

Abstract
This paper describes a simple, rapid and sensitive liquid chromatography–tandem mass spectrometry assay for the determination of duloxetine in human plasma. A duloxetine stable labeled isotope (duloxetine d5) was used as an internal standard. Analyte and the internal standard were extracted from 100 μL of human plasma via solid phase extraction technique using Oasis HLB cartridges. The chromatographic separation was achieved on a C18 column by using a mixture of acetonitrile–5 mM ammonium acetate buffer (83:17, v/v) as the mobile phase at a flow rate of 0.9 mL/min. The calibration curve obtained was linear (r2≥0.99) over the concentration range of 0.05–101 ng/mL. Multiple-reaction monitoring mode (MRM) was used for quantification of ion transitions at m/z 298.3/154.1 and 303.3/159.1 for the drug and the internal standard, respectively. Method validation was performed as per FDA guidelines and the results met the acceptance criteria. A run time of 2.5 min for each sample made it possible to analyze more than 300 plasma samples per day. The proposed method was found to be applicable to clinical studies.
Keywords: Duloxetine in human plasma, Solid-phase extraction (SPE), Liquid chromatography–tandem mass spectrometry, Method validation, Pharmacokinetic studies
1. Introduction
Duloxetine hydrochloride (CAS no.: 136434-34-9), is a balanced selective serotonin and norepinephrine reuptake inhibitor. The drug is used in the treatment for major depression [1], pain related to diabetic peripheral neuropathy [2], [3], and stress urinary incontinence [4]. Duloxetine has a low affinity toward serotonergic, cholinergic, adrenergic, and histamine receptors, and this specificity of action accounts for its greater safety profile with respect to tricyclic antidepressants [5], [6], [7]. Usually duloxetine is administered in the form of capsules dosage form containing 20, 30 or 60 mg of active constituent in enteric-coated pellets and the most common doses for the treatment of major depression are 40–60 mg daily.
As per the literature, numerous analytical methods have been reported for the determination of duloxetine which include liquid chromatography–tandem mass spectrometric methods (LC–MS/MS) [7], [8], [9], [10], [11], [12], liquid chromatography with single-quadrupole mass spectrometric method (LC–MS) [13], [14], gas chromatographic mass spectrometric method [15], capillary electrophoresis method [16] and high performance liquid chromatographic (HPLC) methods [17], [18]. Of all the above, only five methods [8], [9], [12], [13], [14] are comparable with the present work. The method proposed by Ma et al. [13] and Choong et al. [14] described a single-quadrupole mass spectrometry (LC–MS) with selected-ion monitoring (SRM) mode to detect the precursor ion. But in the present method, a triple-quadrupole mass spectrometry (LC–MS/MS) with multiple-reaction monitoring (MRM) mode was used to detect both the precursor ion and fragment ion. It shows that the proposed method is highly specific. Moreover, the method proposed by Choong et al. [14] utilizes multi-step solid-phase extraction with an LLOQ of 2 ng/mL which is not sensitive enough and involving complexity like gradient elution, typical mobile phase consisting of two or more buffers with the pH adjustment, longer chromatographic run time (>13 min). Another method was reported by Selvan et al. [8] for the determination of duloxetine in human plasma with plasma concentration range of 0.1–50 ng/mL by using liquid chromatography with atmospheric pressure ionization–tandem mass spectrometry. This method employs protein precipitation (PP) method for the sample preparation. PP is most likely to cause ion suppression, since this method fails to sufficiently remove endogenous compounds such as lipids, phospholipids, fatty acids, etc. [19], [20], [21]. Recently Reddy et al. [12] reported an LC–MS/MS method for the determination of duloxetine in 300 μL of human plasma with an LLOQ of 0.1 ng/mL, this method employs liquid–liquid extraction (LLE), evaporation, drying and reconstitution for sample preparation. The salient features of chromatographic methods developed for duloxetine in human plasma are summarized in Table 1.
Table 1.
Salient features of LC–MS methods developed for duloxetine in human plasma.
| Sr. no. | Column; mobile phase; flow rate; injection volume | Extraction procedure; sample volume; internal standard; mean recovery (duloxetine) | Detection technique; linear dynamic range; analytical run time; retention time; application | Ref. |
|---|---|---|---|---|
| 1 | Phenomenex C18 (250 mm×4.6 mm i.d., 5 μm); acetonitrile–0.2% formic acid, 5 mM ammonium acetate (55:45, v/v); 0.90 mL/min; 20 μL | PP with acetonitrile; 0.30 mL; fluoxetine; 87.2% | LC–MS/MS; 0.89–106.8 ng/mL; 10 min; 6.6 min; Pharmacokinetic study in 30 healthy Chinese volunteers | [7] |
| 2 | Gemini–C18 (50 mm×4.6 mm i.d., 3 μm); acetonitrile–5 mM ammonium acetate (45:55, v/v, pH 3.5); 0.30 mL/min; 5 μL | PP with methanol; 0.20 mL; Haloperidol; 86.9% | LC–MS/MS; 0.1–50 ng/mL; 5 min; 1.57 min; Pharmacokinetic study in 12 healthy volunteers | [8] |
| 3 | X–terra RP8 (50 mm×4.6 mm i.d., 5 μm); acetonitrile–30 mM ammonium formate (90:10, v/v, pH 5.0); 0.40 mL/min; 20 μL | LLE with MTBE–n-hexane (80:20, v/v); 0.30 mL; fluoxetine; 80.3% | LC–MS/MS; 0.1–100 ng/mL; 3 min; 1.51 min; Pharmacokinetic study in 12 healthy volunteers | [12] |
| 4 | Thermo Hypersil–Hypurity C18 (150 mm×2.1 mm, i.d., 5 μm); acetonitrile–methanol–20 mM ammonium acetate pH 3.5 (42:20:38, v/v/v); 0.24 mL/min; 10 μL | PP with acetonitrile; 0.20 mL; flupentixol; 83.5% | LC–MS; 0.8–100 ng/mL; 4 min; 2.0 min; Pharmacokinetic study in 12 healthy Chinese male volunteers | [13] |
| 5 | Xbridge C18 (100 mm×2.1 mm; i.d., 3.5 μm); acetonitrile–20 mM ammonium acetate (pH 8.1) gradient mode; 0.30 mL/min; 5 μL | SPE with Oasis MCX cartridges; 0.50 mL; remoxipride; 92.0% | HPLC–MS; 2–200 ng/mL; 13 min; 8.4 min; Pharmacokinetic study in 8 patients | [14] |
| 6 | Zorbax SB C18 (50 mm×2.1 mm, i.d., 5 μm); acetonitrile–5 mM ammonium acetate buffer (83:17, v/v); 0.90 mL/min; 10 μL | SPE with Oasis HLB cartridges; 0.10 mL; duloxetine d5; 86.7% | LC–MS/MS; 0.05–101 ng/mL; 2.5 min; 1.1 min; Pharmacokinetic study in 6 healthy male Indian volunteers | PM |
LLE, liquid–liquid extraction; PP, protein precipitation; SPE, solid phase extraction; MTBE, methyl tert butyl ether; PM, present method.
The present work describes a simple, selective and sensitive method, which employs solid phase extraction (SPE) technique (SPE) for sample preparation and liquid chromatography with electrospray ionization–tandem mass spectrometry for quantitation of duloxetine in 100 μL of human plasma. SPE is the most popular sample pre-treatment approach nowadays due to following advantages: high recovery, effective pre-concentration, the need for less organic solvent (compared to LLE), no foaming in the formation of emulsions, ease of operation and greater possibility of automation [19–21]. The method uses duloxetine d5 as an internal standard (IS). The use of stable labeled isotopes of the duloxetine as an IS increases the assay precision and limits variable recovery between the analyte and the IS. Application of this assay method to a clinical pharmacokinetic study in healthy male volunteers following oral administration of duloxetine is described. The authenticity in the measurement of study data is demonstrated through incurred samples reanalysis (ISR).
2. Experimental
2.1. Chemicals
Duloxetine hydrochloride reference standard (99.6% pure) was obtained from Hetero Labs Limited (Hyderabad, India). Duloxetine d5 oxalate (100.0% pure) was employed as an IS and was obtained from Clearsynth Labs Limited (Mumbai, India). Their structures are shown in Fig. 1. HPLC grade acetonitrile was purchased from J.T. Baker (Phillipsburg, USA). Analytical grade formic acid and ammonium acetate were purchased from Merck Ltd (Mumbai, India). Water used for the LC–MS/MS analysis was prepared by using Milli Q water purification system procured from Millipore (Bangalore, India). The control human plasma sample was procured from Deccan's Pathological Lab's (Hyderabad, India).
Fig. 1.
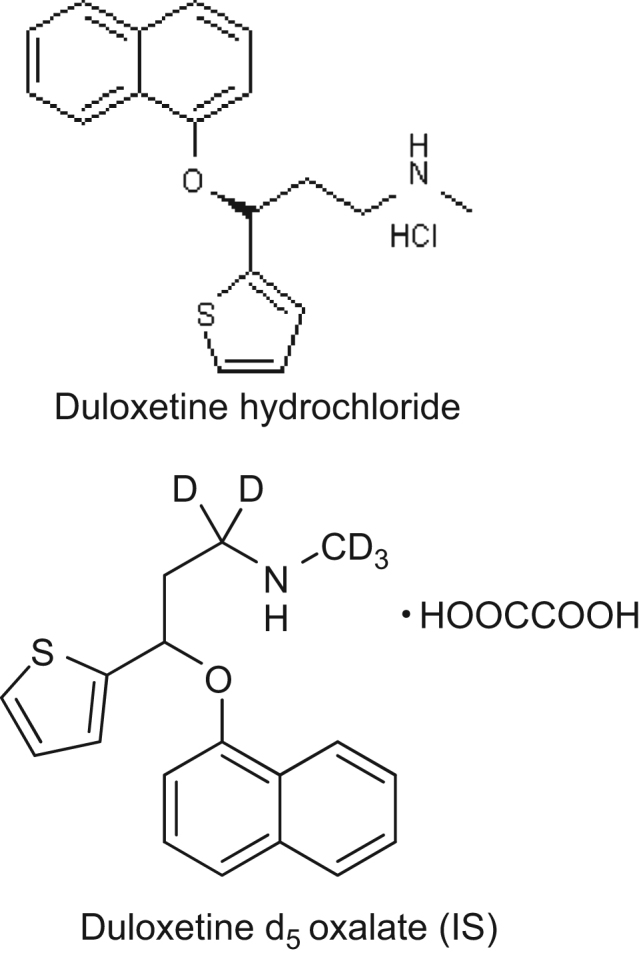
Chemical structures of duloxetine hydrochloride and duloxetine d5 oxalate (IS).
2.2. LC–MS/MS instrument and conditions
An HPLC system (Shimadzu, Kyoto, Japan) consisting of a Zorbax SB C18 column (50 mm×2.1 mm, 5 μm; Agilent Technologies, Santa Clara, CA, USA), a binary LC-20AD prominence pump, an auto-sampler (SIL-HTc) and a solvent degasser (DGU-20A3) was used for the study. Aliquot of 10 μL of the processed samples was injected into the column, which was kept at 40 °C. An isocratic mobile phase consisting of a mixture of acetonitrile–5 mM ammonium acetate buffer (83:17, v/v) was used to separate the analyte from the endogenous components and delivered at a flow rate of 0.9 mL/min into the electrospray ionization chamber of the mass spectrometer. Quantification was achieved with MS–MS detection in positive ion mode for the analyte and the IS using an MDS Sciex API-4000 mass spectrometer (Foster City, CA, USA) equipped with a Turboionspray™ interface at 500 °C. The ion spray voltage was set at 5500 V. The source parameters viz. the nebulizer gas (GS1), auxiliary gas (GS2), curtain gas and collision gas were set at 40, 42, 20, and 6 psi, respectively. The compound parameters viz. the declustering potential (DP), collision energy (CE), entrance potential (EP) and collision cell exit potential (CXP) were 18, 10, 10, 10 V for duloxetine and 18, 9, 10, 9 V for the IS. Detection of the ions was carried out in the MRM mode, by monitoring the transition pairs of m/z 298.3 precursor ion to the m/z 154.1 for duloxetine and m/z 303.3 precursor ion to the m/z 159.1 product ion for the IS. Quadrupoles Q1 and Q3 were set on unit resolution. The analysis data obtained were processed by Analyst Software™ (version 1.4.2).
2.3. Preparation of plasma standards and quality controls
Standard stock solutions of duloxetine and IS (1 mg/mL) were prepared in methanol. Working solutions for calibration and controls were prepared by appropriate dilution in water–methanol (60:40, v/v; diluent). The IS working solution (200 ng/mL) was prepared by diluting its stock solution with diluent.
Calibration samples were prepared by spiking 950 μL of control human plasma with the 50 μL working standard solution of the analyte as a bulk, to obtain duloxetine concentration levels of 0.05, 0.10, 0.51, 2.53, 5.05, 10.1, 20.2, 40.4, 80.9 and 101 ng/mL as a single batch at each concentration. Similarly, quality control (QC) samples were also prepared as a bulk based on an independent weighing of standard drug, at concentrations of 0.05 (LLOQ), 0.15 (low), 15.7 (middle 1), 50.6 (middle 2) and 90.2 ng/mL (high) as a single batch at each concentration. The calibration and control bulk samples were divided into aliquots in micro centrifuge tubes (Tarson, 2 mL) and stored in the freezer at −70±10 °C until analyses.
2.4. Sample processing
A 100 μL aliquot of human plasma sample was mixed with 10 μL of the IS working solution (200 ng/mL of duloxetine d5). To this, 25 μL of 0.1% formic acid solution was added after vortex mixing for 10 s. The sample mixture was loaded onto an Oasis HLB cartridge (30 mg/mL) that was pre-conditioned with 1.0 mL of methanol followed by 1.0 mL of water. The extraction cartridge was washed with 1.0 mL of 0.1% formic acid solution followed by 1.0 mL of 5% methanol. Analyte and IS were eluted with 0.5 mL of mobile phase. Aliquot of 10 μL of the extract was injected into the chromatographic system.
2.5. Bioanalytical method validation
The validation of the above method was carried out as per US FDA guidelines [22]. The parameters determined were selectivity, specificity, sensitivity, matrix effect, linearity, precision, accuracy, recovery, dilution integrity and stability. Selectivity was assessed by comparing the chromatograms of six different batches of blank plasma obtained from six different sources including one lipemic and hemolyzed plasma. Potential interference from acetaminophen, diphenhydramine, pantoprazole, nicotine, ibuprofen, caffeine and pseudoephedrine was evaluated. Sensitivity was determined by analyzing six replicates of plasma samples spiked with the lowest level of the calibration curve concentrations. Matrix effect was checked with six different lots of K2EDTA plasma. Three replicate samples each of LQC and HQC were prepared from different lots of plasma (36 QC samples in total). For checking the linearity standard calibration curves containing at least 10 points (non-zero standards) were plotted. In addition, blank plasma samples were also analyzed to confirm the absence of direct interferences. Intra-day precision and accuracy were determined by analyzing six replicates at five different QC levels on two different days. Inter-day precision and accuracy were determined by analyzing six replicates at five different QC levels of five different runs. Recoveries of analyte and IS were determined by comparing the peak area of extracted analyte standard with the peak area of non-extracted standard. Recoveries of duloxetine were determined at concentrations of 0.15 (low), 50.6 (middle 2) and 90.2 (high) ng/mL, whereas for IS was determined at a concentration of 200 ng/mL. Dilution integrity was performed to extend the upper concentration limit with acceptable precision and accuracy. Six replicates each at a concentration of about 1.7 times of the uppermost calibration standard were diluted two- and four-fold with blank plasma. The diluted samples were processed and analyzed.
Stability tests were conducted to evaluate the analyte stability in stock solutions and in plasma samples under different conditions. The stock solution stability at room temperature and refrigerated conditions (2–8 °C) was performed by comparing the area response of the analyte (stability samples) with the response of the sample prepared from fresh stock solution. Bench top stability (8 h), processed samples stability (autosampler stability for 48 h, wet extract stability for 24 h and reinjection stability for 24 h), freeze–thaw stability (three cycles), long-term stability (58 days) were performed at LQC and HQC levels using six replicates at each level. Samples were considered to be stable if assay values were within the acceptable limits of accuracy (±15% SD) and precision (≤15% RSD).
2.6. Pharmacokinetic study design
A pharmacokinetic study was performed in healthy male subjects (n=6). The Ethics Committee (Hyderabad Independent Ethics Committee, Hyderabad, India) approved the protocol and the volunteers provided with informed written consent. The subjects were fasted 10 h before administration of the drug formulation. Blood samples were collected following oral administration of duloxetine hydrochloride (60 mg) at pre-dose and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.33, 4.67, 5, 5.33, 5.67, 7, 7.5, 8, 9, 10, 12, 24, 36, 48, and 72 h, post-dose in K2EDTA vacutainer collection tubes (BD, Franklin, NJ, USA). The tubes were centrifuged at 3200 rpm for 10 min and the plasma was collected. The collected plasma samples were stored at −70±10 °C till their use. Plasma samples were spiked with the IS and processed as per the extraction procedure described earlier. Along with the clinical samples, the QC samples at low, middle 1, middle 2 and high concentration levels were also assayed in triplicate. Plasma concentration–time profile of duloxetine was analyzed by non-compartmental method using WinNonlin Version 5.1. An incurred sample reanalysis was also conducted by selecting the 12 subject samples (2 samples from each subject) near Cmax and the elimination phase. The percent change in the value should not be more than ±20% [23].
3. Results and discussion
3.1. Method development
To develop a rapid, sensitive and simple assay method for the extraction and quantification of duloxetine during method development different options were evaluated to optimize detection and chromatography parameters. The inherent selectivity of MS/MS detection was also expected to be beneficial in developing a selective and sensitive method. Protonated form of analyte and IS, [M+H]+ ion was the parent ion in the Q1 spectrum and was used as the precursor ion to obtain Q3 product ion spectra. The most sensitive mass transition was observed from m/z 298.3 to 154.1 for duloxetine and from m/z 303.3 to 159.1 for the IS. As earlier publications have discussed the details of fragmentation patterns of duloxetine [12], we are not presenting the data pertaining to this. LC–MRM is a very powerful technique for pharmacokinetic studies since it provides sensitivity and selectivity requirements for analytical methods [24]. Thus, the MRM technique was chosen for the assay development. The MRM state file parameters were optimized at a concentration of 50 ng/mL to maximize the response for the analyte.
Chromatographic conditions, especially the composition of the mobile phase, column type, flow rate and column oven temperature were optimized through several trials to achieve good resolution and increased intensity of the signals of the analyte and IS, as well as for the short run time. Separation was attempted using various combinations of methanol, acetonitrile and buffer with varying contents of each component on different columns like C8 and C18 of different makes like Grace, Chromolith, BDS Hypersil, Hypurity advance, Zorbax, Kromasil, Ace and Intertsil etc. It was found that a mixture of acetonitrile and 5 mM ammonium acetate (83:17, v/v) could achieve this purpose and was finally adopted as the mobile phase. Zorbax SB C18 column (50 mm×2.1 mm, 5 μm) gave good peak shape and response even at lowest concentration level for the analyte and IS. The mobile phase was operated at a flow rate of 0.9 mL/min. The retention time of analyte and the IS were low enough (1.13 min) allowing a small run time of 2.5 min.
Due to high drug protein binding, protein precipitation (PP) was tried initially using acetonitrile and methanol as precipitating agents but the response was inconsistent especially at the LLOQ level. Thus, the simple SPE technique was employed for the sample preparation in this work and provided high recoveries of the drugs. The use of stable labeled isotopes of the analyte as an IS is recommended for bioanalytical assays to increase assay precision and limit variable recovery between analyte and the IS [25], [26]. For an LC–MS/MS analysis, utilization of stable isotope-labeled drugs as IS proves to be helpful when a significant matrix effect is possible. At the initial stages of this work, several compounds were investigated to find a suitable IS and finally duloxetine stable labeled isotope duloxetine d5 was found to be best for the present purpose.
3.2. Selectivity and chromatography
The selectivity of the method was examined by analyzing blank human plasma extract (Fig. 2A) and an extract spiked only with the IS (Fig. 2B). As shown in Fig. 2A, no significant direct interference in the blank plasma traces was observed from endogenous substances in drug-free human plasma at the retention time of the analyte and IS. Similarly, Fig. 2B shows the absence of direct interference from the IS to the MRM channel of the analyte. Fig. 2C depicts a representative ion-chromatogram for the LLOQ sample (0.05 ng/mL). Similarly, no interference was observed from commonly used medications such as acetaminophen, diphenhydramine, pantoprazole, nicotine, ibuprofen, caffeine and pseudoephedrine (data not shown). A representative chromatogram resulting from the analysis of subject blank plasma sample and 5.67 h subject plasma sample after the administration of a 60 mg oral single dose of duloxetine is shown in Fig. 3.
Fig. 2.

Typical MRM chromatograms of duloxetine (left panel) and IS (right panel) in human blank plasma (A), human plasma spiked with IS (B), and an LLOQ sample along with IS (C).
Fig. 3.

MRM chromatograms resulting from the analysis of subject blank plasma sample (A) and 5.67 h subject plasma sample (B), after the administration of a 60 mg oral single dose of duloxetine. The sample concentration was determined to be 48.2 ng/mL.
3.3. Sensitivity
The lowest limit of reliable quantification for the analyte was set at the concentration of the LLOQ. The precision and accuracy of analyte at LLOQ concentration was found to be 7.60% and 97.8%, respectively.
3.4. Matrix effect
Matrix effect assessment was done with the aim to check the effect of different lots of plasma on the back calculated value of QC's nominal concentration. The results found were well within the acceptable limits as shown in Table 2. No significant matrix effect was observed in all the six batches of human plasma for the analyte at low and high quality control concentrations. Also, the extraction method was rugged enough and gave accurate and consistent results when applied to real subject samples.
Table 2.
Matrix effect of duloxetine in human plasma (n=3).
| Plasma lot | LQC (0.15 ng/mL) |
HQC (90.2 ng/mL) |
||
|---|---|---|---|---|
| Concentration found (mean±SD; ng/mL) | Accuracy (%) | Concentration found (mean±SD; ng/mL) | Accuracy (%) | |
| Lot 1 | 0.17±0.01 | 109.67 | 96.20±3.60 | 106.61 |
| Lot 2 | 0.170±0.002 | 113.71 | 96.68±1.71 | 107.14 |
| Lot 3 | 0.160±0.003 | 107.72 | 98.32±1.81 | 108.96 |
| Lot 4 | 0.16± 0.02 | 106.28 | 95.69±0.56 | 106.04 |
| Lot 5 | 0.17±0.01 | 109.84 | 99.45±2.38 | 110.21 |
| Lot 6 | 0.14±0.01 | 95.51 | 97.51±2.45 | 108.05 |
3.5. Linearity
The ten-point calibration curve was found to be linear over the concentration range of 0.05–101 ng/mL for duloxetine. After comparing the two weighting models (1/x and 1/x2), a regression equation with a weighting factor of 1/x2 of the drug to the IS concentration was found to produce the best fit for the concentration–detector response relationship. The mean correlation coefficient of the weighted calibration curves generated during the validation was ≥0.99.
3.6. Precision and accuracy
The results for intra-day and inter-day precision and accuracy in plasma quality control samples are summarized in Table 3. The intra-day and inter-day precision deviation values were all within 15% of the relative standard deviation (RSD) at low, middle 1, middle 2 and high quality control levels, whereas within 20% at LLOQ QCs level. The intra-day and inter-day accuracy deviation values were all within 100±15% of the actual values at low, middle 1, middle 2 and high quality control levels, whereas within 100±20% at LLOQ QCs level. The results revealed good precision and accuracy.
Table 3.
Precision and accuracy data for duloxetine.
| Quality control | Run | Concentration found (mean±SD; ng/mL) | Precision (%) | Accuracy (%) |
|---|---|---|---|---|
| Intra-day variations (n=6) | ||||
| LLOQ | 1 | 0.056±0.002 | 4.20 | 110.48 |
| 2 | 0.057±0.006 | 9.81 | 111.39 | |
| 3 | 0.051±0.004 | 7.19 | 100.62 | |
| 4 | 0.049±0.002 | 4.00 | 96.69 | |
| 5 | 0.051±0.002 | 4.39 | 100.75 | |
| LQC | 1 | 0.147±0.002 | 1.38 | 97.41 |
| 2 | 0.153±0.007 | 4.14 | 101.46 | |
| 3 | 0.152±0.004 | 2.64 | 101.01 | |
| 4 | 0.155±0.004 | 2.32 | 102.80 | |
| 5 | 0.154±0.006 | 4.07 | 102.30 | |
| MQC1 | 1 | 15.17±0.32 | 2.10 | 96.86 |
| 2 | 14.64±0.61 | 4.18 | 93.45 | |
| 3 | 15.79±0.31 | 1.94 | 100.79 | |
| 4 | 15.86±0.13 | 0.82 | 101.26 | |
| 5 | 15.97±0.21 | 1.30 | 101.98 | |
| MQC2 | 1 | 48.86±1.06 | 2.17 | 96.61 |
| 2 | 48.93±1.79 | 3.66 | 96.73 | |
| 3 | 50.36±0.60 | 1.19 | 99.57 | |
| 4 | 50.41±0.76 | 1.51 | 99.67 | |
| 5 | 46.65±0.90 | 1.93 | 92.23 | |
| HQC | 1 | 90.87±3.15 | 3.46 | 100.70 |
| 2 | 92.18±1.64 | 1.78 | 102.16 | |
| 3 | 95.49±0.93 | 0.97 | 105.82 | |
| 4 | 94.97±0.83 | 0.87 | 105.24 | |
| 5 | 94.71±1.28 | 1.35 | 104.95 | |
| Inter-day variations (n=30) | ||||
| LLOQ | 0.053±0.004 | 8.32 | 103.99 | |
| LQC | 0.152±0.005 | 3.53 | 101.00 | |
| MQC1 | 15.49±0.61 | 3.95 | 98.87 | |
| MQC2 | 49.04±1.73 | 3.53 | 96.96 | |
| HQC | 93.64±2.46 | 2.63 | 103.77 | |
Spiked concentrations of LLOQ, LQC, MQC1, MQC2 and HQC are 0.05, 0.15, 15.66, 50.58 and 90.24 ng/mL, respectively.
3.7. Extraction efficiency
Six replicates at low, medium and high quality control concentration for duloxetine were prepared for recovery determination. SPE with HLB cartridges was proved to be robust and provided the cleanest samples. The mean overall recovery of duloxetine was 86.73±1.37% with the precision range of 1.11–3.58% and the recovery of the IS was 85.01% with the precision range of 1.42–1.70%. The recoveries of analyte and IS were good and reproducible. Therefore, the assay has been proved to be robust in high throughput bioanalysis.
3.8. Dilution integrity
The upper concentration limit of duloxetine can be extended to 172 ng/mL for by 1/2 and 1/4 dilutions with screened human blank plasma. The mean back-calculated concentrations for 1/2 and 1/4 dilution samples were within 85–115% of their nominal value. The coefficients of variation (%CV) for 1/2 and 1/4 dilution samples were less than 5%.
3.9. Stability studies
In the different stability experiments carried out viz. bench top stability (8 h), autosampler stability (48 h), freeze–thaw stability (3 cycles), reinjection stability (24 h), wet extract stability (24 h at 2–8 °C) and long–term stability at −70 °C for 58 days the mean % nominal values of the analyte were found to be within ±15% of the predicted concentrations for the analyte at their LQC and HQC levels (Table 4). Thus, the results were found to be within the acceptable limits during the entire validation.
Table 4.
Stability data for duloxetine in plasma (n=6).
| Stability test | QC (spiked concentration, ng/mL) | Mean±SD (ng/mL) | Accuracy/stability (%) | Precision (%) |
|---|---|---|---|---|
| A autosampler stability (at 10 °C for 48 h) | 0.15 | 0.147±0.006 | 97.37 | 4.21 |
| 90.24 | 93.25±0.78 | 103.45 | 0.84 | |
| Wet extract stability (at 2–8 °C for 24 h) | 0.15 | 0.149±0.004 | 98.91 | 2.54 |
| 90.24 | 83.35±1.50 | 92.36 | 1.80 | |
| Bench top stability (8 h at room temperature) | 0.15 | 0.155±0.004 | 103.11 | 2.30 |
| 90.24 | 92.62±0.51 | 102.64 | 0.55 | |
| Freeze–thaw stability (three cycles) | 0.15 | 0.146±0.002 | 96.60 | 1.69 |
| 90.24 | 83.54±1.17 | 92.58 | 1.40 | |
| Reinjection stability (24 h) | 0.15 | 0.140±0.009 | 93.08 | 6.24 |
| 90.24 | 87.61±5.71 | 97.09 | 6.51 | |
| Long-term stability (at −70 °C for 58 days) | 0.15 | 0.156±0.018 | 103.23 | 11.8 |
| 90.24 | 91.52±4.80 | 101.42 | 5.24 | |
Stock solutions of duloxetine and IS were found to be stable for 15 days at 2–8 °C. The percentage stability (with the precision range) of duloxetine and IS was 101% (1.18–1.32%) and 97.7% (0.54–1.96%), respectively.
3.10. Pharmacokinetic study results
In order to verify the sensitivity and selectivity of this method in a real-time situation, the present method was used to test for duloxetine in human plasma samples collected from healthy male volunteers (n=6). The mean plasma concentrations vs. time profile of duloxetine is shown in Fig. 4 and corresponding pharmacokinetic parameters are listed in Table 5. These values were in close proximity when compared with earlier reported values [7].
Fig. 4.

Mean plasma concentration–time profile of duloxetine in human plasma following oral administration of duloxetine hydrochloride (60 mg capsule) to healthy volunteers (n=6).
Table 5.
Pharmacokinetic parameters of duloxetine (n=6, mean±SD).
| Parameter | Estimated value |
|---|---|
| Cmax (ng/mL) | 48.45±8.27 |
| tmax (h) | 5.50±0.86 |
| AUC0−t (ng h/mL) | 988±205 |
| AUC0−inf (ng h/mL) | 1024±221 |
| t1/2 (h) | 13.80±1.99 |
3.11. Incurred sample reanalysis
Since, the FDA has introduced the necessity of incurred sample reanalysis evaluation at the Crystal City III meeting [27], it is necessary to demonstrate assay reproducibility by using dosed subject samples. Incurred sample reanalysis was performed using two plasma samples from each subject and re-assayed in a separate batch run. The differences in concentrations between the incurred sample reanalysis and the initial values for all the tested samples were less than 10% (Table 6), indicating good reproducibility of the present method.
Table 6.
Incurred samples reanalysis data of duloxetine.
| Sample | Initial conc. (ng/mL) | Re-assay conc. (ng/mL) | Differencea (%) |
|---|---|---|---|
| 1 | 46.21 | 45.23 | −2.14 |
| 2 | 0.47 | 0.43 | −7.64 |
| 3 | 39.70 | 38.65 | −2.69 |
| 4 | 5.98 | 6.26 | 4.69 |
| 5 | 54.65 | 55.96 | 2.36 |
| 6 | 2.46 | 2.30 | −6.46 |
| 7 | 40.25 | 42.64 | 5.77 |
| 8 | 4.57 | 4.76 | 4.20 |
| 9 | 49.22 | 48.38 | −1.72 |
| 10 | 2.40 | 2.59 | 7.39 |
| 11 | 33.50 | 36.47 | 8.49 |
| 12 | 0.92 | 0.83 | −9.63 |
Expressed as [(initial conc.−re-assay conc.)/average]×100%.
4. Conclusions
The LC–MS/MS assay presented in this paper is simple, rapid, specific and sensitive for quantification of duloxetine in human plasma and is fully validated according to commonly acceptable FDA guidelines. The method showed suitability for pharmacokinetic studies in humans. The simple SPE method gave consistent and reproducible recoveries for the analytes from plasma. The method provided good linearity. A sample turnover rate of less than 2.5 min makes it an attractive procedure in high-throughput bioanalysis of duloxetine. From the results of all the validation parameters, we can conclude that the developed method can be useful for bioavailability and bioequivalence (BA/BE) studies and routine therapeutic drug monitoring with the desired precision and accuracy.
Acknowledgments
The authors gratefully acknowledge Wellquest Clinical Research Laboratories for providing necessary facilities to carry out this work.
Footnotes
Peer review under responsibility of Xi'an Jiaotong University.

Contributor Information
Jaswanth Kumar Inamadugu, Email: jaswanth.kumarreddy@gmail.com.
Nageswara Rao Pilli, Email: nagr_80@yahoo.co.in.
References
- 1.Hunziker M.E., Suehs B.T., Bettinger T.L. Duloxetine hydrochloride: a new dual-acting medication for the treatment of major depressive disorder. Clin. Ther. 2005;27:1126–1143. doi: 10.1016/j.clinthera.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Turcotte J.E., Debonnel G., Montigny C.D. Assessment of the serotonin and norepinephrine reuptake blocking properties of duloxetine in healthy subjects. Neuropsychopharmacology. 2001;24:511–521. doi: 10.1016/S0893-133X(00)00220-7. [DOI] [PubMed] [Google Scholar]
- 3.Smith T.R. Duloxetine in diabetic neuropathy. Expert Opin. Pharmacother. 2006;7:215–223. doi: 10.1517/14656566.7.2.215. [DOI] [PubMed] [Google Scholar]
- 4.Westanmo A.D., Gayken J., Haight R. Duloxetine: a balanced and selective norepinephrine- and serotonin-reuptake inhibitor. Am. J. Health Syst. Pharm. 2005;62:2481–2490. doi: 10.2146/ajhp050006. [DOI] [PubMed] [Google Scholar]
- 5.Mallinckrodt C.H., Prakash A., Andorn A.C. Duloxetine for the treatment of major depressive disorder: a closer look at efficacy and safety data across the approved dose range. J. Psychiatr. Res. 2006;40:337–348. doi: 10.1016/j.jpsychires.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Hirschfeld R.M., Vornik L.A. Recognition and diagnosis of bipolar disorder. J. Clin. Psychiatry. 2004;65:5–9. [PubMed] [Google Scholar]
- 7.Zhao R.K., Cheng G., Tang J. Pharmacokinetics of duloxetine hydrochloride enteric-coated tablets in healthy Chinese volunteers: a randomized, open-label, single- and multiple-dose study. Clin. Ther. 2009;31:1022–1036. doi: 10.1016/j.clinthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Selvan P.S., Gowda K.V., Mandal U. Determination of duloxetine in human plasma by liquid chromatography with atmospheric pressure ionization-tandem mass spectrometry and its application to pharmacokinetic study. J. Chromatogr. B. 2007;858:269–275. doi: 10.1016/j.jchromb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Lantz R.J., Gillespie T.A., Rash T.J. Metabolism, excretion, and pharmacokinetics of duloxetine in healthy human subjects. Drug Metab. Dispos. 2003;31:1142–1150. doi: 10.1124/dmd.31.9.1142. [DOI] [PubMed] [Google Scholar]
- 10.Chalon S.A., Granier L.A., Vandenhende F.R. Duloxetine increases serotonin and norepinephrine availability in healthy subjects: a double-blind, controlled study. Neuropsychopharmacology. 2003;28:1685–1693. doi: 10.1038/sj.npp.1300209. [DOI] [PubMed] [Google Scholar]
- 11.Suri A., Reddy S., Gonzales C. Duloxetine pharmacokinetics in cirrhotics compared with healthy subjects. Int. J. Clin. Pharmacol. Ther. 2005;23:78–84. doi: 10.5414/cpp43078. [DOI] [PubMed] [Google Scholar]
- 12.Reddy D.C., Bapuji A.T., Rao V.S. Development and validation of a LC/MS/MS method for the determination of duloxetine in human plasma and its application to pharmacokinetic study. E-J. Chem. 2012;9:899–911. [Google Scholar]
- 13.Ma N., Zhang B.K., Li H.D. Determination of duloxetine in human plasma via LC/MS and subsequent application to a pharmacokinetic study in healthy Chinese volunteers. Clin. Chim. Acta. 2007;380:100–105. doi: 10.1016/j.cca.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Choong E., Rudaz S., Kottelat S. Therapeutic drug monitoring of seven psychotropic drugs and four metabolites in human plasma by HPLC–MS. J. Pharm. Biomed. Anal. 2009;50:1000–1008. doi: 10.1016/j.jpba.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Anderson D., Reed S., Lintemoot J. A first look at duloxetine (Cymbalta) in a postmortem laboratory. J. Anal. Toxicol. 2006;30:576–580. doi: 10.1093/jat/30.8.576. [DOI] [PubMed] [Google Scholar]
- 16.Musenga A., Amore M., Mandrioli R. Determination of duloxetine in human plasma by capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. B. 2009;877:1126–1132. doi: 10.1016/j.jchromb.2009.02.059. [DOI] [PubMed] [Google Scholar]
- 17.Johnson J.T., Oldham S.W., Lantz R.J. High performance liquid chromatographic method for the determination of duloxetine and desmethyl duloxetine in human plasma. J. Liq. Chromatogr. Relat. Technol. 1996;19:1631–1641. [Google Scholar]
- 18.Mercolini L., Mandrioli R., Cazzolla R. HPLC analysis of the novel antidepressant duloxetine in human plasma after an original solid-phase extraction procedure. J. Chromatogr. B. 2007;856:81–87. doi: 10.1016/j.jchromb.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 19.Kole P.L., Venkatesh G., Kotecha J. Recent advances in sample preparation techniques for effective bioanalytical methods. Biomed. Chromatogr. 2011;25:199–217. doi: 10.1002/bmc.1560. [DOI] [PubMed] [Google Scholar]
- 20.Van Eeckhaut A., Lanckmans K., Sarre S. Validation of bioanalytical LC–MS/MS assays: evaluation of matrix effects. J. Chromatogr. B. 2009;877:2198–2207. doi: 10.1016/j.jchromb.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Nováková L., Vlcková H. A review of current trends and advances in modern bio-analytical methods: chromatography and sample preparation. Anal. Chim. Acta, 2009;656:8–35. doi: 10.1016/j.aca.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 22.US DHHS, FDA, CDER. Guidance for Industry: Bioanalytical Method Validation, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CV), 2001. Available at: 〈http://www/fda.gov/cder/guidance/index.htm〉.
- 23.Fast D.M., Kelley M., Viswanathan C.T. Workshop report and follow-up-AAPS workshop on current topics in GLP bioanalysis: assay reproducibility for incurred samples—implications of crystal city recommendations. AAPS J. 2009;11:238–241. doi: 10.1208/s12248-009-9100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karra V.K., Pilli N.R., Inamadugu J.K. Simultaneous determination of pioglitazone and candesartan in human plasma by LC–MS/MS and its application to a human pharmacokinetic study. J. Pharm. Anal. 2012;2:167–173. doi: 10.1016/j.jpha.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viswanathan C.T., Bansal S., Booth B. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm. Res. 2007;24:1962–1973. doi: 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- 26.Ponnuru V.S., Challa B.R., Nadendla R. Quantification of sibutramine and its two metabolites in human plasma by LC-ESI-MS/MS and its application in a bioequivalence study. J. Pharm. Anal. 2012;2:249–257. doi: 10.1016/j.jpha.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Boer T., Wieling J. Incurred sample accuracy assessment: design of experiments based on standard addition. Bioanalysis. 2011;3:983–992. doi: 10.4155/bio.11.36. [DOI] [PubMed] [Google Scholar]