

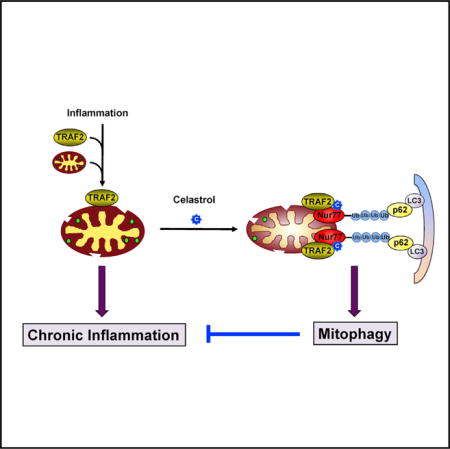
SUMMARY
Mitochondria play an integral role in cell death, autophagy, immunity, and inflammation. We previously showed that Nur77, an orphan nuclear receptor, induces apoptosis by targeting mitochondria. Here, we report that celastrol, a potent anti-inflammatory pentacyclic triterpene, binds Nur77 to inhibit inflammation and induce autophagy in a Nur77-dependent manner. Celastrol promotes Nur77 translocation from the nucleus to mitochondria, where it interacts with tumor necrosis factor receptor-associated factor 2 (TRAF2), a scaffold protein and E3 ubiquitin ligase important for inflammatory signaling. The interaction is mediated by an LxxLL motif in TRAF2 and results not only in the inhibition of TRAF2 ubiquitination but also in Lys63-linked Nur77 ubiquitination. Under inflammatory conditions, ubiquitinated Nur77 resides at mitochondria, rendering them sensitive to autophagy, an event involving Nur77 interaction with p62/SQSTM1. Together, our results identify Nur77 as a critical intracellular target for celastrol and unravel a mechanism of Nur77-dependent clearance of inflamed mitochondria to alleviate inflammation.
Graphical abstract
INTRODUCTION
Nur77 (also called TR3, NGFIB, and NR4A1), an orphan member of the nuclear receptor superfamily and an immediate early response gene, plays a critical role in a plethora of cellular processes in response to diverse stimuli such as mitogens, cytokines, and stress, metabolic, and apoptotic signals (Beard et al., 2015; Evans, 2009; Hamers et al., 2013; Lee et al., 2011; McMorrow and Murphy, 2011; Moll et al., 2006; Zhang, 2007). Recent interest has focused on its potent anti-inflammatory effect in inflammatory diseases and cancer. Genetic studies have revealed a critical role of Nur77 in controlling the inflammatory responses, highlighted by its protective function in atherosclerosis (Hamers et al., 2012; Hanna et al., 2012), obesity (Perez-Sieira et al., 2013), diabetes (Chao et al., 2009), asthma (Kurakula et al., 2015), arthritis (De Silva et al., 2005), and inflammatory bowel disease (Hamers et al., 2015; Wu et al., 2016).
The NF-κB signaling pathway plays a pivotal role in inflammation (Chen, 2012; Hayden and Ghosh, 2008; Karin and Gallagher, 2009). Stimulation of the tumor necrosis factor receptor (TNFR) with the proinflammatory cytokine results in NF-κB activation, which is mediated by members of the TNFR-associated factor (TRAF) family characterized by a highly conserved TRAF domain and a RING domain. TRAF2, the most widely studied TRAF member, serves as a scaffold protein and E3 ubiquitin ligase by binding to numerous TNFR-family proteins to mediate the activation of the inhibitor of κB (IκB) kinase (IKK) and NF-κB (Borghi et al., 2016; Bradley and Pober, 2001). Previous studies have revealed extensive interaction between Nur77 and NF-κB signaling pathways. Ectopic expression of Nur77 in macrophages increases the expression of the inducible inhibitor of NF-κB kinase (Pei et al., 2006), while Nur77 could inhibit inflammatory gene expression (Harant and Lindley, 2004). However, the precise mechanism underlying the potent anti-inflammatory effect of Nur77 remains obscure.
Nur77 can function in the nucleus as a transcriptional factor to modulate target gene transcription (Beard et al., 2015; Evans, 2009; Hamers et al., 2013; Lee et al., 2011; McMorrow and Murphy, 2011; Moll et al., 2006; Zhang, 2007). Regulation of gene transcription by nuclear receptors is mediated through their interaction with coactivators or corepressors via the LxxLL motif present in coactivators or the L/IxxI/VI motif present in corepressors, respectively (Dasgupta et al., 2014; Hermanson et al., 2002; Hsia et al., 2010; Hu and Lazar, 1999; Lonard and O’Malley, 2012; Xu et al., 1999). Recent studies have revealed a critical role of mitochondria in regulating inflammatory processes. The organelle is susceptible to damage from inflammatory signals; it in turn releases danger signals, including reactive oxygen species (ROS), for the assembly and activation of inflammasome (Green et al., 2011; Gurung et al., 2015; Levine et al., 2011; Nunnari and Suomalainen, 2012; Pinton and Kroemer, 2014). We previously showed that Nur77 could translocate from the nucleus to mitochondria to induce cytochrome c release and apoptosis (Kolluri et al., 2008; Li et al., 2000; Lin et al., 2004; Zhang, 2007). Whether Nur77 participates in modulating mitochondrial quality control and mitochondria-mediated inflammatory signaling remains unknown. Identifying agents that bind Nur77 to induce its mitochondrial targeting will facilitate the investigation of the complex role of Nur77 at mitochondria.
Here we report our discovery of celastrol, a triterpenoid quinine methide isolated from the root of Tripterygium wilfordii, which is commonly known as “Thunder God Vine” (Corson and Crews, 2007; Kannaiyan et al., 2011; Liu et al., 2015; Salminen et al., 2010; Wong et al., 2012), as a Nur77 modulator. Our results demonstrate that celastrol confers its anti-inflammatory effect by inducing Nur77 mitochondrial translocation and, subsequently, Nur77-dependent elimination of damaged mitochondria through autophagy.
RESULTS
Celastrol Binds to Nur77
We used surface plasmon resonance (SPR)-based assay to screen natural compounds from marine and terrestrial sources with known anti-inflammatory activity for binding to Nur77 (Figure S1A) and identified celastrol (Figure 1A) as a potent binder with a Kd of 0.29 μM (Figure 1B). The binding was confirmed by circular dichroism (CD) spectroscopy, which showed an altered CD spectrum of the Nur77-LBD by celastrol (Figure S1B). High-performance liquid chromatography (HPLC) analysis also revealed a direct binding of celastrol to purified Nur77-LBD, but not to the LBD of retinoid X receptor (RXRα-LBD) (Figure S1C). In the reporter assay, celastrol inhibited transactivation of Nur77, but not glucocorticoid receptor (GR) (Figure S1D). Molecular docking studies suggested that celastrol binds to a previously identified, hydrophobic groove on the surface of Nur77 protein (Lee et al., 2014; Zhan et al., 2012; Figure S1E).
Figure 1. Celastrol Binding to Nur77 and Their Regulation of Inflammation.
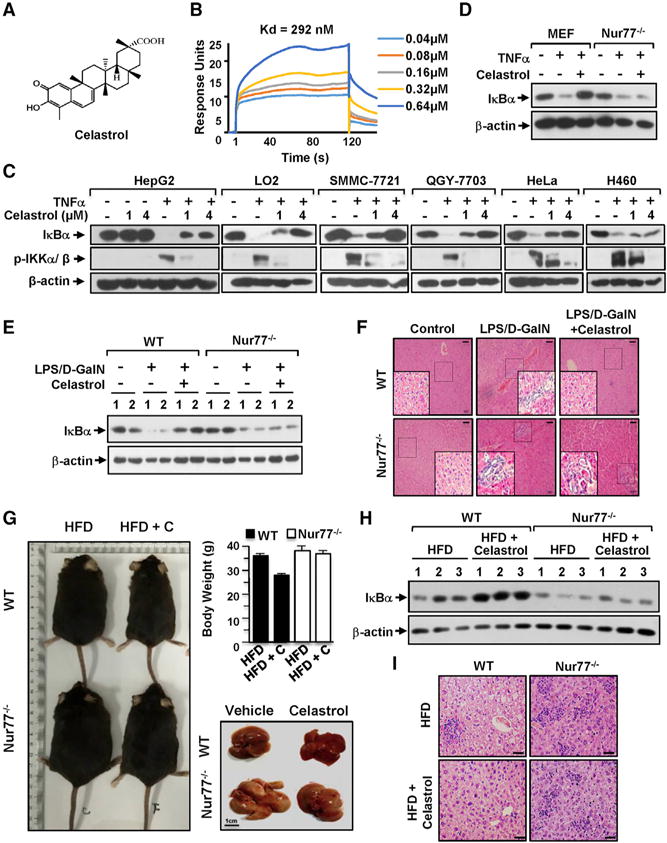
(A) Celastrol structure.
(B) Celastrol binding to Nur77 was illustrated by SPR assay of purified Nur77-LBD with celastrol.
(C) Inhibition of TNFα-induced IKKα/β phosphorylation and IκBα degradation by celastrol. Cells treated with celastrol and/or TNFα (20 ng/mL) for 30 min were analyzed by western blotting (WB).
(D) WB of MEFs and Nur77−/− MEFs treated with celastrol (1 μM) and/or TNFα for 1 hr. Nur77 is required for celastrol inhibition of TNFα-induced IκBα degradation.
(E) Expression of IκBα protein in liver lysates from two representative wild-type and Nur77−/− mice treated with LPS, D-GalN, and celastrol.
(F) Representative images show H&E-stained section of liver tissues from wild-type and Nur77−/− mice treated with LPS, D-GalN, and celastrol (scale bar, 100 μm).
(G) Role of Nur77 in celastrol inhibition of HFD-induced obesity. Body weight and liver morphology (n ≥ 6 per group) of wild-type (WT) and Nur77−/− mice. Celastrol, C. Data were represented as mean ± SEM.
(H) Expression of IκBα in liver lysates from three representative wild-type and Nur77−/− mice.
(I) Representative images show H&E staining of liver tissue from wild-type and Nur77−/− mice (n ≥ 6 per group). Scale bar, 50 μm.
For western blots, one of three similar experiments is shown.
See also Figure S1.
Nur77 Mediates the Anti-inflammatory Effects of Celastrol
Celastrol antagonized the effects of inflammatory cytokine TNFα, including its induction of IκBα degradation, nuclear translocation of the p65 subunit of NF-κB, and NF-κB transactivation. Inhibition of TNFα-induced IκBα degradation by celastrol correlated with its suppression of IKKα/β phosphorylation (Figure 1C), demonstrating that celastrol acts at or upstream of IKK activation. The role of Nur77 was illustrated by data showing that transfection of Nur77 siRNA into HepG2 cells, which inhibited Nur77 expression, abrogated the inhibitory effect of celastrol on TNFα-induced IκBα degradation (Figure S1F), while transfection of RXRα siRNA had no effect (Figure S1G). In addition, celastrol inhibited TNFα-induced IκBα degradation (Figure 1D) in mouse embryonic fibroblasts (MEFs), but not in MEFs lacking Nur77 (Nur77−/− MEFs).
Nur77 Is Required for Celastrol Inhibition of Acute Liver Inflammation
To determine the role of Nur77 in vivo, we examined the effect of celastrol on lipopolysaccharide (LPS) and D-galactosamine (D-GalN)-induced hepatic inflammatory injury using Nur77-null mice. Administration of celastrol to wild-type mice reduced LPS- and D-GalN-induced serum levels of hepatic injury markers alanine aminotransferase (ALT) and aspartate aminotransferase (AST) by 35% and 47%, respectively (Figure S1H). Although Nur77−/− mice had higher serum ALT and AST than wild-type mice when injected with LPS and D-GalN, celastrol showed a much reduced inhibitory effect in these mice. LPS- and D-GalN-induced serum production of the proinflammatory cytokines IL-1β and IL-6 (Figure S1I) and their hepatic mRNA expression (Figure S1J) were significantly inhibited by celastrol in wild-type mice, while such effects were largely attenuated in Nur77−/− mice. Celastrol also alleviated LPS- and D-GalN-induced downregulation of IκBα expression (Figure 1E), destruction of hepatic architecture (Figure 1F), and p65 nuclear translocation (Figure S1K) in wild-type but not Nur77−/− mice. Histological analysis of lung tissue revealed a Nur77-dependent inhibition of LPS- and D-GalN-induced lung inflammation and neutrophil infiltration by celastrol (not shown). Thus, celastrol inhibition of LPS- and D-GalN-induced acute inflammation and NF-κB activation is Nur77 dependent.
Nur77 Is Required for Celastrol Inhibition of Chronic Inflammation in Obese Animals
We next determined the effect of celastrol and Nur77 using the high-fat diet (HFD)-induced obesity animal model, which represents a state of chronic low-grade inflammation (Hotamisligil, 2006; Nathan and Ding, 2010). In agreement with recent results (Liu et al., 2015; Ma et al., 2015), administration of celastrol significantly reduced body weight, adipose tissue mass, the size of adipocytes, and ameliorated fatty liver caused by HFD in wild-type mice (Figures 1G and S1L). Nur77−/− mice exhibited increased susceptibility to HFD-induced obesity, gaining more body weight and fat mass than wild-type mice, consistent with the metabolic role of Nur77 (Chao et al., 2007, 2009; Chen et al., 2015; Perez-Sieira et al., 2013). Importantly, Nur77−/− mice were resistant to the anti-obesity effects of celastrol, as celastrol showed only about 4% inhibition on body weight of Nur77−/− mice, compared to 22% inhibition in wild-type mice (Figure 1G). The ability of celastrol to attenuate HFD-induced adiposity (not shown) and serum production of ALT and AST (Figure S1M) was also compromised in Nur77−/− mice. Furthermore, celastrol administration inhibited the effect of HFD on activating the inflammatory pathway—including the expression and production of IL-1β and IL-6 (Figures S1N and S1O), IκBα expression (Figure 1H), p65 nuclear translocation (Figure S1P), hepatic inflammation (Figure 1I), and accumulation of hepatic neutrophil (Figure S1Q) and macrophage (Figure S1R)—in wild-type, but not Nur77−/− mice. Thus, celastrol inhibition of chronic inflammation in obese animals is also Nur77 dependent.
Nur77 Mediates the Autophagic Effect of Celastrol
Recent developments reveal a crucial role of the autophagy pathway and proteins in regulating inflammation (Green et al., 2011; Levine et al., 2011). We next determined whether the anti-inflammatory effect of celastrol could be attributed to its induction of autophagy. Western blotting (WB) revealed a strong induction of LC3-II, a marker for autophagy, upon treatment of HepG2 cells with celastrol for 6 hr (Figure 2A). Although treatment with TNFα alone had no effect, its combination with celastrol resulted in a stronger induction of LC3-II expression, suggesting a role of inflammation in promoting the autophagic effect of celastrol. The autophagic effect of celastrol was also illustrated by its induction of the formation of punctate green fluorescent protein LC3 (GFP-LC3) (Figure 2B) or red fluorescent protein LC3 (RFP-LC3) (Figure S2A), a hallmark of autophagy induction. Nur77 was essential for the autophagic effect of celastrol, as the induction of LC3-II expression by celastrol was observed in MEFs, but not Nur77−/− MEFs (Figure 2C). In vivo, celastrol administration induced LC3-II expression (Figures 2D and S2B) and aggregated distribution of LC3B (Figures 2E and S2C) in liver tissues from LPS- and D-GalN-injected wild-type mice, but not Nur77−/− mice. Similar induction of aggregated LC3B immunostaining (Figure 2F) and LC3-II expression (Figure 2G) by celastrol was found in HFD-induced wild-type, but not Nur77−/− obese mice.
Figure 2. Role of Nur77 in Celastrol Induction of Autophagy.
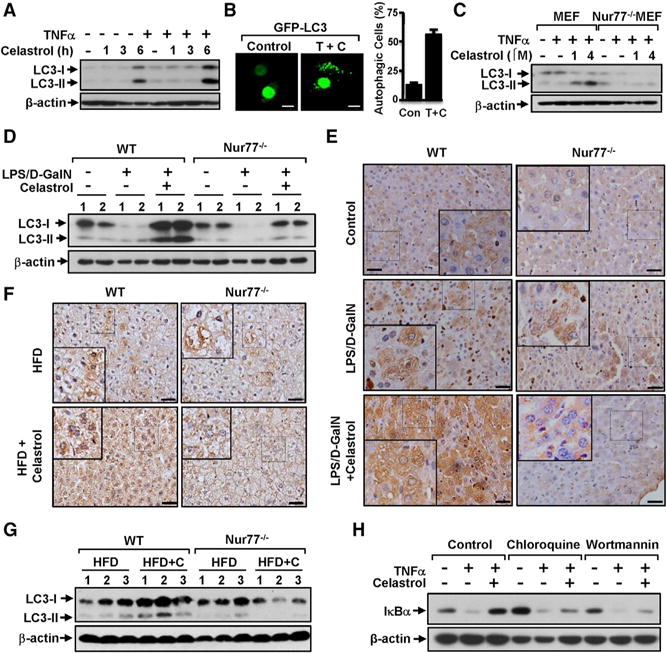
(A) HepG2 cells treated with celastrol (2 μM) were exposed to TNFα (20 ng/mL) for an additional 30 min and analyzed by WB.
(B) Representative images show the formation of GFP-LC3 punctate in HepG2 cells after treatment with celastrol and TNFα. TNFα, T; celastrol, C. About 200 GFP-LC-3 transfected cells were examined for displays of GFP-LC3 punctate and scored. Data were represented as mean ± SEM for three experiments. Scale bar, 10 μm.
(C) Nur77 is required for induction of LC3-II analyzed by WB in MEFs and Nur77−/− MEFs treated with celastrol and TNFα (20 ng/mL) for 4 hr.
(D) Celastrol induces LC3-II expression in liver tissue from wild-type but not Nur77−/− mice treated with LPS and D-GalN, as analyzed by WB.
(E) Representative images show increased LC3B expression by celastrol in liver tissue from wild-type but not Nur77−/− mice treated with LPS and D-GalN, examined by immunohistochemistry staining. Scale bar, 50 μM.
(F) Representative images show increased LC3B expression by celastrol in liver tissue from wild-type but not Nur77−/− mice fed with HFD. Scale bar, 50 μM.
(G) Celastrol induces LC3-II expression in liver tissue from wild-type but not Nur77−/− mice fed with HFD, as analyzed by WB.
(H) Autophagic inhibitors attenuate the inhibitory effect of celastrol on TNFα-induced IκBα degradation. HepG2 cells treated with hydroxychloroquine (20 μM) or wortmannin (100 nM) for 1 hr before exposure to TNFα and celastrol were analyzed by WB.
For WB, one of three or five similar experiments is shown.
See also Figure S2.
The concurrent effects of celastrol on inhibiting inflammation and inducing autophagy suggested that both events might converge. Indeed, treatment of HepG2 cells with the autophagic inhibitors chloroquine, a lysosomotropic agent that prevents fusion of endosomes and lysosomes, and wortmannin, which inhibits both class I phosphoinositide 3-kinase (PI3K) and class III PtdIns3K, impaired the ability of celastrol to inhibit TNFα-induced IκBα degradation (Figure 2H). Thus, Nur77-dependent induction of autophagy by celastrol contributes to its anti-inflammatory function.
Celastrol Induces Colocalization of Nur77 and TRAF2 at Mitochondria
To determine whether mitochondrial targeting of Nur77 accounts for its anti-inflammatory and autophagic functions, we analyzed the effect of celastrol on the subcellular localization of Nur77. Nur77 predominantly resided in the nuclei of MEFs, which were not affected by TNFα treatment (Figure 3A). However, celastrol treatment induced diffused distribution of Nur77 throughout cells. Addition of TNFα led to punctate Nur77 staining, which colocalized extensively with mitochondria (Figure 3A). The combination treatment also resulted in speckled Nur77 staining in the cytoplasm of several cancer cell lines (Figures S3A and S3B). GFP-Nur77, which was found exclusively in the nuclei of control cells, displayed a speckled pattern in the cytoplasm of cells treated with celastrol and TNFα (Figure S3C). In contrast, the combination treatment had no effect on the nuclear localization of transfected Myc-RXRα (Figure S3D), demonstrating a specific effect of the treatment. Furthermore, cellular fractionation assay revealed an accumulation of an upshifted Nur77 band in the mitochondrial fraction prepared from celastrol- and TNFα-treated cells (Figure 3B).
Figure 3. Celastrol Induction of Nur77 Colocalization with TRAF2 at Mitochondria.
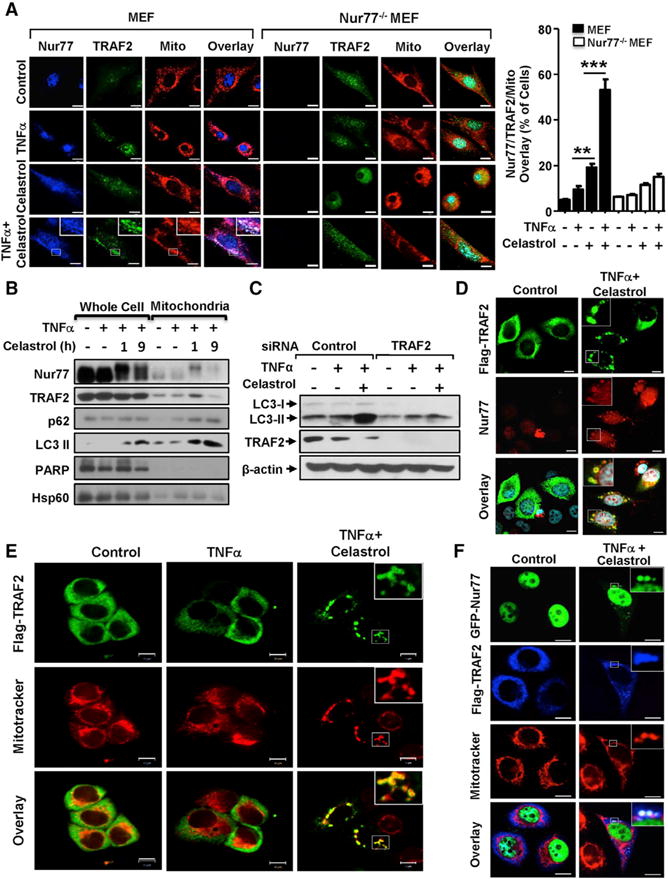
(A) Colocalization of endogenous Nur77 and TRAF2. Representative images show colocalization of endogenous TRAF2 and Nur77 with mitochondria in MEFs and Nur77−/− MEFs after treatment with celastrol (2 μM) and/or TNFα (20 ng/mL) for 1 hr, examined by immunostaining. Statistical data were represented as mean ± SEM of five independent images. **p < 0.01, ***p < 0.001 (Student’s t test).
(B) Celastrol induces coaccumulation of Nur77, TRAF2, p62, and LC3-II at mitochondria. Whole-cell lysates and mitochondrial fractions prepared from HepG2 cells treated with celastrol (4 μM) and/or TNFα (20 ng/mL) were analyzed by WB. One of three similar experiments is shown.
(C) TRAF2-dependent induction of autophagy by celastrol. Lysates from HepG2 cells transfected with control or TRAF2 siRNA were treated with celastrol (2 μM) and TNFα (20 ng/mL) for 6 hr and analyzed by WB.
(D) Representative images illustrate colocalization of endogenous Nur77 with transfected Flag-TRAF2 in HepG2 cells after treatment with celastrol (4 μM) and TNFα (20 ng/mL) for 1 hr, examined by immunostaining.
(E) Representative images show colocalization of transfected Flag-TRAF2 with mitochondria in HepG2 cells after treatment with celastrol and TNFα, analyzed by immunostaining.
(F) Representative images show colocalization of transfected Flag-TRAF2 and GFP-Nur77 with mitochondria in HepG2 cells after treatment with celastrol and TNFα, examined by immunostaining. Scale bars, 10 μm.
See also Figure S3.
Recent studies indicate a critical role of TRAF2 at mitochondria. TRAF2 was recruited to mitochondria by the mitochondrial adaptor protein MAVS or the mitochondrial E3 ligase MULAN (Hou et al., 2011; Liu et al., 2013; Zemirli et al., 2014) to mediate mitochondrion-based inflammatory signaling, including IKK-dependent NF-κB activation. Previous studies (Kim et al., 2010; Yang et al., 2015) and our data (Figure S3E) showed that TNFα could enhance TRAF2 mitochondrial accumulation in a time-dependent manner. Prolonged exposure of HepG2 cells to TNFα (6 hr or longer) resulted in mitochondrial dysfunction, revealed by a decrease in mitochondrial membrane potential (ΔΨm) (Figure S3F) and an altered mitochondrial network (Figure S3G). Transfection of TRAF2 siRNA impaired LC3-II induction by celastrol and TNFα treatment (Figure 3C) similarly to the effect of knocking down Nur77, implying that Nur77 might act in concert with TRAF2 to mediate the autophagic effect of celastrol. Our immunostaining revealed an extensive colocalization of endogenous TRAF2 with Nur77 at mitochondria in MEFs treated with celastrol and TNFα (Figure 3A). Induction of TRAF2 accumulation at mitochondria by celastrol and TNFα was suppressed in Nur77−/− MEFs, revealing a role of Nur77. Nur77 colocalization with TRAF2 was also observed in several other cell lines (Figures S3A and S3B). In addition, transfected Flag-TRAF2 formed punctate structures colocalizing with endogenous Nur77 (Figure 3D) and mitochondria (Figure 3E) in celastrol- and TNFα-treated cells. Cotranslocation of Nur77 and TRAF2 to mitochondria was further illustrated by immunostaining showing extensive colocalization of transfected GFP-Nur77 and Flag-TRAF2 with mitochondria (Figure 3F) and by cellular fractionation assay revealing coaccumulation of TRAF2 and the upshifted Nur77 band in the purified mitochondrial fraction (Figure 3B).
Celastrol Promotes Nur77 Interaction with TRAF2
Celastrol- and TNFα-induced cotranslocation of Nur77 and TRAF2 to mitochondria prompted us to determine whether Nur77 interacted with TRAF2. Coimmunoprecipitation (coIP) assay demonstrated that endogenous Nur77 protein induced by TNFα was specifically coimmunoprecipitated with TRAF2 in a celastrol-dependent manner (Figure 4A). Ectopically expressed Nur77 and TRAF2 also interacted in cells treated with celastrol, but not with TNFα or RXRα ligand CD3254 (Figure 4B). For comparison, celastrol failed to induce Nur77 interaction with either TRAF3 or TRAF5 (Figure S4A), whereas other known Nur77 modulators, including 3-Cl-AHPC (Lin et al., 2004) and DIM-C-pPhOH (Lee et al., 2014) could not promote Nur77 interaction with TRAF2 (Figure S4B). We also conducted GST pull-down assay and showed that purified Nur77-LBD was pulled down by GST-TRAF2, but not GST protein, in the presence of celastrol (Figure 4C). Incubation of lysates containing ectopically expressed Myc-Nur77 with celastrol promoted interaction with GST-TRAF2 (Figure S4C). Thus, Nur77, upon binding celastrol, can directly interact with TRAF2.
Figure 4. Celastrol Promotes Nur77 Interaction with TRAF2.
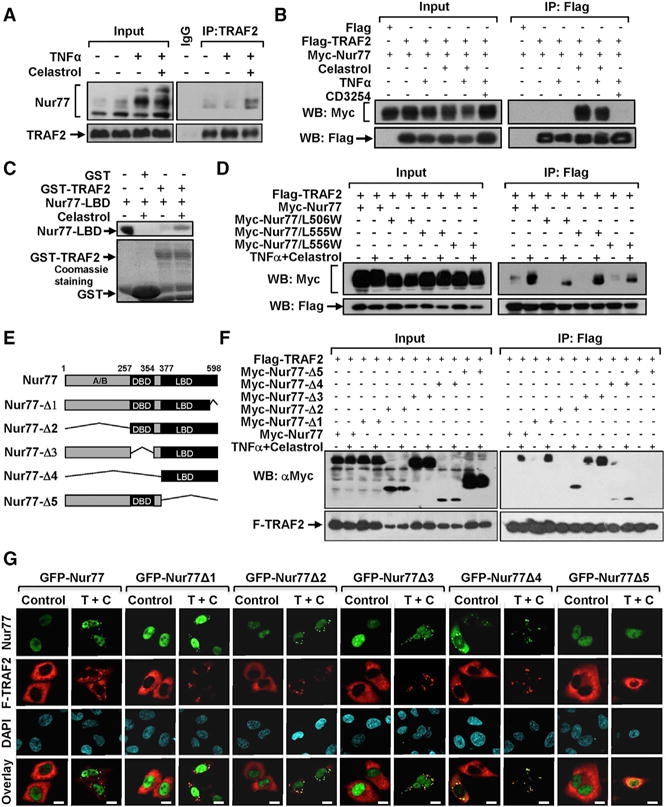
(A, B) Celastrol promotes Nur77 interaction with TRAF2. The interaction of endogenous (A) or transfected (B) Nur77 with TRAF2 in HepG2 cells treated with TNFα (20 ng/mL), celastrol (1 μM), or CD3254 (1 μM) was analyzed by coIP.
(C) Celastrol promotes Nur77-LBD interaction with TRAF2 in vitro. Purified Nur77-LBD incubated with or without 4 μM celastrol was pulled down by GST or GST-TRAF2 protein and analyzed by WB. GST proteins used were shown by Coomassie blue staining.
(D) HepG2 cells transfected with the indicated Flag-TRAF2 and Myc-Nur77 or mutants were treated with celastrol (4 μM) and TNFα (20 ng/mL) for 1 hr and analyzed by coIP.
(E) Schematic representation of Nur77 and mutants. DBD, DNA-binding domain; LBD, ligand-binding domain.
(F) Mutational analysis of Nur77 interaction with TRAF2. HepG2 cells transfected with the indicated Flag-TRAF2 and Myc-Nur77 or mutants were treated with celastrol (4 μM) and TNFα (20 ng/mL) for 1 hr and analyzed by coIP.
(G) Colocalization of TRAF2 with Nur77 or mutants. HepG2 cells transfected with Flag-TRAF2 and Nur77 or mutants were treated with celastrol (4 μM) and TNFα (20 ng/mL) for 1 hr and examined by immunofluorescence staining.
For coIP, one of three or five similar experiments is shown.
See also Figure S4.
To further characterize Nur77 interaction with TRAF2, we examined whether mutation of the celastrol binding site in Nur77 would impact its interaction with TRAF2. Amino acid residues presumably involved in interacting with celastrol (Figure S1E) were mutated, and the resulting mutants were analyzed for their binding to TRAF2. Substitution of Leu506 with Trp (Nur77/L506W) impaired its binding to celastrol (Figure S4D) and markedly reduced its interaction with TRAF2 (Figure 4D). Replacing Leu555 or Leu556 with Trp in the region also reduced the effect of celastrol. Consistently, Nur77/L506W, Nur77/L555W, and Nur77/L556W colocalized only slightly with TRAF2 in celastrol- and TNFα-treated cells (Figure S4E). Thus, celastrol binding is crucial for its induction of Nur77 interaction and colocalization with TRAF2. The fact that TRAF2 coaccumulated with Nur77 but not Nur77/L556W at mitochondria, accompanied with LC3-II induction (Figure S4F), suggests that their interaction is required for their colocalization at mitochondria and the induction of autophagy.
To identify regions in Nur77 responsible for binding TRAF2, several deletion mutants of Nur77 were generated (Figure 4E). Myc-tagged Nur77 or mutants were cotransfected with Flag-TRAF2 into cells and analyzed by coIP assay. Nur77 mutants lacking the C-terminal 20 amino acids (Nur77-Δ1), the N-terminal A/B domain (Nur77-Δ2), or the central DNA-binding domain (DBD)(Nur77-Δ3) could still interact with TRAF2 in the presence of celastrol and TNFα (Figure 4F). In contrast, the Nur77 mutant with LBD deleted (Nur77-Δ5) could not. Consistently, the LBD alone (Nur77-Δ4) was capable of binding TRAF2 (Figure 4F). The importance of the Nur77 LBD was also illustrated by immunostaining showing extensive colocalization of transfected Flag-TRAF2 with Nur77-Δ1, Nur77-Δ2, Nur77-Δ3, and Nur77-Δ4, but not with Nur77-Δ5, in celastrol- and TNFα-treated cells (Figure 4G). Interestingly, GFP-Nur77-LBD was found in the cytoplasm even in the absence of celastrol and TNFα treatment, displaying speckled structures colocalizing with Flag-TRAF2.
An LxxLL Motif in TRAF2 Mediates TRAF2 Interaction with Nur77
To identify regions in TRAF2 that are required for its interaction with Nur77, several TRAF2 deletion mutants were constructed (Figure 5A). CoIP assays revealed that deletion of either the N-terminal RING domain and zinc finger (TRAF2-Δ3) or the C-terminal coiled-coil TRAF-N and β sandwich TRAF-C domains (TRAF2-Δ1) impaired the interaction (Figure 5B). In contrast, TRAF2-Δ2 that contains the RING and zinc finger domains as well as the TRAF-N domain interacted with Nur77 even in the absence of celastrol and TNFα. Thus, the hinge region between zinc finger domain and TRAF-N domain might be important for Nur77 binding. Inspection of the hinge region and the flanking sequences revealed the presence of an LxxLL motif, 245LAMLL249. The LxxLL motifs (NR box) are commonly found in nuclear receptor coactivators to mediate their interaction with nuclear receptors for transcription regulation (Hu and Lazar, 1999; Xu et al., 1999). To determine the role of the LxxLL motif, a peptide (TRAF2-peptide) encompassing the 245LAMLL249 motif and a mutant peptide (TRAF2/AA-peptide) with two C-terminal Leu residues substituted with Ala were synthesized (Figure 5C). SPR assay revealed that the TRAF2 peptide binds to the purified Nur77-LBD protein with a Kd of 4.8 μM (Figure 5D), which is in the range of LxxLL coregulator peptide binding to nuclear receptors (Darimont et al., 1998; Hu and Lazar, 1999). In contrast, the TRAF2/AA mutant peptide failed to bind (Figure 5E). To confirm the significance of the LAMLL motif, two C-terminal Leu residues in the motif were substituted with Ala in TRAF2. The resulting TRAF2 mutant (TRAF2/AA) exhibited reduced interaction with Nur77 as compared to wild-type TRAF2 (Figure 5F). TRAF2/AA also failed to colocalize with Nur77 in cells (Figure 5G). The LxxLL motif in transcriptional coregulators can mediate their interaction with a broad range of nuclear receptors (Dasgupta et al., 2014; Hermanson et al., 2002; Hsia et al., 2010; Hu and Lazar, 1999; Lonard and O’Malley, 2012; Xu et al., 1999). Interestingly, mutation of the LAMLL motif impaired TRAF2 interaction with RXRα-LBD induced by 9-cis-RA, but not by synthetic RXRα ligand CD3254 (Figures S5A and S5B). Together, the LAMLL motif identified in TRAF2 may act in analogy to the LxxLL motif used by the nuclear transcriptional coregulators.
Figure 5. Analysis of the LxxLL Motif in TRAF2 for Binding Nur77.
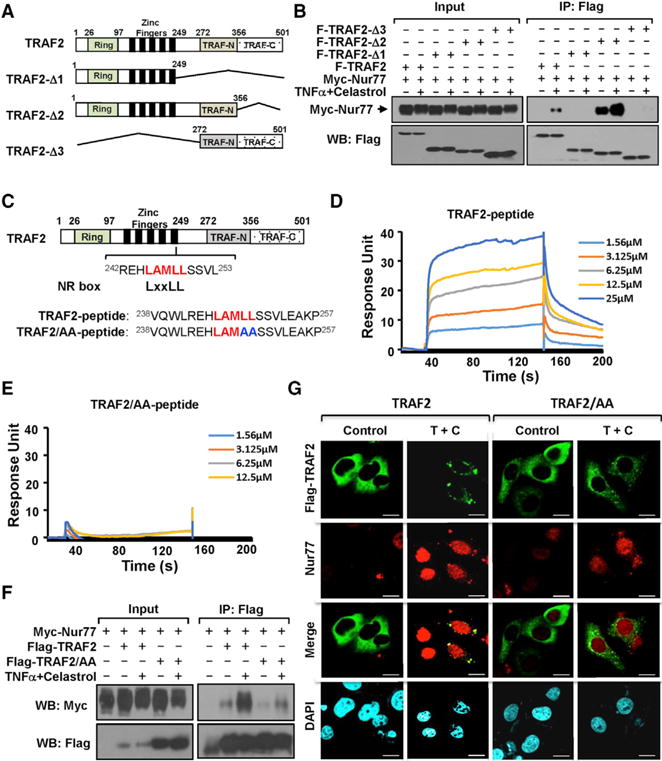
(A) Schematic representation of TRAF2 and mutants.
(B) Mutational analysis of TRAF2 interaction with Nur77. HepG2 cells transfected with Myc-Nur77 and Flag-TRAF2 or mutants were treated with celastrol (4 μM) and TNFα (20 ng/mL) for 1 hr and analyzed by coIP.
(C) Schematic representation of the LAMLL motif in TRAF2. Peptides encompassing the LxxLL motif or mutant are shown.
(D and E) Nur77 binds TRAF2 peptide, but not mutant peptide. TRAF2 peptide (D) or mutant peptide (E) was analyzed for binding to purified Nur77-LBD protein by SPR assay.
(F) Mutation of the LAMLL motif in TRAF2 reduces its interaction with Nur77. HepG2 cells transfected with Myc-Nur77 and Flag-TRAF2 or Flag-Nur77/AA were treated with celastrol (4 μM) and TNFα (20 ng/mL) for 1 hr and analyzed by coIP.
(G) Mutation of the LAMLL motif in TRAF2 impairs its colocalization with Nur77. Representative images illustrate colocalization of endogenous Nur77 with transfected Flag-TRAF2 or Flag-TRAF2/AA in HepG2 cells after treatment with (4 μM) and TNFα (20 ng/mL) for 1 hr were examined by immunostaining. Scale bars, 10 μm.
For coIP, one of three similar experiments is shown.
See also Figure S5.
TRAF2-Nur77 Interaction Inhibits TNFα-Induced Ubiquitination of TRAF2 and RIP1
For classical TNFα activation of the IKK-NF-κB pathway, binding of TNFα to TNFα receptor 1 (TNFR1) triggers recruitment of RIP1 and TRAF2, whose subsequent ubiquitination is crucial for IKK activation (Chen, 2005; Karin and Gallagher, 2009). To determine how celastrol-induced Nur77 interaction with TRAF2 contributed to its anti-inflammatory effect, we first examined whether the interaction inhibited TRAF2 ubiquitination. Cotransfection of Flag-TRAF2 with HA-tagged ubiquitin (HA-Ub) resulted in ubiquitination of Flag-TRAF2, which was enhanced when cells were treated with TNFα and reduced when they were cotreated with celastrol (Figure S6A). The inhibitory effect of celastrol on Flag-TRAF2 ubiquitination was significantly augmented in cells cotransfected with Nur77, but not TRAF2-binding defective mutant Nur77/L506W (Figure 6A), demonstrating a role of Nur77 interaction with TRAF2. We also studied the effect of celastrol on TNFR1 interaction with TRAF2 and RIP1. TNFR1 strongly interacted with TRAF2 and RIP1 upon exposure of cells to TNFα, but this was completely inhibited when cells were cotreated with celastrol (Figure 6B). Similar to its effect on TRAF2 ubiquitination, celastrol attenuated TNFα-induced RIP1 ubiquitination (Figure 6C). Thus, celastrol-induced Nur77 interaction with TRAF2 likely results in inhibition of the classical IKK-NF-κB pathway, providing one molecular explanation for its potent anti-inflammatory activity.
Figure 6. Effect of Nur77 Interaction with TRAF2 on Their Ubiquitination and Mitochondrial Targeting.
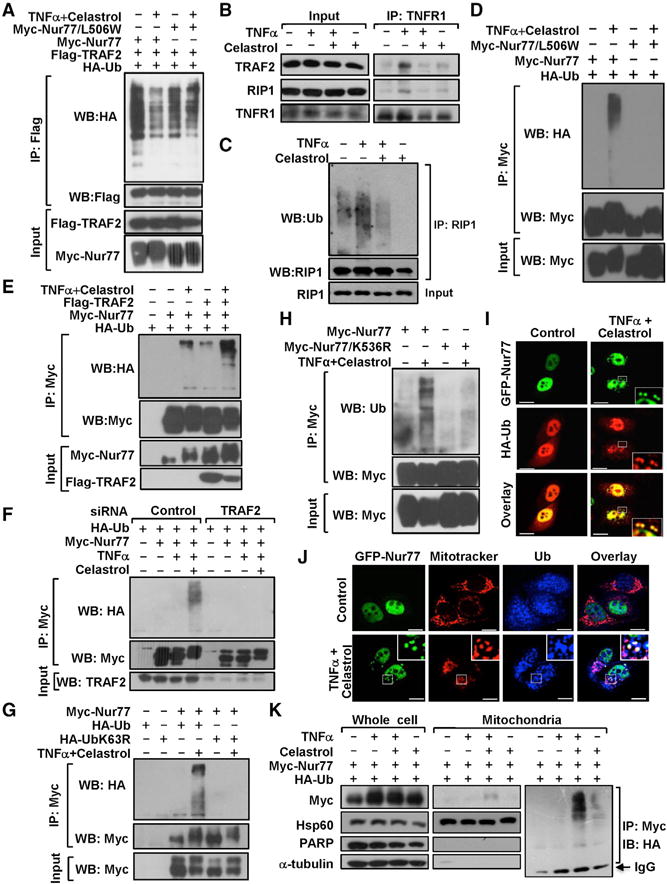
(A) Inhibition of TRAF2 polyubiquitination by celastrol. HepG2 cells transfected with the indicated plasmids were treated with 4 μM celastrol and/or 20 ng/mL TNFα for 1 hr and analyzed using IP followed by WB.
(B) TNFα-induced TNFR1 recruitment of TRAF2 and RIP1 is inhibited by celastrol. HepG2 cells were treated with celastrol (4 μM) and TNFα (20 ng/mL) for 30 min and analyzed by coIP.
(C) Inhibition of TNFα-induced RIP1 ubiquitination by celastrol. HepG2 cells were treated with 4 μM celastrol and/or 20 ng/mL TNFα for 30 min and analyzed using IP followed by WB.
(D–H) Analysis of Nur77 ubiquitination. Shown are (D) effect of celastrol binding, (E) effect of TRAF2 transfection, (F) effect of TRAF2 siRNA transfection, (G) K63-linkage-specific Nur77 ubiquitination, and (H) role of K536 in Nur77 ubiquitination. HepG2 cells transfected with the indicated plasmids were treated with 4 μM celastrol and/or TNFα for 1 hr and analyzed using coIP followed by WB.
(I) Representative images illustrate colocalization of transfected Myc-Nur77 with HA-Ub in HepG2 cells after treatment with 4 μM celastrol and 20 ng/mL TNFα for 1 hr, analyzed by confocal microscopy.
(J) Ubiquitinated Nur77 resides at mitochondria. Representative images illustrate colocalization of transfected GFP-Nur77 with endogenous ubiquitin and mitochondria in HepG2 cells after treatment with 4 μM celastrol and 20 ng/mL TNFα for 1 hr, analyzed by immunostaining.
(K) Ubiquitinated Nur77 resides at mitochondria. HepG2 cells transfected with the indicated plasmids were treated with 4 μM celastrol and/or 20 ng/mL TNFα for 1 hr. Whole-cell lysates and mitochondrial fractions were analyzed by WB. Mitochondrial fractions were also subjected to IP followed by WB.
Scale bars, 10 μm. For ubiquitination assays, one of three or five similar experiments is shown.
See also Figure S6.
Celastrol Induces TRAF2-Dependent Nur77 and Mitochondrial Ubiquitination
The RING domain of TRAF2 possesses E3 Ub ligase activity, promoting K63-linked ubiquitination of its target proteins (Chen, 2012; Karin and Gallagher, 2009). We next determined whether TRAF2 binding to Nur77 resulted in Nur77 ubiquitination. Transfected Myc-Nur77 was significantly ubiquitinated in cells treated with celastrol (Figure S6B). Although TNFα alone had no effect, it enhanced the ability of celastrol to induce Nur77 ubiquitination, suggesting a role of inflammation in Nur77 ubiquitination. The effect of celastrol was likely due to its binding to Nur77, as celastrol and TNFα treatment failed to induce ubiquitination of Myc-Nur77/L506W (Figure 6D). Induction of Nur77 ubiquitination by celastrol and TNFα required TRAF2, as the event was enhanced by cotransfection of Flag-TRAF2 (Figure 6E) but suppressed by TRAF2 siRNA transfection (Figure 6F). Transfection of Lys63-only ubiquitin mutant (UbK63R) failed to incur Nur77 ubiquitination (Figure 6G), indicating a Lys63-linkage-specific Nur77 ubiquitination. Our effort to identify Lys residues responsible for Nur77 ubiquitination showed that, while Nur77 was heavily ubiquitinated by endogenous ubiquitin in celastrol- and TNFα-treated cells, Nur77/K536R, a mutant with Lys536 replaced with Arg, was not (Figure 6H); this demonstrated a critical role of Lys536 in Nur77 ubiquitination.
We next determined whether ubiquitinated Nur77 resided at mitochondria. Immunostaining showed that mitochondrial networks collapsed and were heavily decorated with ubiquitin in celastrol- and TNFα-treated cells (Figure S6C). The celastrol and TNFα treatment also induced the formation of punctate GFP-Nur77 structures in the cytoplasm, colocalizing extensively with ubiquitin (Figure 6I) and mitochondria (Figure 6J). Furthermore, the treatment induced colocalization of Flag-TRAF2 with ubiquitin and mitochondria (Figure S6D). Cellular fractionation assays revealed the presence of ubiquitinated Nur77 in the mitochondrial fraction of celastrol- and TNFα-treated cells (Figure 6K). Thus, TRAF2-dependent ubiquitination of Nur77 contributed to celastrol and TNFα induction of mitochondrial ubiquitination.
Celastrol Induces Mitophagy and Nur77 Interaction with p62/SQSTM1
Ubiquitination of mitochondrial proteins plays a crucial role in their sequestration and degradation within lysosomes through autophagy (Youle and Narendra, 2011). We determined whether mitochondria targeted by ubiquitinated Nur77 were prone to autophagy. Treatment of MEFs, but not Nur77−/− MEFs, with celastrol and TNFα induced aggregated LC3 staining and its colocalization with Nur77 and mitochondria (Figures 7A and S7A). GFP-LC3 also displayed punctate staining overlapping with that of both Nur77 and mitochondria (Figure S7B). Such colocalization of Nur77 with aggregated LC3 and mitochondria depended on levels of TRAF2, because knocking down TRAF2 by TRAF2 siRNA transfection inhibited the colocalization, while ectopic expression of Flag-TRAF2 enhanced it (Figure S7B). Furthermore, transfected Flag-TRAF2 colocalized with mitochondria and speckled GFP-LC3 structures in celastrol- and TNFα-treated cells (Figure 7B). Transfected GFP-Nur77, but not Nur77 ubiquitination-resistant mutant GFP-Nur77/K536R, formed punctate structures colocalizing extensively with RFP-LC3 and mitochondria in celastrol- and TNFα-treated cells (Figure 7C), demonstrating a role of Nur77 ubiquitination. Thus, celastrol and TNFα induction of Nur77 interaction with TRAF2 and subsequent Nur77 ubiquitination serves to prime mitochondria for autophagy.
Figure 7. Celastrol Induces Mitophagy and Nur77 Interaction with p62/SQSTM1.

(A) Celastrol promotes Nur77 colocalization with mitochondria and LC3. Representative images show colocalization of endogenous Nur77 with mitochondria and LC3 in MEFs after treatment with celastrol (2 μM) and TNFα (20 ng/mL) for 1 hr, examined by immunostaining.
(B) Representative images show colocalization of transfected Flag-TRAF2 with GFP-LC3 and mitochondria in HepG2 cells after treatment with celastrol and TNFα, examined by immunostaining.
(C) Representative images show colocalization of transfected GFP-Nur77, but not GFP-Nur77/K536R, with RFP-LC3 and mitochondria in HepG2 cells after treatment with celastrol and TNFα, examined by immunostaining. T, TNFα; C, celastrol. Scale bar, 5 μm.
(D) Celastrol induces Nur77 interaction with p62 in a ubiquitination-dependent manner. HepG2 cells transfected with Myc-Nur77 and Flag-p62 or Flag-p62/ΔUBA were treated with celastrol and TNFα and analyzed by coIP.
(E and F) Colocalization of Nur77 with p62. Representative images illustrate colocalization of transfected endogenous Nur77 (E) or transfected GFP-Nur77 (F) with p62 in HepG2 cells after treatment with 4 μM celastrol and 20 ng/mL TNFα for 1 hr, analyzed by immunostaining. Scale bar, 10 μm.
(G–I) Role of p62 in Nur77-dependent mitophagy. Representative images illustrate colocalization of p62 with ubiquitin (G), mitochondria (H), and LC3 (I) in MEFs, but not in Nur77−/− MEFs, after treatment with 2 μM celastrol and 20 ng/mL TNFα for 1 hr, examined by immunostaining. Scale bar, 20 μm.
See also Figure S7.
To determine the possible mechanism by which mitochondrial Nur77 induced mitophagy, we examined whether it interacted with p62/SQSTM1 (p62), which is implicated in the PINK1/Parkin mitophagic pathway (Geisler et al., 2010) and was more recently shown to mediate NF-κB induction of mitophagy (Zhong et al., 2016). CoIP assays showed that p62 strongly and specifically interacted with the slow-migrating, but not the fast-migrating, Nur77 band in the presence of celastrol and TNFα (Figure 7D). Deletion of the ubiquitin-associated (UBA) domain from p62 abolished the interaction (Figure 7D), suggesting the role of Nur77 ubiquitination. The interaction between Nur77 and p62 was confirmed by extensive colocalization of punctate p62 structures with endogenous Nur77 (Figure 7E) and transfected GFP-Nur77 in celastrol- and TNFα-treated cells. Unlike p62, the p62 mutant, p62/ΔUBA, failed to display punctate staining with Nur77 in celastrol- and TNFα-treated cells (Figure S7C), again suggesting the involvement of Nur77 ubiquitination. Furthermore, treatment of cells with celastrol and TNFα resulted in extensive colocalization of p62 with mitochondria (Figure S7D). Celastrol- and TNFα-induced punctate p62 structures were labeled with ubiquitin (Figure 7G), and they colocalized with mitochondria (Figure 7H) and LC3 (Figure 7I) in wild-type, but not Nur77−/−, MEFs. Cellular fractionation assays revealed a co-accumulation of p62 with LC3, TRAF2, and the slow-migrating Nur77 band in the mitochondrial fraction prepared from cells treated with celastrol and TNFα for 1 hr, while prolonged treatment (9 hr) resulted in reduced levels of Nur77 and TRAF2, likely due to their degradation via mitophagy (Figure 3B). Thus, Nur77 interaction with p62 serves to tether ubiquitin-labeled mitochondria to the LC3-positive phagophore for engulfment.
DISCUSSION
Celastrol has attracted increasing interest recently, especially for its potent anti-inflammatory activities in different inflammatory diseases, including obesity (Liu et al., 2015). However, the mechanism by which celastrol exerts its beneficial effects remains largely unknown. Here, we report that celastrol could exert its anti-inflammatory effect by binding to Nur77, identifying Nur77 as a direct intracellular target of celastrol and providing important insight into the mechanism of celastrol actions. Celastrol inhibition of HFD-induced obesity was suppressed in Nur77−/− mice (Figure 3), revealing a critical role of Nur77 in mediating the anti-obesity effect of celastrol. How Nur77 mediates the leptin-sensitizing activity of celastrol remains unknown. Hypothalamic inflammation is implicated in the development of leptin resistance (Valdearcos et al., 2015). Specific deletion of IKKβ in the medio-basal hypothalamus protected against HFD-induced obesity (Zhang et al., 2008), while pharmacological inhibition of IKKβ/NF-κB improved leptin sensitivity in HFD-fed mice (Yu et al., 2013). Defective hypothalamic autophagy could lead to hypothalamic inflammation by activating the IKKβ/NF-κB pathway (Meng and Cai, 2011). We found that celastrol could also inhibit inflammation and induce autophagy in the hypothalamus (data not shown). As Nur77 is expressed and strongly induced by stress and cytokine in the hypothalamus (Campos-Melo et al., 2013; Murphy and Conneely, 1997), Nur77 may confer the leptin-sensitizing effect of celastrol by regulating hypothalamic inflammation.
Celastrol-induced Nur77 interaction and colocalization with TRAF2 is mediated by an LxxLL motif in TRAF2 (Figure 5). This LxxLL motif was originally discovered in nuclear receptor coregulators that are responsible for interacting with nuclear receptors for transcription regulation. As the LAMLL motif in TRAF2 was also required for 9-cis-RA dependent interaction of RXRα-LBD with TRAF2 (Figure S5), it is conceivable that, in analogy to nuclear receptor coregulators, TRAF2 may serve as a nongenomic coregulator of other nuclear receptors besides Nur77—such as RXRα, which is known to bind TRAF2 (Zeng et al., 2015)— in order to mediate their regulation of inflammatory signaling. It can be also envisioned that the LxxLL motif or similar sequences may be utilized by some other cytoplasmic signaling molecules to mediate the crosstalk between nuclear receptors and cytoplasmic signaling pathways.
Our data demonstrated that celastrol-induced Nur77 interaction with TRAF2 contributes to celastrol inhibition of the classical IKK/NF-κB pathway. Celastrol was shown to act as an inhibitor of the proteasomal CT-like activity to enhance the accumulation of ubiquitinated proteins such as IκBα (Lu et al., 2014). While it cannot be excluded that celastrol targets multiple intracellular proteins, our data showed that celastrol acts upstream of the IκBα degradation; this was revealed by its inhibition of IKKα/β activation in several cell lines examined (Figure 1C). Furthermore, celastrol-induced Nur77 interaction with TRAF2 suppressed TNFα-induced TNFR1 recruitment of TRAF2 and RIP1 and their ubiquitination (Figure 6). Ubiquitination of TRAF2 is subjected to positive or negative regulation by a number of cellular binding partners, such as sphingosine 1-phosphate (Alvarez et al., 2010). Our data, therefore, identified Nur77 as another inhibitor of TRAF2 ubiquitination.
Recent studies indicate that removal of damaged mitochondria by autophagy represents an effective way to control inflammatory responses (Green et al., 2011; Nunnari and Suomalainen, 2012). The mechanism by which dysfunctional mitochondria are recognized and engulfed by autophagosomes involves ubiquitination of mitochondrial proteins followed by interaction with the adaptor proteins connecting ubiquitin with LC3 (Geisler et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010). Our current studies revealed a new, Nur77-dependent mitophagic mechanism in which celastrol-induced Nur77 mitochondrial targeting (Figure 3) and subsequent TRAF2-dependent Nur77 ubiquitination (Figure 6) serve to prime damaged mitochondria for autophagy through interaction with p62 (Figure 7), thereby alleviating inflammation. As the overproduction of ROS from damaged mitochondria is associated with oxidative damage, our finding also provides a plausible explanation for the well-established antioxidant activity of celastrol. Maintaining mitochondrial fidelity is critical to elicit the correct signaling responses to achieve cellular homeostasis, and it represents an effective approach to reverse pathological states in chronic inflammatory diseases (Kotas and Medzhitov, 2015; Levine et al., 2011; Nathan and Ding, 2010). Our finding that Nur77 mediates the crosstalk between inflammation and mitophagy suggests that appropriate expression and function of Nur77 is critical in maintaining homeostasis. Indeed, alterations in Nur77 expression, post-translational modification, and subcellular localization are implicated in the onset and progression of cancer, metabolic disease, and inflammatory disease (Evans, 2009; Hamers et al., 2013; Lee et al., 2011; McMorrow and Murphy, 2011; Moll et al., 2006; Zhang, 2007). As inflammatory mitochondrial dysfunction is implicated in the development of a range of metabolic diseases and cancer, there is an impetus for the development of mitochondrial therapeutics. To this end, our results identify Nur77 as an attractive target and celastrol as a promising lead for this class of therapeutics.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Myc | Santa Cruz | Cat. sc-40; RRID: AB_627268 |
| Anti-Hsp60 | Abcam | Cat. ab46798; RRID: AB_881444 |
| Anti -CD68 | Abcam | Cat. ab955; RRID: AB_307338 |
| Anti -Ly6g | Abcam | Cat. ab25377; RRID: AB_470492 |
| Anti-RIP1 | BD Biosciences | Cat. 551042; RRID: AB_394015 |
| Anti-Nur77 | BD Biosciences | Cat. 554088; RRID: AB_395232 |
| Anti-Ubiquitin | Cell Signal Technology | Cat. 3933S; RRID: AB_394015 |
| Anti-β-actin | Cell Signal Technology | Cat. 4970S; RRID: AB_2223172 |
| Anti-IκBα | Cell Signal Technology | Cat. 4814S; RRID: AB_10693636 |
| Anti-LC3 | Cell Signal Technology | Cat. 4108S; RRID: AB_2137703 |
| Anti-p-Ikkα/Ikkβ | Cell Signal Technology | Cat. 2078s; RRID: AB_2079379 |
| Anti-Parp | Cell Signal Technology | Cat. 9542; RRID: AB_2160739 |
| Anti-TNFR1 | Santa Cruz Biotechnology | Cat. sc-8436; RRID: AB_628377 |
| Anti-Nur77 (M-210) | Santa Cruz Biotechnology | Cat. sc-5569; RRID: AB_653373 |
| Anti-LC3B | Santa Cruz Biotechnology | Cat. sc-28266; RRID: AB_2137719 |
| Anti-p65 | Santa Cruz Biotechnology | Cat. sc-109; RRID: AB_632039 |
| Anti-GST | Santa Cruz Biotechnology | Cat. sc-138; RRID: AB_627677 |
| Anti-TRAF2 | Santa Cruz Biotechnology | Cat. sc-7187; RRID: AB_793340 |
| Anti-IκBα | Santa Cruz Biotechnology | Cat. sc-371; RRID: AB_793340 |
| Anti -FLAG | Sigma | Cat. F1804; RRID: AB_262044 |
| Goat Anti-Mouse IgG F(ab′)2 Secondary Antibody, HRP conjugate | Pierce | Cat. 31436; RRID: BDSC_31436 |
| Goat anti-Rabbit IgG F(ab′)2 Secondary Antibody, HRP conjugate | Pierce | Cat. 31461; RRID: BDSC_31461 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Proteinase K | Amresco | Cat. 0706 |
| Wortmannin | Beyotime | Cat. S1952 |
| mouse IL-1β ELISA kit | Elabscience Biotechnology | Cat. E-EL-M0037c |
| mouse IL-6 ELISA kit | Elabscience Biotechnology | Cat. E-EL-M0044c |
| Collagenase Type IV | GIBCO | Cat. 17104-019 |
| G418 Sulfate | Merck | Cat.345810-5GM |
| Recombinant Human TNFa protein | Peprotech | Cat. 300-01a-50 |
| Lipopolysaccharide | Sigma | Cat. L2630 |
| D-Galactosamine | Sigma | Cat. G0500 |
| Chloroquine | Sigma | Cat. C6628-25G |
| Dexamethasone | Sigma | Cat. D8040 |
| MG132 | Sigma | Cat. C2211-5MG |
| 9-cis-RA | Sigma | Cat. R4643-1MG |
| Nur77 siRNA | Sigma | Cat.SASI_Hs02_0 0333289 |
| TRAF2 siRNA | Sigma | Cat.SASI_Hs01_0 0095954 |
| RXRα siRNA | Sigma | Cat.SASI_HS01_0 0097639 |
| Control siRNA | Sigma | Cat.sc-37007 |
| CD3254 | Santa Cruz Biotechnology | Cat. sc-358786 |
| Critical Commercial Assays | ||
| PageRuler Prest Protein Ladder | Fermantas | Cat. 26617 |
| Protein G Agarose, Fast Flow | Millipore | Cat.16-266 |
| Aspartate aminotransferase Assay Kit Nanjing Jiancheng | Bioengineering Institute | Cat. C010-1 |
| Alanine aminotransferase Assay Kit Nanjing Jiancheng | Bioengineering Institute | Cat. C009-1 |
| FastStart Universal SYBR Green Master(ROX) | Roche | Cat. 4913850001 |
| TianScript RT kit | TianGen Biotechnology | Cat. KR104-02 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat. 23225 |
| Experimental Models: Cell Lines | ||
| Mouse: primary embryonic fibroblasts | This paper | C57BL/6J |
| HepG2 | ATCC | HB-8065 |
| HEK293T | ATCC | CRL-3216 |
| LO2 | Cell Bank, Shanghai Cell Biology Institute | GNHu 6 |
| SMMC-7721 | Cell Bank, Shanghai Cell Biology Institute | TCHu 52 |
| QGY-7703 | Cell Bank, Shanghai Cell Biology Institute | TCHu 43 |
| HeLa | ATCC | CCL2 |
| H460 | ATCC | HTB-177 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Mouse:B6;129S2-Nr4a1tm1Jmi/J | The Jackson Laboratory | Stock No: 006187 |
| pcmv-myc-Nur77 | This study | Kolluri et al., 2008 |
| pcmv-myc-Nur77/L506W | This study | N/A |
| pcmv-myc-Nur77/L555W | This study | N/A |
| pcmv-myc-Nur77/L556W | This study | N/A |
| pcmv-myc-Nur77ΔAF2 | This study | N/A |
| pcmv-myc-Nur77ΔAB | This study | Li et al., 2000 |
| pcmv-myc-Nur77ΔDBD | This study | Li et al., 2000 |
| pcmv-myc-Nur77ΔAB/ΔDBD | This study | Kolluri et al., 2008 |
| pcmvmyc-Nur77ΔLBD | This study | Lin et al., 2004 |
| pEGFP-C1-Nur77 | This study | Li et al., 2000 |
| pET-15b-Nur77/LBD | This study | N/A |
| pFlag-cmv-2-TRAF2 | This study | Zeng et al., 2015 |
| pFlag-cmv-2-TRAF2(1-249) | This study | N/A |
| pFlag-cmv-2-TRAF2(97-356) | This study | N/A |
| pFlag-cmv-2-TRAF2(272-501) | This study | N/A |
| GST-TRAF2 | This study | Zeng et al., 2015 |
| GFP-LC3 | This study | N/A |
| RFP-LC3 | This study | N/A |
| HA-Ub | This study | N/A |
| HA-UbK63R | This study | N/A |
| pBind-Nur77 | This study | N/A |
| pBind-GR/LBD | This study | Zeng et al., 2015 |
| pG5 | This study | Zeng et al., 2015 |
| pFlag-cmv-2-p62 | This study | N/A |
| pFlag-cmv-2-p62-ΔUBA | This study | N/A |
| Sequence-Based Reagents | ||
| IL-6 forward: CCAGAGATACAAAGAAATGATGG | This study | N/A |
| IL-6 reverse: ACTCCAGAAGACCAGAGGAAAT | This study | N/A |
| IL-1β forward: TGGTGTGTGACGTTCCCATT | This study | N/A |
| IL-1β reverse: CAGCACGAGGCTTTTTTGTTG | This study | N/A |
| Software and Algorithms | ||
| LSM-510 confocal laser scanning microscope system (CarlZeiss) | open source | https://www.zeiss.com/global/home.html |
| image analysis system (Olympus, Tokyo, Japan) | open source | http://www.olympus-ims.com/en/microscope/software/ |
| GraphPad Prism | open source | http://www.graphpad.com/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiao-kun Zhang (xzhang@sbpdiscovery.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Wild-type and Nur77−/− mice (male, 4–6 weeks old, C57BL/6 background) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). The protocols for animal studies were approved by the Animal Care and Use Committee of Xiamen University, and all mice were handled in accordance with the “Guide for the Care and Use of Laboratory Animals” and the “Principles for the Utilization and Care of Vertebrate Animals.” Animals were used for isolating cultures of primary cells from mice. All cell lines are described in the key Resources Table.
METHOD DETAILS
Cell Culture Conditions
Human hepatocellular carcinoma cell lines HepG2, SMMC-7721, QGY-7703 were maintained in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum (FBS). Human hepatocellular carcinoma cell line LO2, human lung carcinoma cell line NIH-H460 and human cervical cancer cell line HeLa were cultured in RPMI-1640 supplemented 10% FBS. Primary MEFs and Nur77−/− MEFs were extracted from fetus of 14d-16d wild-type and Nur77−/− pregnant female mice (C57BL/6 background).
Compounds
136 natural compounds with approved anti-inflammatory effect in human were purified from traditional Chinese medicinal plants and used to screen for binding to Nur77. Celastrol was obtained from Thunder of God Vine. The 60% aqueous ethanol (V/V) solution-soluble extract of Thunder of God Vine was extracted with ethyl acetate and the ethyl acetate fraction was separately subjected multiple chromatographic steps on silica gel or Sephadex LH-20. The subfraction was purified through preparative HPLC eluting with aqueous acetonitrile elution to produce celastrol. Celastrol purchased from Sigma (St. Louis, MO, USA) and Chengdu Pufei De Biotech Co., Ltd (Chengdu, Sichuan, China) was used to confirm the results.
Plasmids
Plasmids pcmv-myc-Nur77, pcmv-myc-Nur77/L506W, pcmv-myc-Nur77/L555W, pcmv-myc-Nur77/L556W, pcmv-myc-Nur77ΔAF2, pcmv-myc-Nur77ΔAB, pcmv-myc-Nur77ΔDBD, pcmv-myc-Nur77ΔAB/ΔDBD, pcmv-myc-Nur77ΔLBD, pEGFP-C1-Nur77, pET-15b-Nur77/LBD, pFlag-cmv-2-TRAF2, pFlag-cmv-2-TRAF3, pFlag-cmv-2-TRAF5, pFlag-cmv-2-TRAF2(1-249), pFlag-cmv-2-TRAF2(97-356), pFlag-cmv-2-TRAF2(272-501), GST-TRAF2, GFP-LC3, RFP-LC3, HA-Ub, HA-UbK63R, pBind-Nur77, pBind-GR/LBD, pG5pFlag-cmv-2-p62, Myc-Nur77/K536R, and GFP-Nur77/K536R were described previously (Chen et al., 2014; Kolluri et al., 2008; Li et al., 2000; Lin et al., 2004; Zeng et al., 2015) or were constructed using PCR or the QuikChange mutagenesis kit.
Antibodies and Reagents
Anti-Myc (9E10) (Cat. Sc-40), and anti-Hsp60 (Cat. ab46798) antibodies from Abcam (UK); anti-Ubiquitin (Cat. 3933S), anti-β-actin (Cat. 4970S), anti-IκBα (Cat.4814S), and anti-LC3 (Cat. 4108S) antibodies from Cell Signal Technology (Beverly, MA, USA); anti-Nur77 (M-210) (Cat. sc-5569), anti-LC3B (Santa Cruz sc-28266), anti-p65 (Santa Cruz sc-109) and anti-GST (Cat.sc-138) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA); mouse IL-1β (interleukin 1β, Cat. E-EL-M0037c) and IL-6 (interleukin 6, Cat. E-EL-M0044c) ELISA kits for IL-1β and IL6 from Elabscience Biotechnology (Wuhan, Hubei, China); LPS (Lipopolysaccharide, Cat. L2630) and D-Gal N (D-Galactosamine, Cat. G0500) from Sigma (St. Louis, MO, USA); TianScript RT kit (Cat. KR104-02) from TianGen Biotechnology (Beijing, China); and Nur77, RXRα, and control siRNAs from Sigma were used.
Real-Time Polymerase Chain Reaction
After grinding liver tissue by tissuelyser-24, total RNA was extracted with Trizol. A total of 2 μg RNA was used to prepare cDNA using oligo(dT)12 as a primer. TIANScript RT Kit’s was used for real-time polymerase chain reaction (PCR) analysis. Each sample was run in triplicate. The relative RNA amounts were calculated with the ΔΔCt method by ABI Stepone PCR instrument and normalized with an internal control, β-actin. Primers: IL-6 forward: CCAGAGATACAAAGAAATGATGG, reverse: ACTCCAGAAGACCAGAGGAAAT; IL-1β forward: TGGTGTGTGACGTTCCCATT, reverse: CAGCACGAGGCTTTTTTGTTG.
Biacore Assay
After coupling 50 μg purified ligand-binding domain (LBD) of Nur77 (Nur77-LBD) protein to CM5 of Biacore, over 100 (20 μM) with known anti-inflammatory activity were initially screened for their binding to Nur77-LBD by Biacore T200. The identified celastrol was tested again with a gradient concentration of 0.04 μM, 0.08 μM,0.16 μM,0.32 μM,and 0.64 μM injected through flow cells immobilized with Nur77-LBD. The kinetic profiles were shown.
Circular Dichroism
Circular dichroism (CD) analysis of celastrol binding to Nur77-LBD followed protocol described previously (Kolluri et al., 2008). Celastrol (200 μl, 0.1 mg/mL) was added to the phosphate buffer (10 mM, pH7.4) of purified Nur77-LBD protein (1 mL, 1 mg/mL) extracted and purified from E. coli. After incubation for 3 hr at 4°C, 0.7 mL of the incubation buffer was measured with Jasco J-810 spectropolarimeter, and the CD spectra were obtained from 190 nm to 260 nm. Nur77-LBD alone was used as control.
High-Performance Liquid Chromatography
High-performance liquid chromatography (HPLC) was conducted as described (Zhou et al., 2010). Briefly, celastrol (600 μl, 0.1 mg/mL) was incubated with purified Nur77-LBD protein (5 mL, 1 mg/mL). After incubation for 3 hr at 4°C, the complex of celastrol and Nur77-LBD was captured using Ni beads. The complex was degenerated and the bound celastrol was extracted by organic solvent. The bound celastrol was subjected to ODS column (5 μm, 4.6 3 250 mm) through 0.2% H3PO4 aqueous acetonitrile elution using HPLC spectrometer (Shimadzu LC 20A, Japan). The detection wavelength was 425 nm.
Molecular Simulation
Docking of celastrol to Nur77 (PDB code: 4JGV) was performed using AutoDock V4.2. Celastrol conformations were generated by the Lamarckian genetic algorithm. Grid center was chosen around the reported coordinates (−12.08, 18.29, −4.233) of THPN in Nur77 crystal structure and grid size was set to 40×40×40 (X, Y, Z) grid points with a spacing of 0.375A between grid points. The standard docking protocol in AutoDock was applied: the number of randomly placed individuals was 150, the maximum number of energy evaluations was 2.5 million, the rate of gene mutation was 0.02, the rate of crossover was 0.8, the probability of performing local search on individual was 0.06, the lower bound on rho was 0.01. PyMOL version 0.99 was used for molecular visualization.
Dual-Luciferase Reporter Assay
Reporter assays were conducted as described (Chen et al., 2014; Kolluri et al., 2008; Li et al., 2000; Lin et al., 2004; Zhou et al., 2010). Cells were transfected with the corresponding plasmids for 24 hr and then treated with compounds for 12 hr. Cells were lysed and Lusiferase relative activity was tested by the Dual-Luciferase Reporter Assay System according to the manufacturer’s instructions. Transfection efficiency was normalized to Renilla luciferase activity.
Confocal Microscopy
Confocal microscopy was conducted as described (Kolluri et al., 2008; Li et al., 2000; Lin et al., 2004). Cells mounted on glass slides were permeabilized with PBS containing 0.1% Triton X-100 and 0.1 mol/L glycine for 15 min, and blocked with 1% bovine serum in PBS for 30 min at room temperature, followed with incubation with various primary antibodies at room temperature for 3 hr, and detected by FITC-labeled anti-IgG (1:400), anti-goat IgG conjugated with Cy3(1:400), or Cy5-labeled antibody at room temperature for 1 hr. Cells were costained with 4′,6-diamidino-2-phenylindole (DAPI) (1:10000 dilution) to visualize nuclei. The images were taken under a fluorescent microscope (CarlZeiss) or an LSM-510 confocal laser scanning microscope system (CarlZeiss). HepG2 cells, MEFs, and Nur77−/− MEFs were treated with celastrol (1 μM or 4 μM) for the indicated time before exposed to TNFα (20 ng/mL) for additional 30 min. Mitochondria were marked by Mitotracker (Red) (1:10000 dilution) for 30 min before fixed by 4% buffered formalin/PBS. Hsp60, Ub, p65, TRAF2, and Nur77 were examined by immunostaining.
Coimmunoprecipitation
Coimmunoprecipitation (CoIP) assays were conducted as described before (Kolluri et al., 2008; Lin et al., 2004; Zhou et al., 2010). Cells were harvested in lysis buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 5 mM ethylenediaminetetraacetic acid, containing protease inhibitors). Lysate was incubated with 1 μg antibody at 4°C for 2 hr. Immunocomplexes were then precipitated with 30 μl of protein A/G-Sepharose. After an extensive washing with lysis buffer, the beads were boiled in SDS sample loading buffer and assessed by western blotting (WB). HepG2 cells transfected with 2 μg Myc-Nur77, Flag-TRAF2, or HA-Ub expression vectors in 10-cm dishes were treated with treated with celastrol (4 μM) for 2 hr plus TNFα (20 ng/mL) for additional 30 min as indicated and analyzed by coIP using anti-Myc or anti-Flag antibody.
Western Blotting
WB was performed as described previously (Kolluri et al., 2008; Lin et al., 2004; Zhou et al., 2010). Cell lysates were boiled in sodium dodecyl sulfate (SDS) sample loading buffer, resolved by 10% SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to nitrocellulose. The membranes were blocked in 5% milk in Tris-buffered saline and Tween 20 (TBST; 10 mM Tris–HCl [pH 8.0], 150 mM NaCl, 0.05% Tween 20) for 1 hr at room temperature. After washing twice with TBST, the membranes were incubated with appropriate primary antibodies in TBST for 1 hr and then washed twice, probed with horseradish peroxide-linked anti-immunoglobulin (1:5000 dilution) for 1 hr at room temperature. After three washes with TBST, immunoreactive products were visualized using enhanced chemiluminescence reagents and autoradiography. HepG2 cells, MEFs and Nur77−/− MEFs were treated with Celastrol in 1 μM and 4 μM for 1 hr before exposed to TNFα (20 ng/mL) for additional 30 min. Lysates prepared were analyzed with IκB (1:1000 dilution) and β-actin (1:10000 dilution) antibodies by WB. HepG2 cells, MEFs and Nur77−/− MEFs were treated with celastrol in 2 μM for 9 hr before exposed to TNFα (20 ng/mL) for additional 30 min. Lysates prepared were analyzed with LC3B (1:1000 dilution) and β-actin antibodies by WB.
Animal Studies
Wild-type and Nur77−/− mice (male, 4-6 weeks old, C57BL/6 background) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). The protocols for animal studies were approved by the Animal Care and Use Committee of Xiamen University, and all mice were handled in accordance with the “Guide for the Care and Use of Laboratory Animals” and the “Principles for the Utilization and Care of Vertebrate Animals.” All of the mice were maintained in animal room with 12 hr light/12 hr dark cycles at Laboratory Animal Center in Xiamen University. Celastrol was dissolved in DMSO and diluted with normal saline containing 5.0% (V/V) Tween-80 to a final concentration 0.05 mg/mL. Normal saline with DMSO and 5.0% Tween-80 was employed as the vehicle control. For acute liver inflammation animal studies, mice of 8-10 weeks were intraperitoneally (i.p.) coinjected with LPS (80 μg/kg) and D-GalN (200 mg/kg). After 6 hr, mice were bled from the eye and the concentrations of AST, ALT, IL-1β and IL-6 in blood were measured by ELISA assay. Liver tissues were also collected, and the relative levels of mRNA expression of IL-1β and IL-6 were determined by RT-PCR. To estimate the efficacy of celastrol, mice were intraperitoneally injected with celastrol (0.2 or 0.5 mg/kg body weight) 12 hr before LPS and D-GalN injection. For HFD-induced obese animal studies, wild-type and Nur77−/− lean male mice were placed on 60 kcal% HFD (research diet) at the age of 4 weeks soon after weaning, and maintained on the same diet for 17 weeks before the experiments. All other animals were maintained on chow diet (13.5% from fat calories; lab diet). Animals were housed under 12 hr of light and 12 hr dark cycle with unrestricted access to food and water unless otherwise described. Vehicle or celastrol (0.1 mg/kg) was administered at fixed time (7:00 PM) once a day for two weeks (i.p., once a day). Body weight was monitored daily before drug treatment.
Mouse Tissue Processing and Histological Examination
Mouse tissues for histological examination were fixed in 10% or 4% neutral buffered formalin phosphate (pH 7.0) for periods not exceeding 24 hr, and were subsequently embedded in paraffin or OCT compound, respectively. They were sliced into 4-μm sections for hematoxylin and eosin (HE) staining, immunofluorescence, and for immunohistochemistry. To examine hepatocytes, HE-stained liver sections were analyzed with an image analysis system (Olympus, Tokyo, Japan). For immunostaining, liver sections were incubated with anti-LC3B (1:150 dilution) or anti-p65 (1:100 dilution) antibody. Positive areas were counted and measured, respectively, for 10 low-powered (× 100) fields per slide and analyzed by ImageJ software (NIH). A total of 10 sections in ablation region, each section randomly selected five fields at ×400 magnification and photographed counting the number of cells. The cells of sections independently counted and analyzed by GrapgPad Prism by two pathologists previously uninformed.
Detection of AST, ALT, IL-1β, and IL-6 Levels from Serum
Serum levels of liver enzymes, including AST and ALT, were determined using an automatic analyzer (NanJingjiancheng, China). Serum levels of IL-1β and IL-6 were determined by enzyme-linked immunosorbent assays (ELISA) according the manufacturer’s protocol (Elabscience, China).
Prediction of Potential Ubiquitination Sites on Nur77
Several online programs including UbPred, CPLM 1.0, and BDM-PUB were used to predict potential ubiquitination sites on Nur77. Three sites (Lys32, Lys381, and Lys536) were predicted to be potential ubiquitination sites on Nur77 and were there chosen for mutagenesis studies.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative data are presented as means ± SEM, and statistical significance are reported in the figures and in the figure legends. All experiments were independently repeated at least three times. ANOVA with Tukey’s post-test (One-way ANOVA for comparisons between groups, Two-way ANOVA for comparisons of magnitude of changes between different groups from different cell lines) was used to compare values among different experimental groups using the GraphPad program. For experiments with only two groups, Student’s t test was used as specified in the figure legends. p < 0.05 was considered statistically significant (*), p < 0.01 as highly significant (**), p < 0.001 as extremely significant (***), and ns as not significant.
Supplementary Material
Highlights.
Celastrol binds Nur77 to inhibit inflammation by autophagy
Celastrol induces Nur77 translocation to mitochondria and interaction with TRAF2
An LxxLL motif in TRAF2 mediates its interaction with Nur77
Celastrol promotes Nur77 ubiquitination, p62/SQSTM1 interaction, and mitophagy
Acknowledgments
This study was supported by grants from the National Natural Science Foundation (91429306, U1405229, 81672749, 91129302, 31271453, 31471318, 81502406, 81301705), the Regional Demonstration of Marine Economy Innovative Development Project (14PYY051SF04, 16PYY007SF17, and 12PYY001SF08), the Fujian Provincial Science & Technology Department (2017YZ0002-1), the Xiamen Science and Technology project (3502Z2015007, 3502Z20133008, 3502Z20131015), the Ministry of Education of China (IRT-14R32, 313050, and JA14009), the U.S. Army Medical Research and Material Command (W81XWH-11-1-0677), the National Institutes of Health (CA179379, CA172025, CA192642, and CA030199), and the California Breast Cancer Research Program (20IB-0138).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.03.008.
AUTHOR CONTRIBUTIONS
Conceptualization, X.-k.Z. and M.H.; Methodology, M.H., Q.L., G.A., J.L., F.J., and X.-k.Z.; Investigation, M.H., Q.L., G.A., S.C., C.X., L.X., X.C., D.Z., Y.Z., X.Y., and L.C.; Writing – Original Draft, X.-k.Z, M.H., Q.L., and Y.S.; Writing – Review & Editing, X.-k.Z., M.H., Q.L., Y.S., H.Z., M.T.D.-M., and J.M.; Funding Acquisition, X.-k.Z., X.L., J.L., F.J., H.Z., M.T.D.-M., and J.M.; Resources, Z.W., F.Z., H.C., H.F., S.Y., and X.L.; Supervision, X.-k.Z.
References
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard JA, Tenga A, Chen T. The interplay of NR4A receptors and the oncogene-tumor suppressor networks in cancer. Cell Signal. 2015;27:257–266. doi: 10.1016/j.cellsig.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi A, Verstrepen L, Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem Pharmacol. 2016;116:1–10. doi: 10.1016/j.bcp.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs) Oncogene. 2001;20:6482–6491. doi: 10.1038/sj.onc.1204788. [DOI] [PubMed] [Google Scholar]
- Campos-Melo D, Galleguillos D, Sánchez N, Gysling K, Andrés ME. Nur transcription factors in stress and addiction. Front Mol Neurosci. 2013;6:44. doi: 10.3389/fnmol.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LC, Zhang Z, Pei L, Saito T, Tontonoz P, Pilch PF. Nur77 coordinately regulates expression of genes linked to glucose metabolism in skeletal muscle. Mol Endocrinol. 2007;21:2152–2163. doi: 10.1210/me.2007-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LC, Wroblewski K, Zhang Z, Pei L, Vergnes L, Ilkayeva OR, Ding SY, Reue K, Watt MJ, Newgard CB, et al. Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes. 2009;58:2788–2796. doi: 10.2337/db09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang ZG, Aleshin AE, Chen F, Chen J, Jiang F, Alitongbieke G, Zeng Z, Ma Y, Huang M, et al. Sulindac-derived RXRα modulators inhibit cancer cell growth by binding to a novel site. Chem Biol. 2014;21:596–607. doi: 10.1016/j.chembiol.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu R, Chen HZ, Xiao Q, Wang WJ, He JP, Li XX, Yu XW, Li L, Wang P, et al. Enhancement of hypothalamic STAT3 acetylation by nuclear receptor Nur77 dictates leptin sensitivity. Diabetes. 2015;64:2069–2081. doi: 10.2337/db14-1206. [DOI] [PubMed] [Google Scholar]
- Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769–774. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Lonard DM, O’Malley BW. Nuclear receptor coactivators: master regulators of human health and disease. Annu Rev Med. 2014;65:279–292. doi: 10.1146/annurev-med-051812-145316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva S, Han S, Zhang X, Huston DP, Winoto A, Zheng B. Reduction of the incidence and severity of collagen-induced arthritis by constitutive Nur77 expression in the T cell lineage. Arthritis Rheum. 2005;52:333–338. doi: 10.1002/art.20736. [DOI] [PubMed] [Google Scholar]
- Evans PC. Nur77: orphaned at birth but adopted by the nuclear factor kappaB signaling pathway. Circ Res. 2009;104:707–709. doi: 10.1161/CIRCRESAHA.109.195529. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21:193–201. doi: 10.1016/j.molmed.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers AA, Vos M, Rassam F, Marinković G, Kurakula K, van Gorp PJ, de Winther MP, Gijbels MJ, de Waard V, de Vries CJ. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res. 2012;110:428–438. doi: 10.1161/CIRCRESAHA.111.260760. [DOI] [PubMed] [Google Scholar]
- Hamers AA, Hanna RN, Nowyhed H, Hedrick CC, de Vries CJ. NR4A nuclear receptors in immunity and atherosclerosis. Curr Opin Lipidol. 2013;24:381–385. doi: 10.1097/MOL.0b013e3283643eac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers AA, van Dam L, Teixeira Duarte JM, Vos M, Marinković G, van Tiel CM, Meijer SL, van Stalborch AM, Huveneers S, Te Velde AA, et al. Deficiency of nuclear receptor Nur77 aggravates mouse experimental colitis by increased NFκB activity in macrophages. PLoS ONE. 2015;10:e0133598. doi: 10.1371/journal.pone.0133598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, Hedrick CC. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harant H, Lindley IJ. Negative cross-talk between the human orphan nuclear receptor Nur77/NAK-1/TR3 and nuclear factor-kappaB. Nucleic Acids Res. 2004;32:5280–5290. doi: 10.1093/nar/gkh856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13:55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia EY, Goodson ML, Zou JX, Privalsky ML, Chen HW. Nuclear receptor coregulators as a new paradigm for therapeutic targeting. Adv Drug Deliv Rev. 2010;62:1227–1237. doi: 10.1016/j.addr.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Lee SB, Park JK, Yoo YD. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L) Cell Death Differ. 2010;17:1420–1434. doi: 10.1038/cdd.2010.19. [DOI] [PubMed] [Google Scholar]
- Kolluri SK, Zhu X, Zhou X, Lin B, Chen Y, Sun K, Tian X, Town J, Cao X, Lin F, et al. A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell. 2008;14:285–298. doi: 10.1016/j.ccr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurakula K, Vos M, Logiantara A, Roelofs JJ, Nieuwenhuis MA, Koppelman GH, Postma DS, van Rijt LS, de Vries CJ. Nuclear receptor Nur77 attenuates airway inflammation in mice by suppressing NF-κB activity in lung epithelial cells. J Immunol. 2015;195:1388–1398. doi: 10.4049/jimmunol.1401714. [DOI] [PubMed] [Google Scholar]
- Lee SO, Li X, Khan S, Safe S. Targeting NR4A1 (TR3) in cancer cells and tumors. Expert Opin Ther Targets. 2011;15:195–206. doi: 10.1517/14728222.2011.547481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Li X, Hedrick E, Jin UH, Tjalkens RB, Backos DS, Li L, Zhang Y, Wu Q, Safe S. Diindolylmethane analogs bind NR4A1 and are NR4A1 antagonists in colon cancer cells. Mol Endocrinol. 2014;28:1729–1739. doi: 10.1210/me.2014-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, Lin B, Chen G, Lu J, Lin F, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, Chen ZJ. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife. 2013;2:e00785. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonard DM, O’Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8:598–604. doi: 10.1038/nrendo.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Shi W, Deshmukh RR, Long J, Cheng X, Ji W, Zeng G, Chen X, Zhang Y, Dou QP. Tumor necrosis factor-α sensitizes breast cancer cells to natural products with proteasome-inhibitory activity leading to apoptosis. PLoS ONE. 2014;9:e113783. doi: 10.1371/journal.pone.0113783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, Liu J, Finkel T, Mueller E. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1α transcriptional axis. Cell Metab. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. [DOI] [PubMed] [Google Scholar]
- McMorrow JP, Murphy EP. Inflammation: a role for NR4A orphan nuclear receptors? Biochem Soc Trans. 2011;39:688–693. doi: 10.1042/BST0390688. [DOI] [PubMed] [Google Scholar]
- Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286:32324–32332. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM, Marchenko N, Zhang XK. p53 and Nur77/TR3 - transcription factors that directly target mitochondria for cell death induction. Oncogene. 2006;25:4725–4743. doi: 10.1038/sj.onc.1209601. [DOI] [PubMed] [Google Scholar]
- Murphy EP, Conneely OM. Neuroendocrine regulation of the hypothalamic pituitary adrenal axis by the nurr1/nur77 subfamily of nuclear receptors. Mol Endocrinol. 1997;11:39–47. doi: 10.1210/mend.11.1.9874. [DOI] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Castrillo A, Tontonoz P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol. 2006;20:786–794. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- Perez-Sieira S, Martinez G, Porteiro B, Lopez M, Vidal A, Nogueiras R, Dieguez C. Female Nur77-deficient mice show increased susceptibility to diet-induced obesity. PLoS ONE. 2013;8:e53836. doi: 10.1371/journal.pone.0053836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Kroemer G. Cancer therapy: altering mitochondrial properties. Nat Chem Biol. 2014;10:89–90. doi: 10.1038/nchembio.1440. [DOI] [PubMed] [Google Scholar]
- Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: molecular targets of Thunder God Vine. Biochem Biophys Res Commun. 2010;394:439–442. doi: 10.1016/j.bbrc.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Valdearcos M, Xu AW, Koliwad SK. Hypothalamic inflammation in the control of metabolic function. Annu Rev Physiol. 2015;77:131–160. doi: 10.1146/annurev-physiol-021014-071656. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KF, Yuan Y, Luk JM. Tripterygium wilfordii bioactive compounds as anticancer and anti-inflammatory agents. Clin Exp Pharmacol Physiol. 2012;39:311–320. doi: 10.1111/j.1440-1681.2011.05586.x. [DOI] [PubMed] [Google Scholar]
- Wu H, Li XM, Wang JR, Gan WJ, Jiang FQ, Liu Y, Zhang XD, He XS, Zhao YY, Lu XX, et al. NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis. J Pathol. 2016;238:457–469. doi: 10.1002/path.4670. [DOI] [PubMed] [Google Scholar]
- Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9:140–147. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- Yang KC, Ma X, Liu H, Murphy J, Barger PM, Mann DL, Diwan A. Tumor necrosis factor receptor-associated factor 2 mediates mitochondrial autophagy. Circ Heart Fail. 2015;8:175–187. doi: 10.1161/CIRCHEARTFAILURE.114.001635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wu Y, Szabo A, Wu Z, Wang H, Li D, Huang XF. Teasaponin reduces inflammation and central leptin resistance in diet-induced obese male mice. Endocrinology. 2013;154:3130–3140. doi: 10.1210/en.2013-1218. [DOI] [PubMed] [Google Scholar]
- Zemirli N, Pourcelot M, Ambroise G, Hatchi E, Vazquez A, Arnoult D. Mitochondrial hyperfusion promotes NF-κB activation via the mitochondrial E3 ligase MULAN. FEBS J. 2014;281:3095–3112. doi: 10.1111/febs.12846. [DOI] [PubMed] [Google Scholar]
- Zeng Z, Sun Z, Huang M, Zhang W, Liu J, Chen L, Chen F, Zhou Y, Lin J, Huang F, et al. Nitrostyrene derivatives act as RXRα ligands to inhibit TNFα activation of NF-κB. Cancer Res. 2015;75:2049–2060. doi: 10.1158/0008-5472.CAN-14-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan YY, Chen Y, Zhang Q, Zhuang JJ, Tian M, Chen HZ, Zhang LR, Zhang HK, He JP, Wang WJ, et al. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nat Chem Biol. 2012;8:897–904. doi: 10.1038/nchembio.1069. [DOI] [PubMed] [Google Scholar]
- Zhang XK. Targeting Nur77 translocation. Expert Opin Ther Targets. 2007;11:69–79. doi: 10.1517/14728222.11.1.69. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Liu W, Su Y, Wei Z, Liu J, Kolluri SK, Wu H, Cao Y, Chen J, Wu Y, et al. NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell. 2010;17:560–573. doi: 10.1016/j.ccr.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.