binding partners can influence REC domain phosphorylation and dephosphorylation (Barbieri et al., 2010, Schuster et al., 2001).
An intrinsically disordered region of NepR, FR1, interacts specifically with PhyR
Results from simulations and in vitro structural assays support a model in which PhyR and PhyR~P primarily exist in a closed conformation in which the REC domain is docked against the SL domain (Figures 1 and 2). PhyR phosphorylation mobilizes regions of structure, which correlates with a lowered energetic barrier to the PhyR open state (Figure 1). Our results suggest that an additional molecular component is required to initiate switching to a PhyR conformation that is competent to bind NepR with high affinity and activate GSR transcription. Given the evidence for NepR binding to unphosphorylated PhyR (Figures 4 and 5) and published studies showing that NepR enhances PhyR phosphorylation by either its cognate histidine kinase or by low-molecular-weight phosphoryl donors (Herrou et al., 2015, Kaczmarczyk et al., 2014), we postulated that energy from interaction with NepR or with the NepR–σEcfG complex may contribute to PhyR opening.
When bound to PhyR, NepR is characterized by two conserved central helices, α1 and α2, that are flanked by intrinsically disordered regions (IDR) of structure (Herrou et al., 2012, Herrou et al., 2015, Campagne et al., 2012) termed FR1 and FR2 (Figure 6A). As previously reported (Herrou et al., 2015), C. crescentus NepR binds C. crescentus PhyR in a BTH assay (Figure 3B); however, a truncated NepR construct lacking FR1 and FR2 (NepRSV) does not bind full-length PhyR (Herrou et al., 2012). We observed that NepRSV interacts with the “activating” PhyR α11-β5 loop mutant, T228A, albeit more weakly than does full-length NepR (Figure 6B). Thus, the requirement for the NepR FR1/2 regions in PhyR binding is less stringent when PhyR harbors the activating T228A mutation. The other activating α11-β5 loop mutants shown in Figure 3B (i.e. L227A and E233A) do not bind NepRSV in a BTH assay.
Figure 6. Intrinsically disordered NepR N-terminal sequences confer PhyR-NepR interaction specificity.

(A) C. crescentus (Cc) and B. abortus (Ba) NepR constructs used in bacterial two-hybrid (BTH) interaction assays: SV, a NepR construct lacking both FR1 and FR2; Cc FR1+Ba 1/2 NepR is a chimeric construct containing Cc NepR residues 1–31 fused to Ba NepR residues 28–75; Ba FR1+Cc 1/2 NepR is a chimeric construct containing Ba NepR residues 1–27 fused to Cc NepR residues 32–68; asterisk marks location of residues previously identified as required for PhyR-NepR binding (Campagne et al., 2012). (B) BTH interaction assay between NepR constructs (labelled by color from panel A) and three constitutively active PhyR mutants (labelled on axis). (C) Interaction of wild-type and chimeric NepR constructs (by color from panel A) with wild-type C. crescentus (Cc) or B. abortus (Ba) PhyR. Protein stability controls are presented in Figure S7. Error bars represent standard deviation.
We focused on FR1 to test whether this IDR specifically interacts with PhyR. We made chimeric NepR constructs in which we exchanged FR1 between two Alphaproteobacterial species, B. abortus and C. crescentus (mutants are referred to as Ba and Cc, respectively). NepR orthologs from these species share 3 of the 4 residues in the helical region required for PhyR binding (Figure 6A) (Campagne et al., 2012). Each NepR construct was assessed for interaction with wild-type Cc and Ba PhyR and Cc PhyR mutants in a BTH assay. The specificity of PhyR–NepR interaction in this assay was determined by FR1, and not by the α1-α2 helical domain (Figures 6B,C and S4B). In other words, a conspecific interaction between the NepR FR1 region and PhyR determines PhyR–NepR binding in this assay.
To further investigate the role of FR1 as a determinant of PhyR–NepR interaction specificity, and to define residues required for PhyR–NepR interaction, we prepared a series of Cc NepR truncation mutants, termed T1–T5, and assessed binding to PhyR by BTH. Most of the FR1 region can be deleted without compromising NepR–PhyR interaction. However, binding was completely lost when the approximately 10 residues N-terminal to α1 were deleted (Figure S6, T3 vs T4). To test whether a specific residue within this IDR of NepR structure is necessary for PhyR–NepR interaction, we performed an alanine scan and assayed PhyR interaction by BTH. Several alanine substitutions result in moderately decreased PhyR binding, but no single mutation completely disrupted the PhyR–NepR interaction (Figure S6). This suggests that multiple contacts in this region are responsible for the PhyR–NepR interaction and may help explain FR1 sequence diversity.
Discussion
Accessing the PhyR open state
Previous biochemical and structural analyses of PhyR suggest a model in which phosphorylation of its REC domain results in a structural transition from a ‘closed’ to an ‘open’ and active conformation (Figure 1A) (Campagne et al., 2012, Francez-Charlot et al., 2009, Herrou et al., 2010, Herrou et al., 2012). We directly tested this model of PhyR activation using experimental biophysical, biochemical, genetic, and computational approaches. We provide evidence that phosphorylation of the PhyR REC domain is not sufficient to induce formation of a large fraction of open-state PhyR in vitro, though disruption of a hydrogen bonding network in the α11-β5 loop and solvation of residues at the REC-SL interface (upon phosphorylation) are associated with a lowered energetic barrier to opening (Figures 1–3). This result is consistent with a growing body of data that implicate α11-β5 conformational change in PhyR activation (Campagne et al., 2012, Corrêa & Gardner, 2016, Herrou et al., 2012). Our data support a GSR activation model in which NepR interaction with PhyR, likely via an intrinsically disordered region of NepR polypeptide, supplies free energy to open and activate phospho-PhyR.
Our study identified several C. crescentus PhyR mutants that decoupled NepR binding activity from REC domain phosphorylation in a BTH binding assay, but did not strongly de-repress GSR transcription in vivo. The discrepancy in these results may be explained by the fact that NepR in the C. crescentus cell is largely bound to σEcfG. Phosphorylation dependent conformational changes in other regions of PhyR may be necessary to liberate NepR from σEcfG in vivo and activate GSR transcription. For example, in addition to restructuring of the α11-β5 loop in some simulations, MD trajectories of PhyR~P showed clear extension of the α4 helix of the SL into the solvent. This helix is an important contributor to PhyR-NepR binding that has been reported to interact directly with NepR in the S. melonis FR1 system (Campagne et al., 2012). It is possible that extension of α4 is tightly controlled by phosphorylation in a manner independent of α11-β5 loop conformation.
Probing the role of an intrinsically disordered region (IDR) of NepR in allosteric control of PhyR
NepR comprises two conserved central helices that bind the PhyR SL domain with high affinity (Campagne et al., 2012, Herrou et al., 2012), flanked by IDRs at the N-and C-termini. The IDR at the N-terminus of NepR, termed FR1, does not have a conserved primary structure but is required for C. crescentus NepR to stimulate PhyR phosphorylation and bind to phospho-PhyR (PhyR~P) with high affinity in vitro (Herrou et al., 2015). We have provided evidence for an interaction between NepR and unphosphorylated PhyR (Figure 5B). This interaction has spectroscopic features of a so-called fuzzy complex (Tompa & Fuxreiter, 2008), in which NepR might adopt a partially structured state that modulates the structure and activity of PhyR. Consistent with this possibility, we observed modest changes in 15N-PhyR chemical shifts in the presence of conspecific NepR (Figure 4B). Future studies of PhyR-NepR pairs that form a more stable interaction without phosphorylation, such as those from B. abortus, may provide more in depth structural understanding of specific interactions between NepR FR1 and unphosphorylated PhyR. It is possible that NepR adopts multiple stable conformations in its interaction with PhyR prior to forming a stable, high-affinity PhyR~P/NepR complex.
A ternary complex between PhyR, NepR, and σEcfG underlying partner switching?
We favor a model in which PhyR forms a ternary complex with NepR–σEcfG in the cell, possibly mediated by the FR1 IDR of NepR (Figure 7). In such a model, FR1 may be disordered when NepR is bound to σEcfG (Campagne et al., 2012) and form a structured interaction with PhyR to facilitate formation of a ternary complex. Upon PhyR binding, NepR would promote REC domain phosphorylation and open state formation and be subsequently passed from σEcfG to the PhyR SL domain. This mechanism would avoid high levels of free NepR in the cell, which might be recognized as a disordered polypeptide and be rapidly degraded. We note similarities between the Alphaproteobacterial PhyR–NepR–σEcfG regulatory system and mammalian hypoxia-inducible factor (HIF-1α), which is also regulated by partner switching interactions mediated through an IDR (Dames et al., 2002, De Guzman et al., 2004, Freedman et al., 2003, Freedman et al., 2002). Structural disorder is often advantageous for proteins that perform multiple roles, interact with numerous partners, or rapidly transition between conformational states (Wright & Dyson, 2015). In the case of HIF-1α, protein disorder is proposed to enable efficient protein partner switching even when cellular concentrations of competing partners are similar (Berlow et al., 2017). Whether unstructured regions of NepR enable partner switching between equimolar PhyR and σEcfG remains to be determined and requires additional experimentation.
Figure 7. A model of PhyR-NepR Interaction and GSR activation depicted as a hypothetical reaction coordinate.

The relative energies of intermediates are not known, but partner switching is depicted as an overall downhill reaction. (A) Nascent PhyR–NepR interaction orders features of NepR structure and primes the site of PhyR phosphorylation (white dotted lines). After initial PhyR-NepR encounter, it is not known whether NepR remains associated with PhyR in a PhyR-NepR-σEcfG ternary complex, or if the initial interaction is transient (question marks). (B) Phosphorylation results in α11-β5 loop rearrangement and lowers the energy barrier to form a PhyR~P open state. (C) NepR interaction contributes to PhyR~P opening and NepR partner switching, thereby releasing σEcfG and activating GSR transcription.
High-resolution structural studies of isolated PhyR~P have been impeded by the requirement that NepR be added to the phosphorylation reaction mixture to yield a high fraction of PhyR~P. The three active mutants reported in this study, L227A, T228A, and E233A, have properties of PhyR~P in a BTH binding assay and may be good candidates for further structural studies of activated PhyR. These mutants maintain the requirement for a conspecific NepR FR1 IDR to function in a BTH assay (Figure 6B). We suggest FR1 facilitates formation of the PhyR open state by contacting residues at the SL-REC domain interface, possibly following α11-β5 loop rearrangement. The precise mechanism and specific residues involved in this process merit further exploration. We further note that PhyR REC does not contain residues involved in the so-called Y-T coupling (Cho et al., 2000) or FATGY (Campagne et al., 2016) activation mechanisms and thus provides an excellent model to explore the diversity of REC domain signaling mechanisms.
The data presented herein expand our molecular understanding of the central GSR regulatory proteins PhyR and NepR by demonstrating (1) PhyR and NepR interact prior to phosphorylation and this interaction nucleates NepR structure, (2) REC domain phosphorylation is insufficient to shift a measureable fraction of PhyR to an open conformation as measured by SAXS, and (3) an unstructured region at the NepR amino terminus confers PhyR-NepR binding specificity. Collectively, these results identify new molecular/structural control features of a large transcriptional program required for alphaproteobacterial stress adaptation.
Experimental Procedures
Protein expression and purification
All primers and strains used in this study are outlined in Tables S2 and S3. A 5 mL overnight culture was used to inoculate 1 L lysogeny broth (LB) medium with 50 μg•mL−1 kanamycin (Kan) and grown to an optical density at 600 nm (OD600) of 0.6–0.8 at 37 °C and 210 rpm for unlabeled cultures. For uniformly 15N-labled proteins, the overnight culture was used to inoculate 1 L M9 minimal media (pH 7.4) supplemented with 15NH4Cl (1 g•L−1, Cambridge Isotope Laboratories). Protein expression was induced by adding 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and incubating for an additional ~20 h at 18 °C. Cells were harvested by centrifugation and pellets resuspended in 25 mM Tris-HCl (pH 7.6), 125 mM NaCl, and 10 mM imidazole. Cell suspensions were disrupted by passage through a Microfluidizer (Microfluidics). Cell debris was pelleted by centrifugation at 39,000 g for 30 min at 4 °C. The soluble fraction was then applied to a Ni-NTA resin column (nitrilotriacetic acid; GE Healthcare). The resin was washed sequentially with Tris-NaCl buffer containing 10, 30, and 125 mM imidazole and proteins eluted with 350 and 500 mM imidazole. Elution fractions were concentrated and subjected to SEC using a Superdex S300 26/60 column (GE Healthcare) equilibrated with 25 mM Tris (pH 7.6) and 125 mM NaCl. Fractions containing the protein of interest were pooled and, if necessary, concentrated prior to use. For the B. abortus histidine kinase LovHKΔ8–109, protein eluted from the Ni-NTA column was dialyzed overnight at 4 °C against 25 mM Tris-HCl (pH 7.6), 125 mM NaCl, and 50% (v/v) glycerol. Purified proteins were determined to be >95% pure by 15% SDS-PAGE.
Preparation of B. abortus PhyR~P and PhyR~P:NepR
For B. abortus PhyR~P, phosphorylated protein was generated by incubating PhyR with its cognate histidine kinase, LovHKΔ8–109 (Kim et al., 2014, Willett & Kirby, 2012). PhyR~P used in SEC-SAXS experiments was generated using 490 μM Ba PhyR with 7 μM LovHKΔ8–109 in kinase buffer (25 mM Tris, 50 mM KCl, 1 mM each CaCl2, MgCl2, MnCl2, and dithiothreitol (DTT), pH 7.6) containing 10 mM ATP for 20 h to ensure full phosphorylation. LovHKΔ8–109 kinase activity was confirmed under these conditions by adding 3 μM [γ-32P]-ATP, followed by SDS-PAGE and autoradiography. 32P-labeled PhyR was separated by 12% SDS-PAGE and visualized using a Typhoon Imager (GE). To estimate the percentage of phosphorylated PhyR generated by phosphoryl transfer from LovHKΔ8–109, samples were subjected to SDS-PAGE Phos-tag mobility shift detection system (Kinoshita et al., 2006) using methods previously developed for analyzing aspartyl phosphorylation in response regulators (Barbieri & Stock, 2008, Gao & Stock, 2013). PhyR~P:NepR was used as a representative fully phosphorylated PhyR protein and generated by reacting 25 μM PhyR and 1 mM acetyl phosphate (AcP) in kinase buffer and 50 μM NepR for ~20 h. PhyR~P and PhyR~P:NepR phosphorylation reactions were quenched by addition of SDS loading buffer. Protein samples (1.26 μg) were separated by 9% SDS-PAGE including 50 mM Phos-tag acrylamide and 100 mM MnCl2 (or standard 9% SDS-PAGE). All Phos-tag acrylamide gels were electrophoresed at a constant voltage of 100 V. Gels were stained with Coomassie and analyzed using ImageJ. Comparison of the relative intensities in Phos-tag vs standard gels indicated that PhyR~P generated from LovHKΔ8–109 was >85% phosphorylated. Purified B. abortus PhyR~P protein was used for SAXS, circular dichroism (CD), and SEC experiments.
Preparation of C. crescentus PhyR~P:NepR
The C. crescentus (Cc) PhyR~P:NepR complex was prepared by incubating purified PhyR and NepR with kinase buffer and AcP. For SEC experiments, complexes were prepared using 25 μM PhyR, 50 μM NepR, and 1 mM AcP. For NMR spectroscopy experiments, 300 μM PhyR, 275 μM 15N-NepR, and 10 mM AcP were incubated in kinase buffer for 4 h to ensure complete phosphorylation, followed by buffer exchange by SEC into the indicated NMR buffer and concentrated.
CD and thermal denaturation measurements
CD spectra and thermal denaturation profiles were collected using a Jasco J-1500 CD spectrometer. Samples were buffer exchanged by SEC into 10 mM Tris-HCl (pH 7.6) and 125 mM NaCl. Fractions containing eluent were collected, combined, and diluted to 20 μM and analyzed in a 1.0 mm quartz cuvette. For experiments with NepR alone, 150 μM protein was used due to low signal relative to PhyR. In thermal denaturation experiments, the temperature was gradually increased from 26 to 86 °C at a rate of 2 °C•min−1. A complete CD spectrum was collected at each temperature and CD intensity at 222 nm was monitored for denaturation. CD intensity at 222 nm was plotted and fitted to a two-state model. Thermal denaturation experiments were performed in triplicate.
Analytical size-exclusion chromatography
SEC was performed using an analytical Superdex G200 10/300 column (GE Healthcare). Samples, 20 μM, were injected onto the column pre-equilibrated with the indicated buffer and eluted at a rate of 0.6 mL•min−1 unless otherwise indicated.
SAXS data collection and analysis
All SEC-SAXS data were collected at Sector 18-ID (BioCAT) of the Advanced Photon Source at the Argonne National Laboratory. Purified proteins, either Ba PhyR or PhyR~P, were loaded onto a Superdex G200 10/300 SEC column (GE Healthcare) pre-equilibrated with 25 mM Tris pH 7.6 and 125 mM NaCl at a concentration of approximately 16 mg•mL−1 and eluted at a rate of 0.8 mL•min−1. Data were collected across the SEC elution profile using an Aviex charge coupled device (CCD) detector and analyzed using the ATSAS software package (Petoukhov et al., 2012). The buffer background, i.e. SAXS scattering prior to protein elution, was averaged and subtracted from averaged scattering data containing PhyR using PRIMUS (Konarev et al., 2003). Estimates of the radius of gyration (Rg) were obtained using Guinier approximation from data at low q values. GNOM was used to generate a real space pair distribution function (PDDF or p(r)) with Dmax calibrated until the PDDF curve fell smoothly to zero (Svergun, 1992). Generation of theoretical scattering curves of PhyR models derived from MD simulations and umbrella sampling was performed using CRYSOL (Svergun et al., 1995).
Biomolecular NMR
NMR spectra were acquired at 35 °C on a Bruker AVANCE IIIHD 600 MHz NMR spectrometer equipped with a room temperature TXI probe. HN TROSY (Pervushin et al., 1997) and HSQC (Bax et al., 1990) spectra were acquired for 15N-PhyR and 15N-NepR, respectively. CLEANEX-PM (Hwang et al., 1998) spectra were also acquired with 100 ms mixing time. Spectra were processed in NMRpipe (Delaglio et al., 1995) and viewed in NMRViewJ (Johnson & Blevins, 1994). All experiments using 15N-NepR were performed in 10 mM HEPES pH 7.0, 125 mM NaCl, and 10% (v/v) 2H2O and experiments using 15N-PhyR were performed in 10 mM Bis-Tris pH 6.5, 125 mM NaCl, and 10% (v/v) 2H2O with 400 μM 15N-labeled protein.
C. crescentus transcriptional assays
C. crescentus ΔphyR carrying the plasmid pRKLac290-PsigU (Tetr) was used to assess σT (i.e. σEcfG) transcriptional activity as previously described (Herrou et al., 2010, Herrou et al., 2015). Into this background, we transformed pMT553 (Kanr) carrying various phyR alleles fused to Pvan, to assess σT-dependent transcription in the presence of each phyR mutant. For the HA-tagged PhyR experiments, we transformed, into the same background, pMT585 (Kanr) carrying the indicated phyR alleles also encoding an N-terminal HA-tag fused to Pxyl, to assess σT-dependent transcription (Figure 3D, inset). All C. crescentus strains were grown in peptone-yeast extract (PYE) medium (containing 5 μg•ml−1 Kan and 1 μg•ml−1 Tet) and diluted to a starting OD660 of ~0.05 (30°C, 200 rpm). Hyperosmotic stress was induced by adding 150 mM sucrose to the culture medium. Expression of phyR alleles was induced from the van promoter by adding 0.5 mM vanillate to the culture medium. β-Galactosidase activities were measured at an OD660 of ~0.25 in triplicate.
Western blotting
Overnight cultures of C. crescentus GSR transcriptional reporter strains expressing HA-tagged PhyR were used to inoculate 2 ml PYE medium (containing 5 μg•mL−1 Kan and 1 μg•mL−1 tetracycline (Tet)) to a starting OD660 of ~0.05 (30°C, 200 rpm) and 0.15 % (w/v) D-xylose was added. When an OD660 of ~0.3 was reached, cells were harvested by centrifugation at ~21,000 g for 1 min at room temperature, yielding ~0.5 OD*ml of cells. Samples were then resuspended in lysis buffer (25 mM Tris-HCl (pH 7.6), 125 mM NaCl, 10 mM CaCl2, 10 μg•mL−1 DNase I, 1% (w/v) SDS, 0.1% (v/v) Triton X, and EDTA-free protease inhibitor tablet) and western blotted as described previously (Eaton et al., 2016). A 1:1,500 dilution of monoclonal anti-mouse anti-HA antisera and a 1:4000 dilution of monoclonal goat anti-mouse antibodies conjugated to horseradish peroxidase (HRP) were used (Thermo Scientific). The secondary antibody was detected with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) imaged using a ChemiDoc MP System (Bio-Rad). Bands were detected and quantified using Image Lab software (Bio-Rad).
For western blot analysis of T18-NepR constructs, cultures were grown as described for BTH assays until OD600 reached 0.5 and 1 mL aliquots harvested by centrifugation at ~21,000 g for 1 min. Cell pellets were stored at −80 °C until use. Pellets were resuspended in 100μL lysis buffer and vortexed until the pellet was disrupted. SDS loading buffer (3×) was added and samples heated at 95 °C for 10 min. Western blotting was performed as described (Herrou et al., 2015). T18-NepR constructs were detected with anti-T18 antibody (1:1000 dilution; 3D1, KeraFast) in 10 mL TBST, 5% (w/v) milk for 1 h and visualized by incubation with HRP-conjugated mouse secondary antibody (1:5000 dilution) for 30 min. Visualization and quantitation was performed as described above.
BTH Protein Interaction Assays
Interactions between PhyR and NepR constructs inside bacterial cells were measured using a BTH system (Karimova et al., 1998). Wild-type C. crescentus nepR and phyR genes were previously cloned into pUT18c and pKT25 vectors, respectively (Herrou et al., 2015). The indicated B. abortus nepR allele was cloned into pUT18c between BamHI and EcoRI and the phyR allele into pKT25 between XbaI and KpnI. Chimeric nepR FR1 alleles were cloned into pUT18c at the KpnI restriction site using isothermal assembly (Gibson et al., 2009). All primers used in the study are listed in Table S2. Correct plasmid construction was confirmed by sequencing. BTH experiments were performed largely as previously described (Herrou et al., 2015, Karimova et al., 1998). Briefly, pUT18c and pKT25 plasmids carrying the indicated alleles were co-transformed into chemically competent Escherichia coli reporter strain BTH101 and plated on LB agar containing 100 μg•mL−1 ampicillin (Amp), 50 μg•mL−1 Kan, 1 mM IPTG, and 40 μg•ml−1 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) and grown for 24 h at 30 °C. Single colonies were picked to inoculate 2.5 mL LB broth supplemented with Amp (100 μg•mL−1), Kan (50 μg•mL−1), and IPTG (1 mM). Cultures were grown overnight to saturation (30 °C, 200 rpm) and used to inoculate fresh LB medium (Amp-Kan-IPTG). Once OD600 reached 0.4–0.5, β-galactosidase activity was measured as previously described (Karimova et al., 1998).
Molecular Dynamics Simulations
We constructed a model of full-length PhyR in its closed state using Modeller (Webb & Sali, 2002), with constraints provided by a crystal structure of full-length C. crescentus PhyR (PDB 3N0R) (Herrou et al., 2010). The structural template lacked the Mg2+ ion adjacent to the phosphorylation site required for response regulator aspartyl phosphorylation (Bourret, 2010). The Mg2+ ion and phosphorylation at residue D192 was modeled using parameters in the GAAMP resource (Huang & Roux, 2013). Protein structures were solvated in water with 15 Å of padding and neutralized with KCl using VMD (Humphrey et al., 1996). All simulations were carried out in NAMD 2.9 with the all-atom CHARMM22 + CMAP force field (Phillips et al., 2005, Brooks et al., 2009). Simulation parameters were identical to those previously described (Herrou et al., 2012). Systems were energy minimized for 1000 steps and predynamically equilibrated at 300 K in a constant-temperature, constant-pressure ensemble for 100 ps. A pressure of 1 atm was maintained with a Langevin piston of period 200 fs and a decay time 100 fs. Systems were energy minimized again for 30,000 steps and heated from 0 to 300 K for 300 ps in a periodic constant-temperature, constant-volume ensemble, reassigning temperature every 1 ps. 50 ns production simulations were performed with Langevin dynamics and periodic boundary conditions from prepared systems.
Umbrella Sampling
For two of the PhyR simulations described above (one unphosphorylated, one phosphorylated), the systems were forked into a steered MD simulation at 10 and 50 ns to sample conformations for varying inter-domain distances. Using the NAMD collective variables module (Fiorin et al., 2013), harmonic restraints with force constant of 15 kcal•mol−1•Å−2 were applied to the inter-domain distance defined as the distance between the center of mass of residues 1–120 (SL domain) and the center of mass of residues 140–266 (REC domain). The equilibrium value of harmonic restraints moved smoothly across the reaction coordinate at a rate of ~1 ns per 1 Å in order to sample initialization conformations for umbrella sampling. Conformations sampled from steered MD simulations were used to initialize umbrella sampling of inter-domain distance sampled from its initial value to 34 Å at a resolution of 0.25 Å (Torrie & Valleau, 1977). These initial conformations were heated from 0 to 300 K as described previously. Production simulations carried out with fixed harmonic restraints with a force constant of 15 kcal/mol/Å2. Simulations were run for 6 ns with the inter-domain distance sampled during the last 3 ns. Sampling frequency was 5 fs. These data were then used to generate free energy curves using the Weighted Histogram Analysis Method (WHAM) (Grossfield, Kumar et al., 1992). RMSF, RMSD, and cross-correlation values calculated using the R package bio3d (Grant et al., 2006). All simulations performed on the University of Chicago RCC Midway server. Structure analysis and rendering were carried out in PyMol and VMD (Humphrey et al., 1996, Schrödinger, 2015).
Supplementary Material
Abbreviated Summary.
Receiver (REC) proteins are a diverse class of regulators that control physiological responses in microbes and plants as a function of their phosphorylation state. The conserved REC protein, PhyR, is an anti-anti-σ factor that regulates stress-dependent transcription in Alphaproteobacteria. We have applied a combination of experimental and computational approaches to uncover structural determinants of PhyR activation.
Acknowledgments
This work was supported by the National Institutes of Health grants R01GM087353 and R01AI107159. We are grateful to Dr. Srinivas Chakravarthy and Advanced Photon Source BioCAT beamline 18ID for SEC-SAXS data collection time and assistance with data analysis. We also thank members of the Crosson Laboratory, in particular Jonathan Willett and Julien Herrou, for thoughtful discussion and insight on this project.
Footnotes
Author Contributions
J.L.L., D.S.E, and S.C conceived and designed experiments, analyzed results, and wrote the manuscript. J.L.L. and D.S.E. performed the experiments and J.R.S assisted with NMR experimental design, data collection, analysis, and interpretation. The authors have no conflicts of interest to declare.
References
- Alvarez-Martinez CE, Lourenco RF, Baldini RL, Laub MT, Gomes SL. The ECF sigma factor sigma(T) is involved in osmotic and oxidative stress responses in Caulobacter crescentus. Mol Microbiol. 2007;66:1240–1255. doi: 10.1111/j.1365-2958.2007.06005.x. [DOI] [PubMed] [Google Scholar]
- Ames SK, Frankema N, Kenney LJ. C-terminal DNA binding stimulates N-terminal phosphorylation of the outer membrane protein regulator OmpR from Escherichia coli. Proc Natl Acad Sci USA. 1999;96:11792–11797. doi: 10.1073/pnas.96.21.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. Regulation of response regulator autophosphorylation through interdomain contacts. J Bio Chem. 2010;285:32325–32335. doi: 10.1074/jbc.M110.157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CM, Stock AM. Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal Biochem. 2008;376:73–82. doi: 10.1016/j.ab.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A, Ikura M, Kay LE, Torchia DA, Tschudin R. Comparison of different modes of two-dimensional reverse-correlation NMR for the study of proteins. J Magn Reson. 1990;86:304–318. [Google Scholar]
- Berlow RB, Dyson HJ, Wright PE. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature. 2017;543:447–451. doi: 10.1038/nature21705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret RB. Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol. 2010;13:142–149. doi: 10.1016/j.mib.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux BY, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagne S, Damberger FF, Kaczmarczyk A, Francez-Charlot A, Allain FH-T, Vorholt JA. Structural basis for sigma factor mimicry in the general stress response of Alphaproteobacteria. Proc Natl Acad Sci USA. 2012;109:E1405–E1414. doi: 10.1073/pnas.1117003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagne S, Dintner S, Gottschlich L, Thibault M, Bortfeld-Miller M, Kaczmarczyk AA, et al. Role of the PFXFATG[G/Y] motif in the activation of SdrG, a response regulator involved in the Alphaproteobacterial general stress response. Structure. 2016;24:1237–1247. doi: 10.1016/j.str.2016.05.015. [DOI] [PubMed] [Google Scholar]
- Cho HS, Lee S-Y, Yan D, Pan X, Parkinson JS, Kustu S, Wemmer DE, Pelton JG. NMR structure of activated CheY. J Mol Biol. 2000;297:543–551. doi: 10.1006/jmbi.2000.3595. [DOI] [PubMed] [Google Scholar]
- Corrêa F, Gardner Kevin H. Basis of mutual domain inhibition in a bacterial response regulator. Cell Chem Biol. 2016;23:945–954. doi: 10.1016/j.chembiol.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa F, Ko W-H, Ocasio V, Bogomolni RA, Gardner KH. Blue light regulated two-component systems: enzymatic and functional analyses of light-oxygen-voltage (LOV)-histidine kinases and downstream response regulators. Biochemistry. 2013;52:4656–4666. doi: 10.1021/bi400617y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha /CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guzman RN, Martinez-Yamout MA, Dyson HJ, Wright PE. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J Biol Chem. 2004;279:3042–3049. doi: 10.1074/jbc.M310348200. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Eaton DS, Crosson SD, Fiebig A. Proper control of Caulobacter crescentus cell-surface adhesion requires the general protein chaperone, DnaK. J Bacteriol. 2016;198:2631–2642. doi: 10.1128/JB.00027-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig A, Herrou J, Willett J, Crosson S. General stress signaling in the Alphaproteobacteria. Annu Rev Genet. 2015;49:603–625. doi: 10.1146/annurev-genet-112414-054813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorin G, Klein ML, Hénin J. Using collective variables to drive molecular dynamics simulations. Molec Phys. 2013;111:3345–3362. [Google Scholar]
- Flechard M, Fontenelle C, Blanco C, Goude R, Ermel G, Trautwetter A. RpoE2 of Sinorhizobium meliloti is necessary for trehalose synthesis and growth in hyperosmotic media. Microbiology. 2010;156:1708–1718. doi: 10.1099/mic.0.034850-0. [DOI] [PubMed] [Google Scholar]
- Francez-Charlot A, Frunzke J, Reichen C, Ebneter JZ, Gourion B, Vorholt JA. Sigma factor mimicry involved in regulation of general stress response. Proc Natl Acad Sci USA. 2009;106:3467–3472. doi: 10.1073/pnas.0810291106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francez-Charlot A, Kaczmarczyk A, Fischer HM, Vorholt JA. The general stress response in Alphaproteobacteria. Trends Microbiol. 2015;23:164–171. doi: 10.1016/j.tim.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat Struct Biol. 2003;10:504–512. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA. 2002;99:5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Stock AM. Probing kinase and phosphatase activities of two-component systems in vivo with concentration-dependent phosphorylation profiling. Proc Natl Acad Sci USA. 2013;110:672–677. doi: 10.1073/pnas.1214587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Gourion B, Sulser S, Frunzke J, Francez-Charlot A, Stiefel P, Pessi G, Vorholt JA, Fischer H-M. The PhyR-σEcfG signalling cascade is involved in stress response and symbiotic efficiency in Bradyrhizobium japonicum. Mol Microbiol. 2009;73:291–305. doi: 10.1111/j.1365-2958.2009.06769.x. [DOI] [PubMed] [Google Scholar]
- Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Grossfield A. “WHAM: the weighted histogram analysis method,” version 2.0.9. [Google Scholar]
- Herrou J, Crosson S, Fiebig A. Structure and function of HWE/HisKA2-family sensor histidine kinases. Curr Opin Microbiol. 2017;36:47–54. doi: 10.1016/j.mib.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J, Foreman R, Fiebig A, Crosson S. A structural model of anti-anti-σ inhibition by a two-component receiver domain: the PhyR stress response regulator. Mol Microbiol. 2010;78:290–304. doi: 10.1111/j.1365-2958.2010.07323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J, Rotskoff G, Luo Y, Roux B, Crosson S. Structural basis of a protein partner switch that regulates the general stress response of α-proteobacteria. Proc Natl Acad Sci USA. 2012;109:E1415–E1423. doi: 10.1073/pnas.1116887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J, Willett JW, Crosson S. Structured and dynamic disordered domains regulate the activity of a multifunctional anti-σ factor. mBio. 2015;6:e00910. doi: 10.1128/mBio.00910-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Roux B. Automated force field parameterization for non-polarizable and polarizable atomic models based on ab initio target data. J Chem Theory Comput. 2013;9 doi: 10.1021/ct4003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hwang T-L, Zijl PCMv, Mori S. Accurate quantitation of water-amide proton exchange rates using the Phase-Modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J Biomol NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Kaczmarczyk A, Campagne S, Danza F, Metzger LC, Vorholt JA, Francez-Charlot A. Role of Sphingomonas sp. strain Fr1 PhyR-NepR-σEcfG cascade in general stress response and identification of a negative regulator of PhyR. J Bacteriol. 2011;193:6629–6638. doi: 10.1128/JB.06006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarczyk A, Hochstrasser R, Vorholt JA, Francez-Charlot A. Complex two-component signaling regulates the general stress response in Alphaproteobacteria. Proc Natl Acad Sci USA. 2014;111:E5196–5204. doi: 10.1073/pnas.1410095111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarczyk A, Hochstrasser R, Vorholt JA, Francez-Charlot A. Two-tiered histidine kinase pathway involved in heat shock and salt sensing in the general stress response of Sphingomonas melonis Fr1. J Bacteriol. 2015;197:1466–1477. doi: 10.1128/JB.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-S, Caswell CC, Foreman R, Roop RM, Crosson S. The Brucella abortus general stress response system regulates chronic mammalian infection and is controlled by phosphorylation and proteolysis. J Bio Chem. 2013;288:13906–13916. doi: 10.1074/jbc.M113.459305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-S, Willett JW, Jain-Gupta N, Fiebig A, Crosson S. The Brucella abortus virulence regulator, LovhK, is a sensor kinase in the general stress response signalling pathway. Mol Microbiol. 2014;94:913–925. doi: 10.1111/mmi.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding Tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Konrat R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J Magn Reson. 2014;241:74–85. doi: 10.1016/j.jmr.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosol S, Contreras-Martos S, Cedeño C, Tompa P. Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules. 2013;18:10802. doi: 10.3390/molecules180910802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragelj J, Ozenne V, Blackledge M, Jensen MR. Conformational propensities of intrinsically disordered proteins from NMR chemical shifts. ChemPhysChem. 2013;14:3034–3045. doi: 10.1002/cphc.201300387. [DOI] [PubMed] [Google Scholar]
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- Li G-W, Burkhardt D, Gross C, Weissman Jonathan S. Quantifying Absolute Protein Synthesis Rates Reveals Principles Underlying Allocation of Cellular Resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco RF, Kohler C, Gomes SL. A two-component system, an anti-sigma factor and two paralogous ECF sigma factors are involved in the control of general stress response in Caulobacter crescentus. Mol Microbiol. 2011;80:1598–1612. doi: 10.1111/j.1365-2958.2011.07668.x. [DOI] [PubMed] [Google Scholar]
- Mertens HDT, Svergun DI. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J Struct Biol. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T-2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa EC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader JM, Li GW, Childers WS, Perez AM, Weissman JS, Shapiro L, McAdams HH. Dynamic translation regulation in Caulobacter cell cycle control. Proc Natl Acad Sci U S A. 2016;113:E6859–E6867. doi: 10.1073/pnas.1614795113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader JM, Zhou B, Li GW, Lasker K, Childers WS, Williams B, et al. The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014;10:e1004463. doi: 10.1371/journal.pgen.1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L. The PyMOL Molecular Graphics System, Version 1.8. 2015. [Google Scholar]
- Schuster M, Silversmith RE, Bourret RB. Conformational coupling in the chemotaxis response regulator CheY. Proc Natl Acad Sci USA. 2001;98:6003–6008. doi: 10.1073/pnas.101571298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheftic SR, White E, Gage DJ, Alexandrescu AT. NMR Structure of the HWE Kinase Associated Response Regulator Sma0114 in Its Activated State. Biochemistry. 2014;53:311–322. doi: 10.1021/bi401497h. [DOI] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- Svergun D, Barberato C, Koch MHJ. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- Sycz G, Carrica MC, Tseng TS, Bogomolni RA, Briggs WR, Goldbaum FA, Paris G. LOV histidine kinase modulates the general stress response system and affects the virB operon expression in Brucella abortus. PLoS One. 2015;10:e0124058. doi: 10.1371/journal.pone.0124058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J Comput Phys. 1977;23:187–199. [Google Scholar]
- Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Prot Bioinf. 2002 doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett JW, Kirby JR. Genetic and biochemical dissection of a HisKA domain identifies residues required exclusively for kinase and phosphatase activities. PLOS Genetics. 2012;8:e1003084. doi: 10.1371/journal.pgen.1003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.