

Abstract
Background and Purpose
Cerebral amyloid angiopathy (CAA) is a degenerative disorder characterized by amyloid-β (Aβ) deposition in the blood–brain barrier (BBB). CAA contributes to injuries of the neurovasculature including lobar hemorrhages, cortical microbleeds, ischemia, and superficial hemosiderosis. We postulate that CAA pathology is partially due to Aβ compromising the BBB.
Methods
We characterized 19 patients with acute stroke with “probable CAA” for neurovascular pathology based on MRI and clinical findings. Also, we studied the effect of Aβ on the expression of tight junction proteins and matrix metalloproteases (MMPs) in isolated rat brain microvessels.
Results
Two of 19 patients with CAA had asymptomatic BBB leakage and posterior reversible encephalopathic syndrome indicating increased BBB permeability. In addition to white matter changes, diffusion abnormality suggesting lacunar ischemia was found in 4 of 19 patients with CAA; superficial hemosiderosis was observed in 7 of 9 patients. Aβ40 decreased expression of the tight junction proteins claudin-1 and claudin-5 and increased expression of MMP-2 and MMP-9. Analysis of brain microvessels from transgenic mice overexpressing human amyloid precursor protein revealed the same expression pattern for tight junction and MMP proteins. Consistent with reduced tight junction and increased MMP expression and activity, permeability was increased in brain microvessels from human amyloid precursor protein mice compared with microvessels from wild-type controls.
Conclusions
Our findings indicate that Aβ contributes to changes in brain microvessel tight junction and MMP expression, which compromises BBB integrity. We conclude that Aβ causes BBB leakage and that assessing BBB permeability could potentially help characterize CAA progression and be a surrogate marker for treatment response.
Keywords: amyloid-β, blood–brain barrier, cerebral amyloid angiopathy, neuroimaging
Cerebral amyloid angiopathy (CAA) is a degenerative neurovascular disease that affects most patients with Alzheimer’s disease (AD) and approximately 30% of elderly people.1 The main characteristic of CAA is deposition of amyloid-β (Aβ) proteins in leptomeningeal and cortical blood vessels in the brain that contribute to cortical cerebral brain hemorrhage. Because CAA is a disease of the elderly, it overlaps with a variety of cardiovascular risk factors (eg, hypertension, diabetes, hypercholesterolemia) that could contribute to CAA pathology, in particular white matter lesions.2,3 Recent findings suggest that elevated levels of 2 transcription factors, serum response factor and myocardin, contribute to CAA phenotype. Both serum response factor and myocardin regulate differentiation of mural vascular cells in response to hypoxia and lead to the development of CAA and hypercontractile arterial phenotype with brain hypoperfusion in patients with AD and mouse models, thus aggravating white matter lesions.3,4 Cortical cerebral brain hemorrhage is the main criterion for CAA diagnosis and causes and/or contributes to neurodegeneration with dementia and cognitive impairment that overlaps with AD. However, CAA also exhibits acute phases with neurovascular injury that show symptoms ranging from headache, weakness, sensory loss, visual changes, or speech problems.2
CAA is diagnosed according to the Boston criteria that distinguish 4 categories.5 Diagnoses of “definite CAA” and “probable CAA with supporting pathological evidence” require histopathologic evidence from postmortem tissue or evidence of amyloid deposition in pathological tissue in addition to clinical criteria. Diagnosis of “probable CAA” requires clinical and radiological evidence showing at least 2 lobar hemorrhagic lesions due to CAA. The same criteria apply to “possible CAA,” but radiological evidence of only 1 hemorrhage is required.
A reliable CAA diagnosis solely based on neurological signs and cognition is difficult. However, MRI with gradient-echo sequences now allows detection of small cortical and subcortical hemorrhages and thus, diagnosis of different CAA phenotypes including hemosiderosis (SH), acute cerebral ischemia, and superficial and posterior reversible encephalopathic syndrome (PRES). For the diagnosis of the latter, diffusion-weighted imaging and fluid-attenuated inversion recovery imaging (FLAIR)6–9 are required.
In the present study, we used MRI to classify CAA in 19 patients who were admitted to our stroke unit with acute stroke symptoms, and phenotyped these patients with CAA based on symptoms of neurovascular damage, that is, blood–brain barrier (BBB) leakage. Recent evidence suggests that BBB leakage observed in patients with CAA is, at least in part, due to Aβ accumulation in cerebral microvessels.10,11 To test this hypothesis, we performed additional experiments in which we exposed isolated rat brain microvessels to human Aβ40 and determined the effect of Aβ on proteins of the tight junction (TJ) complex and on matrix metalloproteases (MMPs) that are responsible for degradation of the microvessel basal membrane. We also measured barrier integrity in isolated brain microvessels from 9-month-old transgenic human amyloid precursor protein (hAPP)-overexpressing mice (Tg2576) that are a well-established CAA animal model.12,13
Methods
Animals
Animal protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee and were in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International regulations and the US Department of Agriculture Animal Welfare Act. Male CD IGS Sprague-Dawley rats (275–300 g) were from Charles River Laboratories (Wilmington, MA). Male transgenic hAPP mice (Tg2576 strain, 129S6.Cg-Tg[APPSWE] 2576Kha) and wild-type mice from Taconic Farms (Germantown, NY) were 9 months old at the time of the experiments. Animals were housed under controlled conditions (23°C, 35% relative humidity, 12-hour dark/light cycle) with free access to water and food.
Chemicals
ZO-1, occludin, claudin-1, and claudin-5 antibodies were from Invitrogen (Carlsbad, CA). MMP-2, MMP-9, β-actin antibodies, and MMP-9 protein were from Abcam (Cambridge, MA). Human Aβ40 was from EMD Chemicals (Gibbstown, NJ).
Brain Microvessel Isolation
Brain microvessels were isolated as described previously.14,15 Animals were euthanized with CO2and decapitated. Brains were removed, dissected, and homogenized in phosphate-buffered saline. Ficoll (Sigma, St Louis, MO) was added to a final concentration of 15% and the homogenate was centrifuged (5800 g, 20 minutes, 4°C). After resuspending the pellet in 1% bovine serum albumin, the suspension was passed over a glass bead column. Microvessels were collected in 1% bovine serum albumin, washed with phosphate-buffered saline, and used for experiments.
Brain Microvessel Membrane Isolation
Brain microvessels were homogenized in lysis buffer (Sigma) containing Complete protease inhibitor (Roche, Mannheim, Germany).14,15 Samples were centrifuged at 10 000 g (15 minutes, 4°C) and the resulting supernatants were centrifuged at 100 000 g (90 minutes, 4°C). Brain microvessel membranes were resuspended and stored at −80°C.
Western Blotting
Brain microvessel membranes were analyzed by Western blotting as described previously.14,16 Western blots were performed with the Invitrogen NuPage Bis-Tris electrophoresis/blotting system (Invitrogen, Carlsbad, CA). After protein transfer, membranes were blocked and incubated with primary antibody. Membranes were washed and incubated with horseradish peroxidase-conjugated ImmunoPure secondary IgG (1:15 000; Pierce, Rockford, IL). Proteins were detected with SuperSignal West Pico Chemoluminescent Substrate (Pierce) and visualized using a BioRad Gel Doc 2000system (BioRad, Hercules, CA). Western blots were quantitated by densitometric analysis using QuantityOne 1-D Version 4.6.5 software (BioRad Laboratories). Data were normalized to β-actin loading controls.
Texas Red Efflux
Microvessels were transferred to glass cover slips and loaded with 2 μmol/L Texas red (sulforhodamin101-free acid, 1 hour, 25°C). After washing with phosphate-buffered saline, Texas red efflux from microvessels was monitored with confocal microscopy (Nikon C1, Nikon TE2000 inverted microscope, 40× oil objective, NA 1.3, 543 nm HeNe laser; Nikon Instruments, Melville, NY). Mannitol (100 mmol/L) was used as a positive control for barrier opening and maximum permeability marker. For each treatment, 7 confocal images were acquired and luminal fluorescence was measured using Image J (National Institutes of Health, Bethesda, MD).17 First-order Texas red efflux rate constants were calculated by nonlinear regression (Prism 4; GraphPad Software, San Diego, CA).
Zymography
Brain capillary lysate was incubated with Gelatin Sepharose 4B (GE Healthcare, Piscataway, NJ) for 90 minutes. After centrifugation (500 g, 2 minutes) and washing with phosphate-buffered saline/ Tween20, MMPs were eluted with Novex Tris-Glycine SDS sample buffer (1×). The eluate was loaded on Novex 10% Zymogram gelatin gels (Invitrogen, Carlsbad, CA) and electrophoresis was performed. Gels were stained with BioRad Coomassie Brilliant Blue R-250 staining solution and clear bands indicating protease activity were visualized using a BioRad Gel Doc 2000 system (BioRad, Hercules, CA). Band intensity representing MMP activity was determined by densitometric analysis.
Patient Selection and Phenotyping
Our patient study (study period: June 2010 to March 2011) was approved by the University of Regensburg ethics committee according to the declarations of Helsinki (1995) and Tokyo (1998). All patients admitted to the study were identified in the Stroke Unit of the Bezirksklinikum Regensburg, University of Regensburg, Germany, and underwent routine cerebral CT. Patients presented with atypical intracerebral hemorrhage or SH with symptoms attributed to either direct brain damage or secondary symptoms such as focal epileptic seizures. Patients >55 years with atypical or multiple hemorrhages were screened for CAA. Risk factors for white matter disease, particularly hypertension and diabetes, were documented and classified.18
Magnetic Resonance Imaging
MRI was performed using a 1.5-T MR system (Siemens Symphony, Erlangen, Germany). The following MR sequences were applied: axial T2, T1, FLAIR (repetition time 7530, inversion time 2300, echo time 110), diffusion-weighted imaging, apparent diffusion coefficient, high-resolution sagittal magnetization prepared rapid gradient echo-T1 (MPRAGE-T1), time-of-flight MR angiography, and axial T2* fast low-angle shot sequences (FLASH). The MRI sequences had 21 slices of 5-mm thickness and an interslice gap of 1 mm with an in-plane voxel size of 1×1 and field of view of 250 mm. Sequence details included: axial T2* FLASH=800, echo time=26 ms, and flip angle=20°; FLAIR echo time=110 ms, repetition time=7530 ms, inversion time=2300 ms, and flip angle=180°; diffusion-weighted imaging repetition time=4900 ms, echo time=108 ms, b-value=1000 and flip angle=90°″ T1 pre-/postcontrast repetition time=511 ms, echo time=12 ms, and flip angle=90°. Additional sequences included venous time-of-flight MR angiography and postcontrast MRI (0.01 mmol/kg, Magnevist; Schering, Berlin, Germany) and were applied in cases of contrast agent contraindications or when considered appropriate by the neuroradiologist.
The following criteria were used for MRI image analysis. Using a single reader (F.S.) from FLAIR sequences, white matter changes (WMCs) were categorized into 3 classes according to Verdelho et al19: “mild” WMCs were defined as single lesions <10 mm and/or areas of grouped lesions <20 mm in diameter; “moderate” WMCs were defined as single lesions between 10 and 20 mm and/or areas of grouped lesions >20 mm in diameter; “severe” WMCs were defined as single lesions or confluent areas of hyperintensity ≥20 mm in diameter. Microbleeds were only diagnosed when >2 mm and <10 mm in diameter and localized in cortical/ corticosubcortical regions excluding hemorrhage in basal ganglia, thalamus, pons, or cerebellum. SH was diagnosed when located distal to lobar hemorrhage. PRES was diagnosed from initial FLAIR images when lesions had disappeared on follow-up examinations. Contrast-enhancing lesions suggesting BBB leakage were diagnosed using T1 and postcontrast T1 sequences.
Statistical Analysis
Data are presented as mean± SEM. A 2-tailed unpaired Student t test was used to evaluate differences between groups. Two-way analysis of variance was performed for Texas red efflux rates. Differences were considered statistically significant when P<0.05. Due to the lack of a standardized design and a control patient group, descriptive analysis was performed for the clinical study.
Results
CAA and BBB Dysfunction
Over a period of 1 year, 19 patients fulfilled the criteria for “probable CAA” (9 female, 10 male, average age: 75 years; age range: 61–84 years; 19 patients represent approximately 3% of all admissions to our unit within 12 months). No patient was diagnosed with “definitive or probable CAA with supporting evidence.” Details of patient data are summarized in the Table.
Table.
Summary of Patient Data
| Patient No. | Age, y | Sex | Hypertension | Diabetes Mellitus | Microbleeds | Lobar Bleeds | Additional MRI Findings | WMC | Apolipoprotein E | Additional Information |
|---|---|---|---|---|---|---|---|---|---|---|
| 1* | 84 | F | − | + | >5 | >3 | SH, BBB leakage | Severe | N/P | Rapid-onset dementia, malignant course, angiography negative |
| 2 | 61 | M | + | + | 11 | 2 | 2 acute lacunar ischemia | Severe | N/P | … |
| 3 | 74 | M | + | − | 3 | 1 | Acute lacunar ischemia | Mild | ε3/ε4 | Focal seizures |
| 4 | 73 | F | + | − | 3 | 0 | SH, PRES | Severe | ε3/ε4 | Angiography negative, focal seizures, |
| 5 | 78 | M | + | − | >20 | 1 | SDH | Mild | N/P | Mild cognitive impairment, incidental cerebral and abdominal aneurysm |
| 6* | 71 | M | + | + | >20 | >2 | PRES, BBB leakage | Severe | ε3/ε3 | PRES, BBB leakage Asymptomatic, occipital bleeds/WMC, angiography negative |
| 7 | 75 | M | − | − | >7 | 1 | SH | Severe | N/P | Acute and old severe SH, mild cognitive impairment, complex partial seizures |
| 8 | 78 | F | − | + | 5 | 0 | SH | Mild | ε3/ε3 | Mild cognitive impairment |
| 9 | 85 | F | − | − | >20 | 1 | … | Mild | ε3/ε4 | Occipital bleeds/WMC |
| 10 | 70 | M | + | − | >40 | 0 | … | Severe | N/P | Microbleeds cortical and subcortical, mild hypertension, dementia >7 y |
| 11 | 83 | F | − | − | 0 | 2 | SDH | Moderate | N/P | … |
| 12 | 78 | M | − | − | 2 | 0 | SH | Mild | ε2/ε3 | Focal seizures, additional old basal ganglia hemorrhage |
| 13 | 84 | F | + | − | 0 | 2 | … | Mild | N/P | SH due to severe lobar cortical hemorrhage |
| 14 | 74 | M | + | − | >20 | 3 | … | Moderate | ε3/ε4 | Generalized seizure, status postembolic ischemia due to AF |
| 15 | 66 | M | + | − | 8 | 0 | Acute lacunar infarct | Moderate | ε2/ε4 | Mild cognitive impairment, angiography negative |
| 16 | 80 | F | + | − | 1 | 3 | … | Moderate | N/P | SH due to severe lobar cortical hemorrhage |
| 17 | 65 | F | + | − | 8 | … | Acute lacunar infarct | Mild | N/P | Chemotherapy due to mamma carcinoma |
| 18 | 81 | M | − | − | 7 | 2 | SH | Moderate | N/P | … |
| 19 | 74 | F | + | − | 12 | 2 | SH | Mild | N/P | AF, malignant melanoma, rapid CAA |
WMCs indicates white matter changes; F, female; M, male; SH, superficial hemosiderosis; BBB, blood– brain barrier; PRES, posterior reversible encephalopathic syndrome; SDH, subdural hematoma; N/P, not performed; CAA, cerebral amyloid angiopathy.
Patient 1 is represented in Figure 1; Patient 6 is represented in Figure 2.
Twelve of 19 patients (63%) had a history of hypertension. At the time of discharge, 18 out of 19 patients (95%) were on antihypertensive medication to treat blood pressure-induced cerebral hemorrhage or to minimize the risk of hemorrhage recurrence. Four patients were diabetic. Six patients were diagnosed with severe WMCs, 2 of which were identified with PRES and reversible contrast-enhancing lesions due to BBB leakage. Five other patients were diagnosed with moderate WMCs; the remaining 8 patients had mild WMCs.
The most common findings within the patient population included lobar hemorrhage in 13 of 19 (68%) patients, cortico/corticosubcortical microbleeds in 17 of 19 (89%) patients, SHs in 7 of 19 (37%) patients, acute lacunar ischemic infarcts at atypical sites in 4 of 19 (21%) patients, and both PRES and reversible BBB leakage in 2 of 19 (10%) patients (Figures 1 and 2). In these 2 patients we also observed severe WMCs.
Figure 1.

Imaging data of Patient 1. A, Day 19 after initial presentation: cranial CT showing left frontal small subarachnoid hemorrhage. B, Day 22: postcontrast T1-MRI showing left frontal and small left parietal area of contrast enhancement. C, Day 28: diffusion-weighted imaging (upper) and apparent diffusion coefficient (lower) showing small right cerebellar infarct. D, Day 28: postcontrast T1-MRI showing new area of contrast enhancement, whereas left frontal lesion disappeared.
Figure 2.

Imaging data for Patient 6 with CAA and PRES. A, MRI with T2* FLASH sequences showing multiple microhemorrhages. B, Subtraction image demonstrating BBB leakage around a small micro-hemorrhage. C, FLAIR image with occipital edema. D, FLAIR image 2 months later with new left parietal and no occipital edema. CAA indicates cerebral amyloid angiopathy; PRES, posterior reversible encephalopathic syndrome; FLASH, fast low-angle shot; BBB, blood–brain barrier; FLAIR, fluid-attenuated inversion recovery.
Human Aβ40 Decreases TJ Proteins and Increases MMPs
To test the hypothesis that Aβ compromises BBB integrity, we exposed isolated rat brain microvessels to human Aβ40 (hAβ40) for 6 hours and determined protein expression of 4 major TJ proteins and 2 matrix-degrading MMPs by Western blotting of microvessel membranes. Figure 3A shows that hAβ40 exposure decreased claudin-1 and claudin-5 protein expression in brain microvessel membranes in a concentration-dependent manner (claudin-1: control: 100%, 10 nmol/L Aβ: 115%, 50 nmol/L Aβ: 96%, 100 nmol/L Aβ: 69%; claudin-5: control: 100%, 10 nmol/L Aβ: 118%, 50 nmol/L Aβ: 92%, 100 nmol/L Aβ: 81%; determined by densitometric analysis; data are normalized to β-actin-loading controls). Protein expression of ZO-1 and occludin remained unchanged. In contrast, expression of MMP-2 and MMP-9 was elevated with increasing hAβ40 concentration (Figure 3B; MMP-2: control: 100%, 10 nmol/L Aβ: 175%, 50 nmol/L Aβ: 224%, 100 nmol/L Aβ: 194%; MMP-9: control: 100%, 10 nmol/L Aβ: 120%, 50 nmol/L Aβ: 158%, 100 nmol/L Aβ: 163%). Consistent with this, zymography revealed a 2.4-fold increase of MMP-9 activity in brain microvessels treated with hAβ40 (Figure 3C; control: 100%, hAβ40: 239%±72%); MMP-2 activity was undetectable by zymography.
Figure 3.

Aβ effects on tight junction (TJ) proteins and MMPs in brain microvessels. Western blots showing concentration-dependent (A) reduction of the TJ proteins claudin-1 and claudin-5 and (B) increase of the matrix metalloproteases MMP-2 and MMP-9 in rat brain microvessels exposed to human Aβ40 for 6 hours. Expression of the TJ proteins ZO-1 and occludin remained unchanged. C, Zymogram showing increased MMP-9 activity in isolated brain microvessels that were exposed to 100 nmol/L Aβ40 for 6 hours. Aβ indicates amyloid-β.
We also analyzed TJ protein and MMP expression in brain microvessels from 9-month-old transgenic hAPP mice (Tg2576 strain; hAPP gene with Swedish mutation). hAPP mice overexpress hAPP resulting in Aβ overproduction. At 9 months, hAPP mice exhibit substantial cerebrovascular hAβ accumulation and are therefore a model for CAA.20 Consistent with our data from rat brain microvessels, claudin-1 and claudin-5 expression was decreased in microvessels from hAPP mice compared with microvessels from wild-type mice; ZO-1 and occludin expression were unchanged (Figure 4). Densitometric analysis revealed that claudin-1 expression was decreased by 84%±10% and claudin-5 expression was decreased by 43%±1%. As in our Aβ experiments with microvessels, MMP-2 and MMP-9 expression was increased in microvessels from transgenic hAPP mice compared with those from wild-type mice by 90%±12% and 145%±7%, respectively.
Figure 4.
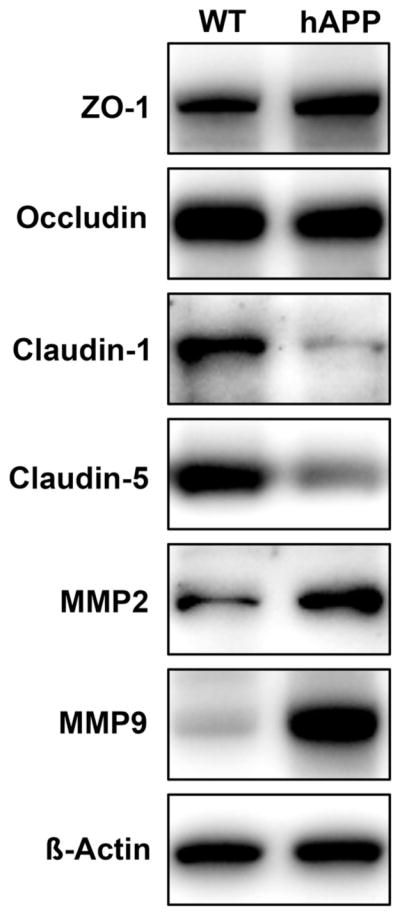
TJ proteins and MMPs in brain microvessels from hAPP mice. Western blots showing reduction of claudin-1 and claudin-5 and increase of MMP-2 and MMP-9 in brain microvessels from hAPP mice. Expression of ZO-1 and occludin remained unchanged. TJ indicates tight junction; MMP, matrix metalloprotease; hAPP, human amyloid precursor protein.
Our results demonstrate that hAβ40 reduces expression of 2 major TJ proteins and increases expression of 2 matrix-degrading enzymes in brain microvessels ex vivo and in brain microvessels of hAPP mice in vivo. These findings suggest that Aβ triggers BBB leakage observed in CAA.
Increased BBB Permeability in Transgenic hAPP Mice
To test the extent to which decreased claudin-1 and claudin-5 expression and increased MMP-2 and MMP-9 expression affect BBB integrity, we measured the permeability of isolated brain microvessels from wild-type and hAPP mice using a previously established assay.15 In this assay, isolated brain microvessels are loaded in a solution of fluorescent Texas red dye (641 Da, 2 μmol/L, 1 hour). After washing with buffer, Texas red efflux from microvessel lumens is monitored by live cell imaging using confocal microscopy; afterward, luminal Texas red fluorescence is quantitated using image analysis software.
As previously described, we used a 100-mmol/L hypertonic mannitol solution as a positive control to osmotically increase microvessel permeability.17 Figure 5 shows that Texas red efflux from microvessels from hAPP mice was faster compared with efflux from microvessels from wild-type mice; addition of mannitol increased Texas red efflux to the maximum effect. Texas red efflux rates over the entire time course were calculated as 0.265±0.04 minutes−1 (wild-type), 0.397±0.05 minutes−1 (hAPP; P<0.001 compared with wild-type), 0.336±0.05 minutes−1 (wild-type with mannitol; P<0.001 compared with wild-type), and 0.356±0.03 minutes−1 (hAPP with mannitol; P<0.001 compared with wild-type). Thus, Texas red efflux in microvessels from hAPP mice was 1.5-fold that of microvessels from wild-type mice indicating higher permeability. Note that luminal Texas red fluorescence in microvessels from hAPP mice did not reach the same levels as in microvessels treated with mannitol (positive control for maximum barrier opening). This observation argues for barrier leakage rather than full disruption.
Figure 5.

Permeability of brain microvessels from hAPP mice. Efflux of the fluorescence dye Texas red (TR) was monitored over 60 minutes in microvessels from wild-type and hAPP mice with/without mannitol used as positive control for barrier opening. Data are mean±SEM for 7 brain microvessels per time point from 1 microvessel isolation from 20 mice; SEM are in the range 3.1 to 11.4. Shown are arbitrary units (0–255). First-order efflux rates were calculated using nonlinear regression. hAPP indicates human amyloid precursor protein.
Discussion
CAA is a multifaceted disorder resulting from Aβ deposition in brain microvessels that causes injury to the neurovascular unit especially in the elderly. Here, we demonstrate in brain microvessels from rats that hAβ40 decreases expression of the TJ proteins claudin-1 and claudin-5 and increases expression and activity of the matrix-degrading enzymes MMP-2 and MMP-9. We confirmed our results in microvessels from transgenic hAPP mice that overproduce human Aβ. Consistent with these findings, we show increased Texas red efflux in brain microvessels from hAPP mice compared with microvessels from wild-type mice.
Our findings suggest that Aβ contributes to loss of BBB integrity that could be responsible for BBB dysfunction, cognitive decline observed in patients with CAA, and additional cerebrovascular pathology including SH, PRES, lacunar cerebral ischemia, and BBB leakage. Indeed, in a group of 19 patients with “probable CAA,” we identified 2 patients with BBB leakage for MRI contrast agents, and 1 of the 2 patients already presented with PRES 10 months earlier. Both contrast leakage and PRES are likely due to increased BBB permeability, which underscores the relevance of our animal data.
Aβ Contributes to Loss of BBB Integrity
The effect of Aβ on BBB integrity has been studied in several cell culture models. Gonzalez-Velasquez et al showed that treatment of cultured human brain endothelial cells with 2.5 to 10 μmol/L Aβ40 triggered the TJ protein ZO-1 to retreat from the plasma membrane, which was accompanied by decreased transendothelial electric resistance.11 Marco and Skaper demonstrated that exposing rat brain endothelial cells to 20 μmol/L Aβ42 triggered ZO-1 and claudin-5 relocation from the plasma membrane, a decrease in occludin expression, and an increase in claudin-1 expression.21 Tai et al demonstrated that Aβ40 activated microtubule-associated protein kinase, which decreased occludin expression and increased permeability in human brain endothelial cell cultures, whereas claudin-5 and ZO-1 remained unchanged.22 Carrano et al analyzed postmortem CAA patient brain slices for TJ protein and observed a loss of occludin, claudin-5, and ZO-1 in brain microvessels.23 We show that hAβ40 decreased rat brain microvessel expression of claudin-1 and claudin-5, whereas ZO-1 and occludin expression was not affected.
The discrepancies in our observations and those mentioned are likely due to several factors. First, the Aβ species were different (hAβ40 versus Aβ40 aggregates versus Aβ42). We used monomeric hAβ40, which is 1 main Aβ species in AD and CAA. Second, the Aβ concentrations were different (nanomolar versus micromolar). We used hAβ40 at 100 nmol/L, which is comparable to Aβ40 levels found in brain tissue from patients with AD, whereas micromolar Aβ concentrations do not reflect AD or CAA and probably lead to acute toxicity.24 Third, the experimental models in the mentioned studies were in vitro cultures of rat, mouse, or immortalized human endothelial cells. In contrast, we used intact, living brain capillaries ex vivo that were isolated from rats and confirmed our data with microvessels from hAPP mice (Tg2576). These mice overexpress the Swedish mutation of the human APP gene (APPswe) and show progressive Aβ accumulation in brain and brain microvessels, Aβ brain aggregates similar to “senile plaques” in human AD, and cognitive deficits that correlate with Aβ brain load.20 In hAPP mice, Aβ also accumulates in brain microvessels, impairs vascular smooth muscle function, and causes cell death, all of which are CAA characteristics.25 Therefore, hAPP mice are a well-established CAA model to study the Aβ effect on the neurovasculature and the mechanisms of these effects.13
Reports also show that expression of matrix-degrading MMPs is changed in the CAA brain and that increased MMPs contribute to BBB dysfunction. Lee et al found Aβ-mediated upregulation of MMP-9 in mouse endothelial cells and a mouse CAA model (APPsw mice).26 These data are consistent with our findings in microvessels exposed to Aβ and microvessels from hAPP mice showing increased MMP-2 expression and increased MMP-9 expression and activity. In this regard, Taskanen et al performed microarrays with human brain tissue and found an association between increased MMP-19 expression and CAA with intracerebral hemorrhage as well as an association between increased MMP-26 expression and CAA alone.27
Together, our data suggest that Aβ contributes to the loss of barrier integrity in CAA by decreasing TJ protein expression and increasing MMP expression and activity. Likely consequences are barrier leakage and cortical cerebral hemorrhage, which are consistent with our observations in patients with CAA.
Amyloid-β Brain Clearance
In the diseased brain, Aβ accumulates in the pericapillary space or in capillary membranes, which is a CAA hallmark. Aβ is mainly produced in neurons and astrocytes and then released into brain interstitial fluid.28 Recent evidence suggests that the BBB is critical for Aβ brain clearance and that this process involves 2 steps.29 Studies indicate that low-density lipoprotein receptor-related protein 1 is responsible for the first step—Aβ uptake from the brain into endothelial cells.30–32 In CAA, lipoprotein receptor-related protein 1 is downregulated by serum response factor and myocardin, which reduces lipoprotein receptor-related protein-mediated clearance of Aβ.33 Both transcription factors, serum response factor and myocardin, have been shown to be overexpressed in patients with AD and mouse models and are thought to be involved in the development of CAA and the hypercontractile arterial phenotype that is associated with brain hypoperfusion.4 The second step—Aβ efflux from the endothelium into blood—seems to be mediated by the efflux transporter P-glycoprotein (P-gp, ABCB1).34,35 This was first demonstrated by Cirrito et al who showed reduced Aβ brain clearance in P-gp-null mice.36 With regard to CAA, Vogel-gesang et al reported P-gp upregulation in human brain microvessels in early CAA suggesting a compensatory mechanism to increase Aβ brain clearance. However, in later stages of CAA, vessels with high Aβ load showed no P-gp expression.37 We recently demonstrated that P-gp is decreased in brain microvessels from hAPP mice. Treating these mice with pregnenolone–carbonitrile restored P-gp, decreased Aβ brain levels, and significantly reduced Aβ deposition in microvessels.14
Our data from brain microvessels suggest that Aβ40 contributes to BBB dysfunction and our patient data indicate that CAA is accompanied by multiple injuries, of which only lobar hemorrhage and cognitive decline attributed to AD are clinically relevant. It has been shown that neurovascular injury in CAA results from amyloid aggregation when cerebrovascular occlusive disease and inflammation are also present.7 In this regard, it has been shown that soluble Aβ40 can cause inflammation with monocyte transmigration through endothelial cells, decreased transendothelial electric resistance, and TJ redistribution, all of which contribute to BBB dysfunction.11 Moreover, it has been demonstrated that soluble Aβ40 generates reactive oxygen species and proinflammatory cytokines, which can induce MMPs that promote cerebral hemorrhage through basal membrane degradation.10
Finally, Aβ40-mediated injury to the neurovasculature unit could have profound effects on neuroendocrine crosstalk among neurons, astrocytes, microglia, pericytes, and endothelial cells ultimately resulting in BBB breakdown. Postmortem analysis of brains from patients with advanced AD demonstrated that thinning and discontinuities of the vascular basal membrane are associated with BBB leakage and brain penetration of the plasma protein prothrombin.38 However, BBB leakage, especially in the elderly, could also be due to other diseases such as hypertension, diabetes, and inflammation.39,40
Clinical Findings
Noninvasive neuroimaging is critical to diagnose CAA. Imaging can be indirect, for example, cerebral CT and MRI, to visualize hemorrhage indicative of CAA, or direct, for example, positron emission tomography, to visualize deposition of Aβ labeled with the Pittsburgh Compound B.41,42 We used MRI to characterize 19 patients with “probable CAA” for neurovascular pathology. We found the following 3 main characteristics.
Superficial Hemosiderosis
SH in the context of CAA appears to be more frequent than previously described and occurred in 7 of 19 patients with CAA, who became symptomatic with acute cephalgia. SH is characterized by distinct symptoms such as absent headache, focal seizures mimicking transient ischemic attacks, and migraine aura-like symptoms.43 SH can only be diagnosed in patients with at least “probable CAA” and after exclusion of vasculitis, arterial dissection, dural arteriovenous fistulas, or cortical venous thrombosis. Finelli reported of 4 patients with CAA with SH and included a summary of 7 previously published cases in the report.41 Raposo et al reanalyzed MRI data from 10 patients with SH according to the Boston criteria and reclassified 8 of 10 patients as SH with probable CAA and 2 of 10 patients as SH with “possible CAA.”8 Note that at this point we cannot rule out if additional factors such as hypertension, diabetes, and hypercholesterolemia could have potentially contributed to CAA in these patients.
Ischemic Lacunar Infarction, WMCs, and CAA
Menon and Kidwell reported on 1 patient with evolving small vessel ischemic injury detected by diffusion-weighted imaging.7 We observed this phenomenon in 4 of 19 patients, none of whom displayed symptoms such as transitory ischemic attacks or stroke. Without autopsy data, one could speculate that endothelial dysfunction leads to small vessel occlusion and silent brain infarcts that could contribute to WMC and cognitive decline, which may contribute to the development of dementia.44 Again, given our patient collective with acute neurological symptoms, the true incidence of these ischemic lesions may be overestimated.
PRES and CAA-Induced Inflammation
Symptoms of PRES are diverse and include headache, confusion, seizures, and vision loss. In 1 patient, Kofler et al linked PRES to CAA based on MRI evidence FLAIR. Additional evidence from a brain biopsy included endothelial cell activation, T-cell trafficking, and increased expression of vascular endothelial growth factor, all indicating inflammation.6 The authors concluded that PRES was triggered by inflammation from an upper respiratory infection the patient had earlier. In contrast, Bernstein and colleagues interpreted a similar finding as CAA-induced inflammation (CAA-I) and stated that the resolution of symptoms was due to successful steroid treatment.45 In a recent review, Chung et al described overlapping CAA-I symptoms with those of PRES, including headaches, cognitive and behavioral changes as well as seizures and focal neurological deficits.9 In addition, histopathologic evidence of CAA-I includes either focal spots with inflammatory cells such as multinucleated giant cells and macrophages or infiltration by epithelioid cells adjacent to amyloid-loaded microvessels. The majority of reports describe CAA-I diagnosis using FLAIR-MRI and only rarely using postcontrast-enhanced MRI.
In our study, PRES was diagnosed in 2 of 19 patients (10%) and reversible BBB leakage was also diagnosed in 2 of 19 patients. With the proposed criteria, all 4 of these patients would be classified as probable CAA-I. In our patients, PRES was found in brain areas with microbleeds that are a potential trigger for edema due to reduced expression of the TJ proteins claudin-1 and claudin-5.
Classic PRES etiology is not fully understood but could result from vasogenic edema with decreased serum albumin levels and increased BBB permeability.46 PRES is often associated with hypertension and medically induced immunosuppression. Note that CAA-I has a similar pathophysiological background and that hemorrhage can also be caused by immunosuppression.47 However, PRES and CAA-I clearly differ in their inflammatory response and endothelial activation. In addition, increased BBB permeability seen in both states could differ and be due to paracellular or transcellular barrier opening. For example, reduced expression of TJ proteins could increase paracellular BBB permeability allowing MRI contrast agents to enter the brain.48 Indeed, Carrano et al recently showed that Aβ reduced claudin-5, occludin, and ZO-1 expression in human brain microvessels.23 We did not observe reduced occludin and ZO-1 expression but found reduced claudin-1 and claudin-5 expression in Aβ-treated rat brain microvessels and in microvessels from hAPP mice. Consistent with this, paracellular BBB permeability was increased in brain microvessels from hAPP mice. Alternatively, edema could be caused by increased transcellular permeability due to changes in plasma membrane proteins that are involved in maintaining central nervous system water/electrolyte homeostasis. Whether or not this is the case in CAA is unclear.
Together, it remains to be shown if PRES and CAA-I are linked and contribute to overall CAA pathology.
Study Limitations
This study has 3 limitations: (1) recruitment of patients with CAA was performed in the stroke unit setting as opposed to a memory clinic with focus on acute therapy and diagnostics. Therefore, a standard stroke MRI protocol was used and contrast agents were only given when deemed necessary by the attending neuroradiologist. Neuropsychological testing or analysis of cerebrospinal fluid was not performed, which is important for dementia work-up but not acute stroke therapy. Patient control groups with other underlying factors (eg, PRES or SH) were not included in our study and brain biopsy or histopathologic verification was not performed. Therefore, the patients with CAA included in our study may differ significantly from patients in other studies. Future prospective and longitudinal studies should link neurological, neuro-radiological, and neuropsychological findings in acute stroke and neurodegenerative neurology to better characterize CAA; (2) the sensitivity of gadolinium-based MRI contrast agents (approximately 600 Da) to detect BBB leakage is dependent on the MRI field strength, the MRI sequence, and the timing after administration of contrast agents; and (3) the animal model used here (hAPP mice) resembles the genetic variants of CAA, and thus, our findings likely do not apply to patients with spontaneous CAA. Small animal MRI with high field strength to visualize cortical microbleeds may further help to characterize hAPP mice with respect to CAA.
Experimental Therapy of AD/CAA
Two strategies that increase Aβ brain clearance and lower Aβ brain levels have been proposed to treat CAA/AD.49,50 One approach, Aβ immunotherapy, capitalizes on antibodies that bind and clear plasma Aβ, thereby creating a peripheral “sink” that accelerates removal of soluble Aβ from the brain.51 In a recent study, passive immunization was used in patients with AD who were treated with the Aβ antibody bapineuzumab,38 and treatment with this antibody at the highest dose led to mild symptomatic and reversible vasogenic edema in 2 of 19 patients (10%). Clinical symptoms were similar to those observed in PRES and CAA-I indicating that the cerebral endothelium is the target of this therapy and thus, likely an interesting target for AD and CAA. This approach—passive immunization with an Aβ antibody—represents a case of “life imitating art” in which therapeutically administered antibodies mimic naturally occurring anti-Aβ antibodies.52 A similar approach is based on using recombinant, soluble lipoprotein receptor-related protein that also binds Aβ has thus far only been tested in mice.49 Another strategy to treat CAA and AD targets the efflux transporter P-gp at the BBB.49 P-gp has been shown to be involved in Aβ brain clearance but its expression is reduced in AD.14,44,53,54 Therefore, in this approach, P-gp is restored through activation of the nuclear receptor PXR to improve Aβ brain clearance. Given that a large number of drugs are P-gp substrates that could act as competitive P-gp inhibitors, modulation of P-gp could result in drug–drug interactions. Such complications should be considered when treating elderly patients with AD using multiple medications and could limit the therapeutic benefit of BBB P-gp induction.
Conclusions
Reduced Aβ brain-to-blood clearance through the BBB, compromised BBB integrity, and increased BBB permeability could all contribute to WMCs and mental decline in CAA/AD. Indeed, our patient and animal data suggest amyloid deposition in the neurovasculature contributes to dynamic multifocal endothelial dysfunction in CAA. Our study further suggests that the Boston criteria for the diagnosis of CAA might need to be refined to include SH, ischemia, and CAA-I with MRI findings of PRES and BBB leakage.
Future studies should focus on serial neuroimaging, BBB and inflammatory biomarkers, other cerebrovascular risk factors, and treatment to identify the events that trigger BBB dysfunction. A prospective and longitudinal study should be conducted to investigate the overall incidence of SH, ischemia, and CAA-I with MRI findings of PRES and BBB leakage in acute and chronic phases of the disease; the findings should be correlated with the Boston criteria.
Together, our findings suggest that the BBB plays a crucial role in CAA/AD pathology. Novel therapeutic strategies that prevent Aβ-mediated BBB leakage and restore BBB function could help improve CAA/AD treatment.
Acknowledgments
We thank Britt Johnson for editorial assistance with the article.
Sources of Funding
This work was supported by University of Minnesota startup funds for A.M.S.H. and B.B.
Footnotes
Disclosures
None.
References
- 1.Love S, Miners S, Palmer J, Chalmers K, Kehoe P. Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front Biosci. 2009;14:4778–4792. doi: 10.2741/3567. [DOI] [PubMed] [Google Scholar]
- 2.Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of cerebral amyloid angiopathy. J Neurol Sci. 2007;257:23–30. doi: 10.1016/j.jns.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 3.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood–brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, et al. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc Natl Acad Sci U S A. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/wnl.56.4.537. [DOI] [PubMed] [Google Scholar]
- 6.Kofler J, Bartynski WS, Reynolds TQ, Lieberman FS, Murdoch GH, Hamilton RL. Posterior reversible encephalopathy syndrome (PRES) with immune system activation, VEGF up-regulation, and cerebral amyloid angiopathy. J Comput Assist Tomogr. 2011;35:39–42. doi: 10.1097/RCT.0b013e3181f31917. [DOI] [PubMed] [Google Scholar]
- 7.Menon RS, Kidwell CS. Neuroimaging demonstration of evolving small vessel ischemic injury in cerebral amyloid angiopathy. Stroke. 2009;40:e675–e677. doi: 10.1161/STROKEAHA.109.552935. [DOI] [PubMed] [Google Scholar]
- 8.Raposo N, Viguier A, Cuvinciuc V, Calviere L, Cognard C, Bonneville F, et al. Cortical subarachnoid haemorrhage in the elderly: a recurrent event probably related to cerebral amyloid angiopathy. Eur J Neurol. 2011;18:597–603. doi: 10.1111/j.1468-1331.2010.03214.x. [DOI] [PubMed] [Google Scholar]
- 9.Chung KK, Anderson NE, Hutchinson D, Synek B, Barber PA. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry. 2011;82:20–26. doi: 10.1136/jnnp.2009.204180. [DOI] [PubMed] [Google Scholar]
- 10.Zipfel GJ, Han H, Ford AL, Lee JM. Cerebral amyloid angiopathy: progressive disruption of the neurovascular unit. Stroke. 2009;40:S16–S19. doi: 10.1161/STROKEAHA.108.533174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez-Velasquez FJ, Kotarek JA, Moss MA. Soluble aggregates of the amyloid-beta protein selectively stimulate permeability in human brain microvascular endothelial monolayers. J Neurochem. 2008;107:466–477. doi: 10.1111/j.1471-4159.2008.05618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han BH, Zhou ML, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J, et al. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J Neurosci. 2008;28:13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158:1065–1071. doi: 10.1016/S0002-9440(10)64053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartz AM, Miller DS, Bauer B. Restoring blood–brain barrier p-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol. 2010;77:715–723. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauer B, Hartz AM, Lucking JR, Yang X, Pollack GM, Miller DS. Coordinated nuclear receptor regulation of the efflux transporter, MRP2, and the phase-II metabolizing enzyme, GSTPI, at the blood–brain barrier. J Cereb Blood Flow Metab. 2008;28:1222–1234. doi: 10.1038/jcbfm.2008.16. [DOI] [PubMed] [Google Scholar]
- 16.Hartz AM, Bauer B, Block ML, Hong JS, Miller DS. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and p-glycoprotein up-regulation at the blood–brain barrier. Faseb J. 2008;22:2723–2733. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartz AM, Bauer B, Fricker G, Miller DS. Rapid regulation of p-glycoprotein at the blood–brain barrier by endothelin-1. Mol Pharmacol. 2004;66:387–394. doi: 10.1124/mol.104.001503. [DOI] [PubMed] [Google Scholar]
- 18.Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, et al. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993;43:1683–1689. doi: 10.1212/wnl.43.9.1683. [DOI] [PubMed] [Google Scholar]
- 19.Verdelho A, Madureira S, Moleiro C, Ferro JM, Santos CO, Erkinjuntti T, et al. White matter changes and diabetes predict cognitive decline in the elderly: the LADIS study. Neurology. 2010;75:160–167. doi: 10.1212/WNL.0b013e3181e7ca05. [DOI] [PubMed] [Google Scholar]
- 20.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 21.Marco S, Skaper SD. Amyloid beta-peptide 1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–224. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 22.Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010;14:1101–1112. doi: 10.1111/j.1582-4934.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid beta induces oxidative stress-mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 24.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at a beta 40 or a beta 42(43) J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 25.Han F, Ali Raie A, Shioda N, Qin ZH, Fukunaga K. Accumulation of beta-amyloid in the brain microvessels accompanies increased hyperphosphorylated tau proteins following microsphere embolism in aged rats. Neuroscience. 2008;153:414–427. doi: 10.1016/j.neuroscience.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 26.Lee JM, Yin K, Hsin I, Chen S, Fryer JD, Holtzman DM, et al. Matrix metalloproteinase-9 in cerebral-amyloid-angiopathy-related hemorrhage. J Neurol Sci. 2005;229–230:249–254. doi: 10.1016/j.jns.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 27.Tanskanen M, Myllykangas L, Saarialho-Kere U, Paetau A. Matrix metalloproteinase-beta 19 expressed in cerebral amyloid angiopathy. Amyloid. 2011;18:3–9. doi: 10.3109/13506129.2010.541960. [DOI] [PubMed] [Google Scholar]
- 28.Utter S, Tamboli IY, Walter J, Upadhaya AR, Birkenmeier G, Pietrzik CU, et al. Cerebral small vessel disease-induced apolipoprotein e leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J Neuropathol Exp Neurol. 2008;67:842–856. doi: 10.1097/NEN.0b013e3181836a71. [DOI] [PubMed] [Google Scholar]
- 29.Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, et al. Amyloid efflux transporter expression at the blood–brain barrier declines in normal aging. J Neuropathol Exp Neurol. 2010;69:1034–1043. doi: 10.1097/NEN.0b013e3181f46e25. [DOI] [PubMed] [Google Scholar]
- 30.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-SS(1-40) peptide from brain by LDL receptor-related protein-1 at the blood–brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 32.Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood–brain barrier. Stroke. 2004;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 33.Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murozono M, Matsumoto S, Okada S, Nagaoka D, Isshiki A, Watanabe Y. Reduction of brain infarction induced by a transient brain ischemia in mdr1a knockout mice. Neurochem Res. 2009;34:1555–1561. doi: 10.1007/s11064-009-9943-6. [DOI] [PubMed] [Google Scholar]
- 35.Miller DS, Bauer B, Hartz AM. Modulation of p-glycoprotein at the blood–brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60:196–209. doi: 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood–brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, et al. The role of p-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2004;1:121–125. doi: 10.2174/1567205043332225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 41.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 42.Greenberg SM, Grabowski T, Gurol ME, Skehan ME, Nandigam RN, Becker JA, et al. Detection of isolated cerebrovascular beta-amyloid with Pittsburgh Compound B. Ann Neurol. 2008;64:587–591. doi: 10.1002/ana.21528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuvinciuc V, Viguier A, Calviere L, Raposo N, Larrue V, Cognard C, et al. Isolated acute nontraumatic cortical subarachnoid hemorrhage. AJNR Am J Neuroradiol. 2010;31:1355–1362. doi: 10.3174/ajnr.A1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bauer B, Hartz AM, Fricker G, Miller DS. Modulation of p-glycoprotein transport function at the blood–brain barrier. Exp Biol Med (Maywood) 2005;230:118–127. doi: 10.1177/153537020523000206. [DOI] [PubMed] [Google Scholar]
- 45.Bernstein RA, Gibbs M, Hunt Batjer H. Clinical diagnosis and successful treatment of inflammatory cerebral amyloid angiopathy. Neurocrit Care. 2011;14:453–455. doi: 10.1007/s12028-010-9497-0. [DOI] [PubMed] [Google Scholar]
- 46.Pirker A, Kramer L, Voller B, Loader B, Auff E, Prayer D. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32:527–531. doi: 10.3174/ajnr.A2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hefzy HM, Bartynski WS, Boardman JF, Lacomis D. Hemorrhage in posterior reversible encephalopathy syndrome: imaging and clinical features. AJNR Am J Neuroradiol. 2009;30:1371–1379. doi: 10.3174/ajnr.A1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weier K, Fluri F, Kos S, Gass A. Postcontrast FLAIR MRI demonstrates blood–brain barrier dysfunction in pres. Neurology. 2009;72:760–762. doi: 10.1212/01.wnl.0000343007.39226.aa. [DOI] [PubMed] [Google Scholar]
- 49.Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6:404–416. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 51.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma a beta clearance and decreases brain a beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenberg SM, Frosch MP. Life imitates art: anti-amyloid antibodies and inflammatory cerebral amyloid angiopathy. Neurology. 2011;76:772–773. doi: 10.1212/WNL.0b013e31820e7bce. [DOI] [PubMed] [Google Scholar]
- 53.Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, et al. MDR1-p-glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-beta peptides—implications for the mechanisms of Abeta clearance at the blood–brain barrier. Brain Pathol. 2007;17:347–353. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loeb MB, Molloy DW, Smieja M, Standish T, Goldsmith CH, Mahony J, et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J Am Geriatr Soc. 2004;52:381–387. doi: 10.1111/j.1532-5415.2004.52109.x. [DOI] [PubMed] [Google Scholar]