

Abstract
Modulation of neuronal circuits is key to information processing in the brain. The majority of neuromodulators exert their effects by activating G protein coupled receptors (GPCR) that control the production of second messengers directly impacting cellular physiology. How numerous GPCRs integrate neuromodulatory inputs while accommodating diversity of incoming signals is poorly understood. In this study we develop an in vivo tool and analytical suite for analyzing GPCR responses by monitoring the dynamics of a key second messenger, cAMP with excellent quantitative and spatio-temporal resolution in various neurons. With this imaging approach in combination with CRISPR/Cas9 editing and optogenetics we interrogate neuromodulatory mechanisms of defined populations of neurons in an intact mesolimbic reward circuit and describe how individual inputs generate discrete second messenger signatures in a cell and receptor specific fashion. This offers a resource for studying native neuronal GPCR signaling in real time.
eTOC
Muntean et al. develop an in vivo reagent to study processing of neurotransmitter GPCRs signals by monitoring real-time dynamics of cAMP responses. They demonstrate application of this approach, in combination with CRISPR/Cas9 gene editing and optogenetics, to interrogate functional organization of a striatal circuit.
INTRODUCTION
Animal behavior is guided by the coordinated action of neuronal circuits that relies on impeccable communication among individual neurons within the network. In addition to rapid neurotransmission neurons also communicate by slowly acting neuromodulators that fine-tune the circuits to generate elaborate activity patterns ultimately endowing the nervous system with vast computational power and plasticity (Bargmann and Marder, 2013; Marder, 2012). The majority of these neuromodulatory inputs are processed by G protein Coupled Receptors (GPCRs), that influence many aspects of neuronal physiology through the mobilization of second messenger cascades (Gainetdinov et al., 2004; Wettschureck and Offermanns, 2005). With an excess of 800 members, GPCRs are capable of exerting unique effects on neuronal activity owning to their prominent heterogeneity in expression across neuronal populations and differences in subcellular localization (Insel et al., 2015; Magalhaes et al., 2012; Regard et al., 2008). Despite the tremendous diversity, GPCR signals converge downstream and multiple receptors modulate a limited set of second messengers. It has been proposed that various GPCRs decode neuromodulatory signals by generating distinct spatio-temporal profiles of second messenger dynamics and that these patterns, in turn, program the individual neuronal circuits (Brinton, 1990; Cropper et al., 2014; Marder, 2012). How individual inputs generate discrete second messenger signatures in a cell and receptor specific fashion within the context of intact circuitry of a mammalian brain is poorly understood largely due to limited availability of appropriate tools and approaches.
Cyclic AMP (cAMP) is one of the key second messengers that integrates signals from the majority of GPCRs and plays a crucial role in such fundamental processes as learning/memory (Davis, 1996; Kandel, 2012), drug addiction (Nestler, 2016) and motor control (Hakansson et al., 2004). The modulation of intracellular cAMP concentration exerts a powerful effect on neuronal physiology by regulating protein kinases and phosphatases, modifying ion channel function, and altering transcription (Greengard et al., 1999; Kandel, 2012; Nestler, 2016). Activation of GPCRs provides a direct path for altering cAMP levels via either inhibitory or stimulatory effects exerted by heterotrimeric G protein subunits on the cAMP generating enzyme, adenylyl cyclase (AC). Multiple AC isoforms are different in their regulatory mechanisms and distribution setting a stage for a distinct spatio-temporal modulation of cAMP signaling (Sadana and Dessauer, 2009). Thus, studying cAMP dynamics offers a window into the mechanisms of neuromodulatory signaling and decoding by GPCRs (Dunn et al., 2006; Nikolaev et al., 2006; Tomchik and Davis, 2009).
The mesolimbic pathway provides an instructive model where convergent interplay between multiple neuromodulatory inputs orchestrated by selective expression of distinct GPCRs process reward signals (Girault, 2012; Lobo and Nestler, 2011). The core of this circuit includes dopaminergic neurons of the ventral tegmental area (VTA) that innervate the medium-sized spiny neurons (MSNs) in the nucleus accumbens (NAc). The neurons of the direct pathway (dMSN) express the D1 dopamine receptor (D1R) that engages Gs/olf to stimulate cAMP production whereas indirect pathway neurons express D2 dopamine receptor (D2R) that couples to Gi/o to inhibit cAMP production. This is translated into bidirectional regulation of various cAMP effectors (e.g. PKA, CREB, DARPP32, FosB) with direct roles in shaping the activity of MSN neurons (Gerfen and Surmeier, 2011; Kreitzer, 2009; Tritsch and Sabatini, 2012). In addition to dopamine, the activity of MSNs is substantially shaped by a variety of other neuromodulatory inputs including adenosine, acetylcholine, and neuropeptides, whose receptors are differentially segregated across neuronal populations and subcellular compartments (Johnson and Lovinger, 2016; Kreitzer, 2009). The scarcity of appropriate tools and approaches makes it also unclear how individual neurotransmitter GPCRs converging on cAMP modulation interact and what the signaling decoding logic is.
In this study we present a tool and analytical framework for quantitative assessment of real-time cAMP dynamics in response to neuromodulatory inputs acting through GPCRs in the context of intact circuitry of the mammalian brain. We apply this approach to study the integration of GPCR signaling in individual genetically defined neurons of the mesolimbic reward circuit with subcellular resolution.
RESULTS
Mouse model for conditional expression of genetically encoded cAMP sensor
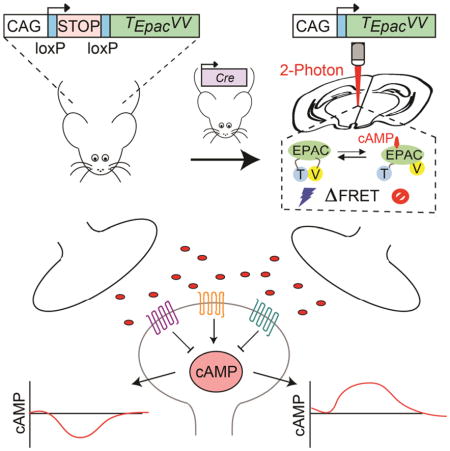
To facilitate real-time monitoring of cAMP dynamics, we generated a reporter mouse line by knocking in a genetically encoded cAMP sensor into the Rosa26 locus (Figure 1). Among several available sensors, we chose to use the latest generation sensor TEpacVV, which reports cAMP binding by changes in Fluorescence Resonance Energy Transfer (FRET) between a mTurquoise donor and tandem Venus acceptor (cp173Venus-Venus) flanking the cAMP binding domain from Epac1 (Klarenbeek et al., 2011). The sensor was placed under the control of a strong CAG promoter driving ubiquitous expression (Niwa et al., 1991) and was preceded by a LoxP-Stop-LoxP cassette to enable conditional expression only in the presence of Cre-recombinase. The resulting cAMP Encoded Reporter (CAMPER) mice were viable and fertile when bred to homozygosity (Figure 1A).
Figure 1. Mouse model for conditional expression of genetically encoded cAMP sensor.

A) Schematic of strategy for imaging cAMP dynamics in primary striatal neurons at DIV14–18.
B) Sample confocal images of primary striatal neurons from CAMPER mice at DIV14. Scale bar is 50 μm.
C) Imaging changes in FRET in response to forskolin (10 μM) and IBMX (100 μM) (top panel) or digitonin (10 μg/μl) (bottom panel).
D) Traces of the FRET change to cAMP standards in digitonin-permeabilized primary striatal neurons. Shading indicates SEM. n ≥ 8 neurons.
E) Scheme of GPCRs coupling to cAMP production in striatal neurons.
F) Pseudocolored images of FRET responses in primary striatal neurons to dopamine (1 μM). Time is shown as minutes following dopamine application (minutes:seconds format). Scale bar is 50 μm. LUT values displayed in ΔFRET.
G) Representative traces to bath application (at time 0) of indicated agonist (1 μM). FRET ratios were converted to cAMP (nM) using the calibration curve shown in panel E. Bidirectional responses were observed for dopamine (52% increased cAMP, 39% decreased cAMP, 9% non-responsive, n=143) and adenosine (55% increased cAMP, 34% decreased cAMP, 11% non-responsive, n=70). Responses to acetylcholine (84% decreased cAMP, 16% non-responsive, n=77) and morphine (6% decreased cAMP, 94% non-responsive, n=215) were always inhibitory.
We initially tested the efficacy of the reporter system using primary cultured neurons. When infected with AAV-Cre virus, striatal neurons exhibited robust fluorescence for both mTurquoise and Venus consistent with the induction of TEpacVV expression (Figure 1B). No fluorescence was observed in non-infected cultures indicating the lack of leaky expression (Figure 1B). Examination of the subcellular distribution of fluorescence revealed that the TEpacVV sensor was broadly distributed across all cellular compartments: cell body, nucleus, endosomes/Golgi, as well as axonal and dendritic processes (Figure S1A). Importantly, application of forskolin together with IBMX, which increase cAMP by activating adenylyl cyclase or inhibiting PDE, respectively, resulted in a robust change in fluorescence of mTurquoise (decrease) and Venus (increase) substantially decreasing the FRET signal (Figures 1C and S1B), which is expected from cAMP binding to TEpacVV sensor (Klarenbeek et al., 2011). Conversely, permeabilizing cells and depleting cAMP resulted in an increase in the FRET (Figures 1C and S1C). This bidirectional response indicated that the sensor is capable of reporting signals that either decrease or increase cAMP production in striatal neurons. We then calibrated the sensor by permeabilizing the neurons, adding cAMP standards and correlating FRET changes with cAMP concentrations (Figures 1D and S1D). We reliably detected cAMP concentrations between 20 nM and 200 μM with EC50 of 3.0 ± 0.5 μM) (Figure 1D), which is in the range of estimated MSN intracellular cAMP concentration (Polito et al., 2015). After reaching maximum the signal remained stable for at least 4 minutes indicating the lack of rapid sensor turnover and/or decay. The maximal amplitude values of the signal were used to generate a calibration curve used to convert ratiometric FRET values to absolute cAMP concentrations reported throughout the study. In summary, neurons from CAMPER mice allow rapid and sensitive detection of changes in absolute cAMP concentration with a broad dynamic range making it suitable for studying both inhibitory and stimulatory neuromodulation.
Diverse neurotransmitter GPCRs selectively modulate cAMP dynamics in a distinct populations of striatal neurons
Striatal neurons respond to several neurotransmitters through GPCRs that are expected to influence cAMP levels by either inhibiting or activating AC activity based on their G protein coupling (Figure 1E). We first characterized the utility of CAMPER for real time monitoring of cAMP dynamics in response to application of several neurotransmitters with key roles in striatal signaling. Treatment of cultures with dopamine revealed bidirectional changes in cAMP observed in most neurons (Figures 1F and S1E). Responses are also displayed in ΔFRET to provide a sense of the experimental response (Figure S1F). A similar bimodal behavior was observed with application of adenosine (Figure 1G). We further observed distinct responses produced by acetylcholine application that only decreased cAMP levels and had a tendency to oscillate when the response reached peak values. Opioid agonist morphine also produced only inhibitory responses that were highly variable in amplitude.
We refined these analyses by employing the genetically encoded driver lines D1R-Cre and D2R-Cre to selectively express Cre recombinase in the dMSN and iMSN, respectively (Figures 2A and S2A). Upon application of dopamine, the majority of neurons showed a detectable change in FRET. Among responsive neurons, all dMSNs increased cAMP whereas all iMSNs decreased cAMP (Figure 2B and Table S1), confirming the segregation of dopamine receptor types. Overall, these experiments indicate that cAMP responses triggered by various neurotransmitters can be studied in genetically identifiable populations of striatal neurons.
Figure 2. Benchmarking dopamine mediated signaling dynamics in cultured striatal neurons.

A) Schematics of strategy to study cAMP dynamics in genetically defined populations of striatal neurons.
B) Population responses to bath applied dopamine (at time=0) to D1R-Cre-CAMPER (dMSNs) and D2R-Cre-CAMPER (iMSNs) primary striatal neurons. 385 out of 462 (83%) of dMSNs responded to dopamine by increasing cAMP. 336 out of 429 (78%) of iMSNs responded by decreasing cAMP level. No dMSNs decreased cAMP and no iMSNs that increased cAMP.
C) Dose response curve of cAMP changes to dopamine. n≥10 neurons.
D) The EC50 values calculated from data in panel C. dMSN cell body (76.5 ± 13.9 nM dopamine), dMSN dendrite (64 ± 9.4 nM dopamine), iMSN dendrites (177.3 ± 26.2 nM dopamine) and iMSN cell body (429.5 ± 53.3 nM dopamine). Error bars indicate SEM values. * = p<0.05, *** = p<0.0001
E) Comparison of kinetic properties at equivalent sensitivity to dopamine. dMSN cell body exhibited an EC66 of 101.9 ± 14.4 nM dopamine whereas iMSN cell body had an EC66 of 1.02 ± 72.3 μM dopamine. Dotted line indicates time to peak.
F) Comparison of kinetic properties at equivalent sensitivity to dopamine. dMSN dendrites had an EC37 of 54.3 ± 10.4 nM dopamine whereas iMSN dendrites had an EC37 of 97.8 ± 12.8 nM dopamine. Normalized percent change in cAMP response was plotted for dMSN dendrite at 50 nM for comparison with iMSN dendrite at 100 nM dopamine. dMSN dendrite required 4.38 ± 0.21 minutes to reach peak response whereas iMSN dendrite required 2.04 ± 0.10 minutes. Dotted line indicates time to peak.
G) Histogram of cAMP amplitude to 100 μM dopamine. There was no difference between dMSN cell body and dMSN dendrite (non-parametric t-test; Kolmogorov-Smirnov; p=0.307) or between iMSN cell body and iMSN dendrite (p=0.2398). The dMSN cell body profile significantly differed from iMSN cell body (p<0.001) and dMSN dendrites were significantly different from iMSN dendrites (p<0.05).
H) Histogram showing population distribution of neurons according to overall change in their cAMP amount in response to 100 μM dopamine. The net flux of cAMP was significantly greater in dMSN dendrite (6333 ± 261 nmol cAMP) compared with dMSN cell body (2063 ± 74 nmol cAMP) (non-parametric t-test; Kolmogorov-Smirnov; p<0.0001). The net flux of cAMP was significantly greater in iMSN cell body (682 ± 32 nmol cAMP) compared with iMSN dendrite (425 ± 17 nmol cAMP) (non-parametric t-test; Kolmogorov-Smirnov; p<0.0001).
I) Responses of striatal neurons to phasic puffs of dopamine (1 μM) applied directly to the neuron being recorded for a brief pulse of 1 second at 5 minute intervals. Representative traces are shown from dMSN and iMSN cell body.
J) Quantification of maximum amplitude of phasic dopamine response. dMSN (838 ± 24.6 nM cAMP) was significantly larger compared with iMSN (444 ± 21.1 nM cAMP). Time to peak was significantly faster in iMSNs (1.19 ± 0.09 minutes) compared with dMSNs (1.51 ± 0.10 minutes). Error bars indicate SEM values. N=9 dMSNs and 11 iMSNs. * = p<0.05, *** = p<0.0001
K) Imaging XYZ planes at each time point during application of a single puff of dopamine (1 second; 1 μM) directly to the dMSN being recorded. Pseudocolored FRET images from one representative experiment are shown and time is displayed in minutes relative to dopamine application. LUT values displayed in ΔFRET.
L) cAMP response from membrane and cytoplasmic compartments of a dMSN following application of a single puff of dopamine (1 second; 1 μM). Data was averaged from nine neurons.
Resolving spatio-temporal profiles of cAMP modulation reveals selectivity in processing dopamine signals by distinct striatal neurons
We next recorded separately from the cell bodies of neurons as well as their distal dendrites (>30 μm from soma) while applying a wide range of doses of dopamine (Figure 2B). Using peak response amplitude as a measure of agonist sensitivity, a dose response curve was plotted for cell bodies and dendrites of both dMSNs and iMSNs (Figure 2C). We detected marked differences in the sensitivity to dopamine across compartments and cell types. Overall, the dMSNs were more sensitive to dopamine as compared to iMSNs. The difference was most pronounced in cell bodies where EC50 for dopamine was ~5 times greater than in iMSNs (Figures 2C and 2D). In addition, the dendrites of dMSN were also more sensitive to dopamine than iMSN dendrites (t-test; p<0.05). Interestingly, we observed that the iMSN dendrites were significantly more sensitive to dopamine than their cell bodies (t-test; p<0.0001). This compartment specific difference in sensitivity was unique to iMSNs and was not seen in dMSN neurons (Figures 2C and 2D).
The kinetics of cAMP accumulation were also distinct between the two types of neurons. The response became faster with increasing dopamine concentration but its cooperativity was more prominent in dMSNs neurons (Figure S2B). In general, iMSNs exhibited twice faster response onset than dMSNs (Figures 2E, 2F, and S2C). However, in dMSN the responses in dendrites were faster than in cell bodies. No such compartment differences were seen in iMSN neurons (Figure S2C). Next, we analyzed the distribution of cAMP amplitudes elicited by a saturating concentration of dopamine (100 μM) (Figure 2G). We found no significant differences in the response amplitudes across compartments in both neuronal populations. However, the response size in both dendrites and cell bodies were significantly larger in dMSN as compared to corresponding compartments of iMSN (Figure 2G). No significant differences in temporal characteristics were observed between compartments and/or cell populations (Figures S2D and S2E). To better understand how different parameters converge at generating the signal, we calculated net cAMP flux produced by saturating dopamine application (Figure 2H). We found that dMSNs produced greater cAMP change relative to iMSN especially in dendrites. In contrast, the dendrites of iMSN had the smallest cAMP flux and the change in cAMP in this compartment was smaller than in the cell bodies.
In addition to slow tonic influx, striatal dopamine is also released in rapid phasic bursts. Thus, we next evaluated MSN responses to phasic application of repeated short puffs of dopamine at the sub-saturating concentration of 1 μM. We observed reliable responses of consistent magnitude and shape recorded from cell bodies of both dMSN and iMSN neurons (Figure 2I). The signals were temporarily resolved and did not show signs of desensitization with repeated application. Similar to tonic application of dopamine, the dMSNs exhibited a greater amplitude and slower onset of peak response compared with iMSNs (Figure 2J). The deactivation kinetics were nearly identical between populations and relatively fast (~1.5 min from peak to baseline) (Figure S2E).
To further resolve spatial cAMP signaling, we analyzed kinetics of cAMP generation at the membrane and intracellular sites by taking single optical planes across the cellular depth over time (Figure 2K). In these experiments, the plasma membrane compartment was identified by using mCherry fused with plasma membrane targeting signal from Ras (Figure S2F). We observed that a phasic puff of dopamine drove initial rapid cAMP production at the plasma membrane compartment, followed by slower but greater cAMP surge from the internal sites (Figures 2L and S2G). In summary these experiments demonstrate that CAMPER approach is capable of resolving complex spatio-temporal profiles of cAMP signaling in genetically defined neurons.
Cell-type specific segregation of receptors contribute to unique effects of adenosine on cAMP modulation
In addition to differential expression of dopamine receptors, adenosine receptor subtype distribution has been proposed to further segregate MSN subpopulations. iMSNs selectively express A2AR which positively couples to cAMP production (Ongini and Fredholm, 1996) whereas segregation of inhibitory A1R and A3R receptors has not been well defined. We first probed adenosine responses in cultured striatal neurons from our D1R- and D2R-CAMPER mice. As with dopamine, we observed bidirectional modulation: cAMP decreased in all dMSNs but increased in all iMSNs (Figure 3A and Table S2). The amplitudes of the stimulatory responses were consistently greater than that of inhibitory responses (Figure 3A). We detected no differences in sensitivity between compartments of dMSN and significantly higher sensitivity of dendrites relative to cell bodies in iMSN (Figures 3B and S3A). Interestingly, the responses recorded from cell bodies of both neuronal populations had similar sensitivity while dendrites of iMSN were more sensitive to adenosine than dendrites of dMSN (Figure 3C). The onset kinetics were similar in both cell bodies and dendrites (Figure S3B) and the only notable difference was prominently faster desensitization of adenosine responses in the dMSN cell bodies (Figure S3C).
Figure 3. Probing modulation of cAMP signaling in cultured striatal neurons by adenosine.

A) Primary striatal neuron response to bath application of adenosine (1 μM). iMSNs (n=33) positively coupled to cAMP production and dMSNs (n=35 neurons) showed an inhibitory effect. Shading indicates SEM range.
B) Dose response curve of cAMP changes to adenosine. n≥10 neurons. Error bars indicate SEM values.
C) The EC50 values calculated from data in panel B. The iMSN dendrites (62 ± 17 nM adenosine) were more sensitive to than dMSN dendrites (125 ± 17 nM adenosine) whereas there was no difference between dMSN and iMSN cell body regions (112 ± 34 and 135 ± 19 nM adenosine, respectively). Error bars indicate SEM values. * = p<0.05
D) Comparison of stimulatory Gαolf-mediated responses. There was no difference in the max response at a saturating concentration (100 μM) between dMSN cell body (5701 ± 451 nM cAMP, n=37) and iMSN cell body (6518 ± 562 nM cAMP, n=34), however dMSN dendrite elicited a greater response (17215 ± 607 nM cAMP, n=33) than iMSN dendrite (14352 ± 654 nM cAMP, n=36). Error bars indicate SEM values. * = p<0.05.
E) Representative cAMP response from saturating concentration of agonist (100 μM) comparing Gαolf signaling.
F) Time to peak at a saturating concentration of agonist (100 μM), dMSN cell body was significantly slower to reach peak response (1.77 ± 0.08 min, n=37) than iMSN cell body (1.47 ± 0.07 min, n=34). Error bars indicate SEM values. * = p<0.05
G) Peak to baseline at a saturating concentration of agonist (100 μM), response termination was significantly faster in dMSN cell body (4.20 ± 0.22 min, n=37) than in iMSN cell body (5.27 ± 0.29 min, n=34). Error bars indicate SEM values. * = p<0.05
H) Comparison of inhibitory Gαi-mediated. There were no differences in the max response elicited at a saturating concentration (100 μM) of agonist between dMSN cell body (711 ± 84 nM cAMP, n=33) and iMSN cell body (702 ± 78 nM cAMP, n=29) or between dMSN dendrite (667 ± 89 nM cAMP, n=36) or iMSN dendrite (711 ± 94 nM cAMP, n=28). Error bars indicate SEM values.
I) Representative cAMP response from saturating concentration of agonist (100 μM) comparing Gai signaling.
J) Time to peak at a saturating concentration of agonist (100 μM), there was a difference in time-to-peak for responses between dMSN (0.99 ± 0.06 min, n=33) cell body and iMSN cell body (0.96 ± 0.05 min, n=29). Error bars indicate SEM values.
K) Peak to baseline at a saturating concentration of agonist (100 μM), response desensitization in dMSN cell body (3.66 ± 0.20 min, n=33) was significantly slower compared with iMSN cell body (3.02 ± 0.17 min, n=29). Error bars indicate SEM values. * = p<0.05.
Reciprocal segregation of stimulatory and inhibitory inputs in MSN subtypes affords a unique opportunity to compare processing of signals via the same G proteins elicited by different receptors. We analyzed stimulatory signaling of Golf by comparing the effect of dopamine on dMSNs with the effect of adenosine on iMSNs (Figure 3D). Notably, dendrites of dMSNs generated significantly more cAMP than those of iMSNs while in cell bodies the response onset was faster for iMSN (Figures 3E and 3F). The response desensitized faster in dMSN (Figure 3G).
Next, we analyzed inhibitory Gi signaling by comparing the effect of dopamine on iMSNs with the effect of adenosine on dMSNs (Figure 3H). No significant differences in either response amplitudes or onset kinetics were found suggesting similar organization and/or regulation of the inhibitory cascades (Figures 3I and 3J). The only notable difference was faster desensitization of responses in iMSN cell bodies (Figure 3K).
In contrast to well-documented exclusive expression of A2AR on iMSN, the molecular identity and segregation of adenosine receptors that provide inhibitory inputs is less clear. To define the receptor mediating inhibitory adenosine input, we first utilized a pharmacological approach targeting two types of Gi/o coupled adenosine receptors: A1R and A3R (Figure 4A). We found that selective A1R agonist 2′MeCCPA efficiently inhibited cAMP modulation in both dMSNs and iMSNs (Figure S4A). In contrast, the A3R selective agonist 2-Cl-IB-MECA did not have an effect in either neuronal population. Activation of the A1R had a stronger effect in dMSN compared with iMSN (Figures 4B and 4C).
Figure 4. Defining molecular identity of adenosine receptor subtypes and their segregation across populations of cultured striatal neurons.

A) Representative traces of cAMP responses to bath application of A1R agonist (2′MeCCPA, 1 μM) in dMSNs (n= 11) and iMSNs (n= 10) or A3R agonist (2-CL-IB-MECA, 1 μM) in dMSNs (n= 36) or iMSNs (n= 39).
B) Dose response curve of cAMP changes to A1R stimulation. n≥7 neurons. Error bars indicate SEM values.
C) Response efficacy and potency. The EC50 values in dMSNs (14.6 ± 4.1 nM 2′MeCCPA) were significantly lower than in iMSNs (142.5 ± 9.1 nM 2′MeCCPA). The max amplitude was greater in dMSNs (715 ± 20 nM cAMP) than in iMSNs (516 ± 26 nM cAMP). Error bars indicate SEM values. * =p<0.05, ** = p<0.001
D) Schematic of the CRISPR/Cas9 editing strategy.
E) Representative cAMP response in putative iMSNs to bath application of adenosine in either A1R wild-type (WT) or A1R knockout (KO). The time to peak was significantly faster in A1R KO (0.922 ± 0.06 minutes) compared with A1R WT (1.82 ± 0.05 mimutes). The peak to baseline timing was significantly faster in A1R WT (4.43 ± 0.27 minutes) compared with A1R KO (5.41 ± 0.13 minutes). n=18 A1R KO and 33 A1R WT. * =p<0.05, ** = p<0.001
We next used a CRISPR/Cas9-based approach to genetically ablate A1R. With this strategy, a construct containing guide RNA targeting A1R, Cas9 and Cre recombinase was delivered to cultured neurons from CAMPER mice by lentiviral transduction (Figure 4D). Given the design of the experimental strategy we could no longer rely on genetic driver lines distinguish between iMSN and dMSN and instead used direction of cAMP response to identify neuronal populations. Application of adenosine elicited only stimulatory responses in approximately half of the neurons recorded (Figure S4B) presumed to be iMSN. Application of 2′MeCCPA to A1R KO elicited response in only 3 out of 46 neurons (6.5%) compared with the 81% responsiveness of WT neurons confirming role of A1R in generation of inhibitory response (Figures S4C and S4D). Thus, the A1R is the dominant receptor type responsible for inhibitory responses of dMSN to adenosine. Because iMSN neurons also responded to specific A1R agonist 2′MeCCPA and no such response was evident upon A1R ablation, we further examined the role of A1R in iMSN. Ablation of A1R substantially accelerated the onset of stimulatory cAMP responses and prolonged response deactivation time, consistent with facilitation of Golf-AC signaling upon removal of competing inhibitory Gi drive on the same neurons (Figure 4E). There were no significant effects on cAMP baseline or response amplitude (Figure S4E). In summary, these findings define contributions of individual adenosine receptors to cAMP signaling in populations of striatal MSN and illustrate the utility of real time cAMP imaging using CAMPER tool in such dissection.
Opioids provide sparse inputs onto cAMP cascade of MSNs subject to paracrine feedback
To analyze the contribution of opioid inputs onto MSNs, we studied the responses to μ-opioid receptor (MOR) agonist morphine which elicited exclusively inhibitory cAMP responses in a limited subset of both dMSNs and iMSNs (Figure 5A). This low level of responsiveness prompted us to hypothesize that MSNs in culture may desensitize to morphine due to chronic exposure to endogenous opioid peptide enkephalin released by iMSN. To test this possibility, a low dose (50 nM) of MOR antagonist naloxone (NLX) was included in the culture. Indeed, we observed that NLX pre-treatment greatly increased responsiveness of both MSN populations to morphine in terms of number of neurons with detectable cAMP signal, response amplitude and its sensitivity (Figures S5A, 5B, and 5C). We next compared the effect of acute opioid stimulation on dMSNs and iMSNs following NLX pre-treatment. We found no significant differences in the amplitudes of cAMP responses elicited by morphine on iMSN and dMSN (Figures 5D and 5E). In contrast, dMSN displayed about 6-fold greater sensitivity to opioid stimulation relative to iMSN. Similar difference in opioid sensitivity between iMSN and dMSN was also seen without NLX pretreatment, suggesting that chronic exposure to opioids scales down responsiveness to opioids across populations proportionately (Figures S5B and S5C).
Figure 5. Modulation of cAMP responses in cultured striatal neurons by opioids.

A) Responses to neurons to morphine in dMSNs (25 out of 302) and iMSNs (22 out 313).
B) Then effect of pretreatment with 50 nM naloxone on subsequent responses to morphine (1 μM).
C) Naloxone pretreatment increased the number of responsive neurons from 7.1% (combined dMSNs and iMSNs, n=615) to 28.4% (combined dMSNs and iMSNs, n=356). No differences in behavior of dMSNs and iMSNs neurons were noted.
D) Dose response curve of cAMP changes to morphine from naloxone pretreated neurons. n≥7 neurons. Error bars indicate SEM values.
E) Response efficacy and potency. Max amplitude of cAMP responses to morphine did not differ between dMSNs and iMSNs (p>0.05) The EC50 was significantly lower for dMSNs (17.4 ± 6.7 nM morphine) than iMSNs (151 ± 29.5 nM morphine). Error bars indicate SEM values. * = p<0.05.
F) Comparison of inhibitory Gαi inputs on dMSN. MOR stimulation produced peak response significantly slower (1.75 ± 0.19 min, n= 19) than A1R stimulation (0.96 ± 0.05 min, n= 33) at a saturating concentration of agonist. The response magnitude of MOR-driven response was significantly smaller (621 ± 26 nM cAMP, n= 19) than that of A1R (715 ± 27 nM cAMP, n= 33). Error bars indicate SEM values. * = p<0.05, ** = p<0.001.
G) Comparison of inhibitory Gαi inputs on iMSNs. MOR stimulation produced peak response significantly slower (1.84 ± 0.20 min, n= 16) than D2R (0.99 ± 0.06 min, n= 29) or A1R stimulation (1.12 ± 0.09 min, n= 17) at a saturating concentration of agonist. There was no difference in activation time between D2R and A1R-mediated responses. D2R stimulation had a significantly greater response magnitude (769 ± 20 nM cAMP, n= 29) compared with MOR (616 ± 29 nM cAMP, n=16) or A1R (514 ± 34 nM cAMP, n= 17). MOR stimulation was significantly greater than A1R stimulation. Error bars indicate SEM values. * = p<0.05
To gain further insight into relationship between inhibitory inputs onto cAMP production in different striatal MSN types we compared kinetics of MOR signaling to that of other Gi-coupled receptors present in the same neurons. In the dMSN neurons at saturating levels of receptor stimulation by respective agonists, MOR responses were considerably slower relative to A1R (1.75 ± 0.19 min morphine vs 0.96 ± 0.05 min adenosine) and produced significantly smaller change in cAMP concentration (621 ± 26 nM cAMP morphine vs. 715 ± 27 nM cAMP adenosine) (Figure 5F). In iMSNs, MOR activation produced greater change in cAMP concentration relative to A1R (616 ± 29 nM cAMP morphine vs. 514 ± 34 nM cAMP adenosine), yet MOR-driven response was still slower than that mediated by A1R (1.84 ± 0.20 min morphine vs 1.12 ± 0.09 min adenosine). When compared to D2R activation (0.99 ± 0.06 min, 769 ± 20 nM cAMP dopamine), MOR was both slower and weaker across the board suggesting that dopamine modulation of iMSNs is dominant relative to MOR (Figure 5G). Overall, these studies reveal differential effects of opioid modulation of striatal MSNs, distinct from other neuromodulatory inputs.
Interrogating real time cAMP dynamics in intact striatal circuit by optogenetics defines integration patterns of dopaminergic inputs
We next sought to investigate how striatal MSNs process information in the context of an intact neuronal circuitry by focusing on in the NAc from the VTA. To evoke dopamine release, neurons in the VTA were stereotaxically injected with AAV-hChR2-mCherry followed by optical stimulation (Figure 6A). Thick sagittal slices were prepared from D1R-Cre:CAMPER and D2R-Cre:CAMPER mice and changes in FRET signals in NAc neurons were recorded by two-photon microscopy while delivering LED impulses of varying temporal patterns (Figure 6B).
Figure 6. Interrogating real time cAMP dynamics of striatal circuitry by optogenetics in acute brain slices.

A) Schematic of target injection site (left) and validation of AAV expression (right) confined to the ventral tegmental area (VTA) and absent from substantia nigra (SN) and fasciculus retroflexus (fr).
B) Schematic of the experimental design in acute slices.
C) Representative trace of responses elicited by varying frequency stimulations (20 flashes, 2-ms duration/flash). n≥15 neurons.
D) The dependence of cAMP response amplitude on stimulation frequency. Error bars are SEM values.
E) dMSNs were more sensitive to lower frequencies of stimulation achieving half maximal response at 9.51 ± 1.16 Hz compared with iMSNs (13.24 ± 1.09 Hz). Error bars are SEM values. * = p<0.05.
F) Peak width at half-maximal response for each frequency. Error bars are SEM values.
G) Quantification of rebound oscillation amplitude in the opposite direction of the initial response. Error bars are SEM values.
H) cAMP response at varying durations of inter-stimulation intervals (ISI). n≥17 neurons.
I) Max amplitude of cAMP responses with increase ISI. Linear regression fit shown as r2. Error bars are SEM values.
J) cAMP response aggregation as a function of inter-stimulation interval duration. Error bars are SEM values.
As expected, about half of the MSNs expressed the cAMP sensor using either D1R-Cre or D2-Cre driver lines (Figure S6A). Neurons in D1R-Cre CAMPER mice responded by elevating cAMP to application of D1R agonist SKF38393 and by lowering upon treatment with A1R agonist 2′MeCCPA. Conversely, neurons from D2R-Cre CAMPER mice elevated cAMP in response to the A2AR agonist CGS21680 and lowered it upon treatment with D2R agonist quinpirole (Figure S6B). Converse experiments, reversing drug applications showed that D1R-Cre and D2R-Cre neurons were unresponsive to treatment with CGS21680 or SKF38393 (Figure S6B), respectively confirming the identity and segregation of MSN subpopulations. Interestingly, we found basal cAMP to be significantly higher in iMSN as compared to dMSN (247 +/− 29 nM vs 146 +/− 17 nM).
We next investigated whether biosensor expression affected intrinsic electrophysiological properties of the MSNs (Figures S7A, S7B, and S7C). We found that both iMSN and dMSN expressing the sensor had resting membrane potential, input resistance, and excitability indistinguishable from the control neurons. We further found that overall behavior of CAMPER mice was normal. When examined in the open field task, commonly used to reveal impact of changes in striatal signaling on motor control, D1R-Cre:CAMPER and D2R-Cre:CAMPER mice showed no difference from their control littermates, in habituation, total distance traveled, or movement velocity (Figure S7D). Together with electrophysiological data, these findings indicate preservation of key properties of striatal neurons in CAMPER mice. Although we did not examine all possible neurophysiological aspects and behavioral reactions, our data suggests that sensor expression does not affect key signaling processes downstream from cAMP. Nonetheless, it is of use to be mindful of such caveats when addressing specific questions using these mice in future studies.
We next studied responses evoked by optogenetic stimulation of VTA neurons using brain slice preparation. We isolated dopamine-mediated responses by supplementing ACSF with NMDA, GABAA, GABAB, and muscarinic antagonists during recordings (Marcott et al., 2014). To study the response to a single stimulation event we delivered a fixed bout of blue light flashes (20 2-ms flashes at 300 μW) at 20 Hz. Using fast scanning cyclic voltammetry we verified that LED pulse stimulation evoked dopamine release and subsequent cAMP responses to this stimulation were ablated in the presence of DR antagonists (Figures S7E and S7F). To understand how VTA activation is translated to downstream signaling in MSNs, we varied the frequency of LED stimulation while recording cAMP response. The lower range of frequencies corresponded to tonic firing patterns of the VTA (<4 Hz) progressing through stimulation range designed to mimic burst firing activity (15–30 Hz) until saturation of the system (at ~ 80 Hz) (Figures 6C and 6D). In dMSN neurons we observed a reliable response at 4 Hz, which saturated at 20 Hz. In contrast, iMSN did not begin to respond until 10 Hz and saturated at 40 Hz (Figure 6D). The lower sensitivity of iMSN responses to dopamine inputs from VTA relative to dMSN were also evident from the significant differences in half-maximal stimulation frequencies (Figure 6E). At saturating stimulation intensities both neuronal populations responded with the same degree of a change in cAMP concentration (~100 nM) suggesting similar constraints that limit cAMP production upon synaptic stimulation. The response cooperativity was greater in dMSN (Hill slope=1.878 ± 0.090) as compared to iMSN (Hill slope=1.428 ± 0.134). Importantly, all dMSN and iMSN in our experiments responded by increasing and decreasing cAMP accumulation, respectively, in response to optogenetic stimulation.
The temporal characteristics of these elemental responses displayed several striking features. First, we noticed that responses took a substantial amount of time to develop, peaking several minutes after the end of the millisecond stimulation and taking a comparable amount of time to fully recover. The entire response duration was significantly shorter in iMSN than in dMSN neurons (Figure 6F). Second, during the recovery, the response continued to develop in the opposite direction past initial baseline and back up again before resting. This oscillatory behavior occurred only in dMSN neurons where it showed prominent rebound decreasing cAMP concentration to the levels of about half of the initial stimulatory response (Figure 6G). The rebound showed similar dependence on stimulus strength – increasing in amplitude with the increase in signaling and saturating at ~80 Hz.
VTA neurons are known to display high frequency multi-burst firing activity in bouts (Juarez and Han, 2016), which we modeled by delivering repeated stimuli while varying the timing interval timing between individual events on the scale of the cAMP response (Figure 6H). Initially the individual cAMP responses were resolved, but with shortening the time between the stimuli individual responses began fusing to form tetanic wave of cAMP surge (dMSN) or depletion (iMSN). We observed significant differences in the ability of MSN populations to integrate stimuli. First, the thresholds triggering tetanic summation of individual responses were different with dMSNs fusing the responses more readily (Figure 6H and 6I). Second, the iMSNs exhibited an oscillatory behavior, responding by a sizable rebound wave of cAMP increase above the baseline following tetanic depression. In both populations the amplitude of the response increased as the inter-stimulus interval decreased indicating that individual responses sum to result in bigger fluctuation of the cAMP (Figures 6H and 6J). These results indicate that two populations of striatal neurons exhibit key differences in temporal tuning allowing them to integrate cAMP signals. From the technical perspective, this additionally reveals the stability of the sensor to measure dynamic changes after a period of sustained cAMP elevations/depressions.
CAMPER mice can be used to examine neuromodulatory control in diverse neuronal populations across brain regions
We further examined the utility of CAMPER mice for measuring cAMP dynamics in other brain regions. We observed that stereotaxic delivery of AAV-Cre virus to cortical and hippocampal regions of CAMPER mice successfully unlocked sensor expression in respective neuronal populations (Figure 7A and 7B). Similar to cultured neurons, viral strategy allowed imaging sparsely labeled neurons with defined soma, processes and spines. Recording baseline FRET signal in the presence of 1 μM TTX revealed that resting cAMP levels in hippocampal pyramidal CA1 neurons (301 ± 11.4 nM cAMP) were more than double the level observed in cortical neurons (155 ± 12.1 nM cAMP) (Figure 7C). To examine the functional cAMP responsiveness of these neurons we stimulated them pharmacologically with dopamine and 2′MeCCPA that engage their dopamine and A1R receptors, respectively. Both types of neurons elicited robust increases in cAMP to dopamine consistent with predominant involvement of D1R type of Gs-coupled dopamine receptors (Figure 7D). Interestingly, the response was significantly larger in hippocampal than in cortical neurons (173 ± 5.7 nM vs. 52 ± 8.7 nM cAMP) (Figure 7E). Both neuronal populations also responded to 2′MeCCPA by decreasing cAMP levels consistent with the activation of Gi-coupled A1-type receptors (Figure 7F). Here we also observed that the cAMP change in hippocampal neurons was significantly larger than in cortical neurons (58 ± 7.4 nM vs. 29 ± 5.1 nM cAMP) (Figure 7G). Collectively, this demonstrates that Cre-mediated sensor unlocking strategy in CAMPER mice can be used for studying cAMP signaling in defined neuronal populations across the brain to reveal key differences in processing of neuromodulatory inputs.
Figure 7. Approach for region-specific control of sensor expression to study endogenous GPCR stimulation across brain regions.

A) Schematic of experimental strategy targeting hippocampus and cortex.
B) TEpacVV fluorescence in CA1 pyramidal neurons in hippocampus (top panel, middle panel) or in cortical neurons (bottom panel). Scale bar in top and bottom panel represents 50 μm whereas scale bar in middle panel represents 10 μm.
C) Quantification of basal cAMP level determined by FRET imaging. Error bars are SEM values. * = p<0.05.
D) Averaged traces of cAMP response to dopamine (10 μM) and forskolin/IBMX (50/200 μM). n>10 neurons.
E) Max cAMP amplitude to dopamine. n>10 neurons. Error bars are SEM values. * = p<0.05.
F) Averaged traces of cAMP response to 2′MeCCPA (1 μM) and forskolin/IBMX (50/200 μM. n>10 neurons.
G) Max cAMP amplitude to 2′MeCCPA. n>10 neurons. Error bars are SEM values. * = p<0.05.
DISCUSSION
In this report, we present a tool and framework for analyzing processing of neuromodulatory inputs by native neurons through studying dynamics of cAMP, a second messenger that mediates the effects of a vast number of GPCRs. The approach is based on the use of a genetically encoded cAMP sensor engineered into the mouse genome to allow conditional expression in defined neuronal populations. We demonstrate the utility of studying neurons in this mouse model for characterizing the effects of various neurotransmitter inputs with temporal precision across subcellular compartments. We found that the particular power of this approach lies in its ability to interrogate neuromodulation in intact neuronal circuits in combination with pharmacological, genetic and optogenetic manipulations to reveal fundamental principles of the signal decoding process.
Since monitoring cAMP provides a more direct assessment of neuromodulatory inputs than PKA-based sensors applied to these neurons (Chen et al., 2014; Goto et al., 2015), the CAMPER approach offers quantitative assessment of both inhibitory and stimulatory signaling with high spatio-temporal resolution. For example, other FRET approaches for cAMP dynamics such as PKA-based sensors (AKAR) have only reported inhibitory cAMP responses after first producing a robust increase in cAMP (Polito et al., 2015). Moreover, the TEPACVV is a single chain sensor compatible with Fluorescence Lifetime IMaging (FLIM) that offer advantages over luciferase-based sensors (Nano-lantern; (Saito et al., 2012) or non-FRET single wavelength reporters (Flamindos; (Kitaguchi et al., 2013)).
Using primary cultured striatal neurons, we were able to assess rapid changes in cAMP dynamics induced by controlled application of neuromodulators. We confirmed segregation of dopamine receptor types across these populations and noted essential differences in their functional properties. The pharmacology of these receptors have been mostly studied in reconstituted systems and yielded substantially varying estimates and therefore our data provide direct evidence for the sensitivity differences in the endogenous system. We further noted a remarkable subcellular segregation of cAMP signaling. Dopamine triggered initial wave of rapid response at the surface of the neuron followed by slower, much larger secondary wave emanating form the intracellular sites. These observations are in agreement with emerging observations that several GPCRs are capable of continuing to signal following their internalization (Irannejad and von Zastrow, 2014; Shenoy and Lefkowitz, 2011). The data suggests that dopamine inputs on MSNs are processed with greater temporal complexity defined by receptor properties that defies their simplistic binary stimulatory/inhibitory binning.
In addition to dopamine, cultured striatal neurons showed differential responsiveness to a variety of inputs including adenosine, opioids, and acetylcholine documenting extensive diversity of inputs into cAMP axis. The adenosine system appears to be of particular interest for its reciprocal effects on cAMP production relative to dopamine inputs: adenosine increases cAMP in all iMSN and decreases in all dMSN. In this study we established that inhibitory effect on cAMP production on dMSN is entirely driven by A1R receptor. Interestingly we further find that A1R is not confined to dMSN and is also present on iMSN where it provides an inhibitory input to adenylyl cyclase thus opposing A2AR action and shaping kinetics of adenosine response in this population.
Taking advantage of the spatial resolution afforded by cultured neurons we characterized propagation of cAMP signals across major cellular and subcellular compartments. Remarkably, we found that parameters of cAMP responses to major neuromodulatory inputs differed substantially between cell bodies and dendritic processes. While the exact mechanisms that underlie these differences remain to be established we think that they are likely related to abundance and composition of the signaling complexes responsible for the production and/or degradation of cAMP. For example, differential dendritic targeting of receptors, adenylyl cyclases and/or phosphodiesterase isoforms and their interactions with scaffolding proteins (Dessauer, 2009; Wong and Scott, 2004) may underpin regional differences in response parameters. Such spatial segregation of cAMP responsiveness along the cell body/dendrite axis may functionally diversify the inputs according to their proximity to cell bodies vs dendrites. Additionally, the need for activity dependent transcriptional programs that require communication with the nucleus may underlie the need for greater cAMP responses in distal neuronal compartments compared with the cell body, thus enabling sufficient activation levels of labile molecules (such as pDARPP-32) to convey information to the nucleus (Li et al., 2015).
One of the key features of the CAMPER mouse is its applicability for revealing regulatory mechanisms and principles of neuromodulation in intact circuitry, which we demonstrate by studying signal processing in MSN neurons of NAc neurons. In this nucleus, a major role is played by dopamine, which has opposing effects on the activity of MSN populations through differential modulation of the cAMP (Goto et al., 2015). We revealed several key features inherent to parallel processing of signals in dMSN and iMSN neurons. For example, these neurons have differential sensitivity and frequency tuning to the same dopamine inputs. We also report differences in their temporal resolution as well as in signal integration thresholds for producing supralinear tetanic response and oscillatory behavior that together underlie unique patterns of cAMP responses produced by these neurons. These observations may underlie the desynchronizaiton of signal processing long believed to be hallmark of signal processing in parallel striatal channels (Albin et al., 1989; Cui et al., 2013; Gerfen and Surmeier, 2011).
Finally, we demonstrated the utility of using CAMPER mice as a universal tool to image cAMP dynamics across multiple brain regions by unlocking sensor expression through stereotaxic delivery of Cre-recombinase encoding viruses. Benefits of this strategy include achieving the anatomical precision, gaining developmental control of sensor expression as well as increasing flexibility of experimental design, e.g. by combining it with other genetic manipulations. One of the interesting observations gained via comparative analysis across brain regions was observed differences in the baseline levels of cAMP. Although undoubtedly influenced by their inputs, transcriptional landscape diversity across neuronal type likely complements basal cAMP levels. It is therefore tempting to speculate how behavioral conditioning or disease models may reflect this homeostatic cAMP set point (Hakansson et al., 2004; Heckman et al., 2015). Therefore CAMPER mice may serve as a valuable reagent for understanding how cAMP gradients and thresholds shape neuronal output and long lasting transcriptional modifications (Kandel, 2012).
In conclusion, we hope that the approach and analytical tools that we describe would be useful for studying the role and mechanisms of neuromodulation across various circuits, thus providing a foundation for uncovering the signaling characteristics of various neurotransmitters while further serving as a guide toward understanding the mechanistic underpinnings of signal regulation. The availability of this tool also simplifies avenues towards FRET imaging approaches in free moving animals through microendoscopy (Goto et al., 2015), cranial windows (Gong et al., 2014) and miniaturized microscopes (Ghosh et al., 2011).
EXPERIMENTAL PROCEDURES
All experiments involving animals were approved by the Institutional Animal Care and Use Committee. Both sexes were used. Standard previously published procedures were followed for primary neuronal cultures, stereotaxic injections, behavioral evaluation, preparation of brain slices, imaging, electrophysiology, and fast scan cyclic voltammetry. For detailed experimental procedures, please see the Supplemental Experimental Procedures.
Quantification and statistical analysis
Student’s t-test and ANOVA were used for pairwise comparisons with the use of asterisks indicating statistical significance (* = p<0.05, ** = p<0.001, *** = p<0.0001). Prism GraphPad software was used to perform statistical analysis. A minimum of three independent biological replicates (cells or animals as stated) was performed for each experiment and data from all subjects/samples was included without exception. Graphs that report mean values also include standard error of the mean.
Supplementary Material
HIGHLIGHTS.
CAMPER reporter mice developed to probe neuromodulation in the endogenous setting
CAMPER mice report modulation of cAMP dynamics by a variety of neurotransmitter GPCRs
Probing real time cAMP flux with optogenetics reports signaling in intact circuits
CAMPER imaging approach reveals principles of dopamine signaling in the striatum
Acknowledgments
We wish to thank Ms. Natalia Martemyanova for producing and maintaining mice examined in this study, the Scripps Research Murine Genetics Core for performing ES cell work, Dr. Seth Tomchik for technical expertise with imaging and critical comments on the manuscript, Tyler Krome for engineering expertise with the LED stimulation apparatus, Dr. Hongkui Zeng (Allen Institute for Brain Science) for Ai9 targeting vector, Dr. Kees Jalink (Netherlands Cancer Institute) for sharing TEPACVV. This work was supported by NIH grants: DA041207 (to B.S.M.), DA036596 (to K.A.M.), and DA026405 (to K.A.M.)
Footnotes
AUTHOR CONTRIBUTIONS
B.S.M. performed all biochemical, cell biological, and imaging experiments described in the paper, analyzed the data and wrote the paper; S.Z. performed electrophysiology, M.T.D. performed viral injections and behavior experiments, H.I. and R.D.B. performed fast scan cyclic voltammetry, C.J. performed viral injections. C.M.M. and R.L.D. generated R26floxstop-TEPACVV mouse model, K.A.M. designed the study, analyzed data, and wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Marder E. From the connectome to brain function. Nat Methods. 2013;10:483–490. doi: 10.1038/nmeth.2451. [DOI] [PubMed] [Google Scholar]
- Brinton RE. Neuromodulation: associative and nonlinear adaptation. Brain research bulletin. 1990;24:651–658. doi: 10.1016/0361-9230(90)90003-i. [DOI] [PubMed] [Google Scholar]
- Chen Y, Saulnier JL, Yellen G, Sabatini BL. A PKA activity sensor for quantitative analysis of endogenous GPCR signaling via 2-photon FRET-FLIM imaging. Front Pharmacol. 2014;5:56. doi: 10.3389/fphar.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cropper EC, Friedman AK, Jing J, Perkins MH, Weiss KR. Neuromodulation as a mechanism for the induction of repetition priming. Curr Opin Neurobiol. 2014;29:33–38. doi: 10.1016/j.conb.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL. Physiology and biochemistry of Drosophila learning mutants. Physiological reviews. 1996;76:299–317. doi: 10.1152/physrev.1996.76.2.299. [DOI] [PubMed] [Google Scholar]
- Dessauer CW. Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn TA, Wang CT, Colicos MA, Zaccolo M, DiPilato LM, Zhang J, Tsien RY, Feller MB. Imaging of cAMP levels and protein kinase A activity reveals that retinal waves drive oscillations in second-messenger cascades. J Neurosci. 2006;26:12807–12815. doi: 10.1523/JNEUROSCI.3238-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, Schnitzer MJ. Miniaturized integration of a fluorescence microscope. Nat Methods. 2011;8:871–878. doi: 10.1038/nmeth.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA. Signaling in striatal neurons: the phosphoproteins of reward, addiction, and dyskinesia. Prog Mol Biol Transl Sci. 2012;106:33–62. doi: 10.1016/B978-0-12-396456-4.00006-7. [DOI] [PubMed] [Google Scholar]
- Gong Y, Wagner MJ, Zhong Li J, Schnitzer MJ. Imaging neural spiking in brain tissue using FRET-opsin protein voltage sensors. Nat Commun. 2014;5:3674. doi: 10.1038/ncomms4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto A, Nakahara I, Yamaguchi T, Kamioka Y, Sumiyama K, Matsuda M, Nakanishi S, Funabiki K. Circuit-dependent striatal PKA and ERK signaling underlies rapid behavioral shift in mating reaction of male mice. Proc Natl Acad Sci U S A. 2015;112:6718–6723. doi: 10.1073/pnas.1507121112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Hakansson K, Lindskog M, Pozzi L, Usiello A, Fisone G. DARPP-32 and modulation of cAMP signaling: involvement in motor control and levodopa-induced dyskinesia. Parkinsonism Relat Disord. 2004;10:281–286. doi: 10.1016/j.parkreldis.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Heckman PR, Blokland A, Ramaekers J, Prickaerts J. PDE and cognitive processing: beyond the memory domain. Neurobiol Learn Mem. 2015;119:108–122. doi: 10.1016/j.nlm.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Insel PA, Wilderman A, Zambon AC, Snead AN, Murray F, Aroonsakool N, McDonald DS, Zhou S, McCann T, Zhang L, et al. G Protein-Coupled Receptor (GPCR) Expression in Native Cells: “Novel” endoGPCRs as Physiologic Regulators and Therapeutic Targets. Mol Pharmacol. 2015;88:181–187. doi: 10.1124/mol.115.098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–116. doi: 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Lovinger DM. Presynaptic G Protein-Coupled Receptors: Gatekeepers of Addiction? Front Cell Neurosci. 2016;10:264. doi: 10.3389/fncel.2016.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez B, Han MH. Diversity of Dopaminergic Neural Circuits in Response to Drug Exposure. Neuropsychopharmacology. 2016;41:2424–2446. doi: 10.1038/npp.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaguchi T, Oya M, Wada Y, Tsuboi T, Miyawaki A. Extracellular calcium influx activates adenylate cyclase 1 and potentiates insulin secretion in MIN6 cells. Biochem J. 2013;450:365–373. doi: 10.1042/BJ20121022. [DOI] [PubMed] [Google Scholar]
- Klarenbeek JB, Goedhart J, Hink MA, Gadella TW, Jalink K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS One. 2011;6:e19170. doi: 10.1371/journal.pone.0019170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Li L, Gervasi N, Girault JA. Dendritic geometry shapes neuronal cAMP signalling to the nucleus. Nat Commun. 2015;6:6319. doi: 10.1038/ncomms7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Nestler EJ. The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat. 2011;5:41. doi: 10.3389/fnana.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes AC, Dunn H, Ferguson SS. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol. 2012;165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Mamaligas AA, Ford CP. Phasic dopamine release drives rapid activation of striatal D2-receptors. Neuron. 2014;84:164–176. doi: 10.1016/j.neuron.2014.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E. Neuromodulation of neuronal circuits: back to the future. Neuron. 2012;76:1–11. doi: 10.1016/j.neuron.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Reflections on: “A general role for adaptations in G-Proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function”. Brain research. 2016;1645:71–74. doi: 10.1016/j.brainres.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res. 2006;99:1084–1091. doi: 10.1161/01.RES.0000250046.69918.d5. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Ongini E, Fredholm BB. Pharmacology of adenosine A2A receptors. Trends in pharmacological sciences. 1996;17:364–372. [PubMed] [Google Scholar]
- Polito M, Guiot E, Gangarossa G, Longueville S, Doulazmi M, Valjent E, Herve D, Girault JA, Paupardin-Tritsch D, Castro LR, et al. Selective Effects of PDE10A Inhibitors on Striatopallidal Neurons Require Phosphatase Inhibition by DARPP-32(1,2,3) eNeuro. 2015:2. doi: 10.1523/ENEURO.0060-15.2015. [DOI] [PMC free article] [PubMed]
- Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135:561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neuro-Signals. 2009;17:5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Chang YF, Horikawa K, Hatsugai N, Higuchi Y, Hashida M, Yoshida Y, Matsuda T, Arai Y, Nagai T. Luminescent proteins for high-speed single-cell and whole-body imaging. Nat Commun. 2012;3:1262. doi: 10.1038/ncomms2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends in pharmacological sciences. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomchik SM, Davis RL. Dynamics of learning-related cAMP signaling and stimulus integration in the Drosophila olfactory pathway. Neuron. 2009;64:510–521. doi: 10.1016/j.neuron.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50. doi: 10.1016/j.neuron.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiological reviews. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.