

Abstract
Different fused-core stationary phase chemistries (C18, Amide, Phenyl-hexyl and Peptide ES-C18) were used for the analysis of 21 structurally representative model peptides. In addition, the effects of the mobile phase composition (ACN or MeOH as organic modifier; formic acid or acetic acid, as acidifying component) on the column selectivity, peak shape and overall chromatographic performance were evaluated. The RP-amide column, combined with a formic acid–acetonitrile based gradient system, performed as best. A peptide reversed-phase retention model is proposed, consisting of 5 variables: log SumAA, log Sv, clog P, log nHDon and log nHAcc. Quantitative structure-retention relationship (QSRR) models were constructed for 16 different chromatographic systems. The accuracy of this peptide retention model was demonstrated by the comparison between predicted and experimentally obtained retention times, explaining on average 86% of the variability. Moreover, using an external set of 5 validation peptides, the predictive power of the model was also demonstrated. This peptide retention model includes the novel in-silico calculated amino acid descriptor, AA, which was calculated from log P, 3D-MoRSE, RDF and WHIM descriptors.
Keywords: Peptides; Fused-core (core-shell, core-enhanced, poro-shell, HALO®) stationary phases; RP-HPLC peptide retention model; In-silico amino acid descriptor
1. Introduction
Peptides are of growing pharmaceutical interest because of their biomedical activity attributed to their great diversity in size, shape and chemical functionalities. They constitute an emerging class of therapeutic agents, possessing greater efficacy and selectivity, as well as an inherent lower toxicity profile compared to the conventional small molecules [1]. For the separation of peptides, reversed-phase high performance liquid chromatography (RP-HPLC) has been most widely employed [2], [3], [4], [5]. In order to identify peptides in complex mixtures, RP-HPLC is combined with mass spectrometry (LC-MS), having an excellent sensitivity and selectivity [6], [7].
Significant progress in RP-HPLC was achieved with the development of smaller, sub-2 μm particles enabling higher resolutions and reduced analysis time [8]. Alternatively, monolithic columns were constructed to speed up the separation and enhance the separating power [9], [10]. Recently, fused-core particles, comprising a 0.5 μm porous outer shell (“HALO®”) fused to a 1.7 μm solid silica core (i.e. fused-core, core-shell or core-enhanced technology) were developed by Kirkland as an alternative to sub−2 μm particles [11], [12], [13]. Different fused-core column chemistries are available: C18, C8, RP-amide, Phenyl-hexyl, Hydrophilic Interaction Liquid Chromatography (HILIC), and most recently Peptide ES-C18 and pentafluorophenyl (PFP) phases (see Table 1).
Table 1.
Properties of the HALO® fused-core stationary phases.
| Phase | Bonded phase | Pore size (Å) | Surface area (m²/g) | Bonding density (μmol/m²) | Endcapped | Base deactivation | Dimensions (l×i.d.) (mm) | pH range |
|---|---|---|---|---|---|---|---|---|
| C18 |
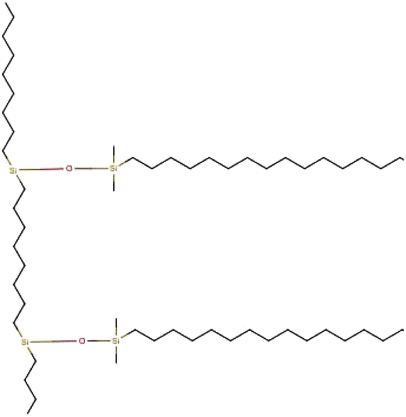 Octadecyldimethylsilane Octadecyldimethylsilane |
90 | 150 | 3.5 | Yes | Yes | 150×4.6 | 2–9 |
| Peptide ES-C18 |
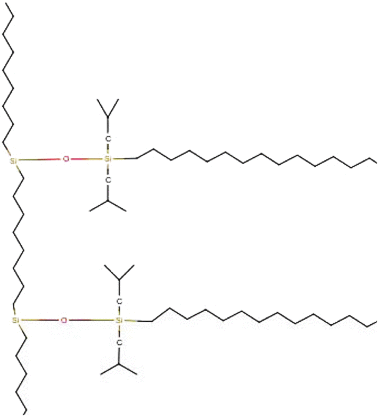 Steric-protected C18, octadecyldiisobutylsilane Steric-protected C18, octadecyldiisobutylsilane |
160 | 80 | 2.0 | No | – | 75×3.0 | 1–9 |
| RP-amide |
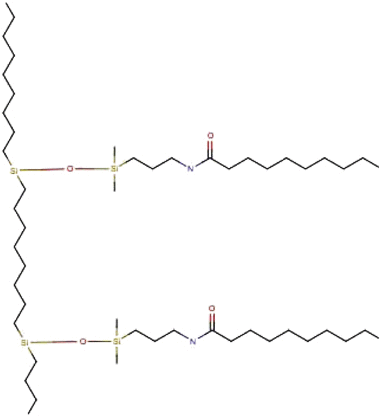 Alkyl amide Alkyl amide |
90 | 150 | 3.0 | Yes | Yes | 150×4.6 | 2–9 |
| Phenyl-hexyl |
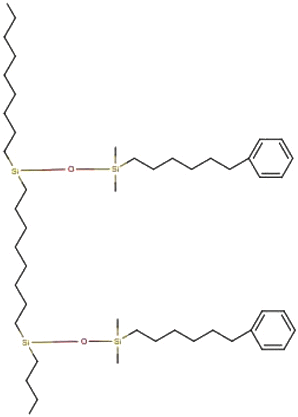 Phenyl-hexyl Phenyl-hexyl |
90 | 150 | 3.4 | Yes | Yes | 75×3.0 | 2–9 |
| HILIC | – | 90 | 150 | – | – | – | 100×3.0 | 2–9 |
Literature survey related to fused-core particle technology demonstrated that most of the reports deal with the kinetic performance evaluation of such columns using classic organic model compounds [14], [15], [16], [17]. Fused-core columns have been characterized based on their Van Deemter curves, demonstrating high plate numbers, reduced mass transfers, and better resolutions [14], [18], [19], [20]. In order to demonstrate the superior efficiency, performance and capacity of the fused-core stationary phases, those columns were compared to UPLC, monolithic and conventional columns [21], [22], [23], [24], [25]. Because of the high resolving power, 2D-HPLC has attracted more attention and has been applied with the fused-core columns in proteomic and metabolomic research [26], [27], [28], [29]. Although the separation of peptides using fused-core columns is scarce in literature, these columns are found to be very promising to reduce analysis time without reducing performance [1]. The limited peptide research using the fused-core columns mostly investigated the influence of temperature, gradient times and flow rate on the retention as well as selectivity and column performance. Due to limited column chemistries currently available for the fused-core stationary phases, lacking e.g. C4 or different polar embedded functionalities, most peptide studies are usually carried out with the classic C18 bonded chemistry. Some research groups compared the performance of the C18 phases as manufactured by four different fused-core suppliers (Advanced Materials Technology for HALO® C18, Phenomenex for Kinetex® C18, Supelco for Ascentis Express® C18 and Agilent for Poroshell® C18) [14], [30], [31], [32], [33]. Only three peptide studies applied the HALO® Peptide ES-C18 fused-core stationary phase for a casein and a tryptic digest, respectively [34], [35], [36], while the HALO® HILIC was only once evaluated [37].
Up till now, no comparative study of the different chemistries in fused-core columns for the analysis of peptides was performed. Moreover, reversed-phase retention modeling using these columns is also missing. In this work, different fused-core column chemistries (HALO® C18, Peptide ES-C18, RP-amide and Phenyl-hexyl) are compared and evaluated using a mixture of synthetic peptides. In addition to the selectivity differences of the stationary phases, the selectivity effects of the mobile phase composition are studied as well [2], [38], [39], [40], [41]. Finally, reversed-phase fused-core peptide retention models were constructed on the different chromatographic systems, containing a novel in-silico calculated amino acid descriptor.
2. Materials and methods
2.1. Reagents
Ultra gradient grade acetonitrile was purchased from Romil (Merelbeke, Belgium). Formic acid was obtained from Acros Organics (Geel, Belgium). Water was purified in-house using an Arium 611 purification system (Sartorius, Göttingen, Germany), yielding ≥18.2 MΩ×cm quality water. Methanol was supplied from Fisher Scientific (Leicestershire, UK) and acetic acid from Merck (Darmstadt, Germany). All synthetic peptides were synthesized by Peptide Protein Research (PPR, Hampshire, UK) with a purity of at least 90%.
2.2. Fused-core (HALO®) columns
Different column chemistries were selected, consisting of C18, RP-amide, Phenyl-hexyl and Peptide ES-C18. Details about the stationary phases, including the bonded phase, dimensions, pore size, surface area and pH-range are given in Table 1.
All HALO® columns have a particle size of 2.7 μm and were supplied by Achrom (Machelen, Belgium).
2.3. Peptide selection
In order to select a limited but representative experimental peptide set, the chemical–structural diversity of 61 peptides given in the Brainpeps® database [42] was visualized using principal component analysis (PCA) and hierarchical cluster analysis (HCA). First, three-dimensional molecular structures were calculated and optimized using Hyperchem 8.0 (Hypercube Inc., Gainesville, FL, USA). Geometry optimization was performed with the molecular mechanics force field method (MM+) using the Polak-Ribière conjugate gradient algorithm with an RMS gradient of 0.1 kcal/(Å.mol), corresponding to 0.4184 kJ/(Å.mol), as stop criterion. The obtained structures were then used to calculate more than 3000 molecular descriptors (Dragon 5.5, Talete, Italy). After removal of constant and correlated, i.e. Pearson correlation coefficient r>0.95, descriptors, PCA and HCA was performed on the normalized descriptors. In total, 21 peptides were selected from the different peptide clusters, showing wide structural variety. In addition, structurally related peptides belonging to the same peptide cluster were selected as well for evaluation of the chromatographic separation of structure analogs. More detailed information regarding the 21 selected peptides, i.e. molecular weight, log P and pI values, is given in Table 2.
Table 2.
Overview of selected peptides (21 model-building and 5 validation peptides).
| Peptide | MW (g/mol) | log P | pI |
|---|---|---|---|
| Adrenomedullin | 5730.46 | −38.53 | 10.39 |
| Amylin | 3921.43 | −26.06 | 10.81 |
| cHP | 234.26 | −1.26 | 8.81 |
| CRH | 4758.50 | −20.78 | 5.59 |
| CTOP | 1062.27 | −1.40 | 9.41 |
| Dermorphin | 802.88 | −1.24 | 9.16 |
| Des-octanoyl ghrelin | 3188.65 | −21.81 | 10.67 |
| DPDPE | 645.79 | 0.87 | 5.70 |
| Endomorphin-1 | 610.71 | 1.76 | 8.61 |
| GALP rat | 6502.44 | −31.07 | 10.17 |
| Kyotorphin | 337.38 | −0.07 | 8.74 |
| LHRH | 1183.29 | −4.14 | 8.08 |
| Mouse Obestatin | 2516.85 | −12.96 | 9.81 |
| MCH | 2386.84 | −5.52 | 8.85 |
| Met5-Enkephalin | 573.66 | −0.69 | 5.82 |
| Orexin A | 3561.14 | −17.35 | 9.71 |
| RC-160 | 1131.38 | 1.50 | 9.73 |
| SB-Aba | 580.68 | 0.05 | 10.28 |
| UCN-I | 4708.04 | −19.32 | 5.70 |
| VDE243 | 671.75 | −0.62 | 5.87 |
| VIP | 3326.83 | −16.19 | 9.71 |
| Endomorphin-2 | 571.68 | 1.99 | 8.61 |
| Neuropeptide Y | 4254.70 | −17.73 | 8.05 |
| Phe13Tyr19-MCH | 2434.88 | −4.71 | 8.85 |
| TAPP | 545.64 | 2.62 | 8.61 |
| Urocortin II | 4153.94 | −14.84 | 10.57 |
2.4. Peptide lyophilization
Prior to analysis, the peptide samples were dissolved in acetonitrile/water 5/95 (v/v) containing 0.1% (w/v) formic acid at a concentration of 1 mg/mL. 100 μL aliquots were dispensed into low-volume polypropylene HPLC vials (Grace Alltech, Deerfield, US) and lyophilized with the in-house developed program using a Christ gamma 1–16 LSC freeze dryer (Qlab, Vilvoorde, Belgium) [43].
2.5. Liquid chromatography and chromatographic properties
The HPLC–PDA–MS apparatus consisted of a Waters Alliance 2695 separations module and a Waters 2996 photodiode array detector with Empower 2 software for data acquisition (all Waters, Milford, MA, USA). The MS apparatus, used for identification of the peptides, consisted of a Finnigan LCQ Classic ion trap mass spectrometer in positive ion mode (all Thermo, San José, CA, USA) equipped with a Waters 2487 dual wavelength absorbance UV detector (Waters, Milford, MA, USA) set at 215 nm and Xcalibur 2.0 software (Thermo, San José, CA, USA) for data acquisition. ESI was conducted using a needle voltage of 4.5 kV. Nitrogen was used as the sheath and auxiliary gas with the heated capillary set at 250 °C. Positive mode mass spectra were obtained in the range of m/z 100 to 2000.
The HALO® columns were thermostated in an oven set at 30 °C, whereas the autosampler temperature was set at 10 °C. Four mobile phase compositions, traditionally employed in peptide analysis, were used:
-
(1)
0.1% w/v formic acid in water (A) and 0.1% w/v formic acid in acetonitrile (B), referred to as FA.
-
(2)
0.1% w/v formic acid in water (A) and 0.1% w/v formic acid in methanol (B), abbreviated as FM.
-
(3)
0.1% w/v acetic acid in water (A) and 0.1% w/v acetic acid in acetonitrile (B), referred to as AA.
-
(4)
0.1% w/v acetic acid in water (A) and 0.1% w/v acetic acid in methanol (B), abbreviated as AM.
The method consisted of a linear gradient from 90:10 v/v A/B to 10:90 v/v A/B, followed by reconditioning with the initial composition 90:10 v/v A/B for 10 min. The peptides were injected as a mixture, each at a concentration of 25 μM, dissolved in acetonitrile/water 5/95 (v/v) containing 0.1% (w/v) formic acid. The injection volume was fixed to 5 μL. Seen the different column dimensions, the gradient time and flow rate were adjusted taking into account the column volume according to the following formula:
where tG,2 and tG,1 are the gradient times of HPLC column 2 and 1, respectively, F is the flow rate and V0 is the dead volume. The flow rate was set to 0.5 mL/min (except for mobile phase system FA, a flow rate of 1 mL/min was used) with a gradient time of 25 min on the C18 and RP-amide column, while for the other two phases, Phenyl-hexyl and Peptide ES-C18, the flow rate was 0.4 mL/min (except for mobile phase system FA, a flow rate of 0.6 mL/min was used) with a gradient time of 10 min. A number of single and multiple chromatographic responses were calculated using the aforementioned 16 different chromatographic systems, including asymmetry factor (As), full width half maximum (FWHM), gradient plate number (Ng) and chromatographic response factor (CRF) [44].
2.6. In-silico amino acid descriptor
Structural descriptors (911) belonging to different classes (i.e. constitutional, topological, topological charge, geometrical, RDF, 3D-MoRSE, Weighed Holistic Invariant Molecular (WHIM), GETAWAY, charge descriptors and molecular properties) were calculated using the optimized three-dimensional structures of the 20 naturally occurring amino acids. After elimination of the constant descriptors, a stepwise multiple linear regression (MLR), as implemented in SPSS 20.0 (P-to-enter ≤0.05 and P-to-remove ≥0.10), was used to model the experimentally obtained retention times of the 20 natural amino acids on a XTerra MS C18 column [45] in function of their calculated structural descriptors.
2.7. Peptide retention model
In order to predict the gradient retention time of peptides on the different fused-core columns, quantitative structure-retention relationships (QSRR) were established for the 16 experimental chromatographic conditions. The peptide retention time is modeled as a function of a limited set of molecular descriptors by means of MLR. Generally, current peptides RP-HPLC QSRR models have the following equation form [46], [47], [48], [49], [50], [51]:
where RT is the peptides gradient RP-HPLC retention time, b0−b3 are regression coefficients estimated by MLR, log SumAA is the logarithm of the sum of the experimentally obtained gradient retention times of the amino acids composing the individual peptide, log VDWvol is the logarithm of the peptide's van der Waals volume and clog P is the logarithm of its theoretically calculated n-octanol–water partition coefficient according to the Ghose–Pritchett–Crippen algorithm.
In our proposed peptide reversed-phase fused-core retention model, similar molecular descriptors were used. log SumAA, however applying the new in-silico calculated descriptor described above replacing the experimentally determined amino acid retention times, log Sv (i.e. van der Waals volume calculated with Dragon software) and clog P. In addition to these three descriptors, the number of donor and acceptor atoms for H-bonding (nHDon and nHAcc, respectively) were added as proposed in RP-HPLC by Du et al. [52]. The descriptor log SumAA is thus no longer experimentally determined, but is calculated in terms of theoretical descriptors, so that the peptide retention can be predicted entirely in-silico, without the need of experiments with each of the amino acids constituting the peptide.
Finally, the predictive power of the newly proposed peptide retention model was demonstrated by calculating the retention times of five validation peptide (Table 2), belonging to the same structural space of the 21 model-building peptides, and comparing these predicted retention times to the experimentally obtained retention times using the 16 different chromatographic systems.
3. Results
3.1. Chromatographic properties of peptides on fused-core stationary phases
A typical chromatogram of UCN-1, MCH and dermorphin on the RP-amide column using FA is shown in Fig. 1. The influence of the mobile phase composition on the column performance was demonstrated by the calculation of the gradient plate number (Ng). The highest plate number was observed for FA on all four columns, with the RP-amide column exceeding the others. The performance of the columns was lowered significantly when using MeOH as organic modifier compared to ACN. Also when looking at FWHM and As, the FA mobile phase composition was generally found to be the best performing mobile phase throughout all columns. When using this mobile phase, i.e. FA, the RP-amide column displayed the best As and second best FWHM, and was thus considered to overall deliver the best peak shapes. When calculating the other chromatographic performance response functions, i.e. resolution product corrected for the retention time of the last eluting peak, separation factor, CRF and peak capacity, the highest values (i.e. better separation of compounds) were again obtained using FA. Also for these chromatographic response factors, the RP-amide column performed the best, followed by C18, Peptide ES-C18 and Phenyl-hexyl.
Fig. 1.

Typical chromatograms: MCH, UCN-1 and dermorphin (from top to bottom) on HALO® RP-amide column, using formic acid–acetonitrile based chromatography.
3.2. In-silico amino acid descriptor
Stepwise MLR, whereby 742 non-constant structural descriptors, derived from the three dimensional amino acid structures, were modeled into a global in-silico amino acid descriptor, describing the reversed-phase retention behavior as given in the literature [45], resulted in following model:
This new in-silico calculated AA descriptor was introduced into the existing peptide retention models, thus replacing the experimentally determined individual amino acids retention times. Linear least squares correlation analysis indicated that this AA model explained 99.4% of the observed amino acid retention variability (R²=0.994).
3.3. Peptide retention model
Sixteen separate QSRR models were developed for each of the sixteen chromatographic conditions, using following general equation:
The QSRR results are summarized in Table 3. Obtained R2 and F values for the prediction models as well as the calculated regression coefficients are tabulated. On average, 85.7% of the peptide retention time variability is explained by our proposed model. Scatter plots of the sixteen chromatographic conditions, displaying the 21 calculated peptide retention times in function of the experimentally obtained retention times are given in Fig. 2.
Table 3.
Retention models obtained on the four fused-core columns using multiple linear regression.
| Column | MPa | Retention models RT=b0+b1 log ∑AA descriptor+b2 clog P+b3 log Sv+b4 log nHDon+b5 log nHAcc |
Experimental vs. predicted model fit predicted RT=a×Experimental RT+b |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R² | F | b0 | b1 | b2 | b3 | b4 | b5 | a | b | ||
| Peptide ES-C18 | FA | 0.818 | 13.448 | −3.698 | 8.352 | 0.075 | −3.030 | −9.095 | 8.096 | 0.818 | 0.694 |
| FM | 0.913 | 31.317 | −5.389 | 13.011 | 0.101 | −3.695 | −10.925 | 9.082 | 0.912 | 0.670 | |
| AA | 0.826 | 14.210 | −3.582 | 9.932 | 0.089 | −3.694 | −9.935 | 8.631 | 0.825 | 0.842 | |
| AM | 0.916 | 32.603 | −7.162 | 12.847 | 0.128 | −2.830 | −12.110 | 10.410 | 0.916 | 0.632 | |
| RP-amide | FA | 0.817 | 13.385 | −7.445 | 14.750 | 0.140 | −5.845 | −17.287 | 16.202 | 0.817 | 1.086 |
| FM | 0.845 | 16.401 | −10.615 | 30.793 | 0.217 | −13.321 | −29.263 | 26.367 | 0.845 | 2.119 | |
| AA | 0.857 | 17.956 | −5.582 | 18.575 | 0.145 | −9.002 | −20.325 | 18.815 | 0.857 | 1.104 | |
| AM | 0.857 | 18.003 | −15.790 | 32.709 | 0.271 | −13.783 | −39.538 | 36.968 | 0.857 | 1.787 | |
| Phenyl-hexyl | FA | 0.796 | 11.736 | −1.215 | 7.356 | 0.044 | −3.315 | −5.332 | 4.209 | 0.797 | 0.707 |
| FM | 0.901 | 27.412 | −3.360 | 14.479 | 0.083 | −5.777 | −10.382 | 8.278 | 0.901 | 0.750 | |
| AA | 0.800 | 11.967 | −2.466 | 9.910 | 0.081 | −4.195 | −9.285 | 7.835 | 0.800 | 0.926 | |
| AM | 0.890 | 24.202 | −6.223 | 14.274 | 0.115 | −4.057 | −13.080 | 10.724 | 0.890 | 0.848 | |
| C18 | FA | 0.834 | 15.023 | −7.006 | 14.159 | 0.113 | −5.683 | −16.758 | 15.644 | 0.833 | 0.970 |
| FM | 0.892 | 24.810 | −10.790 | 29.263 | 0.181 | −11.703 | −29.591 | 26.210 | 0.892 | 1.498 | |
| AA | 0.851 | 17.175 | −6.211 | 18.308 | 0.124 | 8.671 | −21.040 | 19.491 | 0.851 | 1.125 | |
| AM | 0.903 | 27.875 | −14.259 | 30.967 | 0.220 | −12.470 | −35.679 | 32.723 | 0.903 | 1.266 | |
MP=mobile phase, FA=formic acid–acetonitrile, FM=formic acid–methanol, AA=acetic acid–acetonitrile, AM=acetic acid–methanol.
Fig. 2.

Peptide retention model.
The predictive power of the peptide retention model is shown in Fig. 3, whereby the calculated peptide retention time of 5 validation peptides is depicted versus their experimentally obtained retention times, characterized by an average R² value of 0.80.
Fig. 3.

Predictive power of the peptide retention model.
4. Discussion
Application of small, sub−2 μm, fully porous particles results in a higher efficiency, linear velocity and reduced mass transfer, but also requires special instrumentation (Ultra-Performance Liquid Chromatography, UPLC) to cope with the resulting pressure increase [53]. The use of monolithic columns allows to speed up the separation and enhance the separating power [9], [10]. The main drawback of these monolithic columns is the relatively high flow rate required to fully exploit their potential. Alternatively, fused-core particles achieve high separation efficiencies with relative low backpressure, permitting the use of conventional HPLC equipment [1], [11], [54]. Due to their small particle size and limited diffusion path, plate numbers equivalent to UPLC are achieved, minimizing peak broadening, while overall shortening the analysis time [10], [55], [56]. Compared to conventional HPLC columns of the same dimensions, usually packed with 3–5 μm particles, the fused-core columns show a significant gain in performance, expressed as plate number or peak capacity [1].
Multiple fused-core particle chemistries are available (see Table 1). C18 and C8 are used for the separation of hydrophobic compounds whereas the fused-core RP-amide column is a polar-embedded phase, providing enhanced selectivity for samples containing highly water-soluble acidic and basic compounds. Separation on the RP-amide column is affected by hydrophobic interaction with the alkyl chain and hydrogen bonding with the embedded amide group. For the phenyl-hexyl fused-core column, an additional π–π mechanism is described for the separation of aromatic groups. The PFP phase is recommended for the separation of polar bases and halogenated compounds. The primary HILIC retention mechanism is based on hydrophilic partition between the water-rich layer at the surface of the stationary phase and the bulk organic-rich mobile phase [37], [44], [57]. As an extension of the C18 phase, the Peptide ES-C18 phase was specifically designed for the enhanced separation of peptides due to the carefully selected pore size and the use of extra stable (ES) bonding chemistry. Therefore, 100, 120 or 160 Å was selected as the ideal pore size for optimal separation of peptides with a molecular weight of up to 15,000 Da, contrasting a pore size of 90 Å for small molecules. Extra stable bonding was achieved through the use of bulky side chains on the alkylsilanes, providing steric protections of the more labile siloxane bond [34].
Peptide clustering, based on their theoretical descriptors, revealed consistent grouping between HCA and PCA and was used to select a representative peptide set, consisting of 21 peptides from different clusters, for further chromatographic analysis.
These wide structure differences were confirmed by the diverse chromatographic behavior of the 21 selected peptides using the different chromatographic systems. In general it was seen that peptide separation on the RP-amide fused-core column, using the formic acid–acetonitrile based mobile phase, resulted in the best chromatographic responses, thus outperforming the Peptide column. A possible explanation is the additional hydrogen bond interactions between the amide groups of the column and the peptides, which are absent in the Peptide column, resulting in a higher selectivity of the RP-amide column. The acid in the mobile phase serves not only a pH control function, but also an ion-pairing complexation activity with the charged peptide ionic groups and the stationary phase, and will additionally suppress adverse ionic interactions between the peptides and the residual silanol groups on the stationary phase. The use of acetic acid, being more hydrophobic than formic acid, leads to increased peptide retention on the column, which in turn leads to increased resolution [58]. Therefore, hydrophobic ion-pairing agents, e.g. acetic acid, should be used for separation of complex and/or structurally related peptides, whereas more hydrophilic agents, e.g. formic acid, can be used for fast separation of simple peptide mixtures. As such, the acidic mobile phase additives will also influence the selectivity, its extent depending on the stationary phase.
The classic, experimentally obtained individual amino acid retention times descriptor was replaced by an in-silico calculated AA descriptor using a stepwise MLR. This new descriptor is calculated using six structural descriptors. The first two descriptors, i.e. Alog P and Alog P2, give information about the lipophilicity of amino acids whereby Alog P2 is the square of the Alog P value. This Alog P value is calculated using the Ghose-Crippen-algorithm. Mor10v and Mor10e are part of the 3D-Molecule Representation of Structures based on Electron diffraction (3D-MoRSE) descriptors, which provide information derived from the three dimensional coordinates by using the same transformation used in electron diffraction to prepare theoretical scattering curves. These different signals, i.e. indicated by the numeric code, were then weighed by van der Waals volume (v) or by Sanderson electronegativity (e). E1u stands for the unweighed 1st component accessibility directional WHIM index. This is a geometrical descriptor based on statistical indices, calculated from the projections of the atoms along principal axes. The Radial Distribution Function (RDF) descriptors are based on the distance distribution in the geometrical representation of a molecule, and show certain characteristics in common with the 3D-MoRSE descriptors. The RDF descriptors provide information about interatomic distances in which the numeric code indicates an interatomic distance, e.g. 035 corresponding to 3.5 Å, which is the probability of finding an interatomic distance of 3.5 Å. Similar weighing factors as for the 3D-MoRSE descriptors are used [59].
This new in-silico AA descriptor was then introduced into the peptide retention model, of which 16 different QSRR were constructed, modeling the peptide retention times on the 16 different chromatographic conditions used. The proposed model factors differ significantly from the existing reversed-phase peptide retention model factors: our model not only includes the new in-silico amino acid descriptor, but also includes number of hydrogen donors (nHDon) and hydrogen acceptors (nHAcc). The existing models all use the experimentally determined retention time or factor of the individual amino acids as amino acid descriptor and available amino acid descriptors did not include new and/or unnatural amino acids [60]. Our new amino acid descriptor allows the modeling of peptides containing unnatural amino acids including optical isomers (L versus D) as well. The predictive power of our peptide retention models demonstrated the correlation of the predicted retention times versus experimentally obtained for 5 peptides not included in our model building set (mean R²=0.800) (Fig. 3).
5. Conclusion
Fused-core columns are of great interest because of their high performance in combination with a relatively low backpressure allowing the application of these columns on conventional HPLC equipment. Four different column chemistries (Peptide, RP-amide, Phenyl-hexyl and C18) were compared for the separation of 21 selected, structurally diverse, peptides. Highest chromatographic responses were obtained using the RP-amide column and formic acid-acetonitrile based gradient system.
A reversed-phase QSRR retention model was constructed for peptide analysis on the fused-core stationary phases under the sixteen given chromatographic conditions. This model incorporates a novel, in-silico calculated amino acid descriptor, thus rendering the determination of individual amino acid retention times superfluous and allowing the inclusion of new unnatural amino acids in the construction of the QSRR model. The model explained 86% of the observed peptide retention time variability and had a predictive power of 80%.
Acknowledgments
This research was funded by a Ph.D. grant of “Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen)” (No. 091241 for MD and 073402 for SVD) and by the Special Research Fund of the Ghent University (Grant no. BOF 01J22510 for EW and BOF 01D38811 for SS).
Footnotes
Peer review under responsibility of Xi'an Jiaotong University.

Supplementary data associated with this article can be found in the online version at doi:10.1016/j.jpha.2012.11.002.
Appendix A. Supplementary materials
Supplementary Material
References
- 1.Ruta J., Guillarme D., Rudaz S. Comparison of columns packed with porous sub−2 μm particles and superficially porous sub−3 μm particles for peptide analysis at ambient and high temperatures. J. Sep. Sci. 2010;33:2465–2477. doi: 10.1002/jssc.201000023. [DOI] [PubMed] [Google Scholar]
- 2.Riddle L.A., Guiochon G. Influence of mobile phase gradients on the retention and separation of peptides from a cytochrome-c digest by reversed-phase liquid chromatography. Chromatographia. 2006;64:121–127. [Google Scholar]
- 3.Stulik K., Pacakova V., Suchankova J. Stationary phases for peptide analysis by high performance liquid chromatography: a review. Anal. Chim. Acta. 1997;352:1–19. [Google Scholar]
- 4.Hancock W.S., Bishop C.A., Prestidge R.L. Reversed-phase, high-pressure liquid-chromatography of peptides and proteins with ion-pairing reagents. Science. 1978;200:1168–1170. doi: 10.1126/science.206966. [DOI] [PubMed] [Google Scholar]
- 5.Hancock W.S., Bishop C.A., Prestidge R.L. High-pressure liquid-chromatography of peptides and proteins. Use of phosphoric-acid in analysis of un-derivatized pepetides by reversed-phase high-pressure liquid-chromatography. J. Chromatogr. 1978;153:391–398. [Google Scholar]
- 6.Hsieh Y., Duncan C.J.G., Brisson J.-M. Fused-core silica column high-performance liquid chromatography/tandem mass spectrometric determination of rimonabant in mouse plasma. Anal. Chem. 2007;79:5668–5673. doi: 10.1021/ac070343g. [DOI] [PubMed] [Google Scholar]
- 7.De Spiegeleer B., Vergote V., Pezeshki A. Impurity profiling quality control testing of synthetic peptides using liquid chromatography-photodiode array-fluorescence and liquid chromatography-electrospray ionization-mass spectrometry: the obestatin case. Anal. Chem. 2008;376:229–234. doi: 10.1016/j.ab.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 8.van Deemter J.J., Zuiderweg F.J., Klinkenberg A. Longitudinal diffusion and resistance to mass transfer as causes of nonideality in chromatography. Chem. Eng. Sci. 1956;5:271–289. [Google Scholar]
- 9.Issaq H.J., Chan K.C., Blonder J. Separation, detection and quantitation of peptides by liquid chromatography and capillary electrochromatography. J. Chromatogr. A. 2009;1216:1825–1837. doi: 10.1016/j.chroma.2008.12.052. [DOI] [PubMed] [Google Scholar]
- 10.Brice R.W., Zhang X., Colon L.A. Fused-core, sub−2 μm packings, and monolithic HPLC columns: a comparative evaluation. J. Sep. Sci. 2009;32:2723–2731. doi: 10.1002/jssc.200900091. [DOI] [PubMed] [Google Scholar]
- 11.Kirkland J.J., Langlois T.J., DeStefano J.J. Fused core particles for HPLC columns. Am. Lab. 2007;39:18–21. [Google Scholar]
- 12.Kirkland J.J. Development of some stationary phases for reversed-phase high performance liquid chromatography. J. Chromatogr. A. 2004;1060:9–21. [PubMed] [Google Scholar]
- 13.Kirkland J.J., Truskowski F.A., Ricker R.D. Atypical silica-based column packings for high-performance liquid chromatography. J. Chromatogr. A. 2002;965:25–34. doi: 10.1016/s0021-9673(01)01339-5. [DOI] [PubMed] [Google Scholar]
- 14.Badman E.R., Beardsley R.L., Liang Z. Accelerating high quality bioanalytical LC/MS/MS assays using fused-core columns. J. Chromatogr. B. 2010;878:2307–2313. doi: 10.1016/j.jchromb.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 15.Fekete S., Ganzler K., Fekete J. Efficiency of the new sub−2 μm core-shell (Kinetex(TM)) column in practice, applied for small and large molecule separation. J. Pharmaceut. Biomed. 2011;54:482–490. doi: 10.1016/j.jpba.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 16.Olah E., Fekete S., Fekete J. Comparative study of new shell-type, sub−2 μm fully porous and monolith stationary phases, focusing on mass-transfer resistance. J. Chromatogr. A. 2010;1217:3642–3653. doi: 10.1016/j.chroma.2010.03.052. [DOI] [PubMed] [Google Scholar]
- 17.Vaast A., Broeckhoven K., Dolman S. Comparison of the gradient kinetic performance of silica monolithic capillary columns with columns packed with 3 μm porous and 2.7 μm fused-core silica particles. J. Chromatogr. A. 2011;1228:270–275. doi: 10.1016/j.chroma.2011.07.089. [DOI] [PubMed] [Google Scholar]
- 18.Gritti F., Guiochon G. Comparison between the loading capacities of columns packed with partially and totally porous fine particles what is the effective surface area available for adsorption? J. Chromatogr. A. 2007;1169:125–138. doi: 10.1016/j.chroma.2007.10.076. [DOI] [PubMed] [Google Scholar]
- 19.Fekete S., Fekete J., Ganzler K. Shell and small particles: evaluation of new column technology. J. Pharm. Biomed. 2009;49:64–71. doi: 10.1016/j.jpba.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Kaczmarski K., Guiochon G. Modeling of the mass-transfer kinetics in chromatographic columns packed with shell and pellicular particles. Anal. Chem. 2007;79:4648–4656. doi: 10.1021/ac070209w. [DOI] [PubMed] [Google Scholar]
- 21.Gritti F., Cavazzini A., Marchetti N. Comparison between the efficiencies of columns packed with fully and partially porous C-18-bound silica materials. J. Chromatogr. A. 2007;1157:289–303. doi: 10.1016/j.chroma.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 22.Salisbury J.J. Fused-core particles: a practical alternative to sub−2 μm particles. J. Chromatogr. Sci. 2008;46:883–886. doi: 10.1093/chromsci/46.10.883. [DOI] [PubMed] [Google Scholar]
- 23.Song W., Pabbisetty D., Groeber E.A. Comparison of fused-core and conventional particle size columns by LC-MS/MS and UV: application to pharmacokinetic study. J. Pharm. Biomed. 2009;50:491–500. doi: 10.1016/j.jpba.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y., Wang X., Mukherjee P. Critical comparison of performances of superficially porous particles and sub−2 μm particles under optimized ultra-high pressure conditions. J. Chromatogr. A. 2009;1216:4597–4605. doi: 10.1016/j.chroma.2009.03.071. [DOI] [PubMed] [Google Scholar]
- 25.Zheng J., Patel D., Tang Q. Comparison study of porous, fused-core, and monolithic silica-based C(18) HPLC columns for celestoderm-V ointment. J. Pharm. Biomed. 2009;50:815–822. doi: 10.1016/j.jpba.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 26.Alexander A.J., Ma L. Comprehensive two-dimensional liquid chromatography separations of pharmaceutical samples using dual fused-core columns in the 2nd dimension. J. Chromatogr. A. 2009;1216:1338–1345. doi: 10.1016/j.chroma.2008.12.063. [DOI] [PubMed] [Google Scholar]
- 27.Li X., Stoll D.R., Carr P.W. Equation for peak capacity estimation in two-dimensional liquid chromatography. Anal. Chem. 2009;81:845–850. doi: 10.1021/ac801772u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoll D.R., Li X., Wang X. Fast, comprehensive two-dimensional liquid chromatography. J. Chromatogr. A. 2007;1168:3–43. doi: 10.1016/j.chroma.2007.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fairchild J.N., Horvath K., Guiochon G. Approaches to comprehensive multidimensional liquid chromatography systems. J. Chromatogr. A. 2009;1216:1363–1371. doi: 10.1016/j.chroma.2008.12.073. [DOI] [PubMed] [Google Scholar]
- 30.Jandera P., Hajek T., Cesla P. Effects of the gradient profile, sample volume and solvent on the separation in very fast gradients, with special attention to the second-dimension gradient in comprehensive two-dimensional liquid chromatography. J. Chromatogr. A. 2011;1218:1995–2006. doi: 10.1016/j.chroma.2010.10.095. [DOI] [PubMed] [Google Scholar]
- 31.Marchetti N., Cavazzini A., Gritti F. Gradient elution separation and peak capacity of columns packed with porous shell particles. J. Chromatogr. A. 2007;1163:203–211. doi: 10.1016/j.chroma.2007.06.046. [DOI] [PubMed] [Google Scholar]
- 32.Ruta J., Zurlino D., Grivel C. Evaluation of columns packed with shell particles with compounds of pharmaceutical interest. J. Chromatogr. A. 2011;1228:221–231. doi: 10.1016/j.chroma.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 33.Staub A., Zurlino D., Rudaz S. Analysis of peptides and proteins using sub−2 μm fully porous and sub–3 μm shell particles. J. Chromatogr. A. 2011;1218:8903–8914. doi: 10.1016/j.chroma.2011.07.051. [DOI] [PubMed] [Google Scholar]
- 34.Schuster S.A., Wagner B.M., Boyes B.E. Wider pore superficially porous particles for peptide separations by HPLC. J. Chromatogr. Sci. 2010;48:566–571. doi: 10.1093/chromsci/48.7.566. [DOI] [PubMed] [Google Scholar]
- 35.D'Hondt M., Demaré W., Van Dorpe S. Dry heat stress stability evaluation of casein peptide mixture. Food Chem. 2011;128:114–122. doi: 10.1016/j.foodchem.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Schuster S.A., Boyes B.E., Wagner B.M. Fast high performance liquid chromatography separations for proteomic applications using fused-core(r) silica particles. J. Chromatogr. A. 2012;1228:232–241. doi: 10.1016/j.chroma.2011.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chirita R.-I., West C., Finaru A.-L. Approach to hydrophilic interaction chromatography column selection: application to neurotransmitters analysis. J. Chromatogr. A. 2010;1217:3091–3104. doi: 10.1016/j.chroma.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y., Mant C.T., Hodges R.S. Selectivity differences in the separation of amphipathic α-helical peptides during reversed-phase liquid chromatography at pHs 2.0 and 7.0. Effects of different packings, mobile phase conditions and temperature. J. Chromatogr. A. 2004;1043:99–111. doi: 10.1016/j.chroma.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 39.Barbosa J., Toro I., Berges R. Retention behavior of peptides, quinolones, diuretics and peptide hormones in liquid chromatography. Influence of ionic strength and pH on chromatographic retention. J. Chromatogr. A. 2001;915:85–96. doi: 10.1016/s0021-9673(01)00641-0. [DOI] [PubMed] [Google Scholar]
- 40.Sanz-Nebot V., Toro I., Barbosa J. Separation of potentially therapeutic peptide hormones by liquid chromatography. Optimisation of composition and pH of the mobile phase. J. Chromatogr. A. 2000;870:335–347. doi: 10.1016/s0021-9673(99)01154-1. [DOI] [PubMed] [Google Scholar]
- 41.Espinosa S., Bosch E., Roses M. Change of mobile phase pH during gradient reversed-phase chromatography with 2,2,2-trifluoroethanol–water as mobile phase and its effect on the chromatographic hydrophobicity index determination. J. Chromatogr. A. 2002;954:77–87. doi: 10.1016/s0021-9673(02)00165-6. [DOI] [PubMed] [Google Scholar]
- 42.Van Dorpe S., Bronselaer A., Nielandt J. Brainpeps: the blood–brain barrier peptide database. Brain Struct. Funct. 2011;217:687–718. doi: 10.1007/s00429-011-0375-0. [DOI] [PubMed] [Google Scholar]
- 43.Pezeshki A., Vergote V., Van Dorpe S. Adsorption of peptides at the sample drying step: influence of solvent evaporation technique, vial material and solution additive. J. Pharm. Biomed. 2009;49:607–612. doi: 10.1016/j.jpba.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Van Dorpe S., Vergote V., Pezeshki A. Hydrophilic interaction LC of peptides: columns comparison and clustering. J. Sep. Sci. 2010;33:728–739. doi: 10.1002/jssc.200900476. [DOI] [PubMed] [Google Scholar]
- 45.Bodzioch K., Baczek T., Kaliszan R. The molecular descriptor log Sum(AA) and its alternatives in QSPR models to predict the retention of peptides. J. Pharm. Biomed. 2009;50:563–569. doi: 10.1016/j.jpba.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Bodzioch K., Durand A., Kaliszan R. Advanced QSPR modeling of peptides behavior in RPLC. Talanta. 2010;81:1711–1718. doi: 10.1016/j.talanta.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 47.Baczek T., Temporini C., Perani E. Identification of peptides in proteomics supported by prediction of peptide retention by means of quantitative structure-retention relationships. Acta Chromatogr. 2007;18:72–92. [Google Scholar]
- 48.Baczek T., Wiczling P., Marszall M. Prediction of peptide retention at different HPLC conditions from multiple lineair regression. J. Proteome Res. 2005;4:555–563. doi: 10.1021/pr049780r. [DOI] [PubMed] [Google Scholar]
- 49.Kaliszan R., Baczek T., Cimochowska A. Prediction of high-performance liquid chromatography retention of peptides with the use of quantitative structure-retention relationships. Proteomics. 2005;5:409–415. doi: 10.1002/pmic.200400973. [DOI] [PubMed] [Google Scholar]
- 50.Michel M., Baczek T., Studzinska S. Comparative evaluation of high-performance liquid chromatography stationary phases used for the separation of peptides in terms of quantitative structure-retention relationships. J. Chromatogr. A. 2007;1175:49–54. doi: 10.1016/j.chroma.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Kaliszan R. QSPR: quantitative structure-chromatographic-retention relationships. Chem. Rev. 2007;107:3212–3246. doi: 10.1021/cr068412z. [DOI] [PubMed] [Google Scholar]
- 52.Du H., Wang H., Zhang X. Prediction of retention times of peptides in RPLC by using radial basis function neural networks and projection pursuit regression. Chemometr. Intell. Lab. Syst. 2008;92:92–99. [Google Scholar]
- 53.Chen Y.X., Mant C.T., Hodges R.S. Temperature selectivity effects in reversed-phase liquid chromatography due to conformation differences between helical and non-helical peptides. J. Chromatogr. A. 2003;1010:45–61. doi: 10.1016/s0021-9673(03)00877-x. [DOI] [PubMed] [Google Scholar]
- 54.Cunliffe J.M., Maloney T.D. Fused-core particle technology as an alternative to sub-2-alpha m particles to achieve high separation efficiency with low backpressure. J. Sep. Sci. 2007;30:3104–3109. doi: 10.1002/jssc.200700260. [DOI] [PubMed] [Google Scholar]
- 55.Donato P., Dugo P., Cacciola F. High peak capacity separation of peptides through the serial connection of LC shell-packed columns. J. Sep. Sci. 2009;32:1129–1136. doi: 10.1002/jssc.200800639. [DOI] [PubMed] [Google Scholar]
- 56.D’Hondt M., Vangheluwe E., Van Dorpe S. Stability of extemporaneously prepared cytarabine, methotrexate sodium, and methylprednisolone sodium succinate. Am. J. Health-Syst. Pharm. 2012;69:232–240. doi: 10.2146/ajhp110208. [DOI] [PubMed] [Google Scholar]
- 57.Alpert A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic-acids and other polar compounds. J. Chromatogr. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 58.Shibue M., Mant C., Hodges R. Effect of anionic ion-pairing reagent hydrophobicity on selectivity of peptide separations by reversed-phase liquid chromatography. J. Chromatogr. A. 2005;1080:68–75. doi: 10.1016/j.chroma.2005.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Todeschini R., Consonni V. Wiley-VCH; Weinheim: 2001. Handbook of Molecular Descriptors. [Google Scholar]
- 60.Sandberg M., Eriksson L., Jonsson J. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998;41:2481–2491. doi: 10.1021/jm9700575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material