

Abstract
Permethylation is a common derivatization method for MS-based glycomic analyses. Permethylation enhances glycan ionization efficiency in positive MS analysis and improves glycan structural stability. Recent biological glycomic studies have added to the growing body of knowledge and suggest the need for complete structural analysis of glycans. However, reverse phase LC analysis of permethylated glycans usually results in poor isomeric separation. To achieve isomeric separation of permethylated glycans, a porous graphitic carbon (PGC) column was used. PGC columns are well known for their isomeric separation capability for hydrophilic analyses. In this study, we have optimized temperature conditions to overcome the issues encountered while separating permethylated glycans on a PGC column and found that the highest temperature examined, 75°C, was optimal. Additionally, we utilized tandem MS to elucidate detailed structural information for the isomers separated. Glycan standards were also utilized to facilitate structural identifications through MS/MS spectra and retention time comparison. The result is an efficient and sensitive method capable of the isomeric separation of permethylated glycans. This method was successfully applied for the isomeric characterization of N-glycans released from the breast cancer cell lines MDA-MB-231 and MDA-MB-231BR (brain seeking). A total of 127 unique glycan structures were identified with 39 isobaric structures, represented as 106 isomers, with 21 non-isomeric glycans. Thirty seven structures exhibited significant differences in isomeric distribution (P < 0.05). Additionally, alterations in the distribution of isomeric sialylated glycans, structures known to be involved in cell attachment to the blood-brain barrier during brain metastasis, were observed.
Graphical Abstract
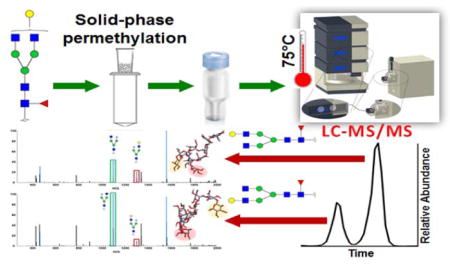
Introduction
Because glycans participate in a variety of biological processes, the importance of glycomics has been well recognized by researchers in recent decades.1,2 Moreover, glycan expression has also been found to be related to several mammalian diseases. For example, cancer cell surface glycans mediate cancer migration and progression.3 Hence, a reliable isomeric glycan quantitation method will serve to assist our obtaining an understanding of glycan biofunctions and their relationships to disease. The development of high-resolution mass spectrometry (HRMS) has facilitated reliable glycan identification based on molecular weight. MALDI-MS and LC-ESI-MS have become the methods of choice for quantitative glycomics, presently. Derivatization is still required for MS-based quantitative glycomics, due to the low ionization efficiency of native glycans in positive-ion mode.
A widely used glycan derivatization method is permethylation. It was first reported for glycan analysis by Ciccanu and Kerek.4 Mechref and coworkers introduced solid-phase permethylation in 2005,5 which improved upon the sample throughput of the original permethylation protocol. Although permethylation protocols are more complex than other derivatization methods commonly used, such as reducing end labeling strategies, the method possesses the advantages of enhancing MS signal and stabilizing sialylated and fucosylated glycan structures, by preventing loss and migration. Due to these advantages, it has become one of the most popular derivatization methods used in MALDI-MS glycomics profiling. However, in the case of LC-ESI-MS analysis, the drawbacks of permethylation can be revealed during the LC separation process. For native and reducing end labeled glycans, which are regularly separated on HILIC and PGC columns, good peak shape and high isomeric resolution can often be achieved. In order to separate permethylated glycans, which are very hydrophobic after derivation, reverse phase LC (RPLC) is a common choice. However, a number of problematic issues arise, the main issue for separating permethylated glycans on RPLC is peak broadening for highly branched complex glycans, which we have recently addressed.6–10 Because intramolecular interactions are increased after permethylation, due to the high-density of methyl groups, the masking of structural differences between isomers occurs. This results in the issue of poor isomeric resolution, which is the problem addressed in this study.
Partial isomeric separation was attained on reversed-phase columns at elevated temperatures. 6 However, such isomeric separation was not adequate. To improve isomeric separation of permethylated glycans on LC, a more structurally sensitive LC column packing material should be used. Porous graphitized carbon (PGC), retention mechanism is considered to be a result of a combination of hydrophobic and electrostatic interactions, is a suitable material for isomeric separation of permethylated glycans. It was first reported as a new column packing material for GC/LC in 198211 and was first utilized in separating glycans and glycopeptides in 1994.12 The influence of electrosorption, solvent, temperature and ion polarity on the performance of acidic glycan separation on a PGC platform has been systematically studied and applied for the isomeric separation of glycans from bovine fetuin and IgG.32,33 The fact that retention of reduced glycans on PGC increased at higher temperature was highlighted.13 More recently, a PGC chip was utilized, coupled with MS, as a platform for comprehensive glycomic profiling. A database was built and has been employed for the analysis of various types of glycan samples, such as glycans from dry blood spots,14 human milk,15,16 cell lines17 and blood serum.18
All of the aforementioned work was conducted with a focus on native glycans, and for a while, it was believed that PGC material was only suited to separate hydrophilic analytes. Utilizing a PGC column for separating permethylated glycans seemed challenging. However, in 2007, a study reported the use of a PGC column for the separation of permethylated glycans.19 Nonetheless, baseline resolution of the high mannose glycan isomers was not demonstrated, and the peak shapes were not of acceptable quality. Additionally, sialylated glycans separation was not efficient. In this study, we have optimized the separation conditions to achieve efficient and sensitive isomeric separation of permethylated glycans using our experience in high-temperature RPLC-MS analysis. Linkage isomers and monosaccharide positional isomers were baseline-resolved for both neutral and sialylated glycans, in our strategy, while MS/MS spectra were utilized to characterize isomeric structures.
Material and Methods
Reagents
HPLC grade solvents, including water, acetonitrile, methanol and isopropanol alcohol, were purchased from Fisher Scientific (Pittsburgh, PA). Sodium hydroxide beads (20–40 mesh, 97%), iodomethane, acetic acid, formic acid, dimethyl sulfoxide (DMSO) and borane-ammonia complex were obtained from Sigma-Aldrich (St. Louis, MO). Empty spin columns were purchased from Harvard Apparatus (Holliston, MA). PNGase F and digestion buffer were from New England Biolabs (Ipswich, MA). The glycoprotein samples, including ribonuclease B, fetuin, α acidic glycoprotein and pooled human blood serum, were bought from Sigma-Aldrich (St. Louis, MO). The standard glycan samples were purchased from Chemily Glycoscience (Atlanta, GA).
Sample Preparation
N-Glycans were released using PNGase F from model glycoproteins, human blood serum and breast cancer cell lines using our laboratory’s routine protocols.7,10,20 Glycans were purified by ice-cold ethanol precipitation. In the case of the cell line samples, dialysis was also utilized to remove sodium deoxycholate (SDC), a detergent used in cell lysis. After purification, cells were reduced 20,21 and permethylated 5,22,23 utilizing protocols previously reported in the literature. Derivatized glycans were dried and resuspended in a 20% aqueous acetonitrile solution, containing 0.1% formic acid, prior to LC-MS analysis. Detailed descriptions of the separation protocols utilized are provided in the supporting information.
Instrument Method
An UltiMate 3000 nanoLC system (Thermo Scientific, Sunnyvale, CA, USA) was used for LC separation in this study. All the separations were conducted on a HyperCarb PGC column (75 μm * 150 mm, 5 μm particle size, Thermo Scientific, Pittsburgh, PA). A HyperCarb PGC trap column (75 μm * 20 mm, 5 μm particle size, Thermo Scientific, Pittsburgh, PA) was also employed for sample loading and online purification. Mobile phase A consisted of 2% acetonitrile, 98% water and 0.1% formic acid and mobile phase B was acetonitrile with 0.1% formic acid. The flow rate was set to 750 nL/min, and the gradient was as follows. In the first 10 minutes mobile phase B was kept at 20%, from 10 to 30 minutes, the percentage of B was increased to 60%, B was further elevated from 60% to 95% in the following 10 minutes, finally 95% B was maintained for 20 minutes to elute highly sialylated glycans. The outlet of the LC column was coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, San Jose, CA) through a nanoESI source with a 1.6 kV ESI voltage. The full MS was set to a resolution of 15,000 with a 500–2000 m/z scan range. Data-dependent acquisition (DDA) was employed to select the top 4 most intense precursor ions for collision induced dissociation (CID) and higher collisional energy dissociation (HCD) MS2. Collision energy during CID was set to 30% of the normalized energy value with a 15 ms activation time, and activation Q was set to 0.250.
Data Processing
Extracted ion chromatograms (EIC) of the first monoisotopic peak of permethylated glycans generated by Xcalibur were used to display the LC peaks for selected glycan ions. The EIC peak areas were recorded to represent the abundance of each glycan. EICs were also utilized to obtain the corresponding retention times of isomers, which facilitated the assignment of MS2 spectra for each isomer. Additionally, the correlation between the retention time of glycans on the PGC column and temperature was presented using Van’t Hoff plots. Retention times of reduced and permethylated N-glycans, derived from RNase B and bovine fetuin, at 25°C, 40°C, 55°C and 75°C were selected for plotting. The MS2 spectra were elucidated by manual assignment. All theoretical fragments for permethylated glycans were calculated using GlycoWorkbench, ± 0.5 Dalton mass tolerance was used for CID MS2 generated and detected in the linear ion trap (LTQ).
Molecular Modeling
Quantum chemical calculations based on density functional theory (DFT) were performed to optimize the 3D structures of two F1A2G1 isomers. The purpose of this 3D modeling is to better understand retention times and tandem MS of such isomers. The geometries were computed using the Perdew–Burke–Ernzerhof (PBE)24–26 functional and the single valence polarization (SVP)27 basis-set without the inclusion of environment effects. Dispersion interactions were taken into account using a revised version of the general empirical dispersion correction proposed by Grimme.28 The revised empirical formalism used in this work is designated as DFT-D3. All calculations were carried out for isolated systems by means of the Turbomole29 program suite. The BIOVIA Discovery Studio Visualizer (Accelrys, San Diego, CA) was used for visualization.30
Results and Discussion
We initially analyzed glycans released from ribonuclease B and fetuin, which are model glycoproteins the glycan structures of which are fully characterized.31,32 Glycans released from ribonuclease B and fetuin were reduced and permethylated following the protocols described above. Initially, LC separation on the PGC column was conducted at ambient temperature, 25°C. Despite numerous attempts being made to adjust the LC gradient, the resolution for the permethylated glycans derived from ribonuclease B resembled what was previously reported19 and was not satisfactory (Figures 1A, 1B, and S-1, 25°C traces). Additionally, no efficient separation could be achieved in the case of sialylated glycans derived from fetuin with wide overlapping peaks observed for all structures (Figures 1C, 1D, and S-2, 25°C traces). This observation is in agreement with previous publications, which attempted to separate permethylated glycans on PGC. It was concluded in this work that PGC is not suitable for sialylated glycans.19
Figure 1.

Extracted ion chromatograms (EICs) of reduced and permethylated glycan structures of (A) Man 7, (B) Man 8 (C) Hex5HexNAc4NeuAc2 and (D) Hex6HexNAc5NeuAc2 derived from Ribo B (A and B) and bovine fetuin (C and D), which were separated using a PGC column at ambient temperature (25°C), 40°C, 55°C, and 75°C. Symbols:
 , N-acetylglucosamine (GlcNAc);
, N-acetylglucosamine (GlcNAc);
 , Galactose (Gal);
, Galactose (Gal);
 , Fucose (Fuc);
, Fucose (Fuc);
 , Mannose (Man);
, Mannose (Man);
 , N-acetylneuraminic acid (NeuAc/Sialic Acid)
, N-acetylneuraminic acid (NeuAc/Sialic Acid)
In our previous study, involving RPLC-MS of permethylated glycans, inefficient separations were also observed for sialylated glycans.10 The observation of broad peaks was due to the presence of different conformers with different hydrophobicity for each glycan structure. Such difference in hydrophobicity among the different conformers of the same structure attributes to broad chromatographic peaks.10 Broad peaks were eliminated by performing the separation at high temperatures.21,33,34 Hence, in this study, we also evaluated the effect of elevating the column temperature on improving the separation on PGC column. LC separations were carried out at temperatures of 40, 55 and 75°C, the latter of which is the upper limit of our LC system.
The separation efficiency and resolution of permethylated high mannose glycans were improved with increasing temperature, and the most optimal separation was achieved at 75°C (lower traces of Figures 1A, 1B, and S-1). At this elevated temperature, isomers were baseline resolved, in contrast to the partially resolved peaks that were observed when the analysis was conducted at 25°C and temperatures <75°C (Figures 1A, 1B and S-1). The separation efficiencies calculated for the non-isomeric separation of Man5 and Man9 at 25°C were 14,799 and 25,883 (plate/m), respectively (Figures S-1A and C, 25°C traces). The values increased to 45,006 and 78,341 (plate/m), respectively, when the column was heated to 75°C (Figures S-1A and C, 75°C traces). For the isomeric separation of Man7 (Figure 1A, 25°C trace) the average plate number was 28,767 m−1, at 25°C, whereas at 75°C an average efficiency of 88,516 (plate/m) was achieved (Figure 1A, 75°C trace). Therefore, the separation efficiency improved by a factor of two, upon increasing the temperature from 25°C to 75 °C. Because PGC separations were carried out using a gradient, it must be stated that separation efficiency calculations are only representative estimates.
As shown in Figures 1A, 1B and S-1B, the EICs of three high mannose structures depicted multiple peaks. Man 6 has two baseline resolved peaks at 27.4, and 29.8 minutes (Figure S-1B, 75°C trace), Man 7 has three peaks at 27.1, 28.2 and 28.9 minutes (Figure 1A, 75°C trace) and Man 8 has three peaks at 27.4, 28.1 and 29.7 minutes (Figure 1B, 75°C trace). The ratio of each isomeric glycan’s abundance agrees with previously reported PGC19 and CE results.35 Additionally, the resolution observed in this study is much higher than that of the previous PGC study performed at ambient temperature. Costello and co-workers reported the resolution between peaks 1 and 2 and peaks 2 and 3, for Man7, to be 0.33 and 0.27, respectively. 19 The resolution for Man 8 isomers was 0.67 and 0.92. 19 Here, the PGC-LC-MS analysis conducted at 75°C improved the chromatographic resolution of Man 7 isomers to 0.75 and 0.46. The chromatographic resolution in the case of Man 8 was improved to 0.78 and 1.45. Generally, there was a 1.2 to 2.3 fold increase in resolution for the permethylated high mannose glycan isomers at high separation temperatures, prompting baseline resolution for all isomers. We also noticed that as the temperature increased, there was a shift from protonated adducts to ammoniated adducts in high mannose samples derived from RNase B (Figure S-3). However, the explanation was still under discussing. Data illustrating the relative abundance of all high mannose glycans released from RNase B are summarized in Table S-1.
Separation efficiency was also improved significantly at each incremental increase of temperature, with the greatest separation efficiency and resolution achieved at the highest temperature examined, 75°C, when complex-type sialylated glycans released from fetuin were evaluated. In the case of bi-antennary bi-sialylated glycan, for example, the overlapping 10-minute group of peaks observed at 25°C, was reduced at 75°C to three baselines resolved isomeric peaks at 34.1, 37.8 and 42.2 minutes (Figure 1C and Table S-2). There was more than 10 fold increase in chromatographic resolution when the column temperature was elevated from 25° to 75°C. In the case of RPLC separation, this increase in resolution is approximately 3 fold.33 This is due to the fact that PGC prompts better interactions with closely related structures (e.g., glycan isomers).
It can be concluded that as column temperature increases the spatial configuration of permethylated glycans becomes more fixed and does not represent a multitude of confirmations, which results in the observation of decreased peak width on the structurally selective PGC column. Meanwhile, the spatial and structural differences, which define isomers remain at high separation temperatures, facilitating the efficient resolution of isomeric peaks. Another physical process that may explain the observed resolution enhancement is an increase in temperature may result in increased mobile phase interactions and decreased intramolecular hydrophobic interactions, resulting in greater solute interaction with the PGC material.
Van’t Hoff plots for permethylated glycans derived from ribonuclease B and fetuin are shown in Figures S-4A and B and described in Tables S-3 and S-4, respectively. According to Figure S-4A and B, all of the plots are linear with R2 values better than 0.94 (R2 values and linear fit data are summarized in Tables S-3 and S-4). The retention times of permethylated glycans increased with increasing temperature, which was demonstrated by negative slopes in the Van’t Hoff plots. The nearly parallel lines of Van’t Hoff plots (Figure S-4A and B) suggested comparable enthalpy associated with the separation of different glycans. The positive entropy compensated for the change in enthalpy, resulting in an overall negative Gibbs free energy, which is in agreement with a previously reported study of reduced native glycans separated on a PGC column.13
The EIC shown in Figure 2A depicts two peaks for F1A2G1 glycan, which is a standard glycan. The peak at retention time 29.2 minutes represents a structure where fucose resides on a branch of the glycan while the peak at 32.7 minutes corresponds to a glycan structure with core fucosylation. The resolution between the two peaks representing core and branched fucosylation was about 3.4, demonstrating the capability to differentiate fucose site isomers using PG-LC-MS at 75°C. Structural information was confirmed by CID tandem MS (Figure 2B and C). Figure 2B displays the tandem mass spectrum of the isomer that eluted at 29.2 minutes. There are unique ions at m/z 432.47 and 638.5; both ions are indicative of branched fucose structures. The ion at m/z 432.47 is a fragment that contains one Hex, one HexNAc, and two cleavage sites. Confident assignment of the number of cleavage sites is facilitated by permethylation. The ion at m/z 638.5 represents a fragment that possesses one Hex, one HexNAc, and one fucose. In the higher m/z range, there are three unique ions for this branch-fucosylated glycan structure. The ion at m/z 1740.95 is generated by the loss of a reducing end HexNAc. The reducing end HexNAc was confirmed because the glycans in this study were reduced and permethylated, thus providing a 16.0313 (1 C plus 4 H) Dalton mass difference between reducing end and branched HexNAc residues. A fucose residue was associated with the ion at m/z 1740.95, further confirming that the fucose of this glycan structure was located on the branch rather than the reducing end.
Figure 2.

(A) EIC of glycan F1A2G1 from mixed standard glycan samples, which were chemically synthesized structures. (B) CID MS/MS spectrum of branch fucosylated glycan that was eluted at 29.2 min. (C) CID MS/MS spectrum of core fucosylated glycan that eluted at 32.7 min. Symbols: as in Figure 1.
Similarly, ions at m/z values of 1481.95 and 1552.94 also reflect a structure with branched fucosylation. Figure 2C is the tandem mass spectrum of the structure eluting at 32.7 minutes; unique fragment ions were observed at m/z 468.36 and 1566.86. The fragment ion at m/z 468.36 consists of a reducing end HexNAc and a fucose residue; indicating a core fucosylation of this glycan structure. The fragment ion at m/z 468.36 is a diagnostic ion for all core fucosylated glycans.21 Confident assignment of the fucosylation sites was made possible because of the absence of fucose migration, which is eliminated by permethylation.
Figure 3A is the EIC of F1A2G1 glycan released from human blood serum. There are two peaks observed at retention times 32.8 and 34.1 minutes, with a resolution of 1.2. The isomers, in this case, do not represent different fucosylation sites, since fucose site isomers commonly demonstrate larger resolution values, > 3, as shown above. Figure 3B depicts the spectrum of the peak eluting at 32.8 minutes while Figure 3C represents the spectrum of the second peak eluting at 34.1 minutes. The diagnostic ion at m/z 468.42, observed in both spectra, indicated that both glycans were core fucosylated. Hence, in this case, the isomers originate from galactose residues residing on different branches (α3 or α6 branches). However, no unique m/z diagnostic ions could be used to discriminate between these two isomers, since most fragments in CID MS2 for permethylated glycans are generated from the dissociation of glycosidic bonds. Contributing to this issue is the fact that the α3 and α6 branches are isobaric.
Figure 3.

(A) EIC of glycan F1A2G1 with galactose attached to different branches, which were released from human blood serum (B) CID MS/MS spectrum of isomeric structure eluted at 32.8 min, with molecular modeling of the structure. (C) CID MS/MS spectrum of isomeric structure eluted at 34.1 min, with molecular modeling of the structure. Symbols: as in Figure 1.
Although no unique fragments were found, fragment ion intensity distributions were quite different. The ratio between ions at m/z 1103.7 (marked as green in Figure 3B and C) versus 1307.8 (marked as red in Figure 3B and C) was about 1.6 in the case of the first peak and was about 10 in the case of the second peak. To clarify the relationship between galactose branch location and retention time, the tandem MS of a synthesized glycan standard was used. For the glycan standard, it is already known that the galactose is located on the α6 branch, the ratio between ions at m/z 1103.7 and 1307.8 is about 1.3 (Figure S-5), which is closer to the ratio observed in the spectrum of the first eluting isomer. This assumption is further supported by computer modeling, performed as described in the materials and methods section, which indicated that when the galactose residue is located on the α6 arm the molecule is more compact and the fucose residue is close to the galactose-containing antennae, thus possibly increasing bond energy. Conversely, when the galactose is on the α3 arm, of the F1A2G1 structure, the distance between the fucose and galactose residues is considerably greater (≈14.1 compared to 6.7 Å), increasing the molecular surface area and preventing the stabilization of the galactose-containing antennae (insets of Figure 3B and C). These modeling data not only helped to explain the increased abundance of the m/z 1103.7 fragment ion in the MS/MS spectra of the structure where galactose resided on the α3 arm but also increased the retention time of the structure. Further, the peak observed in the EIC of the glycan standard (Figure 3 inset) overlapped with the first isomer peak, indicating that the earlier eluting isomer likely has the same structure as the glycan standard. Hence, we can draw the conclusion that the first peak is representative of a α6 branch galactosylated structure while the second peak represents the α3 branch galactosylated structure.
Not only can glycan monosaccharide site isomers be separated by PGC-LC-MS analysis of permethylated glycans at 75°C, but linkage isomers can also be resolved. The EIC in Figure S-6A represents the bi-antennary bi-sialylated glycans released from fetuin. There were a total of four peaks found at 34.1, 37.8, 40.4 and 42.2 minutes, representing a structure with two α2, 3 linked sialic acids, two structures with one α2, 3 and one α2, 6 linked sialic acid and one structure with two α2, 6 linked sialic acids, respectively. Similarly, no unique fragments were found for linkage isomers in the spectrum, due to most cleavages occurring at glycosidic bonds. To identify the specific structures for the four peaks in Figure S-6A, the ratio of the intensity of each of the peaks was compared. According to a previously published structural NMR study of fetuin N-glycans, the abundances of the structures with two α2, 3 linked sialic acids, the two structures with one α2, 3 and one α2, 6 linked sialic acid on different branches and the structure with two α2, 6 linked sialic acids are 10.7%, 57.0%, less than 1.0% and 32.2% for the, respectively.36
In the EICs shown in Figure S-6A, the peak area ratio of these four peaks were 3.6%, 67.0%, 1.4% and 28.0%, which were highly comparable to the NMR data (see Table S-2). We confirmed that the earlier eluting peak possesses more α2, 3 linked sialic acid while the later eluting peak contains more α2, 6 linked sialic acid by analyzing glycan standards using PGC-LC-MS. A pair of bi-antennary mono-sialylated glycan standards were employed, one having α2, 3 linked sialic acid while the second possesses a α2, 6 linked sialic acid residue. The MS/MS spectra shown in Figure S-7A and B, for the standards with α2, 3 linked sialic acid and α2, 6 linked sialic acid, respectively, only allowed general confirmation of standard structure but did not provide enough information to distinguish between the two isomers. The EIC for these two standards demonstrated that the structure with α2, 3 linked sialic acid eluted about 3 minutes earlier than the α2, 6 linked structure (Figure S-6B). With the previously reported NMR data, 36 used in conjunction with our analysis of glycan standards, the principle of sialic acid linkage isomer separation on a PGC column is confirmed.
The reliable identification of sialic acid linkage isomers is important for biomarker discovery studies. It has been reported that sialic acid linkage can influence and reflect cancer progression.37 Breast cancer brain metastasis is considered a serious problem for breast cancer patients due to its high mortality and the lack of effective therapeutic approaches.38 The key event of brain metastasis is the penetration of breast cancer cells through the blood-brain barrier (BBB).39 Many studies have investigated alterations in glycan patterns during this process,40,41 but few have focused on changes in isomeric structures. Herein, we investigated the isomeric alteration of reduced and permethylated N-glycans released from two breast cancer cell lines (MDA-MB-231 (231) and MDA-MB-231BR (231BR)). 231 is considered the most invasive breast cancer cell line. However, it can migrate to many organs such as bone, liver, kidney and the brain. Although, the brain is not the first metastatic target of 231. 231BR is a sub-line of 231 that results in brain metastasis 100% of the time and cannot migrate to any other organs. The specific brain metastatic capacity of the 231BR cell line suggests that there are modifications in 231BR that contribute to the metastatic process that takes place in the brain. Therefore, we chose to compare the N-glycans of the 231 and 231BR cell lines using the high-temperature PGC-LC-MS/MS method described above.
Overall a total of 127 unique glycan structures were identified, in the two cell lines. The total glycan pool was composed of 39 isobaric structures, represented as 106 isomers (Table S-5), with 21 non-isomeric glycans. Among them, 37 structures exhibited significant differences in isomeric distribution (P < 0.05). Sialylation has been demonstrated to be important in the attachment of cells to the BBB, in brain metastasis.42 Alterations in the distribution of isomeric sialylated glycans were observed when 231 was compared with 231BR. Examples of the isomeric separation of bi- and tri- sialylated glycans can be seen in Figure 4A and Figure S-8A, respectively. For the bi-sialylated structures, the 231 cell line contained significantly more of the structure containing two α2, 3 sialic acids. Whereas the 231BR cell line contained more structures with a mixture of α2, 3 and α2, 6 linkages and the structure containing two α2, 6 linkages (Figure 4A). For the tri-sialylated glycans, the 231 cell line contained significantly more of the structure corresponding to the earliest eluting peak, labeled 1 (Figure S-8A), which presumably contains more α2, 3 linked sialic acid based on our previous work shown in Figure S-6B. The 231BR cell line, on the other hand, contained significantly more of the most abundant peak, labeled 2, that represents a structure containing a mixture of α2, 3 and α2, 6 linkages (Figure S-8A). The difference in abundance observed in the latest eluting peak in Figure S-8A, labeled 3, was not deemed statistically significant. For both di- and tri-sialylated structures a trend of increased α2, 6 linked sialic acid was observed in the 231BR cell line when compared to the 231 cell line. Therefore it is reasonable to postulate that this type of linkage may contribute to breast cancer brain metastasis.
Figure 4.

EICs of (A) bi-sialylated glycans and (B) Hex3HexNAc3 glycans released from the 231 cell line and the 231BR cell line. Distributions of isomeric glycans in the 231 and 231BR cell lines exhibited significant differences (P<0.05). Symbols: as in Figure 1.
With regards to non-sialylated structures, statistically significant differences in the quantities of isomers of the glycans A1 and Man 4 were detected in the 231 and 231BR cells lines. The 231 cell line contained significantly more of the isomers represented by peaks 1 and 2 in Figure 4B, while the 231BR cell line contained significantly more of the isomer represented by the peak labeled 2. Additionally, we have reported the observation of Man 4 in the 231 and 231BR cell lines in a previous publication 43, and herein, we investigated changes in the isomeric distribution of Man 4 in these cells. The 231 cell line contained more of the earlier eluting Man 4 isomer, labeled peak 1, while the 231BR cell line contained significantly more of the later eluting Man 4 isomer, labeled peak 2 in Figure S-8B. Based on these data, it is clear that glycomic profiling conducted using a PGC-LC-MS platform at elevated temperatures provides more separation efficiency and resolution of reduced and permethylated N-glycan structures than has previously been demonstrated.
Conclusion
In this study, we developed a strategy for analyzing reduced and permethylated N-glycans on a PGC column at elevated temperatures. This platform retained all benefits provided by permethylation, including MS signal enhancement and the stabilization of glycan structures; and provided sensitive isomeric separation for all glycan species. This is the first report of efficient isomeric separation for permethylated sialylated glycans. With this strategy, isomeric structures can be defined by examining MS/MS spectra for diagnostic ions, comparing the ratio of select fragment ions and the matching of retention times.
Supplementary Material
Figure S-1. EIC of reduced and permethylated (A) Man 5, (B) Man 6 and (C) Man 9 N-glycan released from RNase B. The insets depicted total intensities changes at different analyze temperatures when considering different charge states and adducts.
Figure S-2. EIC of a reduced and permethylated sialylated complex N-glycans, (A) Hex5HexNAc4NeuAc1, (B) Hex6HexNAc5NeuAc3 and (C) Hex6HexNAc5NeuAc4, released from fetuin. The insets depicted total intensities changes at different analyze temperatures when considering different charge states and adducts.
Figure S-3. Full MS spectrums of high mannose structures derived from RNase B. As shown in the figure, the formation of adducts differs with different temperatures. As the column temperature increases, it forms more ammonium adduct. Symbols as in Table S-1.
Figure S-4. Van’t Hoff plots of glycans released from (A) RNase B and (B) fetuin, separated on PGC-LC at ambient temperature (25°C), 40°C, 55°C, and 75°C.
Figure S-5. MS/MS spectrum of core fucosylated complex N-glycan standard F1A2G1, the insert represents the EIC of the standard glycan F1A2G1.
Figure S-6. (A) EIC of permethylated bi-antennary bi-sialylated glycans released from fetuin, (B) EICs of glycan standard with (1) an α2, 3 linked sialic acid and (2) an α2, 6 linked sialic acid.
Figure S-7. MS/MS spectra of mono-sialylated biantennary N-glycan standards with (A) α2, 3 linked and (B) α2, 6 linked sialic acid.
Figure S-8. EICs of (A) tri-sialylated glycans and (B) Man 4 glycans released from the 231 cell line and the 231BR cell line.
Table S-1. Relative abundance of glycan isomers in RNase B.
Table S-2. Relative abundance of bi-antennary di-sialylated glycan isomers in bovine fetuin.
Table S-3. N-glycan structures released from RNase B and used for Van’t Hoff plot.
Table S-4. N-glycan structures released from bovine fetuin and used for Van’t Hoff plot.
Table S-5. Relative abundance of N-glycan isomers in 231 and 231BR breast cancer cell lines.
Acknowledgments
This work was supported by NIH grant (1R01GM112490-01) and CPRIT (RP130624).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1.Freeze HH, Esko JD, Parodi AJ. Essentials of Glycobiology. Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]
- 2.Molinari M. Nat Chem Biol. 2007;3:313–320. doi: 10.1038/nchembio880. [DOI] [PubMed] [Google Scholar]
- 3.Varki A, Kannagi R, Toole BP. Essentials of Glycobiology. Cold Spring Harbor (NY): 2009. [Google Scholar]
- 4.Ciucanu IKF. Carbohydr Res. 1984;131:209–217. [Google Scholar]
- 5.Kang P, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2008;22:721–734. doi: 10.1002/rcm.3395. [DOI] [PubMed] [Google Scholar]
- 6.Zhou S, Hu Y, Mechref Y. Electrophoresis. 2016;37:1506–1513. doi: 10.1002/elps.201500568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desantos-Garcia JL, Khalil SI, Hussein A, Hu Y, Mechref Y. Electrophoresis. 2011;32:3516–3525. doi: 10.1002/elps.201100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Desantos-Garcia JL, Mechref Y. Rapid Commun Mass Spectrom. 2013;27:865–877. doi: 10.1002/rcm.6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Y, Zhou S, Khalil SI, Renteria CL, Mechref Y. Anal Chem. 2013;85:4074–4079. doi: 10.1021/ac400106x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y, Shihab T, Zhou S, Wooding K, Mechref Y. Electrophoresis. 2016;37:1498–1505. doi: 10.1002/elps.201500560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert MT, Knox JH, Kaur B. Chromatographia. 1982;16:138–148. [Google Scholar]
- 12.Hounsell EF. Mol Biotechnol. 1994;2:45–60. doi: 10.1007/BF02789289. [DOI] [PubMed] [Google Scholar]
- 13.Pabst M, Altmann F. Anal Chem. 2008;80:7534–7542. doi: 10.1021/ac801024r. [DOI] [PubMed] [Google Scholar]
- 14.Ruhaak LR, Miyamoto S, Kelly K, Lebrilla CB. Anal Chem. 2012;84:396–402. doi: 10.1021/ac202775t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ninonuevo MR, Park Y, Yin H, Zhang J, Ward RE, Clowers BH, German JB, Freeman SL, Killeen K, Grimm R, Lebrilla CB. J Agric Food Chem. 2006;54:7471–7480. doi: 10.1021/jf0615810. [DOI] [PubMed] [Google Scholar]
- 16.Wu S, Grimm R, German JB, Lebrilla CB. J Proteome Res. 2011;10:856–868. doi: 10.1021/pr101006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hua S, Lebrilla C, An HJ. Bioanalysis. 2011;3:2573–2585. doi: 10.4155/bio.11.263. [DOI] [PubMed] [Google Scholar]
- 18.Aldredge D, An HJ, Tang N, Waddell K, Lebrilla CB. J Proteome Res. 2012;11:1958–1968. doi: 10.1021/pr2011439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costello CE, Contado-Miller JM, Cipollo JF. J Am Soc Mass Spectrom. 2007;18:1799–1812. doi: 10.1016/j.jasms.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou S, Hu Y, DeSantos-Garcia JL, Mechref Y. J Am Soc Mass Spectrom. 2015;26:596–603. doi: 10.1007/s13361-014-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong X, Zhou S, Mechref Y. Electrophoresis. 2016;37:1532–1548. doi: 10.1002/elps.201500561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mechref Y, Kang P, Novotny MV. Methods Mol Biol. 2009;534:53–64. doi: 10.1007/978-1-59745-022-5_4. [DOI] [PubMed] [Google Scholar]
- 24.Perdew JP, Wang Y. Phys Rev B Condens Matter. 1992;45:13244–13249. doi: 10.1103/physrevb.45.13244. [DOI] [PubMed] [Google Scholar]
- 25.Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 26.Perdew JP, Ernzerhof M, Burke K. J Chem Phys. 1996;105:9982–9985. [Google Scholar]
- 27.Schäfer A, Horn H, Ahlrichs R. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 28.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 29.Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C. Chemical Physics Letters. 1989;162:165–169. [Google Scholar]
- 30.Dassault Systèmes. BIOVIA, Discovery Studio Modeling Environment, Release 2017. Dassault Systèmes; San Diego: 2016. [Google Scholar]
- 31.Fu D, Chen L, O’Neill RA. Carbohydr Res. 1994;261:173–186. doi: 10.1016/0008-6215(94)84015-6. [DOI] [PubMed] [Google Scholar]
- 32.Green ED, Adelt G, Baenziger JU, Wilson S, Van Halbeek H. J Biol Chem. 1988;263:18253–18268. [PubMed] [Google Scholar]
- 33.Zhou S, Hu Y, Mechref Y. Electrophoresis. 2016;37:1506–1513. doi: 10.1002/elps.201500568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y, Zhou S, Zhu J, Lubman D, Mechref Y. Electrophoresis. 2017 doi: 10.1002/elps.201700025. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guttman A, Chen F-TA, Evangelista RA, Cooke N. Anal Biochem. 1996;233:234–242. doi: 10.1006/abio.1996.0034. [DOI] [PubMed] [Google Scholar]
- 36.Green ED, Adelt G, Baenziger JU, Wilson S, Van Halbeek H. J Biol Chem. 1988;263:19253–19268. [PubMed] [Google Scholar]
- 37.Alley WR, Jr, Novotny MV. J Proteome Res. 2010;9:3062–3072. doi: 10.1021/pr901210r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arshad F, Wang L, Sy C, Avraham S, Avraham HK. Patholog Res Int. 2010;2011:920509. doi: 10.4061/2011/920509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abbott NJ, Ronnback L, Hansson E. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 40.Saldova R, Reuben JM, Abd Hamid UM, Rudd PM, Cristofanilli M. Ann Oncol. 2011;22:1113–1119. doi: 10.1093/annonc/mdq570. [DOI] [PubMed] [Google Scholar]
- 41.Kolbl AC, Andergassen U, Jeschke U. Front Oncol. 2015;5:219. doi: 10.3389/fonc.2015.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zacharias LG, Hartmann AK, Song E, Zhao J, Zhu R, Mirzaei P, Mechref Y. J Proteome Res. 2016;15:3624–3634. doi: 10.1021/acs.jproteome.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S-1. EIC of reduced and permethylated (A) Man 5, (B) Man 6 and (C) Man 9 N-glycan released from RNase B. The insets depicted total intensities changes at different analyze temperatures when considering different charge states and adducts.
Figure S-2. EIC of a reduced and permethylated sialylated complex N-glycans, (A) Hex5HexNAc4NeuAc1, (B) Hex6HexNAc5NeuAc3 and (C) Hex6HexNAc5NeuAc4, released from fetuin. The insets depicted total intensities changes at different analyze temperatures when considering different charge states and adducts.
Figure S-3. Full MS spectrums of high mannose structures derived from RNase B. As shown in the figure, the formation of adducts differs with different temperatures. As the column temperature increases, it forms more ammonium adduct. Symbols as in Table S-1.
Figure S-4. Van’t Hoff plots of glycans released from (A) RNase B and (B) fetuin, separated on PGC-LC at ambient temperature (25°C), 40°C, 55°C, and 75°C.
Figure S-5. MS/MS spectrum of core fucosylated complex N-glycan standard F1A2G1, the insert represents the EIC of the standard glycan F1A2G1.
Figure S-6. (A) EIC of permethylated bi-antennary bi-sialylated glycans released from fetuin, (B) EICs of glycan standard with (1) an α2, 3 linked sialic acid and (2) an α2, 6 linked sialic acid.
Figure S-7. MS/MS spectra of mono-sialylated biantennary N-glycan standards with (A) α2, 3 linked and (B) α2, 6 linked sialic acid.
Figure S-8. EICs of (A) tri-sialylated glycans and (B) Man 4 glycans released from the 231 cell line and the 231BR cell line.
Table S-1. Relative abundance of glycan isomers in RNase B.
Table S-2. Relative abundance of bi-antennary di-sialylated glycan isomers in bovine fetuin.
Table S-3. N-glycan structures released from RNase B and used for Van’t Hoff plot.
Table S-4. N-glycan structures released from bovine fetuin and used for Van’t Hoff plot.
Table S-5. Relative abundance of N-glycan isomers in 231 and 231BR breast cancer cell lines.