

Abstract
Introduction
The eIF2α kinase heme-regulated inhibitor (HRI) is one of four well-described kinases that phosphorylate eIF2α in response to various cell stressors, resulting in reduced ternary complex formation and attenuation of mRNA translation. Although HRI is well known for its role as a heme sensor in erythroid progenitors, pharmacologic activation of HRI has been demonstrated to have anti-cancer activity across a wide range of tumor sub-types. Here, the potential of HRI activators as novel cancer therapeutics is explored.
Areas covered
We provide an introduction to eIF2 signaling pathways in general, and specifically review data on the eIF2α kinase HRI in erythroid and non-erythroid cells. We review aspects of targeting eIF2 signaling in cancer and highlight promising data using HRI activators against tumor cells.
Expert opinion
Pharmacologic activation of HRI inhibits tumor growth as a single agent without appreciable toxicity in vivo. The ability of HRI activators to provide direct and sustained eIF2α phosphorylation without inducing oxidative stress or broad eIF2α kinase activation may be especially advantageous for tolerability. Combination therapy with established therapeutics may further augment anti-cancer activity to overcome disease resistance.
Keywords: Cancer, eIF2, HRI, translation, ER stress
1. Introduction
Protein synthesis is a complex multistep process, which includes peptide chain initiation, elongation, termination, and recycling [1,2]. Translation initiation is often rate limiting for protein synthesis and requires the formation and recycling of the ternary complex formed by eIF2, GTP, and Met-tRNAi. The eIF2.GTP.Met-tRNAi ternary complex is required for recognition of the translation start codon and 80S assembly. eIF2 is a trimeric guanine nucleotide-binding protein complex that recycles between GTP-bound active and GDP-bound inactive forms. The complex has higher affinity for GDP, which coupled with high intracellular GDP to GTP ratio necessitates activity of a guanine nucleotide exchange factor for successful recycling of eIF2.GDP to eIF2.GTP complex. This guanine nucleotide exchange is catalyzed by a five subunit eIF2B complex whose activity is essential for formation of the ternary complex. Phosphorylation of eIF2α simultaneously increases affinity of eIF2 for eIF2B and blocks its guanine nucleotide exchange activity. This reduces ternary complex abundance and attenuates translation initiation (Figure 1)[3].
Figure 1.
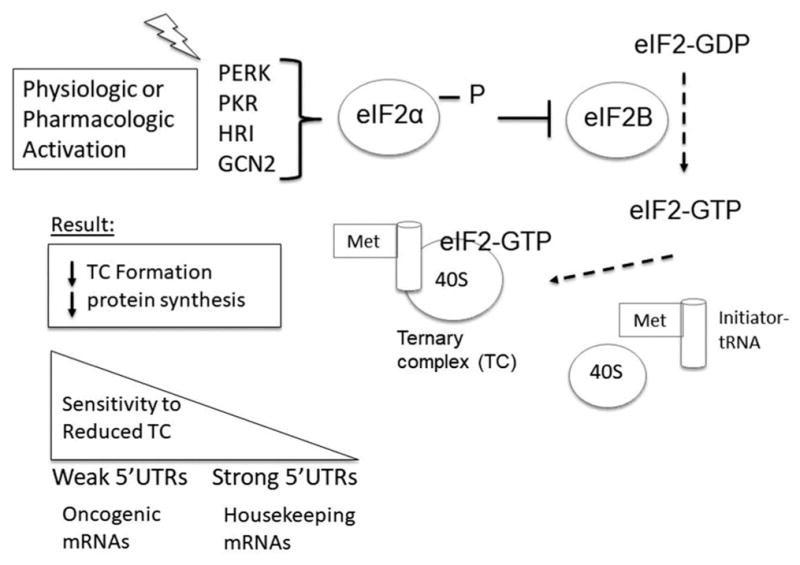
Phosphorylation of eIF2α modulates protein synthesis. Phosphorylation of eIF2α on serine 51 may result from physiologic or pharmacologic activation of eIF2α kinases. There are four well-described eIF2α kinases: PERK, PKR, HRI, and GCN2 (see text for full names) each activated by a diverse set of cellular stressors. Phosphorylation of eIF2α results in attenuated guanine nucleotide (GDP/GTP) exchange activity as a result of inhibitory effects on the guanine nucleotide exchange factor (eIF2B). As a result, less eIF2-GTP-met-tRNAi ternary complex (TC) is able to be formed. Since ternary complexes are required for translation initiation, the end result is a general repression of protein synthesis. mRNAs with long or structured 5′ untranslated regions (UTRs), including many oncogenic mRNAs (e.g. growth factors, transcription factors) may be especially vulnerable to reduced TC abundance (weak 5′UTRs), compared to housekeeping mRNAs with more efficient 5′UTR sequences (strong 5′UTRs). 40S = 40S small ribosomal subunit.
Phosphorylation of eIF2α on serine 51 is mediated by four upstream eIF2 kinases: PKR-like ER kinase (PERK), protein kinase RNA-activated (PKR), general control non-derepressible 2 (GCN2), and heme-regulated inhibitor (HRI). Though these kinases converge on eIF2α phosphorylation, they may be activated by distinct cellular stresses and phosphorylate a diverging set of additional substrates. For example, PERK is activated by endoplasmic reticulum (ER) stress, while PKR is activated in response to viral infection, and GCN2 via nutrient (amino acid) depletion. In contrast, HRI acts primarily as a heme sensor [4]. Heme interacts directly with and inactivates HRI. In the setting of heme deficiency, HRI is activated to attenuate globin translation in erythroid precursors [5]. Certain stressors (such as oxidative stress or proteasome inhibition) may activate different eIF2 kinases depending on the cellular and experimental context [6–9]. In addition, when specific eIF2 kinases are deleted experimentally, an alternative eIF2 kinase may be activated, though usually to a lesser degree, demonstrating potential for redundant functions [10].
While phosphorylation of eIF2α results in a general repression of mRNA translation, some mRNAs are more sensitive to reduced ternary complex formation. mRNAs containing long or highly structured 5′ untranslated region (UTR) sequences, which includes growth factors and cell cycle proteins, may be particularly affected [11]. Reduced ternary complex formation may also promote increased translation of a subset of mRNAs. This group of mRNAs incudes those regulated by multiple upstream open reading frames (uORFs) in the 5′ UTR [12]. In the setting of reduced ternary complex availability, the scanning ribosome may bypass inhibitory uORFs upstream of the bonafide start codon, resulting in a de-repression of mRNA translation (Figure 2). Among these mRNAs, the most well characterized of which is activating transcription factor 4 (ATF4), many are involved in mediating cellular stress responses as part of an integrative stress response [13]. An example of this coordinated response is seen in the setting of HRI activation in erythroid cells, where the attenuation of globin translation is thus paired with upregulation of ATF4, which helps reduce oxidative stress and promote erythroid survival [7,14]. Although this integrated stress response can aid in protecting cells from various stressors, phosphorylation of eIF2α may also lead to proapoptotic outcomes [15]. A notable example is seen with the transcription factor C/EBP homologous protein (CHOP), which is upregulated in response to eIF2α phosphorylation [16]. In early erythroid cells, the induction of CHOP in response to oxidative stress may serve a protective role, possibly by aiding translation recovery via feedback inhibition of eIF2α phosphorylation [7,17]. However, the induction of CHOP can also act to promote apoptosis [8,18]. The intensity and duration of stress likely plays a role in facilitating a switch toward a proapoptotic response, though the precise mechanisms remain to be elucidated [19].
Figure 2.
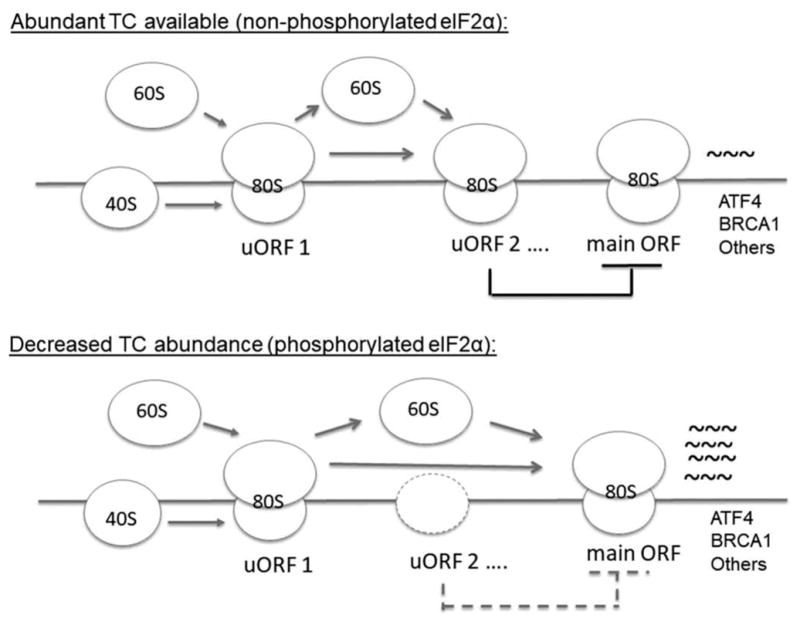
eIF2α phosphorylation results in translational de-repression of select mRNAs. When eIF2α is in a non-phosphorylated state, as is seen during robust translational periods, there is abundant ternary complex (TC) availability for protein synthesis. For specific mRNAs with multiple upstream open reading frames (uORFs), ribosomal occupancy of inhibitory uORFs upstream of the start codon (main ORF) can lead to low levels of protein expression. In the setting of phosphorylated eIF2α, reduced availability of ternary complexes can lead to ribosomal bypass of inhibitory uORFs leading to translational de-repression and increased protein expression of select mRNAS. Notable examples of mRNAs that are regulated in this manner include activating transcription factor 4 (ATF4) and BRCA1. A simplified schematic is demonstrated for illustrative purposes. 60S = large ribosomal subunit. 40S = small ribosomal subunit. 80S = joined 80S ribosome.
Recent manuscripts have reviewed the structure and function of eIF2 kinases and the role of HRI in erythroid cells [4,5]. This review will highlight data on targeting HRI as an anticancer strategy.
1.1. HRI background
HRI (EIF2AK1) is best known for its role in coupling heme availability to globin synthesis in early erythroid cells [5]. Indeed, initial studies characterizing HRI tissue distribution in rabbits noted a restricted expression pattern in reticulocytes and bone marrow but not in other tissues [20,21]. Subsequently, an eIF2α kinase was isolated from rat brain and mouse liver with 82–83% sequence homology to rabbit reticulocyte HRI and similarly containing conserved heme regulatory motifs [22,23]. Using a cDNA probe, mRNA expression for rat and mouse HRI was identified across a wide range of tissues suggesting ubiquitous expression. Nonetheless, Berlanga et al. noted that HRI expression was variable, with higher levels in the liver and spleen, and lower levels in the kidney, brain, and lung [23]. While it was convincingly demonstrated that non-erythroid HRI demonstrates heme-sensitivity [23], it is also known that HRI can be activated by heme-independent mechanisms. For example, HRI from mouse reticulocytes can be activated in response to arsenite, heat shock, or osmotic stress [24]. While reactive oxygen species are important for arsenite-induced activation of HRI, the chaperone molecules Hsp90 and Hsc70 are required for HRI activation in response to multiple stressors, as blockade of either molecule disrupts HRI activation in intact reticulocytes [24].
Modulation of HRI also results in physiologic changes in tissues beyond the erythroid lineage. For example, in mouse hepatocytes, knockout of HRI results in increased ER stress [25], while pharmacologic activation of HRI reduces ER-stress-induced hepatic steatosis and glucose intolerance in mouse models [26]. HRI activation is associated with increased hepatic expression of fibroblast growth factor 21 (FGF21), an important protein for attenuating ER stress [27]. Deletion of HRI in mouse models also results in reduced expression of hepcidin in the liver, impaired macrophage maturation, and reduced erythrophagocytosis [28]. In neuronal cells, HRI has been found to mediate the translation of GluN2B [29], a subunit for the N-methyl-D-aspartate (NMDA) receptor important for neuronal activity [30]. Inhibition of HRI in mice impairs memory retrieval [29], while HRI-mediated translation of β-site APP cleaving enzyme-1 (BACE1) may have a physiologic role in memory consolidation [31]. The precise role of HRI, distinct from other eIF2 kinases, in tissues beyond the erythroid lineage continues to be an area of active investigation.
In summary, HRI has demonstrated the capacity for heme-dependent and heme-independent activation in erythroid and non-erythroid tissues (Table 1).
Table 1.
Modulation of HRI across tissue types.
| Tissue | HRI Deletion/Inhibition | HRI Activation |
|---|---|---|
| Erythroid | Increased ROS; Impaired erythroid differentiation and survival [7,14] | Reduced globin translation [7,14], Increased ratio of fetal to adult globin [71] |
| Macrophage | Impaired maturation and function [28] | Enhanced inflammatory response [28] |
| Hepatic | Increased ER stress [25] Decreased hepcidin [28] | Increased FGF21/Decreased fatty liver changes [26] |
| Neuronal | Impaired memory retrieval [29] | Increased BACEl [31] |
| Cancer | Not reported | Inhibition of tumor growth; apoptosis [32–34] |
The deletion/inhibition or activation of HRI results in tissue-specific effects. The consequence of HRI modulation in each cell type (where known) is reviewed with the relative references. HRI: heme regulated inhibitor; ROS: reactive oxygen species; ER: endoplasmic reticulum; FGF: fibroblast growth factor; BACE: beta-site amyloid precursor protein cleaving enzyme 1.
1.2. EIF2α Phosphorylation and HRI activation in cancer
The phosphorylation of eIF2α has an important role in regulating cell growth and maintaining normal cellular homeostasis. Thus, it has been hypothesized that dysregulated eIF2 signaling may promote malignant transformation. Experimentally, dominant negative PKR mutants, or eIF2α mutants that cannot be phosphorylated on serine 51, have been found to induce malignant transformation in NIH 3T3 cells [35]. In many cancers, eIF2α expression is increased when compared to normal tissue [32,36,37] and likely reflects the heightened translational demand in tumor cells. In some early-stage cancers, an increase in eIF2α kinase activity is observed and is associated with improved survival [38]. The phosphorylation of eIF2α in this setting may initially serve a protective role, in part by acting as a tumor suppressor [39]. However, over time, this initial anti-oncogenic response may be overridden by an adaptive ER stress response favorable to tumor growth. In the tumor microenvironment, and indeed any condition associated with chronic stress, the induction of a temporally and spatially regulated integrated ER stress response allows the cell to adapt to chronic ER stress through the attenuation of protein translation, the upregulation of ER-chaperone proteins and the production of antioxidative molecules, among other factors [13]. This coordinated cellular response is associated not only with the activation of eIF2α kinase PERK, but also with the activation of additional ER proteins including inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6). The activation of IRE1α leads to splicing of X-box-binding protein 1 (XBP1) mRNA, and in concert with ATF6, to the expansion of the ER as part of an adaptive prosurvival stress response [40].
In contrast to the highly coordinated temporally and spatially regulated adaptive cellular responses seen with chronic ER stress, the therapeutic activation of eIF2α kinases, such as HRI, by pharmacologic means does not trigger similar global cellular changes. This is not surprising because unlike chronic ER stress, pharmacologic activation of eIF2α phosphorylation does not activate other unfolded protein response (UPR) pathways such as IRE1/XBP1 or ATF6. Furthermore, unlike UPR-induced eIF2α phosphorylation which is transient, the pharmacological activation of eIF2α phosphorylation can be of higher intensity and much longer duration. As a result, the primary outcome of direct and sustained eIF2α kinase activation is attenuation of translation initiation as a result of reduced ternary complex formation, often leading to cell death [33]. Cancer cells, which adapt to persistent stress through the temporal and spatial induction of UPR pathways [41] may be particularly sensitive to perturbations in ternary complex homeostasis caused by sustained eIF2α phosphorylation. This hypothesis has been supported experimentally as inhibiting ternary complex formation through induction of eIF2α phosphorylation reduces proliferation across a wide range of cancer cell lines in vitro and restricts tumor growth in vivo [42,43].
Despite this potential, few agents designed to specifically target eIF2 signaling have made it to the clinic, in contrast to those targeting other translational regulators such as mechanistic target of rapamycin (mTOR) [44–47]. Nonetheless, several novel agents targeting PERK and other aspects of the ER stress response are in development [48–52]. It is also appreciated that many chemotherapy drugs used clinically do induce eIF2α phosphorylation indirectly to various capacities. A notable example is seen with proteasome inhibition with bortezomib [53,54]. Salubrinal, an inhibitor of eIF2α de-phosphorylation, has also been found to promote apoptosis and overcome drug resistance in cancer cells [55–57]. Direct activators of HRI provide an alternative approach by targeting a specific kinase upstream of eIF2α phosphorylation without globally affecting the downstream activity of all eIF2α kinases (Figure 3). Recent biochemical evidence has demonstrated that a group of specific N,N′-diarylurea molecules directly activate HRI distinctly from other eIF2α kinases without causing oxidative stress [33]. In support of their specificity in targeting HRI, knockdown of PERK, PKR, GCN2 (or all three in combination) has no affect in modulating N,N′-diarylurea drug activity. Anti-proliferative activity of targeting HRI has been demonstrated across several cancer cell lines in vitro including lung, prostate, and melanoma cells. Further, a candidate N,N′-diarylurea, BTdCPU, inhibits tumor growth in breast xenograft models and demonstrates in vivo phosphorylation of eIF2α [33]. Prolonged treatment with BTdCPU at therapeutic doses does not result in appreciable organ toxicity or impact on blood parameters in these mouse models.
Figure 3.
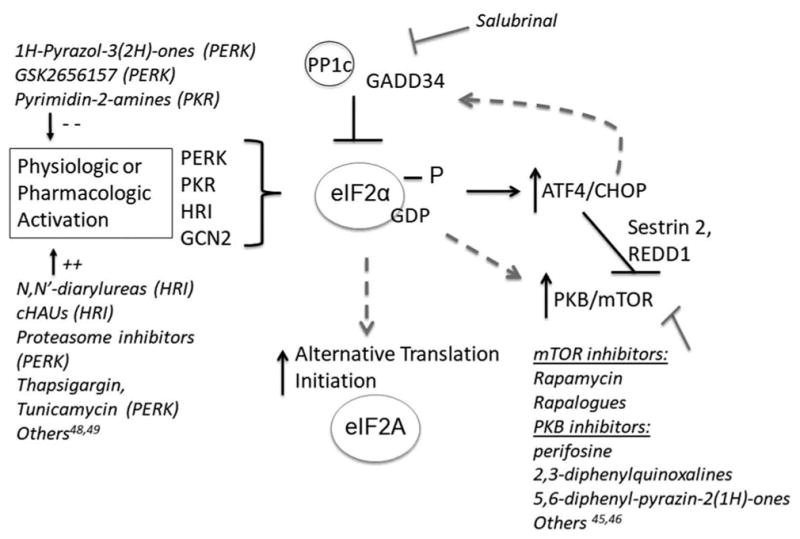
Potential feedback loops and cross-talk following eIF2α phosphorylation. Activation of eIF2α phosphorylation by upstream kinases results in up-regulation of ATF4 and CHOP. Potential feedback loops that could attenuate eIF2α phosphorylation have been described, most notably through GADD34 and PP1c. In addition, activation of eIF2α kinases can be associated with up-regulation of PKB and mTOR signaling, as well as promotion of alternative translation initiation mechanisms such as those that utilize eIF2A. Dashed arrows are used to imply potential avenues of resistance, since feedback loops and cross-talk mechanisms may depend on the type of stress and cell system involved. Pharmacologic agents targeting upstream or downstream aspects of eIF2 signaling are annotated in italics. PP1c = protein phosphatase 1; GADD34 = protein phosphatase 1 regulatory subunit 15A; ATF4 = activating transcription factor 4; CHOP = DNA damage inducible transcript 3; eIF2 = eukaryotic translation initiation factor; PKB = protein kinase B; mTOR = mechanistic target of rapamycin; PERK/PKR/HRI/GCN2: See text for full names.
In a separate study, BTdCPU also demonstrated activity in multiple myeloma (MM) cells in vitro. In this study, BTdCPU treatment lead to the rapid phosphorylation of eIF2α, expression of CHOP, and induction of apoptosis in both dexamethasone-sensitive and -resistant cells [32]. Proteasome inhibitors, which have potent clinical activity in MM, also induce eIF2α phosphorylation and expression of CHOP in MM cells [53,58]. Resistance to proteasome inhibition in MM is associated with attenuated cellular eIF2α phosphorylation and can be overcome using methods that enhance eIF2α phosphorylation, including treatment with salubrinal [57].
The anticancer effects of HRI activation likely go beyond the upregulation of ATF4 and CHOP. In addition to ATF4, there are other mRNAs with upstream ORFs that are susceptible to leaky ribosome scanning and translational de-repression in the setting of reduced ternary complex formation (Figure 2) [59]. Treatment with agents that phosphorylate eIF2α in breast cancer cells result in translational upregulation of BRCA1 protein, an important tumor suppressor molecule. This effect is lost when the uORFs of BRCA1 are disrupted [60]. As discussed above, phosphorylation of eIF2α also significantly suppresses translation initiation, and as a result, mRNAs with highly structured 5′ UTRs (including growth factors, transcription factors, and other oncogenic mRNAs) may be especially impacted [61]. A notable example is the cell cycle protein cyclin D1. Treatment with small molecules that phosphorylate eIF2α and inhibit tumor growth in vivo have been shown to reduce cyclin D1 expression in various tissues [42]. Similar results have now been corroborated in vitro and in vivo by using another chemotype, 1-((1,4-trans)-4-arylox-ycyclohexyl)-3-arylureas (cHAUs) that directly activate HRI. Lead cHAUs similarly activate HRI, induce eIF2α phosphorylation, and inhibit cancer cell proliferation in vitro and melanoma xenograft growth in vivo [34,62].
Phosphorylation of eIF2α is also associated with modulation of PI3K-Akt and mTOR pathways, although this cross talk is thought to primarily serve a protective role [63]. For example, PKR activation results in induction of PI3K signaling, which antagonizes PKR-mediated apoptosis [64]. Thus, while the induction of eIF2α phosphorylation via HRI is a viable anticancer strategy, the inhibition of eIF2α phosphorylation to augment effectiveness of PI3K-Akt blockade has also been proposed as a potential therapeutic combination [65]. Whether HRI activation results in similar cross talk is less clear, though in one study inhibition of mTOR augmented anticancer efficacy in combination with HRI activation in vitro [32]. The upregulation of ATF4 via eIF2α phosphorylation may itself serve to attenuate mTOR activity through the induction of mTOR repressors including Sestrin2 and REDD1 [66–68]. In addition, phosphorylation of eIF2α may promote alternative translational mechanisms, such as those utilizing eIF2A [69] (Figure 3).
Further research is needed to better understand the interplay of HRI with other translational pathways to develop rationale therapeutic combinations.
2. Expert opinion
Pharmacologic activation of the eIF2α kinase HRI is an emerging anticancer strategy. Though activation of HRI may have broad clinical applicability, some cancers such as MM and certain breast cancer subtypes may be particularly susceptible to reduced ternary complex formation caused by sustained eIF2α phosphorylation [32,33,42]. While HRI activators demonstrate single-agent activity in vivo, it will be especially helpful to elucidate how HRI activators pair with other therapeutics used clinically. In MM, for example, the induction of eIF2α phosphorylation can significantly enhance myeloma cell death in combination with proteasome inhibition. [57] Another rational combination may be to pair HRI activators with mTOR inhibitors or other inhibitors of protein synthesis, to promote synergistic effects.
While additional on-target effects beyond the cancer cell are likely, in vivo toxicity profiles have been favorable [33]. No target-dependent toxicities have emerged with direct HRI activation [33,34], while there are reports of secondary benefits of HRI activation in models of human disease including beta-thalassemia and fatty liver disease [26,70,71]. Other small-molecule compounds that phosphorylate eIF2α in vivo and reduce tumor growth are known to be well tolerated, such as eicosapentaenoic acid, a main component of fish oils [42]. Proteasome inhibitors, which induce robust eIF2α phosphorylation, are used extensively in MM and other cancers with manageable toxicity profiles [72]. The specificity of HRI activators to induce eIF2α phosphorylation without promoting oxidative stress or broadly activating eIF2α kinases may be helpful for improving the toxicity profile of these drugs compared to stress-targeting agents in development [48,49]. Although inhibition of eIF2α phosphorylation is also associated with anticancer activity in vivo, this approach suffers from severe target-dependent toxicity which may not be surmountable for clinical development [73]. These target-dependent toxicities include pancreatic injury as well as behavioral inflexibility, which is a hallmark of autism spectrum disorders [74,75]. Inhibition of eIF2α phosphorylation may also exacerbate motor neuron disease [76,77].
For HRI activators, and indeed any therapeutic strategy aimed at inducing eIF2α phosphorylation, it will be important to consider theoretical effects on promoting tumor growth and metastasis. As prefaced above, stress-induced activation of eIF2α phosphorylation can direct adaptation to cellular stress and a prosurvival outcome [15]. Despite these concerns, in vivo studies of specific HRI activators, including N,N′-diarylurea and cHAU compounds [33,34] demonstrate that pharmacologic induction of eIF2α phosphorylation can inhibit tumor growth at nontoxic doses. The ability of HRI activators to provide intense and sustained eIF2α phosphorylation to trigger an apoptotic response is likely critical to an anticancer outcome. Indeed, the efficacy of HRI activators also appears to be correlated with expression of HRI protein in cancer cells [33]. Thus, incorporation of sensitive and accurate methods for quantifying HRI tissue expression will be important in early-phase trials examining HRI activators in patients.
Future studies will provide additional insights into activating HRI in specific biologic and genetic contexts to optimize clinical development and early-phase trials in this promising area.
Article highlights.
eIF2α phosphorylation by upstream eIF2 kinases reduces ternary complex formation
eIF2 kinases can be activated by cellular stressors or by pharmacologic activation
Pharmacologic activation of the eIF2α kinase HRI inhibits tumor growth in vivo
Phosphorylation of eIF2α augments anti-cancer activity when used in combination with established therapeutics
Activation of HRI is not associated with appreciable organ toxicity at anti-cancer doses
This box summarizes key points contained in the article.
Acknowledgments
Funding
This work was supported by NIH grant [R01 CA152312 (BHA) and K12 Career Development Award/Multiple Myeloma Opportunities for Research and Education Award (NB)].
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Spilka R, Ernst C, Mehta AK, et al. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013;340(1):9–21. doi: 10.1016/j.canlet.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 2.Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol. 2012;4(7):a013706. doi: 10.1101/cshperspect.a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr. 2012;3(3):307–321. doi: 10.3945/an.112.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnelly N, Gorman AM, Gupta S, et al. The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JJ. Translational control by heme-regulated eIF2alpha kinase during erythropoiesis. Curr Opin Hematol. 2014;21(3):172–178. doi: 10.1097/MOH.0000000000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajesh K, Krishnamoorthy J, Kazimierczak U, et al. Phosphorylation of the translation initiation factor eIF2alpha at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 2015;6:e1591. doi: 10.1038/cddis.2014.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Suragani RN, Zachariah RS, Velazquez JG, et al. Heme-regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood. 2012;119(22):5276–5284. doi: 10.1182/blood-2011-10-388132. Experimental evidence demonstrating a critical role for HRI in erythroid differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280(14):14189–14202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 9.Yerlikaya A, Kimball SR, Stanley BA. Phosphorylation of eIF2alpha in response to 26S proteasome inhibition is mediated by the haem-regulated inhibitor (HRI) kinase. Biochem J. 2008;412(3):579–588. doi: 10.1042/BJ20080324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhan K, Narasimhan J, Wek RC. Differential activation of eIF2 kinases in response to cellular stresses in schizosaccharomyces pombe. Genetics. 2004;168(4):1867–1875. doi: 10.1534/genetics.104.031443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mignone F, Gissi C, Liuni S, et al. Untranslated regions of mRNAs. Genome Biol. 2002;3(3):REVIEWS0004. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 13.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 14.Han AP, Yu C, Lu L, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20(23):6909–6918. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34(Pt 1):7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 16.Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem. 2011;286(13):10939–10949. doi: 10.1074/jbc.M110.216093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novoa I, Zeng H, Harding HP, et al. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153(5):1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teske BF, Fusakio ME, Zhou D, et al. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013;24(15):2477–2490. doi: 10.1091/mbc.E13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urra H, Dufey E, Lisbona F, et al. When ER stress reaches a dead end. Biochim Biophys Acta. 2013;1833(12):3507–3517. doi: 10.1016/j.bbamcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Pal JK, Chen JJ, London IM. Tissue distribution and immunoreactivity of heme-regulated eIF-2 alpha kinase determined by monoclonal antibodies. Biochemistry. 1991;30(9):2555–2562. doi: 10.1021/bi00223a037. [DOI] [PubMed] [Google Scholar]
- 21.Crosby JS, Lee K, London IM, et al. Erythroid expression of the heme-regulated eIF-2 alpha kinase. Mol Cell Biol. 1994;14(6):3906–3914. doi: 10.1128/mcb.14.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22••.Mellor H, Flowers KM, Kimball SR, et al. Cloning and characterization of cDNA encoding rat heminsensitive initiation factor-2 alpha (eIF-2 alpha) kinase. evidence for multitissue expression. J Biol Chem. 1994;269(14):10201–10204. Characterization of ubiquitous HRI expression in diverse tissues. [PubMed] [Google Scholar]
- 23••.Berlanga JJ, Herrero S, de Haro C. Characterization of the heminsensitive eukaryotic initiation factor 2alpha kinase from mouse nonerythroid cells. J Biol Chem. 1998;273(48):32340–32346. doi: 10.1074/jbc.273.48.32340. Demonstration of heme-sensitive eIF2 kinase activity in tissues beyond the erythroid lineage. [DOI] [PubMed] [Google Scholar]
- 24.Lu L, Han AP, Chen JJ. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol. 2001;21(23):7971–7980. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Acharya P, Chen JJ, Correia MA. Hepatic heme-regulated inhibitor (HRI) eukaryotic initiation factor 2alpha kinase: a protagonist of heme-mediated translational control of CYP2B enzymes and a modulator of basal endoplasmic reticulum stress tone. Mol Pharmacol. 2010;77(4):575–592. doi: 10.1124/mol.109.061259. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26••.Zarei M, Barroso E, Leiva R, et al. Heme-regulated eIF2alpha kinase modulates hepatic FGF21 and is activated by PPARbeta/delta deficiency. Diabetes. 2016;65(10):3185–3199. doi: 10.2337/db16-0155. In vivo evidence demonstrating that pharmacologic activation of HRI can modulate disease phenotype in fatty liver disease and glucose intolerance. [DOI] [PubMed] [Google Scholar]
- 27.Jiang S, Yan C, Fang QC, et al. Fibroblast growth factor 21 is regulated by the IRE1alpha-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J Biol Chem. 2014;289(43):29751–29765. doi: 10.1074/jbc.M114.565960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Liu S, Suragani RN, Wang F, et al. The function of heme-regulated eIF2alpha kinase in murine iron homeostasis and macrophage maturation. J Clin Invest. 2007;117(11):3296–3305. doi: 10.1172/JCI32084. Experimental evidence demonstrating an important role for HRI in mediating macrophage maturation, inflammatory responses, and erythrophagocytosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos-Fernandez E, Tajes M, Ill-Raga G, et al. Glutamatergic stimulation induces GluN2B translation by the nitric oxide-heme-regulated eIF2alpha kinase in cortical neurons. Oncotarget. 2016;7(37):58876–58892. doi: 10.18632/oncotarget.11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storey GP, Opitz-Araya X, Barria A. Molecular determinants controlling NMDA receptor synaptic incorporation. J Neurosci. 2011;31(17):6311–6316. doi: 10.1523/JNEUROSCI.5553-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ill-Raga G, Tajes M, Busquets-Garcia A, et al. Physiological control of nitric oxide in neuronal BACE1 translation by heme-regulated eIF2alpha kinase HRI induces synaptogenesis. Antioxid Redox Signal. 2015;22(15):1295–1307. doi: 10.1089/ars.2014.6080. [DOI] [PubMed] [Google Scholar]
- 32.Burwick N, Zhang MY, de la Puente P, et al. The eIF2-alpha kinase HRI is a novel therapeutic target in multiple myeloma. Leuk Res. 2017;55:23–32. doi: 10.1016/j.leukres.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33••.Chen T, Ozel D, Qiao Y, et al. Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol. 2011;7(9):610–616. doi: 10.1038/nchembio.613. Initial report identifying N,N′-diarylureas as unique inhibitors of ternary complex formation, and specific activators of HRI with anticancer activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.Yefidoff-Freedman R, Fan J, Yan L, et al. Development of 1-((1,4-trans)-4-aryloxycyclohexyl)-3-arylurea activators of the heme regulated inhibitor as selective activators of eucaryotic translation initiation factor 2 alpha (eIF2alpha) phosphorylation arm of the integrated endoplasmic reticulum stress response. J Med Chem. 2017;60(13):5392–5406. doi: 10.1021/acs.jmedchem.7b00059. Development of cHAUs as in vivo compounds that selectively and potently activate HRI with anticancer activity at nontoxic doses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donze O, Jagus R, Koromilas AE, et al. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995;14(15):3828–3834. doi: 10.1002/j.1460-2075.1995.tb00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lobo MV, Martin ME, Perez MI, et al. Levels, phosphorylation status and cellular localization of translational factor eIF2 in gastrointestinal carcinomas. Histochem J. 2000;32(3):139–150. doi: 10.1023/a:1004091122351. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, Rosenwald IB, Hutzler MJ, et al. Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-hodgkin’s lymphomas. Am J Pathol. 1999;155(1):247–255. doi: 10.1016/s0002-9440(10)65118-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He Y, Correa AM, Raso MG, et al. The role of PKR/eIF2alpha signaling pathway in prognosis of non-small cell lung cancer. PLoS One. 2011;6(11):e24855. doi: 10.1371/journal.pone.0024855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mounir Z, Krishnamoorthy JL, Robertson GP, et al. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal. 2009;2(102):ra85. doi: 10.1126/scisignal.2000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oakes SA. Endoplasmic reticulum proteostasis: a key checkpoint in cancer. Am J Physiol Cell Physiol. 2017;312(2):C102. doi: 10.1152/ajpcell.00266.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692–706. doi: 10.1016/j.cell.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aktas BH, Qiao Y, Ozdelen E, et al. Small-molecule targeting of translation initiation for cancer therapy. Oncotarget. 2013;4(10):1606–1617. doi: 10.18632/oncotarget.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen L, Aktas BH, Wang Y, et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget. 2012;3(8):869–881. doi: 10.18632/oncotarget.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhat M, Robichaud N, Hulea L, et al. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14(4):261–278. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
- 45.Benjamin D, Colombi M, Moroni C, et al. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 46.Pal SK, Reckamp K, Yu H, et al. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;19(11):1355–1366. doi: 10.1517/13543784.2010.520701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindsley CW, Zhao Z, Leister WH, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15(3):761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Garg AD, Maes H, van Vliet AR, et al. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol Cell Oncol. 2014;2(1):e975089. doi: 10.4161/23723556.2014.975089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schonthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem Pharmacol. 2013;85(5):653–666. doi: 10.1016/j.bcp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Smith AL, Andrews KL, Beckmann H, et al. Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK) J Med Chem. 2015;58(3):1426–1441. doi: 10.1021/jm5017494. [DOI] [PubMed] [Google Scholar]
- 51.Axten JM, Romeril SP, Shu A, et al. Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med Chem Lett. 2013;4(10):964–968. doi: 10.1021/ml400228e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bryk R, Wu K, Raimundo BC, et al. Identification of new inhibitors of protein kinase R guided by statistical modeling. Bioorg Med Chem Lett. 2011;21(13):4108–4114. doi: 10.1016/j.bmcl.2011.04.149. [DOI] [PubMed] [Google Scholar]
- 53.Chauhan D, Tian Z, Zhou B, et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011;17(16):5311–5321. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yerlikaya A, DoKudur H. Investigation of the eIF2alpha phosphorylation mechanism in response to proteasome inhibition in melanoma and breast cancer cells. Mol Biol (Mosk) 2010;44(5):859–866. [PubMed] [Google Scholar]
- 55.Teng Y, Gao M, Wang J, et al. Inhibition of eIF2alpha dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis. 2014;5:e1060. doi: 10.1038/cddis.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeon YJ, Kim JH, Shin JI, et al. Salubrinal-mediated upregulation of eIF2alpha phosphorylation increases doxorubicin sensitivity in MCF-7/ADR cells. Mol Cells. 2016;39(2):129–135. doi: 10.14348/molcells.2016.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57••.Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009;69(4):1545–1552. doi: 10.1158/0008-5472.CAN-08-3858. Elucidation of eIF2 alpha dephosphorylation as a critical mechanism of resistance to bortezomib that can be overcome using strategies that hyperphosphorylate eIF2 alpha. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Obeng EA, Carlson LM, Gutman DM, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinnebusch AG. Translational regulation of yeast GCN4. A window on factors that control initiatortrna binding to the ribosome. J Biol Chem. 1997;272(35):21661–21664. doi: 10.1074/jbc.272.35.21661. [DOI] [PubMed] [Google Scholar]
- 60.Aktas BH, Bordelois P, Peker S, et al. Depletion of eIF2.GTP.met-tRNAi translation initiation complex up-regulates BRCA1 expression in vitro and in vivo. Oncotarget. 2015;6(9):6902–6914. doi: 10.18632/oncotarget.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kozak M. An analysis of vertebrate mRNA sequences: intimations of translational control. J Cell Biol. 1991;115(4):887–903. doi: 10.1083/jcb.115.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen T, Takrouri K, Hee-Hwang S, et al. Explorations of substituted urea functionality for the discovery of new activators of the heme-regulated inhibitor kinase. J Med Chem. 2013;56(23):9457–9470. doi: 10.1021/jm400793v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koromilas AE, Mounir Z. Control of oncogenesis by eIF2alpha phosphorylation: implications in PTEN and PI3K-akt signaling and tumor treatment. Future Oncol. 2013;9(7):1005–1015. doi: 10.2217/fon.13.49. [DOI] [PubMed] [Google Scholar]
- 64.Kazemi S, Mounir Z, Baltzis D, et al. A novel function of eIF2alpha kinases as inducers of the phosphoinositide-3 kinase signaling pathway. Mol Biol Cell. 2007;18(9):3635–3644. doi: 10.1091/mbc.E07-01-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mounir Z, Krishnamoorthy JL, Wang S, et al. Akt determines cell fate through inhibition of the PERK-eIF2alpha phosphorylation pathway. Sci Signal. 2011;4(192):ra62. doi: 10.1126/scisignal.2001630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem Biophys Res Commun. 2009;379(2):451–455. doi: 10.1016/j.bbrc.2008.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saveljeva S, Cleary P, Mnich K, et al. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget. 2016;7(11):12254–12266. doi: 10.18632/oncotarget.7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye J, Palm W, Peng M, et al. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 2015;29(22):2331–2336. doi: 10.1101/gad.269324.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sendoel A, Dunn JG, Rodriguez EH, et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature. 2017;541(7638):494–499. doi: 10.1038/nature21036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest. 2005;115(6):1562–1570. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hahn CK, Lowrey CH. Eukaryotic initiation factor 2alpha phosphorylation mediates fetal hemoglobin induction through a post-transcriptional mechanism. Blood. 2013;122(4):477–485. doi: 10.1182/blood-2013-03-491043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14(7):417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scheper W, Hoozemans JJ. A new PERKspective on neurodegeneration. Sci Transl Med. 2013;5(206):206fs37. doi: 10.1126/scitranslmed.3007641. [DOI] [PubMed] [Google Scholar]
- 74.Atkins C, Liu Q, Minthorn E, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73(6):1993–2002. doi: 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- 75.Trinh MA, Kaphzan H, Wek RC, et al. Brain-specific disruption of the eIF2alpha kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep. 2012;1(6):676–688. doi: 10.1016/j.celrep.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Way SW, Podojil JR, Clayton BL, et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun. 2015;6:6532. doi: 10.1038/ncomms7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Das I, Krzyzosiak A, Schneider K, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348(6231):239–242. doi: 10.1126/science.aaa4484. [DOI] [PMC free article] [PubMed] [Google Scholar]