

Abstract
Background
Founder mutations in the two breast cancer genes, BRCA1 and BRCA2, have been described in many populations, among these are Ashkenazi-Jewish, Polish, Norwegian and Icelandic. Founder mutation testing in patients with relevant ancestry has been a cost-efficient approach in such populations. Four Norwegian BRCA1 founder mutations were defined by haplotyping in 2001, and accounted for 68% of BRCA1 mutation carriers at the time. After 15 more years of genetic testing, updated knowledge on the mutation spectrum of both BRCA1 and BRCA2 in Norway is needed. In this study, we aim at describing the mutation spectrum and frequencies in the BRCA1/2 carrier population of the largest clinic of hereditary cancer in Norway.
Methods
A total of 2430 BRCA1 carriers from 669 different families, and 1092 BRCA2 carriers from 312 different families were included in a quality of care study. All variants were evaluated regarding pathogenicity following ACMG/ENIGMA criteria. The variants were assessed in AlaMut and supplementary databases to determine whether they were known to be founder mutations in other populations.
Results
There were 120 different BRCA1 and 87 different BRCA2 variants among the mutation carriers. Forty-six per cent of the registered BRCA1/2 families (454/981) had a previously reported Norwegian founder mutation. The majority of BRCA1/2 mutations (71%) were rare, each found in only one or two families. Fifteen per cent of BRCA1 families and 25% of BRCA2 families had one of these rare variants. The four well-known Norwegian BRCA1 founder mutations previously confirmed through haplotyping were still the four most frequent mutations in BRCA1 carriers, but the proportion of BRCA1 mutation carriers accounted for by these mutations had fallen from 68 to 52%, and hence the founder effect was weaker than previously described.
Conclusions
The spectrum of BRCA1 and BRCA2 mutations in the carrier population at Norway’s largest cancer genetics clinic is diverse, and with a weaker founder effect than previously described. As a consequence, retesting the families that previously have been tested with specific tests/founder mutation tests should be a prioritised strategy to find more mutation positive families and possibly prevent cancer in healthy relatives.
Keywords: BRCA1, BRCA2, Founder mutations, Genetic testing
Background
Breast cancer genes 1 and 2, BRCA1/2 have been very well studied since their discovery in 1994 and 1995. Disease-causing mutations in these genes give a high lifetime risk of both breast and ovarian cancer [1–3]. An increased risk of aggressive prostate cancer for male BRCA2 mutation carriers has been described [4], as well as elevated risk of pancreatic cancer [5]. Risk for other cancers is less evident [5, 6]. Preventive measures such as prophylactic mastectomy and oophorectomy or surveillance with breast MRI seem to improve survival for BRCA mutation carriers without significantly reducing quality of life [7–10]. More recently also cancer treatment choices are influenced by BRCA-status, especially for ovarian cancer [11].
Founder mutations in BRCA1 and 2 have been described in many populations, i.e. the Ashkenazi-Jewish, Polish, Norwegian, and Icelandic [12–14]. Therefore, founder mutation testing in patients with relevant ancestry and family history has been a cost-efficient approach during the years with limited access to sequence analysis. A founder mutation may be defined as “a genetic alteration observed with high frequency in a group that is or was geographically or culturally isolated, in which one or more of the ancestors was a carrier of the altered gene”. Founder effect is frequently defined as “the loss of genetic variation that occurs when a new population is established by a very small number of individuals from a larger population” [15]. Different historical, societal and geographic factors may influence development of a founder effect including bottle neck phenomenon, genetic drift, selective mating /inbreeding, and high reproduction.
One of the first studies carried out on BRCA epidemiology in Norway by Moller et al. in 2001, showed that 68% of the mutation carriers had one of the four most frequent Norwegian founder mutations in BRCA1 [16], c.1016dup, c.1556del, c.3328_3229del, c.697_698del, all demonstrated to be true founder mutations through haplotyping [13]. The variant c.1016dup was shown to originate in the south-eastern part of the country, while the other three originated from the south-western part of the country, before the Bubonic plague. Later, in 2007, four more BRCA1 variants and two BRCA2 variants c.3847_3848del and c.2808_2811del were published as frequent mutations in the Norwegian population, but no haplotype study has been carried out to establish a true founder origin in these [12, 17].
Founder mutation testing and MLPA (multiplex ligation-dependent probe amplification) have a lower sensitivity compared to sequencing of the entire genes and MLPA especially when used on a population with mixed genetic background. This has become increasingly obvious in our clinical practice over the years. Due to the multicultural population served by Oslo University Hospital (OUH), as well as the falling costs of testing, sequencing and MLPA as initial test has been chosen over founder mutation testing when BRCA-testing is indicated. Following this, sequencing and MLPA have become the standard test since January 2014. This practice is in line with the fact that genetic variation in any gene is abundant, and rare, pathogenic variants in any gene are expected to exist [18].
The knowledge on frequencies and spectrum of disease-causing variation in BRCA1/2 both nationally and locally is however incomplete. The aim of this study has been to describe the results from the BRCA testing during the last 15 to 20 years. This will give necessary overview of mutation frequencies in our region, and the results may give directions for both future research and serve as an evaluation of the current testing practice.
Methods
Study design
The study was carried out in the Section of Hereditary Cancer, Department of Medical Genetics, Oslo University Hospital, OUH, and was approved by the Data Protection Officer at OUH as a quality of care study. The study group was the full mutation carrier population registered in the clinic. Data collection was done in May 2016, and mutation carriers registered in the clinic before 5th of May 2016 were included. The study subjects were both men and women tested over the years, affected with cancer or not. Families registered with a positive mutation test were included. The lowest number of mutation carriers in a family was set to one. In this study, “family” was defined by the practice of giving an index patient a separate family number if he/she did not already have family members registered in the clinic. A thorough job looking up relatives have been done in each case and if relatives were found, the person have been included in the already registered family. Genetic testing was performed both diagnostically and predictively. All activities fulfilled the requirements of genetic counselling, information and consent stated by the Norwegian Act on Biotechnology, www.lovdata.no. All clinical information was registered in the electronic patient journals at OUH. Close to all positive mutation tests were confirmed in a separate blood sample. On the basis of the selection criteria, 2430 BRCA1 mutation carriers from 669 different families, and 1092 BRCA2 carriers from 312 different families were included in the study.
Genetic testing and testing strategies
Our cancer genetics clinic has offered both diagnostic and predictive testing to individuals fulfilling criteria for BRCA1/2 testing given by the health authorities. Initially our clinic served the whole country with genetic counselling and BRCA testing. Since the late 90s, the department has mainly served the South-Eastern part of Norway. The south-eastern part of Norway contains 2.9 million people, which is a little more than half of the Norwegian population of 5.2 mill.
From around 1995 and onwards, the laboratories performing BRCA analysis used various techniques. Initially, by using techniques such as denaturing gradient gel electrophoresis and sequencing methods, four recurrent BRCA1 mutations were identified in Norwegian families (c.1556del, c.3328_3229del, c.697_698del and c.1016dup) [13]. Eventually other cost-efficient/affordable tests, such as multiplex PCR fragment analysis and sequencing of shorter fragments were used to screen larger groups of individuals, as well as to detect mutations already found in the family. When new frequent mutations were identified these were included in the fragment analysis tests.
Sequencing of BRCA1 and 2 genes has increasingly been offered to our high-risk cancer families since 2000 and 2002 respectively, and MLPA analysis since 2002. Fragment analysis and sequencing/MLPA were used interchangeably in the work-up of these patients until January 2014, when Sanger or high throughput sequencing (HTS) methods have been used combined with MLPA. It should be noted that patients from families with a known genetic mutation have only been tested for this specific mutation except when more than one mutation is suspected. This applied to fourteen families where two mutations in BRCA1 /BRCA2 were identified.
Founder mutation search method
To establish whether the variants found in our cohort were described as founder mutations elsewhere, we used the following strategy: All variants were described with HGVS standard nomenclature (BRCA1 NM_007294.3 and BRCA2 NM_000059.3). A search was performed in Alamut Visual per variant, first with default settings, then adding “founder” to the variant search terms. Alamut searches automatically after all known notations of the variant, mainly in Google. Depending on the search results, the variants were termed either F = Founder, when documentation of this was retrieved, NF = Not founder, when the variant was previously reported but not shown to be a founder anywhere, or NPR = Not previously reported if there were no documents retrievable on the variant. A double check on all variants initially classified as NPR was performed in databases ClinVar, HGMD, UMD, LOVD and BRCA Exchange.
Classification
The original laboratory reports were from different time periods with different routines for variant interpretation and reporting. In order to ensure up to date quality of the variant classification, we reevaluated all mutations reported in the BRCA1/2 carrier population as part of this quality of care study. Variants were interpreted according to the recommendations of the American College of Medical Genetics [19], and ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) using the five-class system: pathogenic (class 5), likely pathogenic (class 4), variant of uncertain significance (class 3), likely benign (class 2), or benign (class 1). A disease-causing mutation was defined as a class 4 or 5 variant according to ACMG/ ENIGMA criteria. The types of mutations for both genes are listed in Table 1, all classified as 4 or 5. The majority of variants were straight forward to classify as they introduce stop or frameshift, or constitute rearrangements or alter splicing. The splicing mutations were either in canonical +/−1 or 2 splice sites or analyzed in functional test by us or others. The missense mutations were identified in the well-known domains, RING and BRCT in BRCA1 and DNA binding domain in BRCA2. Published multifactorial likelihood scores and/ or functional studies were part of the evidence in these cases. Disease-causing missense mutations were found to constitute 9% of BRCA1 and 5% of BRCA2 mutations in this study. Others have suggested that approximately 7% of the load of pathogenic sequence variants in BRCA1 is attributable to missense substitutions [20, 21]. Variants of unknown significance (VUS) were not included in this study.
Table 1.
Types of mutations
| Type of mutation | BRCA1 | BRCA2 |
|---|---|---|
| Frame shift | 49 | 45 |
| Stop | 38 | 25 |
| Rearrangement | 12 | 4 |
| Missense | 11 | 4 |
| Splice variant | 9 | 7 |
| Start loss | 1 | 1 |
| In frame deletion | 0 | 1 |
| 120 | 87 |
For the purpose of this study, a founder mutation was defined as a variant previously reported as such, and this may include common ancestry proven by haplotype studies. A recurrent mutation was defined as a variant to occur in one mutational hot spot as separate events, this term is however used synonymously with frequent variant in many publications. In this study, a frequent mutation was defined as a mutation found in three or more families and subdivided into three categories for systematic purposes. Mutations found in >30 different families each were termed highly frequent, mutations found in 10–30 families were termed moderately frequent. Mutations found in 3–9 families each were termed less frequent. A rare mutation was defined as a mutation found in one or two families. A frequent mutation from any of the three groups may in some cases be considered a founder candidate, depending on e.g. the geographical origin of the families. Any mutation, both frequent and rare in our study, may be known as founder mutations in a specific population.
Mutation frequencies
Throughout this study we have calculated mutation frequencies both as number of mutation carriers per variant, and number of different families per variant. The fraction of mutation carriers carrying the four well-known BRCA1 founder mutations are directly comparable to the numbers obtained in the previous studies done on the subject. The number of families per variant would be indicative of possible new founder mutations, which again may be of separate interest for future studies. A calculation of number of mutation carriers per family was included in the work-up for each variant. Establishing a carrier frequency for the population on the whole was beyond the scope of this study.
Results
The BRCA1 results are shown in Fig. 1 and Table 2, the BRCA2 results are shown in Fig. 2 and Table 3. There were 120 different BRCA1 variants and 87 different BRCA2 variants found among the mutation carriers, 669/981 families had a BRCA1 mutation (68%), and 312/981 had a BRCA2 mutation (32%). Forty-six per cent of the registered BRCA1/2 families (454/981) had a previously known Norwegian founder mutations, identified through the founder search in Alamut. There were five BRCA1 variants and one BRCA2 variant among the six most frequent BRCA1/2 variants (Table 4). These six variants accounted for 47% (1643/3522) of the mutation carriers. In total, 70 % of BRCA1/2 mutation carriers (2466/3522) had a moderately or highly frequent variant (found in more than 10 families). Sixteen per cent (577/3522) had a less frequent variant found in 3–9 families. Fourteen per cent of BRCA1/2 carriers (479/3522) had a rare mutation.
Fig. 1.
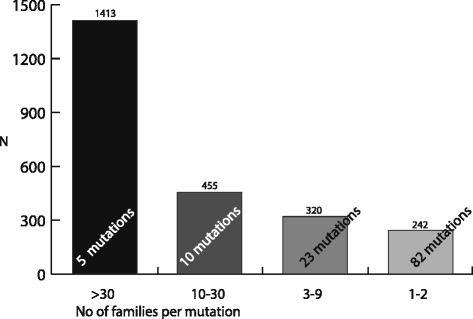
Proportions of BRCA1 mutation carriers vs frequency of mutations (N = 2432)
Table 2.
BRCA1 variants
| No of families | HGVS | Type of mutation | No.of ind. | No. of fam. | Average no of carr./fam | Percentage of carriers/families | Previous reports* | |
|---|---|---|---|---|---|---|---|---|
| >30 | c.1016dup | p.Val340Glyfs*6 | fs | 471 | 111 | 4.2 | 58% | Norwegian, Danish and Swedish |
| c.1556del | p.Lys519Argfs*13 | fs | 399 | 95 | 1413/2430 | Norwegian | ||
| c.3228_3229del | p.Gly1077Alafs*8 | fs | 214 | 45 | Italian, Norwegian | |||
| c.697_698del | p.Val233Asnfs*4 | fs | 182 | 44 | 51% | Norwegian | ||
| c.3178G > T | p.Glu1060* | stop | 147 | 46 | 341/669 | Norwegian | ||
| 10–30 | c.1A > G | p.Met1Val | start codon | 69 | 21 | 3.5 | 19% | Norwegian |
| c.3048_3052dup | p.Asn1018Metfs | fs | 44 | 16 | 455/2430 | Swedish founder | ||
| c.5266dup | p.Gln1756Profs*74 | fs | 30 | 16 | European, Russian | |||
| c.3084_3094del | p.Asn1029Argfs*5 | fs | 43 | 13 | Norwegian | |||
| dup exon 13/c.(4185 + 1_41861)_(4357 + 1_4358–1)dup | p.? | rearr | 41 | 11 | 19% | Norwegian and British | ||
| dup exon 22 /c.(5332 + 1_5333–1)_(5406 + 1_5407–1)del | p.? | rearr | 29 | 11 | 128/669 | Dutch | ||
| c.4745del | p.Asp1582Alafs*19 | fs | 78 | 10 | Norwegian | |||
| c.2351_2357del | p.Ser784Trpfs*6 | fs | 54 | 10 | Norwegian | |||
| del exon 8–13 / c.(441 + 1_442–1)_(4357 + 1_4358–1)del | p.? | rearr | 37 | 10 | British, European founder | |||
| c.3607C > T | p.Arg1203* | stop | 30 | 10 | Greek founder, Belgian, Korean recurrent | |||
| 3–9 | c.1072del | p.Leu358Cysfs*16 | fs | 22 | 8 | 3.2 | 13% | NF |
| c.68_69del | p.Glu23Valfs*17 | fs | 13 | 7 | 320/2430 | Ashkenazi, Polish, Italian, Spanish | ||
| c.4065_4068del | p.Asn1355Lysfs*10 | fs | 12 | 7 | British and German | |||
| c.5047G > T | p.Glu1683* | stop | 39 | 6 | NF | |||
| c.3319G > T | p.Glu1107* | stop | 16 | 8 | 15% | Danish | ||
| c.5075-2A > C | p.? | splice var | 39 | 5 | 101/669 | Norwegian | ||
| c.2475delC | p.Asp825Glufs*21 | fs | 15 | 5 | Swedish and Danish | |||
| c.3700_3704del | p.Val1234Glnfs*8 | fs | 7 | 5 | Rec. Greek, Czech | |||
| c.3331_3334del | p.Gln1111Asnfs*5 | fs | 21 | 4 | Hispanic, Portuguese founder | |||
| c.2591C > G | p.Ser864* | stop | 16 | 4 | NF | |||
| c.3966delA | p.Lys1322Asnfs*3 | fs | 15 | 4 | NF | |||
| del exon 3–16/ c.(80 + 1_81–1)_(4986 + 1_4987–1)del | p.? | rearr | 14 | 4 | NF | |||
| c.130 T > A | p.Cys44Ser | missense | 8 | 4 | NF | |||
| c.1450G > T | p.Gly484* | stop | 18 | 3 | NF | |||
| c.5513 T > G | p.Val1838Gly | missense | 13 | 3 | NF | |||
| c.3756_3759del | p.Ser1253Argfs*10 | fs | 10 | 3 | Recurrent Polish | |||
| c.5251C > T | p.Arg1751* | stop | 10 | 3 | Finnish | |||
| c.1687C > T | p.Gln563* | stop | 6 | 3 | European, Austrian, Slovanian founder | |||
| c.3710del | p.Ile1237Asnfs*27 | fs | 6 | 3 | Danish, Swedish, rec. Polish | |||
| c.4035del | p.Glu1346Lysfs*20 | fs | 6 | 3 | Slovenian, Polish, Latvian, Lithuanian | |||
| c.5309G > T | p.Gly1770Val | missense | 6 | 3 | Moroccan founder | |||
| c.181 T > G | p.Cys61Gly | missense | 5 | 3 | Central and eastern European founder | |||
| c.843_846del | p.Ser282Tyrfs*15 | fs | 3 | 3 | NF | |||
| 1–2 | c.5511G > A | p.Trp1837* | stop | 18 | 2 | 2.4 | 10% | NF |
| c.2869C > T | p.Gln957* | stop | 14 | 2 | 242/2430 | NF (?) | ||
| c.66dupA | p.Glu23Argfs*18 | fs | 12 | 2 | NF | |||
| c.1058G > A | p.Trp353* | stop | 10 | 2 | 15% | NF | ||
| c.5407-25 T > A | p.? | splice var | 9 | 2 | 99/669 | NF | ||
| c.1292dup | p.Leu431Phefs*5 | fs | 8 | 2 | NF | |||
| c.794_795del | p.Ser265Cysfs*21 | fs | 8 | 2 | NF | |||
| c.2558ins356 | p.? | stop | 7 | 2 | NPR | |||
| c.5503C > T | p.Arg1835* | stop | 6 | 2 | Pakistani founder | |||
| c.2681_2682del | p.Lys894Thrfs*8 | fs | 5 | 2 | Scottish | |||
| c.2989_2990dup | p.Asn997Lysfs*4 | fs | 5 | 2 | NF | |||
| c.4689C > G | p.Tyr1563* | stop | 5 | 2 | NF | |||
| c.5153G > C | p.Trp1718Ser | missense | 5 | 2 | NF | |||
| del exon 1–3 / c.(?_1–1)_(134 + 1_135–1)del | p.? | rearr | 4 | 2 | Norwegian founder | |||
| c.1287del | p.Ile429Metfs*12 | fs | 2 | 2 | NPR | |||
| c.3937C > T | p.Gln1313* | stop | 1 | 1 | NF | |||
| c.5095C > T | p.Arg1699Trp | missense | 1 | 2 | NF | |||
| c.3874del | p.Ser1292Leufs*15 | fs | 7 | 1 | Danish founder | |||
| del exon 1–13/ c.(?_1–1)_(4357 + 1_4358–1)del | p.? | rearr | 5 | 1 | Norwegian founder | |||
| c.5534del | p.Glu1346Lysfs*20 | fs | 5 | 1 | NPR | |||
| c.1793 T > G | p.Leu598* | stop | 4 | 1 | NF | |||
| c.115 T > G | p.Cys39Gly | missense | 2 | 1 | Greenlandic/Danish founder | |||
| del exon 5–7/c.(134 + 1_135–1)_(441 + 1_442–1)del | p.? | rearr | 3 | 1 | NF | |||
| del exon 8 c.(441 + 1_442–1)_(547 + 1_548–1)del | p.? | rearr | 3 | 1 | European founder | |||
| c.848 T > A | p.Leu283* | stop | 3 | 1 | NF | |||
| c.929del | p.Gln310Argfs*4 | fs | 3 | 1 | NF | |||
| c.1434_1435del | p.Glu479Lysfs*10 | fs | 3 | 1 | NF | |||
| c.2257dup | p.Ser753Lysfs*9 | fs | 3 | 1 | NPR | |||
| c.3770_3771del | p.Glu1257Glyfs*9 | fs | 3 | 1 | Spanish founder | |||
| c.4612C > T | p.Gln1538* | stop | 3 | 1 | NF | |||
| c.457_458ins21 | p.? | stop | 6 | 2 | NPR | |||
| del exon 18–24/c.(5074 + 1_5075–1)_(5592 + 1_?-1)del | p.? | rearr | 2 | 1 | NPR | |||
| c.1059G > A | p.Trp353* | stop | 2 | 1 | NF | |||
| c.1360_1361del | p.Ser454* | stop | 2 | 1 | Italian | |||
| c.1695dup | p.Lys566Glufs*4 | fs | 1 | 1 | NF | |||
| c.65 T > C | p.Leu22Ser | missense | 2 | 1 | NF | |||
| c.2138C > G | p.Ser713* | stop | 1 | 1 | NF | |||
| c.2389G > T | p.Glu797* | stop | 2 | 1 | NF | |||
| c.2438dup | p.Leu814Thrfs*9 | fs | 2 | 1 | NF | |||
| c.3477_3479delinsC | p.Lys1160Glyfs*4 | fs | 2 | 1 | NF | |||
| c.3689 T > G | p.Leu1230* | stop | 2 | 1 | NF | |||
| c.3835del | p.Ala1279Hisfs*28 | stop | 2 | 1 | NF | |||
| c.4186C > T | p.Gln1396* | stop | 2 | 1 | NF | |||
| c.4484G > A | p.Ala1453Glyfs*10 | splice var. | 2 | 1 | NF | |||
| c.386del | p.Gly129Alafs*34 | fs | 2 | 1 | NPR | |||
| c.4932_4933dup | p.Arg1645Lysfs*14 | fs | 2 | 1 | NF | |||
| c.4972delA | p.Thr1658Profs*19 | fs | 2 | 1 | NPR | |||
| c.4986 + 1G > T | p.? | splice var | 2 | 1 | NF | |||
| c.5407-2A > G | p.? | splice var | 2 | 1 | NF | |||
| c.445G > T | p.Glu149* | stop | 2 | 1 | NPR | |||
| c.510del | p.Ile171Tyrfs*63 | fs | 2 | 1 | NPR | |||
| del exon 11/c.(670 + 1_671–1)_(4096 + 1_4097–1)del | p.? | rearr | 1 | 1 | NPR | |||
| del exon 16/c.(4675 + 1_4676–1)_(4986 + 1_4987–1)del | p.? | rearr | 1 | 1 | NF | |||
| del exon 20–24/c.(5193 + 1_5194–1)_(5592 + 1_?-1)del | p.? | rearr | 1 | 1 | NF | |||
| c.1175_1214del | p.Leu392Glnfs*5 | fs | 1 | 1 | NF | |||
| c.1674dup | p.Gly559Argfs*2 | fs | 1 | 1 | NPR | |||
| c.1823_1826del | p.Lys608Ilefs*3 | fs | 1 | 1 | NF | |||
| c.1961dup | p.Tyr655Valfs*18 | fs | 1 | 1 | NF | |||
| c.2019del | p.Glu673Aspfs*28 | fs | 1 | 1 | NF | |||
| c.2185G > T | p.Glu729* | stop | 1 | 1 | NF | |||
| c.2293G > T | p.Glu765* | stop | 1 | 1 | NF | |||
| c.140G > T | p.Cys47Phe | missense | 1 | 1 | NF | |||
| c.2727_2730del | p.Asn909Lysfs*90 | fs | 1 | 1 | NF | |||
| c.2864C > A | p.Ser955* | stop | 1 | 1 | Hispanic, Californian | |||
| c.188 T > A | p.Leu63* | stop | 1 | 1 | Japanese | |||
| c.2981_2982del | p.Cys994* | stop | 1 | 1 | NF | |||
| c.3005del | p.Asn1002Thrfs*22 | fs | 1 | 1 | NF | |||
| c.213-5 T > A | p.? | splice var | 1 | 1 | NF | |||
| c.3400G > T | p.Glu1134* | stop | 1 | 1 | NF | |||
| c.241C > T | p.Gln81* | stop | 1 | 1 | NF | |||
| c.3544C > T | p.Gln1182* | stop | 1 | 1 | NF | |||
| c.3644_3648del | p.(Asn1215Ilefs*2) | fs | 1 | 1 | NPR | |||
| c.3813dupT | p.(Asn1272*) | stop | 1 | 1 | NF | |||
| c.3817C > T | p.Gln1273* | stop | 1 | 1 | NF | |||
| c.4146_4155dup | p.Ser1386Leufs*8 | fs | 1 | 1 | NF | |||
| c.5074 + 2 T > C | p.? | splice var. | 1 | 1 | NF | |||
| c.5030_5033del | p.Thr1677Ilefs*2 | fs | 1 | 1 | French | |||
| c.5212G > A | p.Gly1738Arg | missense | 1 | 1 | Greek | |||
| c.5193 + 2del | p.? | splice var | 1 | 1 | NF | |||
| c.5434C > G | p.Gly1803Glnfs*11 | splice var. | 1 | 1 | NF | |||
| c.514C > T | p.Gln172* | stop | 1 | 1 | NF | |||
| c.5213G > A | p.Gly1738Glu | missense | 1 | 1 | Danish, Iranian |
*Founders are reported with indication of origin, NF not founder through search, NPR not previously reported
Fig. 2.
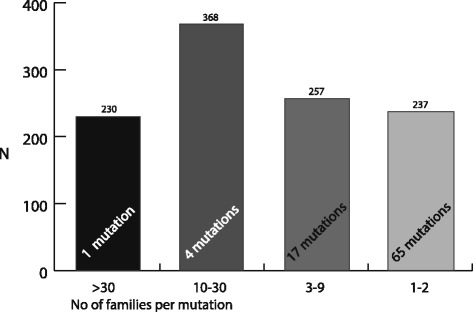
Proportions of BRCA2 mutation carriers vs frequency of mutations (N = 1092)
Table 3.
BRCA2 variants
| No of families | HGVS | Type of mutation | No.of ind. | No. of fam. | Average no of carrier/ fam | Percentage of carriers/families | Previous reports | |
|---|---|---|---|---|---|---|---|---|
| >30 | c.5217_5223del | p.Tyr1739* | stop | 230 | 61 | 3,8 | 21% | NF |
| 230/1092 19% 61/312 |
||||||||
| 10–30 | c.4821_4823delTGAinsC | p.Glu1608Aspfs*6 | fs | 126 | 25 | 3,9 | 34% | NF |
| c.2808_2811del | p.Ala938Profs*21 | fs | 89 | 26 | 368/1092 | Norwegian founder | ||
| c.8331 + 2 T > C | p.? | splice variant | 71 | 29 | Danish founder | |||
| c.3847_3848del | p.Val1283Lysfs*2 | fs | 82 | 14 |
30%
94/312 |
Norwegian, Iranian founder | ||
| 3–9 | dup exon 20/ c.(8487 + 1_8488–1)_(8632 + 1_8633–1)dup | p.? | rearr | 34 | 9 | 3,2 | 23% | NF |
| c.9118-2A > G | p.? | splice variant | 44 | 8 | 257/1092 | Finnish founder, recurrent Polish | ||
| c.5723_5722del | p.Leu1908Argfs*2 | fs | 23 | 8 | NF | |||
| c.2047_2050del | p.Ser683Argfs*46 | fs | 28 | 6 |
26%
80/312 |
NPR | ||
| c.8229_8243del | p.Arg2744_Gly2748del | in frame del | 18 | 5 | NF | |||
| c.5946del | p.Ser1982Argfs*22 | fs | 11 | 5 | Ashkenazi, Hungarian | |||
| c.771_775del | p.Asn257Lysfs*17 | fs | 6 | 5 | Finnish, Icelandic | |||
| c.1905_1909del | Asp635Glufs*15 | fs | 22 | 4 | NF | |||
| c.5576_5573del | p.Ile1859Lysfs*5 | fs | 11 | 4 | NF | |||
| c.6059_6062del | p.Glu2020Valfs*19 | fs | 9 | 4 | NF | |||
| c.9117G > A | p.Val2985Glyfs*4 | splice variant | 5 | 4 | NF | |||
| c.171C > G | p.Tyr57Ter* | stop | 15 | 3 | NF | |||
| c.8177A > G | p.Tyr2726Cys | missense | 9 | 3 | NF | |||
| c.2830A > T | p.Lys944Ter* | stop | 8 | 3 | NF | |||
| c.3847_3848del | p.Val1283Lysfs*2 | fs | 8 | 3 | NF | |||
| c.7069_7070del | p.Leu2357Valfs*2 | fs | 3 | 3 | NF | |||
| c.7480C > T | p.Arg2494* | stop | 3 | 3 | Finnish founder | |||
| 1–2 | del exon 3 | p.? | rearr | 8 | 2 | 3,1 | 22% | NF |
| c.3G > A | p.Met1? | start loss | 6 | 2 | 237/1092 | NF | ||
| c.5157_5161del | p.Asn1719Lysfs*6 | fs | 2 | 2 | 25% | NF | ||
| c.6373dup | p.Thr2125Asnfs*4 | fs | 5 | 2 | 77/312 | NF | ||
| c.6486_6489del | p.Lys2162Asnfs*5 | fs | 2 | 2 | Danish founder | |||
| c.7558C > T | p.Arg2520* | stop | 14 | 2 | NF | |||
| c.7617 + 1G > A | p.? | splice | 2 | 2 | Danish founder | |||
| c.8130del | p.Ser2710Argfs*23 | fs | 9 | 2 | NF | |||
| c.8323dup | p.Met2775Asnfs*7 | fs | 10 | 2 | NF | |||
| c.631 + 4A > G | p.? | splice | 11 | 2 | NF | |||
| c.9154C > T | p.Arg3052Trp | missense | 9 | 2 | NF | |||
| c.9699_9702del | p.Cys3233Trpfs*15 | fs | 12 | 2 | NF | |||
| del exon 19–21/c.(8331 + 1_8332–1)_(8754 + 1_8755–1)del | p.? | rearr | 1 | 1 | NPR | |||
| del exon 20/c.(8487 + 1_8488–1)_(8632 + 1_8633–1)del | p.? | rearr | 1 | 1 | NF | |||
| c.1296_1297del | p.Asn433Glnfs*18 | fs | 1 | 1 | NF | |||
| c.1429del | p.His477Ilefs*8 | fs | 10 | 1 | NPR | |||
| c.1456C > T | p.Gln486* | stop | 5 | 1 | NF | |||
| c.1642C > T | p.Gln548* | stop | 3 | 1 | NF | |||
| c.1658 T > G | p.Leu553* | stop | 4 | 1 | NF | |||
| c.1945C > T | p.Gln649* | stop | 4 | 1 | NF | |||
| c.2636_2637del | p.Ser879* | stop | 2 | 1 | NF | |||
| c.3158 T > G | p.Leu1053* | stop | 1 | 1 | NF | |||
| c.3307_3308dup | p-Leu1103Phefs*2 | fs | 1 | 1 | NPR | |||
| c.3545_3546del | p.Phe1182* | stop | 1 | 1 | NF | |||
| c.3596_3599del | p.Asp1199Valfs*9 | fs | 8 | 1 | NF | |||
| c.3720_3721del | p.Phe1241* | stop | 2 | 1 | NPR | |||
| c.3751dup | p.Thr1251Asnfs*14 | fs | 2 | 1 | NF | |||
| c.171del | p.Tyr57* | stop | 4 | 1 | NPR | |||
| c.3860del | p.Asn1287Ilefs*6 | fs | 1 | 1 | Austrian founder | |||
| c.196C > T | p.Gln66* | stop | 2 | 1 | NF | |||
| c.4095 T > A | p.Cys1365* | stop | 3 | 1 | NF | |||
| c.4258del | p.Asp1420Ilefs*28 | fs | 2 | 1 | Swedish founder | |||
| c.4710del | p.GLu1571Argfs*8 | fs | 1 | 1 | NF | |||
| c.4794_4797del | p.Asn1599Metfs*17 | fs | 1 | 1 | NF | |||
| c.5073dup | p.Trp1692Metfs*3 | fs | 1 | 1 | NF | |||
| c.316 + 1G > T | p.? | splice variant | 5 | 1 | NF | |||
| c.5577del | p.Val1862* | stop | 1 | 1 | NF | |||
| c.5645C > A | p.Ser1882* | stop | 7 | 1 | French founder | |||
| c.5682C > G | p.Tyr1894* | stop | 4 | 1 | NF | |||
| c.6034dup | p.Ser2012Phefs*6 | fs | 3 | 1 | NPR | |||
| c.6084_6088del | p.Glu2029Tyrfs*18 | fs | 5 | 1 | NPR | |||
| c.407del | p.Asn136 Ilefs*16 | fs | 1 | 1 | NF | |||
| c.6611dup | p.Val2205Cysfs*20 | fs | 2 | 1 | NF | |||
| c.6591_6592del | p.Glu2198Asnfs*4 | fs | 3 | 1 | NF | |||
| c.469_470del | p.Lys157Valfs*25 | fs | 3 | 1 | NF | |||
| c.7024C > T | p.Gln2342* | stop | 1 | 1 | NF | |||
| c.517-2A > G | p.? | splice variant | 2 | 1 | NF | |||
| c.7234del | p.Thr2412Leufs*57 | fs | 4 | 1 | NF | |||
| c.7673_7674del | p.Glu2558Valfs*7 | fs | 3 | 1 | NF | |||
| c.7680dup | p.Gln2561Serfs*5 | fs | 1 | 1 | NF | |||
| c.7753G > A | p.Gly2585Arg | missense | 1 | 1 | NF | |||
| c.7829dupT | p.Asp2611Glyfs*7 | fs | 1 | 1 | NF | |||
| c.7878G > C | p.Trp2626Cys | missense | 3 | 1 | NF | |||
| c.7913_7917del | p.Phe2638* | stop | 1 | 1 | Czech founder | |||
| c.8090_8105del | p.Ser2697Lysfs*31 | fs | 2 | 1 | NPR | |||
| c.8396dup | p.Pro2800Thyfs*12 | fs | 9 | 1 | NPR | |||
| c.658_659del | p.Val220Ilefs*4 | fs | 1 | 1 | Lithuanian founder | |||
| c.8878C > T | p.Gln2960* | stop | 1 | 1 | Korean | |||
| c.614delG | p.Ser205Ilefs*6 | fs | 4 | 1 | NPR | |||
| c.9127G > T | p.Glu3043* | stop | 1 | 1 | NF | |||
| c.9227del | p.Gly3076Aspfs*7 | fs | 2 | 1 | NF | |||
| c.9253dup | p.Thr3085Asnfs*26 | fs | 10 | 1 | NF | |||
| c.9382C > T | p.Arg3128* | stop | 1 | 1 | Jewish founder | |||
| c.9403del | p.Leu3135Phefs*28 | fs | 1 | 1 | NF | |||
| c.9523G > T | p.Glu3175* | stop | 3 | 1 | NF |
*Founders are reported with indication of origin, NF not founder through search, NPR not previously reported
Table 4.
BRCA1/2 variants found in more than 30 families
| Gene | Variant | No. of individuals | No. of families | |
|---|---|---|---|---|
| BRCA1 | c.1016dup | p.Val340Glyfs*6 | 471 | 111 |
| BRCA1 | c.1556del | p.Lys519Argfs*13 | 399 | 95 |
| BRCA2 | c.5217_5223del | p.Tyr1739* | 230 | 61 |
| BRCA1 | c.3178G > T | p.Glu1060* | 147 | 46 |
| BRCA1 | c.3228_3229del | p.Gly1077Alafs*8 | 214 | 45 |
| BRCA1 | c.697_698del | p.Val233Asnfs*4 | 182 | 44 |
| Total | 1643 | 403 |
BRCA1
Each of the four well-known founder mutations in BRCA1, c.1556del, c.3328_3229del, c.697_698del and c.1016dup, were found in more than 30 different families each and classified as highly frequent (Table 2, Fig. 1). These four mutations accounted for 52% (1266/2430) of BRCA1 carriers in this study, or 44% of BRCA1 families (295/669). The variant c.1016dup was the most frequent mutation with 471 mutation carriers from 111 families. Together with the fifth highly frequent mutation, c.3178 G > T, also found in more than 30 families, the top five BRCA1 mutations accounted for 58% (1413/2430) of the mutation carriers, or 51% of the families (341/669).
Twenty-seven per cent (33/120) of the variants were classified as moderately frequent (10 variants) and less frequent (23 variants). These accounted for 32% of mutation carriers (775/2430) or 34% of BRCA1 mutation families (229/669).
Sixty-eight per cent (82/120) of the BRCA1 variants were rare. Ten per cent of the BRCA1 mutation carriers (242/2430), 15% of the BRCA1 families (99/669), had one of these mutations.
BRCA2
The single most frequent BRCA2 variant, c.5217_5223del, was found in 230 individuals from 61 different families. This variant accounted for 21% (230 /1092) of BRCA2 carriers, or 19% of families (61/312). It was also the third most prevalent variant when both BRCA1/2 were taken together (Table 4), but it was not found to be reported as a founder from the Alamut search.
Four moderately frequent mutations were found, (c.4821_4823delTGAinsC, c.2808_2811del, c.8331 + 2 T > C and c.3847_3848del), and they accounted for 34% (368/1092) of the BRCA2 mutation carriers, or 30% (94/312) of the families.
Twenty per cent (17/87) of the BRCA2 variants were classified as less frequent, accounting for 23% (257/1092) of the mutation carriers, or 26% (80/312) of the families.
Seventy-five per cent (65/87) of the BRCA2 variants were considered rare, found in 1–2 families each. Twenty-two per cent (237/1092) of the mutation carriers, (25% (77/312) of the families) had one of these rare BRCA2 mutations.
Founder mutation search
Among the variants found in more than ten families each, ten out of fifteen BRCA1 variants and two out of five BRCA2 variants were previously reported as founder mutations in Norway, including the four demonstrated by haplotyping. Another three BRCA1 variants were reported as Norwegian founders, and these were found in the less frequent or rare category. The remaining highly frequent variants were either described as founder mutations in neighbouring/European countries, or previously reported in other countries, but not as founders, which was the case with the two most frequent BRCA2 variants (c.5217_5223del and c.4821_4823delTGAinsC). There were 14 founder mutations among the 23 less frequent mutations in BRCA1 (61%), mainly Central-European, one Norwegian and three Swedish/Danish. There were four founder mutations among the 17 less frequent BRCA2 variants (23.5%), none of them were Norwegian. Seventeen per cent of the rare BRCA1 variants (14/82) and 15.4% of BRCA2 variants (10/65) were previously described as founders. Details on founder mutation origin are listed in Tables 2 and 3.
Variants not previously reported (NPR) were found mainly among the rare variants for both genes. Thirteen BRCA1 and 10 BRCA2 variants (8.2 and 11.5% of variants, respectively) were not previously described in the Alamut search or in available databases. The BRCA2 variant c.2047_2050del, found in six families, was the only frequent variant not previously described.
Discussion
We aimed at describing the BRCA1/2 mutation distribution in the largest genetic clinic in Norway after many years of BRCA testing. Over the last 10 years, the total number of mutation carriers (N = 3522) is almost 2.5-fold and the total number of mutations has almost tripled (N = 207), compared to 2007 when 1300 carriers and 75 distinct mutations were identified [17].
There are three main findings. Firstly, the distribution of both BRCA1 and BRCA2 mutations is quite extreme: A few mutations are very frequent and many mutations are very rare. The four proven BRCA1 founder mutations by Møller et al. in 2001 are still the most common variants among BRCA1 mutation carriers, but these variants account for a smaller proportion of carriers than previously described. Secondly, there is an increasing amount of moderately and less frequent variants in both genes, among which many are considered to be founders. This is especially true for the BRCA2 variant c.5217_522del which is not previously described as a founder, but is shown to be the third most frequent mutation in the BRCA1/2 carrier population as a whole. Thirdly, 71% (148/208) of the BRCA1/2 variants are rare and found in only one or two individuals/families.
Even though the four most common BRCA1 mutations are the exact same in 2016 as in 2001 [13], the proportion of BRCA1 mutation carriers accounted for by the four founders has fallen from 68% in 2001 to 52% in the present study. In the 2001- study, 82 patients who contracted breast cancer prospectively after being recommended breast cancer screening based on their family history, were BRCA1/2 tested. No BRCA2- mutation were found. The patients were included for breast cancer screening based on selection criteria similar to traditional testing criteria. The present study has a retrospective method, and a much higher number of patients compared to the previous study, and any rare mutation will be easier to detect.
As expected when testing more patients, some of the rare/less frequent variants described in 2001 are shown to be frequent, as is the case with the BRCA1 variant c.3178G > T [16]. On the other hand, the BRCA1 variants c.794_795del, c.2558ins356, c.2869C > T and c.5511G > A were all identified in 2001, and have not turned out to be frequent in the patient population over time. It remains to be seen from future testing which of the rare variants in 2016 that remains rare.
The second main finding is that a substantial number of carriers have moderately or less frequent mutations, many of which are founder candidates. The laboratories have, as specified in Method section, offered specific testing for the frequent mutations that have been detected over time. Finding a mutation in more than three families is a liberal but well recognized threshold of suspecting a founder candidate [12, 22]. Both the well-known Icelandic/Finnish founder, BRCA2 c.771_775del, some of the European recurrent mutations, i.e. BRCA1 c.5266dup and, c.181 T > G as well the Ashkenazi Jewish founders are all present in the groups of low or moderately frequent mutations in our study. A recently identified Moroccan BRCA1 variant has been demonstrated in three families at OUH and have been shown to share the same haplotype as in a series of Moroccan patients [23]. Systematic collection of information on geographic or ethnic origin per individual/family was beyond the scope and permits of this study, but was performed in 2007, where it was found to be an apparent geographical connection for some of the frequent mutations in BRCA1 and BRCA2 [17].
The third important finding is that 71% of the BRCA1/2 variants are classified as rare, and that 14% of the mutation carriers in total have one of these rare variants, 10% of BRCA1 carriers and 22% of BRCA2 carriers. Some of these variants are actually reported as founders, mainly from other populations (15 and 17% of variants for BRCA1 and BRCA2 respectively), and some are not previously reported in the available databases (8.2% (BRCA1) and 11.5% (BRCA2)). These NPR variants may possibly represent unique variants in our population, or they are simply not reported to international databases from other laboratories yet.
The amount of rare mutations found in our study may be similar in other countries assumed to have a strong founder effect. In a recent study from Bulgaria, 200 individuals from breast/ovarian cancer families were genotyped with sequencing, and comparable results were found [24]. Two new, and five previously known mutations were identified in BRCA2, while two new and six previously known mutations were identified in BRCA1. In a Danish study on BRCA1/2 founder mutations, a majority of the mutations identified were found one individual or family, which is similar to our study [25]. A rate of 7–13% rare, non-founder mutations has been described in Ashkenazi-Jewish BRCA1/2 carriers [26]. In the Polish population, 10 % of breast/ovarian cancer patients that previously tested negatively for the Polish founder mutations were found to carry other recurrent or founder candidate variants [27]. The questions following from this are what should the indication for BRCA1/2 testing be, and which method for testing will have sufficient sensitivity. Founder mutation testing alone will, according to this, even in founder populations have lower sensitivity than favourable.
To establish the frequencies of rare pathogenic BRCA1/2 mutations is very important due to their significance in cancer prevention [28], also when a broader testing approach for other breast cancer genes with lower penetrance is applied through gene panels [26, 29]. Founder mutations and their effect will dilute in a multi-cultural society as described in this study. If presymptomatic population screening should be discussed in Norway, as it has been piloted for Ashkenazi Jews in the United States, such knowledge is nevertheless crucial [30]. When discussing screening for any disease, rare or common, establishing test sensitivity and specificity is central [31]. If a similar offer of voluntary founder testing in subgroups of the Norwegian society should be planned for, these data can be used to establish the expected false negative rate. On the other hand, if sequencing/MLPA is considered a better choice because of higher detection rate of pathogenic variants, the rate of detecting VUS and the practice of reporting these variants and reevaluation over time must be considered. To establish the frequency of VUS in the patient database was outside the scope of this study, but after the conversion by the laboratory to full gene tests by sequencing combined with MLPA in January 2014, the rate of VUS in diagnostic testing of BRCA1/2 has been 4.9% [32].
Knowing the local mutation spectrum also makes it possible to plan for future epidemiological studies in the larger population, haplotype studies and possibly genotype/phenotype studies. Norwegian founder mutations have previously been considered to have somewhat lower penetrance and lower cancer risk per year than the rarer mutations in the population regarding both breast and ovarian cancer [33]. The issue of possible genotype-phenotype effects in BRCA1/2 mutation carriers have been explored in several studies [22, 34, 35]. Rebbeck et al. presented in 2015 one of the largest studies performed on the subject, confirming the existence of areas with relative variation in breast and ovarian cancer risk. The results await appropriate validation before findings may be transferred to clinical counselling practice.
There are some selection biases due to the changing practice of both patient inclusion and testing over the years. During the first 10 years of BRCA testing in OUH, patients were mainly tested for the known founder mutations. Family members of identified mutation carriers were informed by their relatives of the possibility of predictive testing, and over time, quite large families with founder mutations were identified. An overall larger percentage of carriers than of families for the most frequent mutations illustrate this, as well as the rate of mutation carriers per family (stated in Tables 2 and 3). The rarer mutations in BRCA1 have a larger fraction of families compared to the fraction of mutation carriers, and therefore a lower number of mutation carriers per identified family. The high number of different families per founder mutation may however indicate that this family testing strategy is not the sole reason for the high variant frequency, but rather confirm what is known about these mutations already. The variants are old, present before the historical event of the Bubonic plague in fourteenth Century. The carriers have been the object of high selection i.e. through bottle neck phenomenon, non-random mating/inbreeding, as well as other historical factors favouring establishing large families [13, 16]. Over time the families have grown so large that the descendants loose contact, and hence the number of seemingly unrelated families increase. For BRCA2, the average number of mutation carriers per family is quite similar between the frequent and the rare mutations. This may be due to a true, but weaker founder effect for the most frequent BRCA2 variant, c.5217_5223del, i.e. a younger age of this variant than the BRCA1 founder variants, and hence a true, lower frequency in the population. This may in turn be caused by less selection favouring the mutation, e.g. lower degree of inbreeding, smaller families and other historical factors. However, the numbers may also simply reflect a shorter time span both since the most frequent mutation, c.5217_5223del, was identified in our clinic.
Defining a mutation as rare when identified in two families, and as “less frequent” when found in three families or individuals may seem a bit arbitrary, and even misleading. As shown in Tables 2 and 3, variants found in i.e. three small families consisting of 1–2 persons each may really also represent a rare mutation, and if counted as such it would lead to a higher number of rare mutation carriers. The material represents more than half of the Norwegian population, but are not representative for the nation as such. There are well-known local, founder effects present in both the Western and Northern part of the country that will influence on the national frequencies of founder mutations. Lower inclusion rate of patients especially from Western Norway in the later years may also bias the result presented here towards a lower proportion of these mutations in our patient cohort.
In sum, we find that while the well-known founder effect in Norway is still present, it is weaker than previously described. Several frequent mutations detected over the last 15 years are considered founder candidates, and previously described founders from other populations are also found among rare variants in our population. Due to the significant presence of rare mutations we suggest that in order to identify as many BRCA1/2 mutation positive families as possible one should consider to systematically offer retesting with sequencing and MLPA to individuals and families that have previously only been tested with a limited, founder mutation test. The study also supports the continuation of the introduced testing practice of using sequencing and MLPA as initial test in individuals fulfilling testing criteria. Such a testing practice will over time allow detection of variants, both rare and frequent, that otherwise would be missed. Cost-efficiency of such a test approach will vary among health care systems. However, a similar practice has been shown to be cost- efficient in a recent UK study, especially when allowing healthy mutation positive relatives to be identified before they contract cancer [36].
Conclusions
The mutation spectrum of BRCA1 and BRCA2 mutations in the largest hereditary cancer clinic in Norway is diverse. The four BRCA1 founder mutations identified in 2001, are still the most frequent BRCA1 mutations, but account now for 52% of BRCA1 mutation carriers, compared to 68% in 2001. In total, 46 % of the registered BRCA1/2 families (454/981) had one of the previously known Norwegian founder mutations, identified through the founder search in Alamut. Moreover, several frequent mutations have been identified during the last 15 years, many of which are considered founders in the Norwegian population. Lastly, a majority of mutations are rare, but as a group these rare mutations account for 15% of BRCA1 and 25% of BRCA2 mutation families. The results presented therefore support the current practice of using sequencing and MLPA over limited testing for only founder mutation in our patient population. Only through this strategy will new BRCA1/2 mutations, both rare and frequent be identified. Families and individuals who previously have tested negative for founder mutations should systematically be offered retesting with sequencing and MLPA in order to identify healthy BRCA1/2 carriers and enable them to prevent cancer.
Acknowledgements
We are very grateful for illustration work done by Pernille Frese, Norwegian National Advisory Unit on Women’s Health.
Funding
This project is part of a PhD-grant for Cecilie Heramb (M.D.) given by the Norwegian Women’s Public Health Association/ the Norwegian Foundation for Health and Rehabilitation.
Availability of data and materials
May be made available upon request, but such release of data must also be approved by Data Protection Office.
Abbreviations
- ACMG
American College of Medical Genetics
- BRCA1/2
Breast cancer gene 1 / 2
- BRCT-domain
BRCA1 - C terminus domain
- ClinVar
Public Archive of variants, hosted by National Center of Biotechnology Information
- ENIGMA
Evidence-based Network for the Interpretation of Germline Mutant Alleles
- F
Founder mutation
- HGMD
Human gene mutation database
- HGVS
Human genome variation society
- HTS
High throughput sequencing
- LOVD
Leiden open (Source) variation database
- MLPA
Multiplex ligation-dependent probe amplification
- MRI
Magnetic resonance imaging
- NF
Not founder mutation
- NPR
Not previously reported
- OUH
Oslo University Hospital
- RING-finger domain
Really interesting new gene - finger domain
- UMD/BRCAShare
Universal mutation database - BRCA
- VUS
Variants of unknown significance
Authors’ contributions
CH and LOM designed the study, CH performed the data management work. TW, SL, SLA and CH did the variant searches, TW, SL, SLA classified the variants. All authors contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was carried out in the Section of Hereditary Cancer, Department of Medical Genetics, Oslo University Hospital, OUH, and was approved by the Data Protection Officer at OUH as a quality of care study. All activities fulfilled the requirements of genetic counselling, information and consent stated by the Norwegian Act on Biotechnology, as part of the health service.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Cecilie Heramb, Email: ceherl@ous-hf.no.
Teresia Wangensteen, Email: uxtell@ous-hf.no.
Eli Marie Grindedal, Email: ELIGR@ous-hf.no.
Sarah Louise Ariansen, Email: sarianse@ous-hf.no.
Sheba Lothe, Email: shelot@ous-hf.no.
Ketil Riddervold Heimdal, Email: kheimdal@ous-hf.no.
Lovise Mæhle, Email: LOM@ous-hf.no.
References
- 1.Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2007;96(1):11–15. doi: 10.1038/sj.bjc.6603535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105(11):812–822. doi: 10.1093/jnci/djt095. [DOI] [PubMed] [Google Scholar]
- 4.Edwards SM, Evans DG, Hope Q, Norman AR, Barbachano Y, Bullock S, et al. Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br J Cancer. 2010;103(6):918–924. doi: 10.1038/sj.bjc.6605822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012;107(12):2005–2009. doi: 10.1038/bjc.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: results from a follow-up study. Br J Cancer. 2014;110(2):530–534. doi: 10.1038/bjc.2013.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304(9):967–975. doi: 10.1001/jama.2010.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finch A, Metcalfe KA, Chiang J, Elit L, McLaughlin J, Springate C, et al. The impact of prophylactic salpingo-oophorectomy on quality of life and psychological distress in women with a BRCA mutation. Psycho-Oncology. 2013;22(1):212–219. doi: 10.1002/pon.2041. [DOI] [PubMed] [Google Scholar]
- 9.Ludwig KK, Neuner J, Butler A, Geurts JL, Kong AL. Risk reduction and survival benefit of prophylactic surgery in BRCA mutation carriers, a systematic review. Am J Surg. 2016;212(4):660–669. doi: 10.1016/j.amjsurg.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Kotsopoulos J, Huzarski T, Gronwald J, Singer CF, Moller P, Lynch HT, et al. Bilateral Oophorectomy and breast cancer risk in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst. 2017;109(1). doi: 10.1093/jnci/djw177. [DOI] [PMC free article] [PubMed]
- 11.Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i40–ii4. doi: 10.1093/annonc/mdw094. [DOI] [PubMed] [Google Scholar]
- 12.Janavicius R. Founder BRCA1/2 mutations in the Europe: implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010;1(3):397–412. doi: 10.1007/s13167-010-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moller P, Heimdal K, Apold J, Fredriksen A, Borg A, Hovig E, et al. Genetic epidemiology of BRCA1 mutations in Norway. Eur J Cancer (Oxford, England: 1990) 2001;37(18):2428–2434. doi: 10.1016/S0959-8049(01)00299-4. [DOI] [PubMed] [Google Scholar]
- 14.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science (New York, NY) 2003;302(5645):643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 15.Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007;7(12):937–948. doi: 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 16.Moller P, Borg A, Heimdal K, Apold J, Vallon-Christersson J, Hovig E, et al. The BRCA1 syndrome and other inherited breast or breast-ovarian cancers in a Norwegian prospective series. Eur J Cancer (Oxford, England: 1990) 2001;37(8):1027–1032. doi: 10.1016/S0959-8049(01)00075-2. [DOI] [PubMed] [Google Scholar]
- 17.Moller P, Hagen AI, Apold J, Maehle L, Clark N, Fiane B, et al. Genetic epidemiology of BRCA mutations--family history detects less than 50% of the mutation carriers. Eur J Cancer (Oxford, England: 1990) 2007;43(11):1713–1717. doi: 10.1016/j.ejca.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 18.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29(11):1342–1354. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81(5):873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen HR, Nilbert M, Petersen J, Ladelund S, Thomassen M, Pedersen IS, et al. BRCA1/BRCA2 founder mutations and cancer risks: impact in the western Danish population. Familial Cancer. 2016;15(4):507–512. doi: 10.1007/s10689-016-9875-7. [DOI] [PubMed] [Google Scholar]
- 23.Quiles F, Teule A, Martinussen Tandstad N, Feliubadalo L, Tornero E, Del Valle J, et al. Identification of a founder BRCA1 mutation in the Moroccan population. Clin Genet. 2016;90(4):361–365. doi: 10.1111/cge.12747. [DOI] [PubMed] [Google Scholar]
- 24.Dodova RI, Mitkova AV, Dacheva DR, Hadjo LB, Vlahova AI, Hadjieva MS, et al. Spectrum and frequencies of BRCA1/2 mutations in Bulgarian high risk breast cancer patients. BMC Cancer. 2015;15:523. doi: 10.1186/s12885-015-1516-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomassen M, Hansen TV, Borg A, Lianee HT, Wikman F, Pedersen IS, et al. BRCA1 and BRCA2 mutations in Danish families with hereditary breast and/or ovarian cancer. Acta Oncol (Stockholm, Sweden) 2008;47(4):772–777. doi: 10.1080/02841860802004974. [DOI] [PubMed] [Google Scholar]
- 26.Rosenthal E, Moyes K, Arnell C, Evans B, Wenstrup RJ. Incidence of BRCA1 and BRCA2 non-founder mutations in patients of Ashkenazi Jewish ancestry. Breast Cancer Res Treat. 2015;149(1):223–227. doi: 10.1007/s10549-014-3218-x. [DOI] [PubMed] [Google Scholar]
- 27.Kluska A, Balabas A, Paziewska A, Kulecka M, Nowakowska D, Mikula M, et al. New recurrent BRCA1/2 mutations in polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med Genet. 2015;8:19. doi: 10.1186/s12920-015-0092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King MC, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker award. JAMA. 2014;312(11):1091–1092. doi: 10.1001/jama.2014.12483. [DOI] [PubMed] [Google Scholar]
- 29.Walsh T, Mandell JB, Norquist BM, Casadei S, Gulsuner S, Lee MK, et al. Genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi Jewish women. JAMA Oncol. 2017; [DOI] [PMC free article] [PubMed]
- 30.Wiesman C, Rose E, Grant A, Zimilover A, Klugman S, Schreiber-Agus N. Experiences from a pilot program bringing BRCA1/2 genetic screening to theUS Ashkenazi Jewish population. Genet Med. 2017;19(5):529-36. [DOI] [PubMed]
- 31.Andermann A, Blancquaert I, Beauchamp S, Dery V. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ. 2008;86(4):317–319. doi: 10.2471/BLT.07.050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grindedal EM, Heramb C, Karsrud I, Ariansen SL, Maehle L, Undlien DE, et al. Current guidelines for BRCA testing of breast cancer patients are insufficient to detect all mutation carriers. BMC Cancer. 2017;17(1):438. doi: 10.1186/s12885-017-3422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moller P, Maehle L, Engebretsen LF, Ludvigsen T, Jonsrud C, Apold J, et al. High penetrances of BRCA1 and BRCA2 mutations confirmed in a prospective series. Hered Cancer Clin Pract. 2010;8(1):2. doi: 10.1186/1897-4287-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA. 2015;313(13):1347–1361. doi: 10.1001/jama.2014.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006;98(23):1694–1706. doi: 10.1093/jnci/djj465. [DOI] [PubMed] [Google Scholar]
- 36.Slade I, Hanson H, George A, Kohut K, Strydom A, Wordsworth S, et al. A cost analysis of a cancer genetic service model in the UK. J Commun Genet. 2016;7(3):185–194. doi: 10.1007/s12687-016-0266-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
May be made available upon request, but such release of data must also be approved by Data Protection Office.