

Abstract
The microtubule binding protein tau is strongly implicated in multiple neurodegenerative disorders, including frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), which is caused by mutations in tau. In vitro, FTDP-17 mutant versions of tau can reduce microtubule binding and increase the aggregation of tau, but the mechanism by which these mutations promote disease in vivo is not clear. Here we take a combined biochemical and in vivo modeling approach to define functional properties of tau driving neurotoxicity in vivo. We express wild-type human tau and five FTDP-17 mutant forms of tau in Drosophila using a site-directed insertion strategy to ensure equivalent levels of expression. We then analyze multiple markers of neurodegeneration and neurotoxicity in transgenic animals, including analysis of both males and females. We find that FTDP-17 mutations act to enhance phosphorylation of tau and thus promote neurotoxicity in an in vivo setting. Further, we demonstrate that phosphorylation-dependent excess stabilization of the actin cytoskeleton is a key phosphorylation-dependent mediator of the toxicity of wild-type tau and of all the FTDP-17 mutants tested. Finally, we show that important downstream pathways, including autophagy and the unfolded protein response, are coregulated with neurotoxicity and actin cytoskeletal stabilization in brains of flies expressing wild-type human and various FTDP-17 tau mutants, supporting a conserved mechanism of neurotoxicity of wild-type tau and FTDP-17 mutant tau in disease pathogenesis.
SIGNIFICANCE STATEMENT The microtubule protein tau aggregates and forms insoluble inclusion bodies known as neurofibrillary tangles in the brain tissue of patients with a variety of neurodegenerative disorders, including Alzheimer's disease. The tau protein is thus widely felt to play a key role in promoting neurodegeneration. However, precisely how tau becomes toxic is unclear. Here we capitalize on an “experiment of nature” in which rare missense mutations in tau cause familial neurodegeneration and neurofibrillary tangle formation. By comparing the biochemical activities of different tau mutations with their in vivo toxicity in a well controlled Drosophila model system, we find that all mutations tested increase phosphorylation of tau and trigger a cascade of neurotoxicity critically impinging on the integrity of the actin cytoskeleton.
Keywords: Alzheimer's, Drosophila, tau
Introduction
The neuronal protein tau is a key player in multiple human neurodegenerative disorders. Alzheimer's disease, the most common neurodegenerative disorder, is defined neuropathologically by the combined presence of extracellular Aβ peptides aggregated into plaques and intracellular neurofibrillary tangles comprised of phosphorylated and misfolded tau. While Alzheimer's disease does not show clear genetic linkage to the TAU locus, the familial neurodegenerative tauopathy, frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), is caused by dominant missense or splicing mutations in the TAU gene (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). Thus, primary abnormalities in tau can cause progressive neurodegeneration. To date, 53 pathogenic tau mutations have been reported (Cruts et al., 2012; Ghetti et al., 2015). These mutations alter either the splicing of TAU mRNA or create missense mutations or small deletions in the tau protein, and should provide substantial insight into the mechanisms by which alterations in tau cause disease. Indeed, the majority of disease-causing tau mutations cluster near the C-terminal microtubule binding domains of the protein, and a number of FTDP-17 mutations impair the ability of tau to bind microtubules in vitro (Hasegawa et al., 1998; Hong et al., 1998). These finding are consistent with longstanding models of tauopathy pathogenesis in which the inability of tau to bind microtubules promotes neurotoxicity through lack of appropriate microtubule stabilization and increased availability of cytosolic tau to form neurotoxic aggregates (Ghetti et al., 2015; Bodea et al., 2016).
However, not all disease-linked mutations appear to influence microtubule binding (DeTure et al., 2000; Combs and Gamblin, 2012). Given the neuropathological prominence of fibrillary inclusion bodies in tauopathies, significant effort has been expended by our and other laboratories to determine whether, alternatively, abnormal aggregation of mutant tau may underlie neurotoxicity of the proteins. Indeed, a number of FTDP-17 mutations do promote in vitro fibrillation of tau (Hong et al., 1998; Gamblin et al., 2000). However, recent experiments have raised the possibility that large fibrillar aggregates may not necessarily underlie tau neurotoxicity in vivo. Santacruz et al. (2005) used a repressible mouse model of tauopathy to dissociate memory deficits with neurofibrillary tangle density. Similarly, human tau can be neurotoxic in Drosophila without the formation of significant numbers of large fibrillary inclusions (Wittmann et al., 2001), and when present the number of large inclusions can be dissociated from ongoing neurodegeneration (Colodner and Feany, 2010). These data are consistent with a larger body of evidence supporting the pathogenicity of smaller, or oligomeric, aggregates of Aβ, α-synuclein, and other toxic aggregating proteins (Luheshi et al., 2007; Karpinar et al., 2009). Thus, the propensity of tau to form a particular oligomeric species might correlate better with toxicity than fibril formation. To address these issues, we have here expressed a selected series of biochemically characterized FTDP-17 mutant tau variants in Drosophila to correlate toxicity in a well controlled in vivo setting with biochemical features of the protein. Our results suggest that toxicity in vivo correlates best with a cascade of actin cytoskeletal pathology triggered by excessive phosphorylation of tau.
Materials and Methods
Fly stocks, genetics.
The elav-GAL4, elav-GeneSwitch, and repo-GAL4 drivers were obtained from Bloomington Drosophila Stock Center at Indiana University, as was puc-lacZ (pucE69). The following fly stocks were provided by the indicated investigators: UAS-CD8-PARP-Venus, Darren Williams (Kings College, London, United Kingdom) (Williams et al., 2006); UAS-Atg8a-GFP, Thomas Neufeld (University of Minnesota, Minneapolis, Minnesota) (Juhász et al., 2008); and UAS-Xbp1-EGFP, Hyung Don Ryoo (NYU School of Medicine, New York, New York) (Ryoo et al., 2007). All fly crosses and experiments were performed at 25°C. 0N4R wild-type (WT) and mutant tau cDNAs were transferred from the pT7C vector into the pENTR entry vector and inserted via recombination into the pWALIUM10-roe Drosophila expression vector. Transgenic flies were created using the attP40 landing site (BestGene). To analyze tau turnover, transgenes were controlled with the elav-GeneSwitch driver. Tau was induced by feeding flies 10 mg/ml mifepristone (RU486, Sigma-Aldrich) in instant food (Carolina Biological) for 2 d. Flies were analyzed after the 2 d feeding period (see Fig. 2C, Day 1), and then at 10 and 20 d after drug withdrawal. Full genotypes for all experiments are presented in Extended Data.
Figure 2.

Phosphorylation and turnover of tau in transgenic animals. A, Western blots showing the phosphorylation of tau at proline-directed sites in flies expressing tau using the pan-neuronal elav-GAL4 driver. Tau-1 recognizes nonphosphorylated tau. The blot was reprobed with a phosphorylation-independent antibody (total tau) to assess total (phosphorylated and unphosphorylated) tau and with an antibody to GAPDH to illustrate equivalent protein loading. All blots were repeated a minimum of three times. B, Quantitative analysis of tau phosphorylation at proline-directed sites reveals significant increases in phosphorylation of tau at AT270 and PHF1 in flies expressing R5L, G389R, or R406W mutant tau compared with flies expressing wild-type tau (p < 0.05, Tukey's HSD). Control is elav-GAL4/+. Full genotypes are provided in Figure 2-1. Flies are 1 d old. C, Tau turnover following conditional expression of tau variants in adult neurons using the elav-GeneSwitch driver. Full genotypes are provided in Figure 2-1. The blot was reprobed with an antibody to GAPDH to illustrate equivalent protein loading. All blots were repeated four times. D, Quantitative analysis of tau turnover reveals that WT, S320F, and S352L are less stable than R406W and E14 (p < 0.05, Tukey's HSD). Full statistical analysis is presented in Figure 2-1.
Sectioning, immunostaining and fluorescence.
Adult flies were fixed in formalin at 30 d of age and embedded in paraffin. Serial frontal 4 μm sections including the entire brain were prepared. Antigen retrieval was performed by microwaving in sodium citrate buffer. Immunostaining was performed with the avidin–biotin–peroxidase complex method (Vector Laboratories). Primary antibodies included the following: anti-human poly-(ADP-ribose)-polymerase (PARP; 1:100,000; E51, Abcam; RRID:AB_777102); anti-β-galactosidase (1:500; Promega; RRID:AB_2313752); AT8 (1:10,000, Invitrogen; RRID:AB_223647); and anti-GFP (1:20 to 1: 5000; N86/8, UC Davis/NIH NeuroMab Facility, Davis, CA; RRID:AB_2313651). For quantification of cleaved caspase (UAS-CD8-PARP-Venus), JNK activation (puc-lacZ), and activation of the unfolded protein response (UAS-Xbp1-EGFP), the number of positive cells in the entire brain was counted. For quantification of Atg8a-positive puncta, since the number of puncta was too large to count in the entire brain, the number of puncta in one anatomically consistent section through the calyx of the mushroom body was counted. The mushroom body has been implicated in learning and memory in Drosophila. To quantify the number of inclusions, AT8-immunopositive puncta in five anatomically consistent consecutive sections of the anterior medulla were counted. The medulla is a preferential site of inclusion formation in glial tau transgenic flies (Colodner and Feany, 2010). At least six brains were analyzed per genotype; statistical significance was established using ANOVA with Tukey's HSD.
For the assessment of phalloidin fluorescence, brains from 30-d-old flies were dissected in PBS and fixed in 4% PFA on ice for 30 min. After a 10 min incubation in 0.3% Triton-X, the brains were stained with Acti-stain 555 phalloidin (catalog #PHDH1, Cytoskeleton) at a concentration of 14 nm for 30 min in the dark. Brains were then washed three times in PBS for 60 min each time, mounted, and imaged using confocal microscopy. For quantification of fluorescence, the average pixel density from two-dimensional projections of z-stacks for the entire brain was computed using ImageJ. Three brains were analyzed per genotype; statistical significance was established using ANOVA with Tukey's HSD.
RNA extraction and real-time quantitative PCR.
RNA was isolated from the heads of 1-d-old flies using TRIzol solution as per the suggestion of the manufacturer with an additional centrifugation (12,000 × g, for 10 min) to remove debris before the addition of chloroform. RNA quantity and quality were evaluated on a NanoDrop 1000 spectrophotometer. One microgram of total RNA was used in the reverse-transcription reaction (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems). Primers were designed using Primer Express version 3.0 (Applied Biosystems) and were used at a final concentration of 150 nm. PCRs were set up in a 16 μl reaction volume using SYBR Green PCR Master Mix (Applied Biosystems). PCR amplifications were performed with an Applied Biosystems Step One machine. The RpL32 ribosomal protein L32 transcript was used as an endogenous control. Primers were as follows: tau forward, GAT CAC GCT GGG ACG TAC GG; tau reverse, GGT CAC GTG ACC AGC AGC T; RpL32 forward, GAC CAT CCG CCC AGC ATA C; and RpL32 reverse, CGG CGA CGC ACT CTG TT. Experiments were run in triplicate, and the experiment was repeated four times. Statistical significance was assessed using ANOVA with Tukey's HSD. Significant differences were not detected (p = 0.5).
Western blots.
Fly heads were homogenized in 2× Laemmli buffer (Sigma-Aldrich). Samples were boiled for 10 min, briefly centrifuged, and subjected to SDS-PAGE in a 10% gel. Proteins were transferred to nitrocellulose membranes (Bio-Rad), blocked in 2% milk in PBS with 0.05% Tween 20, and immunoblotted using the following phosphorylation-dependent and phosphorylation-independent anti-tau antibodies: AT270 (1:10,000; Invitrogen; RRID:AB_223651); AT8 (1:10,000; Invitrogen; RRID:AB_223647); Tau-1 (1:1000; catalog #IHCR1015-6, Millipore; RRID:AB_2139842); AT180 (1:10,000; Invitrogen; RRID:AB_223649); PHF1 (1:1000; a gift from Peter Davies, Feinstein Institute for Medical Research, North-Shore Long Island Jewish Health System, Manhasset, New York); and total tau (1:50,000; Dako; RRID:AB_10013724). Blots were reprobed with an anti-GAPDH antibody (1:100,000; Invitrogen; RRID:AB_10977387) to illustrate equivalent protein loading. For quantitative analysis, densitometry was performed and the relative level of tau phosphorylation was assessed at each phosphoepitope. The fold change (see Fig. 2B) was calculated by setting wild-type tau to a value of 1 and calculating levels for the mutants relative to wild type. Each phosphoepitope on each form of tau was assessed at least three times. Statistical significance was established using one-way ANOVA with post hoc test for multiple comparisons (Tukey's HSD).
F-actin ELISA.
Freshly dissected brains from 30-d-old flies of the indicated genotypes were homogenized in 50 μl of actin stabilization buffer from the G-actin/F-actin in vivo assay kit (catalog #BK037, Cytoskeleton). Ten microliters of the homogenate was used in the F-actin ELISA assay (catalog #MBS702018, MyBioSource) and the assay was performed according to the manufacturer protocol. The remainder of the homogenate was used in Western blotting to ensure equal levels of total actin and the input. Each experiment was performed with two technical replicates. The experiments were repeated three times. Statistical significance was assessed using ANOVA with supplementary Tukey's HSD.
Protein expression and purification.
All WT and FTDP-17 mutant proteins were expressed containing a 6×-histidine tag and purified by immobilized metal affinity chromatography followed by size exclusion chromatography, as described previously (Rankin et al., 2005). The FTDP-17 mutations were created using the Quikchange Site-Directed Mutagenesis Kit from Agilent. Protein concentrations were determined using the Pierce BCA protein assay kit (catalog #23225, Thermo Fisher Scientific) with bovine serum albumin (BSA) as a control following the manufacturer protocol. The following mutations were generated in a full-length 0N4R tau background contained in a pT7C vector: R5L, S320F, S352L, G389R, and R406W.
Actin-bundling assay.
Non-muscle actin binding protein biochem kit (catalog #BK013) was purchased from Cytoskeleton. Tau actin-bundling activity was determined by a low-speed sedimentation assay following the manufacturer protocol to determine whether a protein of interest can bundle F-actin (method 3). F-actin (17 μm) was incubated in the presence or absence of tau protein (3.7 μm) for 30 min at room temperature. Samples were then centrifuged at 14,000 × g for 10 min at room temperature to separate actin bundles that pellet from actin filaments that remain in the supernatant. Equal volumes of supernatants and pellets were analyzed by SDS-PAGE, and protein was detected by Coomassie Blue staining. Gels were imaged using a ChemiDoc-It2 Imager from UVP. The density of the bands was determined using the histogram feature of Adobe Photoshop CS6.
Arachidonic acid-induced polymerization.
Recombinant wild-type and mutant tau protein, at a final concentration of 2 μm, was incubated in buffer containing 0.1 mm EDTA; 5 mm dithiothreitol; 10 mm HEPES buffer, pH 7.64; and 200 mm NaCl. Polymerization was induced by the addition of arachidonic acid (catalog #181198-100MG, Millipore) from a 2 mm stock solution in ethanol to a final concentration 75 μm (resulting in a final concentration of 3.75% ethanol vehicle). Ethanol-only reactions with tau (2 μm) were performed as negative controls. Two hundred microliter reactions in 1.5 ml Eppendorf tubes were allowed to proceed overnight at 25°C.
Thioflavin S fluorescence.
The total amount of aggregation was measured using the binding of thioflavin S (catalog #T1892) from Sigma-Aldrich; 150 μl of each reaction mixture was added to separate wells in a 96-well, white, flat-bottom plate. Thioflavin S was diluted in water to a concentration of 0.224 g/ml, and 6 μl was added to the well. The plate was shaken for 30 s followed by incubation in the dark for 20 min. The fluorescence shift was measured by using a Varian Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies) with an excitation wavelength of 440 nm and an emission wavelength of 520 nm. The photomultiplier tube voltage was set to 650 V. Readings from control reactions with 2 μm protein and without arachidonic acid (ethanol vehicle only) were used as a blank and were subtracted from the reading for each reaction.
Transmission electron microscopy.
The arachidonic acid-induced polymerization reaction mixtures were diluted 1:10 in polymerization buffer and 2% glutaraldehyde (catalog #16120, Electron Microscopy Sciences). After a 5 min incubation, a Formvar-coated copper grid (Electron Microscopy Sciences) was placed on top of a 10 μl drop of the diluted sample for 1 min. The grid was then blotted on filter paper, placed on a drop of water, blotted with filter paper, placed on a drop of 2% uranyl acetate, and blotted dry. The grid was then placed on another drop of 2% uranyl acetate for 1 min and blotted dry for a final time. The grids were examined using a TECNAI F20 XT electron microscope (FEI). Images were collected with the Gatan Digital Micrograph Imaging System at a magnification of 3600×. Five images were collected from each grid and analyzed. The amount of aggregated tau in each of the images was quantified by using Image-Pro Plus version 6.0 (Media Cybernetics). Filaments were highlighted using the threshold feature, and the total number of filaments per field was determined. The perimeter of each filament was measured and divided by 2 to estimate the length of the filament. Filaments with a length of <15 nm were excluded because it was more difficult to distinguish objects smaller than this from the background. The average filament length was determined by calculating the mean length of all filaments in a given image. The total amount of aggregated material in each image was calculated by summing the lengths of all filaments in an image. The number of filaments, average length of filaments, and total length of filaments per image was averaged for five different images and reported with the SD of the averages.
Experimental design and statistical analysis.
For animal model studies, all experiments were performed on six animals per genotype and time point (histological analyses) or at least three independent replicates (quantitative real time PCR, Western blotting, and actin stabilization studies). Males and females were used. For in vitro biochemical studies, experiments were repeated at least twice (actin bundling) or were quantified as described in detail above (in vitro tau aggregation). Statistical analysis was performed using ANOVA with supplementary Tukey's HSD. Full statistical analyses are presented in the Extended Data.
Results
To define the biochemical properties of FTDP-17 mutant forms of tau that determine in vivo neurotoxicity, we selected a variety of tau mutants for expression in a well characterized, aging-dependent Drosophila model of tauopathy (Wittmann et al., 2001; Khurana et al., 2006; Fulga et al., 2007). Mutants were chosen initially based on our prior data demonstrating a range of in vitro aggregation characteristics, microtubule binding profiles (Gamblin et al., 2000, 2003; Combs and Gamblin, 2012), and in vivo neurotoxicity (Wittmann et al., 2001; Fulga et al., 2007); and include R5L, S320F, S352L, G389R, and R406W. We also attempted to express P301L mutant human tau in flies but were unable to obtain transgenic animals expressing mutant human tau, perhaps reflecting excessive toxicity of the P301L mutant protein in vivo. All isoforms of tau were expressed in the 0N4R (no N-terminal inserts, four microtubule binding repeat) form of tau because we have previously observed toxicity of wild-type, R406W, and V337M 0N4R human tau when expressed in Drosophila (Wittmann et al., 2001; Khurana et al., 2006; Fulga et al., 2007). Since neurotoxicity of tau is dose dependent (Wittmann et al., 2001), we used a site-directed insertion strategy (Bateman et al., 2006; Bischof et al., 2007) to ensure that expression levels of the various tau isoforms were equivalent. We inserted our tau transgenes into the attP40 landing site (Markstein et al., 2008) and used the bipartite UAS-GAL4 expression system (Brand and Perrimon, 1993) and the pan-neuronal driver elav-GAL4 (Wittmann et al., 2001) to express human wild-type and FTDP-17-linked mutant forms of tau. As predicted by our site-directed insertion strategy, levels of tau expression were similar in heads from animals expressing wild-type and mutant transgenes, as determined by quantitative real-time PCR (Fig. 1).
Figure 1.
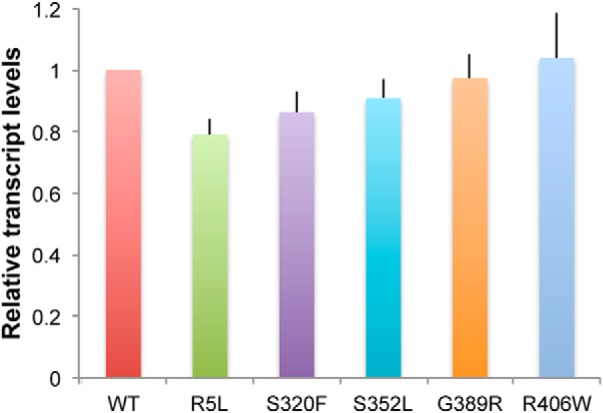
Expression of tau in transgenic animals. Quantitative real-time PCR reveals equivalent levels of tau transcripts in flies expressing 0N4R wild-type and FTDP-17-linked forms of tau under the control of the pan-neuronal elav-GAL4 driver. Values are normalized to wild type. Experiments were run in triplicate and the experiment was repeated at least three times. There were no significant differences in relative transcript levels (p = 0.5, ANOVA). Flies are 1 d old. Full genotypes are provided in Figure 1-1.
Full genotypes. Download Figure 1-1, DOC file (47.5KB, doc)
In contrast, levels of tau protein differed among the different forms of tau (Fig. 2A,B). Most strikingly, levels of tau phosphorylated at the human disease-associated serine-proline (SP) or threonine-proline (TP) motifs, AT270, AT8, AT180, and PHF1, were increased in the R5L, G389R, and R406W variants. Western blotting with the Tau1 antibody, which preferentially recognizes tau lacking phosphorylation between amino acids 189 and 207 (Szendrei et al., 1993; Steinhilb et al., 2007a), is consistent with enhanced phosphorylation at the AT8 site. The phosphospecific antibody AT8 recognizes tau phosphorylated at serine 202 and threonine 205.
Full genotypes and statistical analyses. Download Figure 2-1, DOC file (153KB, doc)
Although increases in phosphorylation were greater than changes in total tau levels, there was a trend for total tau (phosphorylated and nonphosphorylated tau) levels to be higher in flies expressing R5L, G389R, or R406W mutant tau compared with wild-type tau (Fig. 2A,B and Fig. 2-1). To determine whether the stability of tau was altered by the mutants studied, the GeneSwitch (Osterwalder et al., 2001) conditional expression system and the elav-GeneSwitch pan-neuronal driver were used to temporally regulate the expression of wild-type and mutant tau variants. At day 1 following eclosion, flies were placed on medium containing mifepristone (RU486), which induces transcriptional activation in the GeneSwitch system. Flies were maintained on drug-embedded food for 2 d, at which time heads were taken for Western blot analysis (Fig. 2C, Day 1). Flies were then aged for 10 or 20 d and heads were analyzed by Western blot (Fig. 2C, Day 10 and Day 20). Flies grown on food containing the ethanol vehicle only were used as controls (Fig. 2C, No drug). Quantitative Western blot analysis revealed a trend toward greater stability of tau in flies expressing R5L, G389R, and R406W variants (Fig. 2D and Fig. 2-1). To determine whether proline-directed phosphorylation might drive altered stability of FTDP-17 tau variants, we used a mutant form of tau in which 14 SP and TP sites have been mutated to glutamate to mimic phosphorylation [Fig. 2C,D, E14]. All 14 Ser-Pro and Thr-Pro kinase target sites were mutated to glutamate (tauE14). We have previously demonstrated that E14 mutant tau is substantially more toxic that wild-type tau (Khurana et al., 2006; Dias-Santagata et al., 2007; Fulga et al., 2007). The E14 protein was significantly more stable in vivo than wild-type, S320F, or S352L mutant tau (Fig. 2D and Fig. 2-1), which is consistent with a role for phosphorylation in regulating the stability of tau.
To determine whether changes in tau phosphorylation correlated with neurotoxicity in our transgenic animals, we next assessed the toxicity of various tau isoforms. We began by using a well characterized transgenic reporter of caspase activity, which correlates well with cell death in our tau transgenic Drosophila (Loewen and Feany, 2010; Frost et al., 2014). These reporter flies carry a transgenic construct in which the extracellular and transmembrane domains of mouse CD8 is fused to 40 aa from human PARP, including the caspase cleavage site of PARP (UAS-CD8-PARP-Venus; Williams et al., 2006). Endogenous Drosophila caspases cleave the reporter at the PARP cleavage site. Caspase activation is then assessed experimentally using an antibody that is specific for cleaved human PARP. The transgenic caspase reporter provides a robust and quantitative method to monitor endogenous caspase activity and apoptotic cell death in adult Drosophila models of human neurodegenerative disease (Hegde et al., 2014; Wang et al., 2015, 2016). When we examined the brains of 30-day-old flies (flies live for ∼60 d under our culture conditions) expressing wild-type or FTDP-17 mutant forms of human tau in neurons, we observed significantly greater numbers of neurons with activated caspase in flies expressing R5L, G389R, or R406W mutant tau compared with flies expressing wild-type tau (Fig. 3A,B and Fig. 3-1). Note that the cells identified are neurons because the reporter construct is expressed only in neurons under the control of the elav-GAL4 driver, which is consistent with the morphology of the labeled cells (Fig. 3A). Consistent with these findings, animals homozygous (females) or hemizygous (males, the elav-GAL4 driver is on the X-chromosome) for the elav-GAL4 driver and homozygous for the R5L, G389R, or R406W tau transgenes were not viable and were thus not recovered. In contrast, adult flies homozygous (female) or hemizygous (male) for the elav-GAL4 driver and homozygous for the wild-type, S320F, or S352L tau transgenes were viable.
Figure 3.
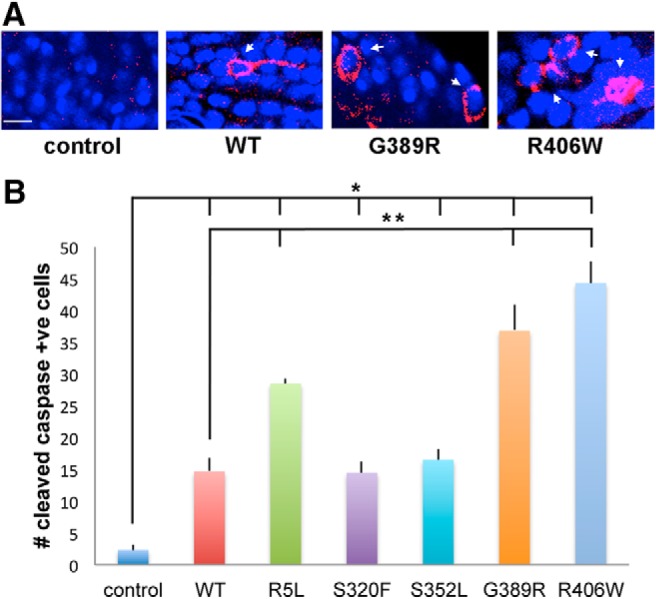
Caspase cleavage in tau transgenic animals. A, Neurons with activated caspase as monitored by cleavage of the transgenic reporter UAS-CD8-PARP (40 aa at caspase cleavage site)-Venus in flies expressing tau using the pan-neuronal elav-GAL4 driver. Arrows indicate cells with caspase activation. Scale bar, 3 μm. B, Quantitative analysis of the number of neurons with activated caspase in the entire brains of control animals or in flies expressing human wild-type or FTDP-17 mutant forms of tau. Six brains were analyzed per genotype. Control is elav-GAL4/+; UAS-CD8-PARP-Venus/+. Full genotypes are provided in Figure 3-1. Flies are 30 d old. **p < 0.01, *p < 0.05, ANOVA with Tukey's HSD. Full statistical analysis is presented in Figure 3-1.
Full genotypes and statistical analysis. Download Figure 3-1, DOC file (75KB, doc)
We have previously demonstrated an important role for oxidative stress in Drosophila models of tauopathy (Dias-Santagata et al., 2007). The induction of stress-activated protein kinase cascades, such as the JNK signaling pathway, is one of the best characterized responses to oxidative damage (Martindale and Holbrook, 2002; Zhu et al., 2003). To determine whether JNK pathway activation correlated with tau-induced neurotoxicity in our transgenic animals, we used the puckered-lacZ (puc-lacZ) reporter system, which is commonly used to monitor JNK pathway activation in Drosophila. The puc gene encodes a phosphatase that inactivates JNK and other MAPK family members and is a well established transcriptional target of JNK signaling in Drosophila (Martín-Blanco et al., 1998). Brains from transgenic flies carrying the puc-lacZ reporter alone or with various tau transgenes expressed in neurons were immunostained at 30 d after eclosion with an antibody against β-galactosidase. Reporter activity was minimal in control flies, while multiple cortical cells demonstrated staining for β-galactosidase in tau transgenic animals (Fig. 4A,B), with the greatest activation occurring in flies expressing R5L, G389R, and R406W mutant forms of tau. We have previously demonstrated that cells with JNK activation are predominantly neurons, consistent with the morphology of the cells labeled (Dias-Santagata et al., 2007).
Figure 4.
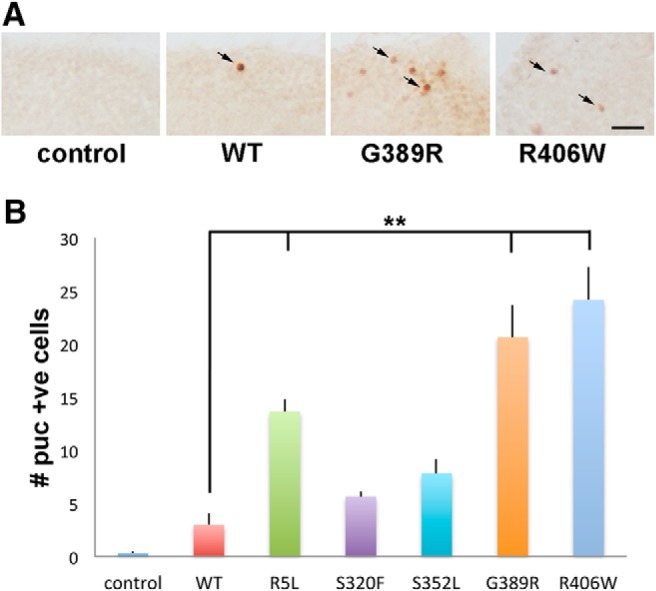
Stress pathway activation in tau transgenic animals. A, Neurons with activated JNK signaling as monitored by the puc-lacZ reporter and immunostaining for β-galactosidase, which is directed to the nucleus (Bier et al., 1989), in flies expressing tau using the pan-neuronal elav-GAL4 driver. Arrows indicate positive nuclei. Scale bar, 10 μm. B, Quantitative analysis of the number of neurons with activated JNK in the entire brains of control animals, or in flies expressing human wild-type or FTDP-17 mutant forms of tau. Six brains were analyzed per genotype. Control is elav-GAL4/+; puc-lacZ/+. Full genotypes are provided in Figure 4-1. Flies are 30 d old. **p < 0.01, ANOVA with Tukey's HSD. Full statistical analysis is presented in Figure 4-1.
Full genotypes and statistical analysis. Download Figure 4-1, DOC file (75KB, doc)
Having demonstrated that increased phosphorylation of tau correlates with established markers of neurotoxicity in our transgenic fly models, we next assessed aggregation of tau. Large inclusions are infrequent in our neuronal tauopathy model (Wittmann et al., 2001). However, when we express human tau in Drosophila glia using the pan-glial driver repo-GAL4, we observe numerous inclusions comprised of filamentous tau at the electron microscopic level (Colodner and Feany, 2010), as can be seen in human glial tauopathies (Feany and Dickson, 1995, 1996; Kovacs et al., 2016). We therefore expressed wild-type and disease-linked mutant versions of tau in fly glia and examined the brains of 30-day-old flies for the presence of tau aggregates using immunostaining for the anti-tau antibody AT8 (Colodner and Feany, 2010). The number of large tau inclusions was significantly increased in flies expressing S320F or S352L mutant tau, compared with animals expressing wild-type, R5L, G389R, or R406W mutant tau (Fig. 5A,B and Fig. 5-1), while the expression (Fig. 5C) and toxicity (Fig. 5D) of tau were similar to neuronal and glial expression.
Figure 5.

In vivo and in vitro aggregation of tau. A, Inclusions as identified by AT8 immunostaining in tissue sections from flies expressing tau using the pan-glial repo-GAL4 driver. Arrows indicate inclusions. Scale bar, 10 μm. B, Quantitative analysis of the number of inclusions in sections from the midportion of the medulla neuropil in control animals or in flies expressing human wild-type or FTDP-17 mutant forms of tau. Six brains were analyzed per genotype. C, Western blot analysis showing the expression of tau expressed in glia. The blot is reprobed for GAPDH to illustrate equivalent protein loading. Control is repo-GAL4/+ in A–C. D, Quantitative analysis of the number of neurons with activated caspase in the entire brains of control animals or in flies expressing human wild-type or FTDP-17 mutant forms of tau. Six brains were analyzed per genotype. Control is UAS-CD8-PARP-Venus/repo-GAL4. Full genotypes are provided in Figure 5-1. Flies are 30 d old in A–D. E, Polymerization of tau measured by thioflavin S staining. F, Electron micrographs of polymerization reaction mixtures. G–I, Quantification of polymerized tau protein in electron micrographs. The graphs display the total number of tau filaments per image (G), the mean length of filaments (in nanometers; H), and the total length of filaments (I). Images in the electron micrographs (F) were quantified using Image-Pro Plus version 6.0, as described in the Materials and Methods. ***p < 0.001, ANOVA with Tukey's HSD. Full statistical analysis is presented in Figure 5-1.
Full genotypes and statistical analysis. Download Figure 5-1, DOC file (178KB, doc)
We have previously shown that S352L mutant tau displays enhanced polymerization in vitro (Combs and Gamblin, 2012), potentially correlating with increased inclusion formation in vivo (Fig. 5A,B). However, these experiments were performed in the 2N4R tau backbone. We have demonstrated that the specific isoform of tau can play an important role in determining key biochemical characteristics of the protein, including aggregation potential (Carlson et al., 2007; Voss et al., 2012; Mutreja and Gamblin, 2017). To correlate more directly with our in vivo work, we thus expressed mutant forms of tau in the context of the 0N4R tau isoform, purified protein from bacterial lysates, and assessed aggregation of the wild-type and mutant tau isoforms. Using 2 μm tau in the presence of 75 μm arachidonic acid to promote polymerization (Carlson et al., 2007), we observed a trend toward increased aggregation of S320F and S352L mutant tau, as measured by thioflavin S fluorescence, compared with wild-type tau and the other mutant forms of tau modeled (Fig. 5E and Fig. 5-1). To examine the aggregates formed by each of the mutants in more detail, we performed electron microscopy. By electron microscopy, wild-type tau formed a mixture of longer filaments and smaller aggregates. Interestingly, although both S320F and S352L mutant tau showed more aggregation than wild-type tau, S320F formed more (Fig. 5F,G), relatively smaller filaments, while S352L promoted the formation of longer filaments (Fig. 5F,H,I). The other mutants studied, R5L, G389R, and R406W, had an aggregation profile more similar to wild type, which is consistent with a primary effect of the mutations on the phosphorylation, not the intrinsic aggregation, of tau.
We have previously demonstrated that phosphorylation-dependent stabilization of actin by tau is a critical downstream mediator of neurotoxicity (Fulga et al., 2007; DuBoff et al., 2012; Frost et al., 2016). We therefore assessed the degree of F-actin polymerization in brains from flies expressing tau transgenes in neurons using the elav-GAL4 driver. Staining for F-actin with fluorescently labeled phalloidin revealed a trend toward increased levels of F-actin in flies expressing human tau, including wild-type tau and all the FTDP-17 linked forms of mutant tau modeled. In addition, R5L, G389R, or R406W mutant tau had a significant increase in phalloidin staining compared with wild-type tau (Fig. 6A,B and Fig. 6-1). To assess actin stabilization using a different method, we used an F-actin ELISA, which uses a monoclonal antibody specific for F-actin. Similar to the results with phalloidin fluorescence, we demonstrated a trend toward increased levels of F-actin in brains from animals expressing R5L, G389R, or R406W using the F-actin ELISA (Fig. 6C and Fig. 6-1).
Figure 6.

Interaction of tau with actin in vivo and in vitro. A, F-actin staining with fluorescent phalloidin in whole fly brains from flies expressing tau using the pan-neuronal elav-GAL4 driver. Scale bar, 50 μm. B, Quantitative analysis of phalloidin fluorescence in the brains of control animals or flies expressing human wild-type or FTDP-17 mutant forms of tau. Three brains were analyzed per genotype. C, F-actin ELISA in homogenates of brains from control animals or flies expressing human wild-type or FTDP-17 mutant forms of tau. Each experiment was performed with two technical replicates. The experiments were repeated three times. *p < 0.05, ANOVA with Tukey's HSD. Full statistical analysis is presented in Figure 6-1. Control is elav-GAL4/+. Full genotypes are provided in Figure 6-1. Flies are 30 d old. D–F, Increased amounts of F-actin are recovered in the pellet following incubation with α-act as a positive control or with human wild-type or FTDP-17 mutant forms of tau (D), as assessed by quantitative analysis of the amount of pelleted actin (E) or protein bound in the pellet (F). BSA is used as a negative control. The experiment was repeated twice.
Full genotypes and statistical analysis. Download Figure 6-1, DOC file (100.5KB, doc)
Although previous data from our laboratory have suggested that phosphorylation of tau is a key determinant of actin-stabilizing activity in vivo (Fulga et al., 2007), FTDP-17-linked variants might additionally or alternatively have altered intrinsic ability to bind and stabilize F-actin. To address this possibility directly, we incubated purified F-actin filaments with tau in vitro and used low-speed centrifugation to pellet actin filaments (Fulga et al., 2007). We used α-actinin (α-act), a major actin cross-linking protein (Foley and Young, 2014), as a positive control, and BSA as a negative control. As expected, we observed increased amounts of pelleted actin following incubation with α-act and no significant bundling using BSA, and confirmed our prior observation (Fulga et al., 2007) of bundling activity of wild-type human tau in vitro (Fig. 6D–F). The amounts of actin recovered in the pellet (Fig. 6D,E) and protein bound in the pellet (Fig. 6D,F) were similar when comparing wild-type tau with the FTDP-17 mutant forms of tau studied here, which is consistent with phosphorylation of tau as the key determinant of actin binding and stabilization.
Proteostasis, or protein homeostasis, is a critical cellular response to the accumulation of misfolded and aggregated proteins and is controlled by autophagy and the unfolded protein response (Labbadia and Morimoto, 2015). We have previously demonstrated that both autophagy and the endoplasmic reticulum unfolded protein response are activated in response to human tau expression in aging fly neurons (Loewen and Feany, 2010; Merlo et al., 2014). To determine whether expression of the range of mutant forms of tau modeled in the current study similarly perturbs pathways controlling proteostasis, we used transgenic markers of autophagy and the unfolded protein response. The UAS-Atg8a-GFP reporter is commonly used to assess the activation of autophagy in Drosophila (Klionsky et al., 2016). Atg8a is the fly homolog of LC3. When we expressed UAS-Atg8a-GFP along with wild-type or mutant tau and stained the brains of aged flies for GFP, we observed increased numbers of GFP-immunoreactive puncta in flies expressing tau in neurons (Fig. 7A,B and Fig. 7-1), which is consistent with our prior observations (Merlo et al., 2014). Of note, the three most toxic versions of tau, R5L, G389R, and R406W, resulted in the greatest activation of autophagy, as monitored by our transgenic reporter.
Figure 7.
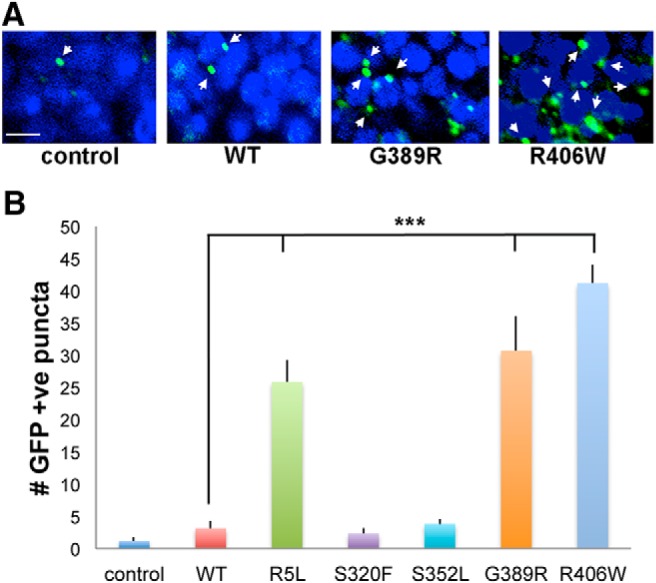
Autophagy in tau transgenic animals. A, Activation of autophagy in flies expressing tau using the pan-neuronal elav-GAL4 driver as monitored by the accumulation of the transgenic reporter UAS-Atg8a-GFP and immunostaining for GFP. Arrows indicate GFP-positive puncta. Scale bar, 3 μm. B, Quantitative analysis of the number of puncta in a section through the calyx of the mushroom body in the brains of control animals, or flies expressing human wild-type or FTDP-17 mutant forms of tau. Six brains were analyzed per genotype. Control is elav-GAL4/+; UAS-Atg8a-GFP/+. Full genotypes are provided in Figure 7-1. Flies are 30 d old. ***p < 0.001, ANOVA with Tukey's HSD. Full statistical analysis is presented in Figure 7-1.
Full genotypes and statistical analysis. Download Figure 7-1, DOC file (74.5KB, doc)
To assess the activity of the unfolded protein response in our transgenic flies, we used a UAS-Xbp1-EGFP reporter system developed by Ryoo et al. (2007). Xbp1 is a central transcription factor controlling the expression of a variety of genes integral to the expression of the unfolded protein response. In the reporter construct, EGFP is located after the IRE-1 splice site in Xbp1 such that EGFP is only in frame after Xbp1 has been spliced by IRE-1. Thus, the UAS-Xbp1-EGFP transgene acts as a reporter for the activation of the unfolded protein response because the expression of EGFP indicates that Xbp1 has been cleaved by IRE-1. When we expressed UAS-Xbp1-EFGP with human tau, we found induction of the reporter (Fig. 8A,B and Fig. 8-1) with expression of wild-type or mutant human tau in neurons, which is consistent with our prior results (Loewen and Feany, 2010). Further, we observed that variants of tau with enhanced neurotoxicity, R5L, G389R, and R406W, were most effective in promoting the activation of the unfolded protein response. Note that for the analysis of autophagy and the unfolded protein response, we monitored pathway activity in neurons because the neuronal-specific driver elav-GAL4 was used to express the reporter transgenes. Neuronal activation of the reporters is consistent with cell morphology (Figs. 7A, 8A) and with prior double-labeled immunofluorescence analyses (Loewen and Feany, 2010; Merlo et al., 2014).
Figure 8.
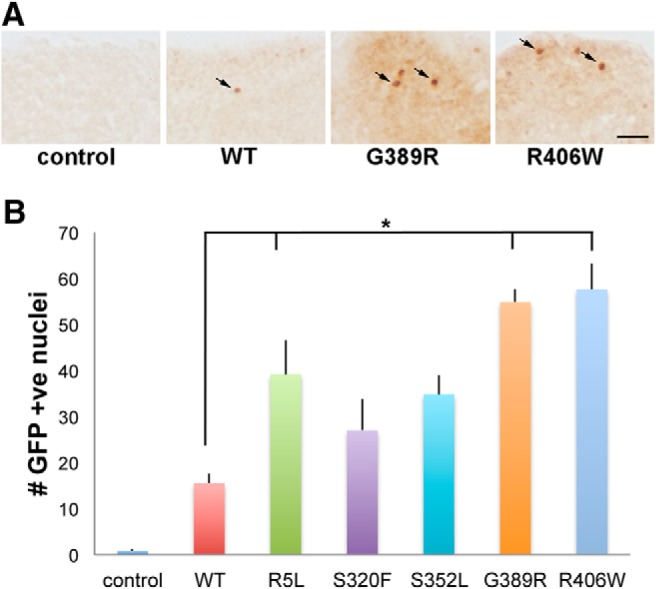
Unfolded protein response in tau transgenic animals. A, Neurons with activation of the unfolded protein response in flies expressing tau using the pan-neuronal elav-GAL4 driver as monitored by the transgenic reporter UAS-Xbp1-EGFP and immunostaining for GFP. GFP is directed to the nucleus via the Xbp1 nuclear localization sequence (Ryoo et al., 2007). Arrows indicate GFP-positive nuclei. Scale bar, 10 μm. B, Quantitative analysis of the number of neurons with activation of the unfolded protein response in the entire brains of control animals or flies expressing human wild-type or FTDP-17 mutant forms of tau. Six brains were analyzed per genotype. Control is elav-GAL4/+; UAS-Xbp1-EGFP/+. Full genotypes are provided in Figure 8-1. Flies are 30 d old. *p < 0.05, ANOVA with Tukey's HSD. Full statistical analysis is presented in Figure 8-1.
Full genotypes and statistical analysis. Download Figure 8-1, DOC file (74.5KB, doc)
Discussion
The 53 known mutations in tau that cause neurodegeneration and neurofibrillary tangle formation in the context of the inherited and severe form of frontotemporal dementia, FTDP-17, should provide important mechanistic clues into the biophysical and cell biological determinants of tau neurotoxicity. Such insights would be of direct relevance to the pathogenesis of FTDP-17 and might also inform our understanding of more common tauopathies, including Alzheimer's disease, which are characterized by the deposition of wild-type tau into insoluble aggregates. However, while studies to date have tended to provide broad support for the loss of microtubule binding and increased aggregation in mediating the toxic effects of FTDP-17-linked missense mutations in tau (Ghetti et al., 2015), not all mutations have clear effects on microtubule binding or aggregation of tau in vitro. Thus, another specific feature of the protein might drive toxicity in vivo. Alternatively, there may be multiple mechanisms by which mutations in tau promote neurodegeneration. To address these possibilities experimentally, we have used an in vivo model of tau toxicity that allows the expression of multiple tau variants at the same levels and the assessment of relevant neuropathological and biochemical phenotypes in parallel.
We find a striking correlation of neurotoxicity with phosphorylation of tau in FTDP-17 mutants (Figs. 2, 3). These findings fit well with prior results in Drosophila tauopathy models demonstrating that altering expression of kinases and phosphatases modifies tau neurotoxicity (Jackson et al., 2002; Shulman and Feany, 2003; Steinhilb et al., 2007b). In addition, the mutation of SP and TP sites to alanine to block phosphorylation reduces tau neurotoxicity, while the mutation of the same SP and TP sites to glutamate to mimic phosphorylation strongly promotes tau neurotoxicity in our transgenic Drosophila (Fulga et al., 2007; Steinhilb et al., 2007a, 2007b). Our results are also consistent with the analysis of patient tissue demonstrating increased phosphorylation of tau in postmortem brain from patients with FTDP-17 (Reed et al., 1997, 2001; Miyasaka et al., 2001). Interestingly, FTDP-17 mutant tau tends to be less phosphorylated compared with wild-type tau in cell culture (Mack et al., 2001; DeTure et al., 2002), which may reflect differences between in vivo and in vitro conditions, and emphasizes the role of in vivo modeling to complement biochemical and cell culture studies. It is not currently clear how the mutations we studied promote the phosphorylation of tau. Increased propensity for phosphorylation at disease-linked sites may be an intrinsic property of FTDP-17 mutant tau. Alonso Adel et al. (2004) demonstrated a substantially increased rate and steady-state in vitro phosphorylation of FTDP-17 tau variants, including the R406W mutant studied here. Although the G389R and R406W mutations are in the C-terminal portion of the tau protein, near the proline-directed phosphorylation sites we study, the R5L mutation is in the N terminus of the protein. Thus, structural interactions between the N and C termini of tau, as are known to occur (Jeganathan et al., 2006), may influence kinase access and efficiency. Alternatively, FTDP-17 mutations may alter another aspect of structure, localization, or activity that secondarily influences phosphorylation.
In addition to a strong effect on increasing tau phosphorylation, FTDP-17 variants showed a trend toward altering total levels of tau (Fig. 2A,B and Fig. 2-1). Since mRNA expression of tau variants was similar (Fig. 1), as expected given the site-directed transgenesis approach, we used the GeneSwitch-regulated expression system to determine whether the stability of the mutant tau proteins was altered in neurons. Consistent with results from Western blot analysis (Fig. 2A), we found a trend toward increased stability of the mutant tau variants (R5L, G389R, and R406W) that also tended to have the higher levels of total tau (Fig. 2A,B and Fig. 2-1). The significantly increased stability of the pseudohyperphosphorylated E14 tau mutant (Fig. 2A,B and Fig. 2-1) supports a role for proline-directed serine and threonine phosphorylation of tau in promoting the stability of the protein in vivo. Further, the enhanced stability of the E14 variant is consistent with a primary effect of FTDP-17 mutants on phosphorylation with consequent altered stability. Interestingly, our finding that tau turns over in ∼6 d in the fly brain is similar to the half-life of ∼10 d reported in a transgenic mouse model of tauopathy (Yamada et al., 2015) and is significantly greater than the half-life of tau typically observed in cell culture (David et al., 2002; Lim et al., 2008).
In contrast to flies expressing R5L, G389R, or R406W mutant tau, animals expressing S320F or S352L mutant tau showed less neurotoxicity and activation of downstream pathways in our in vivo models. The S320F mutation has been linked to frontotemporal dementia of relatively early onset in multiple studies (Rosso et al., 2002, 2003; Rohrer et al., 2009, 2010). Interestingly, the S352L mutation is apparently manifest only in the homozygous state, with heterozygous carriers being normal (Nicholl et al., 2003), which is possibly consistent with a more modest clinical effect of the mutation. Strikingly, both S320F and S352L mutant tau aggregate more readily than the other FTDP-17 variants we studied into large inclusions in transgenic flies (Fig. 5A,B). The enhanced in vivo propensity for aggregate formation correlates well with in vitro aggregation as measured by thioflavin S fluorescence (Fig. 5E). Of note, although total aggregation is enhanced in vitro in both mutants, S320F mutant tau forms more and shorter aggregates, while S352L mutant tau aggregates into long filaments (Fig. 5F–I). Our transgenic data suggest that both types of aggregates deposit effectively into large inclusions in vivo. Further, since the propensity toward increased aggregation in vitro is associated with relatively less neurotoxicity and the formation of large inclusion bodies in vivo, our finding raises the possibility that the therapeutic approaches aimed at promoting the incorporation of smaller, toxic aggregates into larger, less toxic, or inert inclusion bodies may be beneficial. Prior work in our laboratory (Wittmann et al., 2001; Colodner and Feany, 2010) and by other investigators (Santacruz et al., 2005; de Calignon et al., 2010) is consistent with the dissociation of neurofibrillary tangles from neurotoxicity.
The degree to which different FTDP-17-linked mutations activate the same downstream cascade of neurotoxicity, and the relationship of the mechanism of toxicity of mutant tau to that of the wild-type protein, have been unclear. Here we significantly expand our prior work to provide evidence for shared mechanisms of toxicity. We have previously shown that wild-type and R406W mutant human tau activate common downstream mechanisms of toxicity (Khurana et al., 2006; Dias-Santagata et al., 2007; Fulga et al., 2007; Loewen and Feany, 2010; Frost et al., 2014, 2016). We now confirm and extend these findings by demonstrating that F-actin is stabilized by wild-type tau and by all the mutant forms of tau we study, both in vivo (Fig. 6A,B) and in vitro (Fig. 6D–F), and that stabilization of actin correlates well with neurotoxicity. A separate study by another group suggests that two additional FTDP-17 variants, P301L and V337M, likewise promote tau neurotoxicity in vivo through excess stabilization of F-actin (Zhou et al., 2017). The increased in vivo stabilization of the actin cytoskeleton observed with the expression of the most toxic forms of tau most likely reflects hyperphosphorylation of these mutants. We have previously demonstrated that phosphorylation is upstream of, and strongly promotes, stabilization of actin by tau (Fulga et al., 2007). In our current work, we see no increase of in vitro bundling of tau with more toxic versus less toxic variants of tau (Fig. 6D–F), which is consistent with a primary effect of phosphorylation on actin stabilization. Other downstream pathways modulating tau neurotoxicity are similarly activated by all the forms of tau we study here, in proportion with in vivo toxicity including stress pathway activation (Fig. 4), increased autophagy (Fig. 7) and induction of the unfolded protein response (Fig. 8). Thus, despite differences in the relative toxicity of different isoforms, studying the pathogenesis of a single form of mutant tau, as is often done experimentally, is likely to be highly relevant to the larger group of tauopathies, including Alzheimer's disease and other tauopathies characterized by the aggregation and deposition of wild-type tau. These results may have important therapeutic implications because treatments that are effective for one tauopathy may work in a wide range of disorders characterized by abnormal tau deposition.
Footnotes
This work was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant R01-NS-083391. Stocks obtained from the Bloomington Drosophila Stock Center were used in this study. We thank Darren Williams, Thomas Neufeld, and Hyung Don Ryoo for providing fly stocks; and Peter Davies for sharing the PHF1 antibody.
The authors declare no competing financial interests.
References
- Alonso Adel C, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K (2004) Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem 279:34873–34881. 10.1074/jbc.M405131200 [DOI] [PubMed] [Google Scholar]
- Bateman JR, Lee AM, Wu CT (2006) Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 173:769–777. 10.1534/genetics.106.056945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E, Vaessin H, Shepherd S, Lee K, McCall K, Barbel S, Ackerman L, Carretto R, Uemura T, Grell E (1989) Searching for pattern and mutation in the Drosophila genome with a P-lacZ vector. Genes Dev 3:1273–1287. 10.1101/gad.3.9.1273 [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104:3312–3317. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodea LG, Eckert A, Ittner LM, Piguet O, Götz J (2016) Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J Neurochem 138:71–94. 10.1111/jnc.13600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415. [DOI] [PubMed] [Google Scholar]
- Carlson SW, Branden M, Voss K, Sun Q, Rankin CA, Gamblin TC (2007) A complex mechanism for inducer mediated tau polymerization. Biochemistry 46:8838–8849. 10.1021/bi700403a [DOI] [PubMed] [Google Scholar]
- Colodner KJ, Feany MB (2010) Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J Neurosci 30:16102–16113. 10.1523/JNEUROSCI.2491-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs B, Gamblin TC (2012) FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry 51:8597–8607. 10.1021/bi3010818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C (2012) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33:1340–1344. 10.1002/humu.22117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG (2002) Proteasomal degradation of tau protein. J Neurochem 83:176–185. 10.1046/j.1471-4159.2002.01137.x [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464:1201–1204. 10.1038/nature08890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTure M, Ko LW, Yen S, Nacharaju P, Easson C, Lewis J, van Slegtenhorst M, Hutton M, Yen SH (2000) Missense tau mutations identified in FTDP-17 have a small effect on tau-microtubule interactions. Brain Res 853:5–14. 10.1016/S0006-8993(99)02124-1 [DOI] [PubMed] [Google Scholar]
- DeTure M, Ko LW, Easson C, Yen S-H (2002) Tau assembly in inducible transfectants expressing wild-type or FTDP-17 tau. Am J Pathol 161:1711–1722. 10.1016/S0002-9440(10)64448-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB (2007) Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest 117:236–245. 10.1172/JCI28769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B, Götz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75:618–632. 10.1016/j.neuron.2012.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146:1388–1396. [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Dickson DW (1996) Neurodegenerative disorders with extensive tau pathology: a comparative study and review. Ann Neurol 40:139–148. 10.1002/ana.410400204 [DOI] [PubMed] [Google Scholar]
- Foley KS, Young PW (2014) The non-muscle functions of actinins: an update. Biochem J 459:1–13. 10.1042/BJ20131511 [DOI] [PubMed] [Google Scholar]
- Frost B, Hemberg M, Lewis J, Feany MB (2014) Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci 17:357–366. 10.1038/nn.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Bardai FH, Feany MB (2016) Lamin dysfunction mediates neurodegeneration in tauopathies. Curr Biol 26:129–136. 10.1016/j.cub.2015.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB (2007) Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9:139–148. 10.1038/ncb1528 [DOI] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, Berry RW, Binder LI (2000) In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry 39:6136–6144. 10.1021/bi000201f [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI (2003) Tau polymerization: role of the amino terminus. Biochemistry 42:2252–2257. 10.1021/bi0272510 [DOI] [PubMed] [Google Scholar]
- Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M (2015) Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging: MAPT mutations and FTD. Neuropathol Appl Neurobiol 41:24–46. 10.1111/nan.12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437:207–210. 10.1016/S0014-5793(98)01217-4 [DOI] [PubMed] [Google Scholar]
- Hegde VR, Vogel R, Feany MB (2014) Glia are critical for the neuropathology of complex I deficiency in Drosophila. Hum Mol Genet 23:4686–4692. 10.1093/hmg/ddu188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282:1914–1917. 10.1126/science.282.5395.1914 [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34:509–519. 10.1016/S0896-6273(02)00706-7 [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff H.-J., and Mandelkow E (2006) Global hairpin folding of tau in solution. Biochemistry 45:2283–2293. 10.1021/bi0521543 [DOI] [PubMed] [Google Scholar]
- Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, Neufeld TP (2008) The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol 181:655–666. 10.1083/jcb.200712051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinar DP, Balija MBG, Kügler S, Opazo F, Rezaei-Ghaleh N, Wender N, Kim HY, Taschenberger G, Falkenburger BH, Heise H, Kumar A, Riedel D, Fichtner L, Voigt A, Braus GH, Giller K, Becker S, Herzig A, Baldus M, Jäckle H, et al. (2009) Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J 28:3256–3268. 10.1038/emboj.2009.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB (2006) TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol 16:230–241. 10.1016/j.cub.2005.12.042 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12:1–222. 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, Cairns NJ, Crary JF, Duyckaerts C, Ghetti B, Halliday GM, Ironside JW, Love S, Mackenzie IR, Munoz DG, Murray ME, Nelson PT, Takahashi H, Trojanowski JQ, Ansorge O, et al. (2016) Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131:87–102. 10.1007/s00401-015-1509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84:435–464. 10.1146/annurev-biochem-060614-033955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, Finn G, Pastorino L, Lee VM, Lu KP (2008) Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest 118:1877–1889. 10.1172/JCI34308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen CA, Feany MB (2010) The unfolded protein response protects from tau neurotoxicity in vivo. PloS One 5:e13084. 10.1371/journal.pone.0013084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luheshi LM, Tartaglia GG, Brorsson AC, Pawar AP, Watson IE, Chiti F, Vendruscolo M, Lomas DA, Dobson CM, Crowther DC (2007) Systematic in vivo analysis of the intrinsic determinants of amyloid beta pathogenicity. PLoS Biol 5:e290. 10.1371/journal.pbio.0050290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack TG, Dayanandan R, Van Slegtenhorst M, Whone A, Hutton M, Lovestone S, Anderton BH (2001) Tau proteins with frontotemporal dementia-17 mutations have both altered expression levels and phosphorylation profiles in differentiated neuroblastoma cells. Neuroscience 108:701–712. 10.1016/S0306-4522(01)00434-1 [DOI] [PubMed] [Google Scholar]
- Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N (2008) Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat Genet 40:476–483. 10.1038/ng.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12:557–570. 10.1101/gad.12.4.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192:1–15. 10.1002/jcp.10119 [DOI] [PubMed] [Google Scholar]
- Merlo P, Frost B, Peng S, Yang YJ, Park PJ, Feany M (2014) p53 prevents neurodegeneration by regulating synaptic genes. Proc Natl Acad Sci U S A 111:18055–18060. 10.1073/pnas.1419083111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka T, Morishima-Kawashima M, Ravid R, Heutink P, van Swieten JC, Nagashima K, Ihara Y (2001) Molecular analysis of mutant and wild-type tau deposited in the brain affected by the FTDP-17 R406W mutation. Am J Pathol 158:373–379. 10.1016/S0002-9440(10)63979-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja Y, Gamblin TC (2017) Optimization of in vitro conditions to study the arachidonic acid induction of 4R isoforms of the microtubule-associated protein tau. In: Methods in cell biology (Feinstein S.C., LaPointe N.E., eds.), pp 65–88. Amsterdam: Elsevier. [DOI] [PubMed] [Google Scholar]
- Nicholl DJ, Greenstone MA, Clarke CE, Rizzu P, Crooks D, Crowe A, Trojanowski JQ, Lee VM, Heutink P (2003) An English kindred with a novel recessive tauopathy and respiratory failure. Ann Neurol 54:682–686. 10.1002/ana.10747 [DOI] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H (2001) A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A 98:12596–12601. 10.1073/pnas.221303298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825. 10.1002/ana.410430617 [DOI] [PubMed] [Google Scholar]
- Rankin CA, Sun Q, Gamblin TC (2005) Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Brain Res Mol Brain Res 138:84–93. 10.1016/j.molbrainres.2005.04.012 [DOI] [PubMed] [Google Scholar]
- Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, Van Hoesen GW, Schelper RL, Talbot CJ, Wragg MA, Trojanowski JQ (1997) Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 42:564–572. 10.1002/ana.410420406 [DOI] [PubMed] [Google Scholar]
- Reed LA, Wszolek ZK, Hutton M (2001) Phenotypic correlations in FTDP-17. Neurobiol Aging 22:89–107. 10.1016/S0197-4580(00)00202-5 [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, Mackenzie IR, Warren JD, de Silva R, Holton J, Revesz T, Hardy J, Mead S, Rossor MN (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456. 10.1212/WNL.0b013e3181bf997a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, Rossor MN, Warren JD (2010) Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53:1070–1076. 10.1016/j.neuroimage.2009.12.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SM, van Herpen E, Deelen W, Kamphorst W, Severijnen LA, Willemsen R, Ravid R, Niermeijer MF, Dooijes D, Smith MJ, Goedert M, Heutink P, van Swieten JC (2002) A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick's disease. Ann Neurol 51:373–376. 10.1002/ana.10140 [DOI] [PubMed] [Google Scholar]
- Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, de Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, van Duijn CM, Heutink P, van Swieten JC (2003) Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 126:2016–2022. 10.1093/brain/awg204 [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Domingos PM, Kang MJ, Steller H (2007) Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J 26:242–252. 10.1038/sj.emboj.7601477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, Feany MB (2003) Genetic modifiers of tauopathy in Drosophila. Genetics 165:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Bird TD, Ghetti B (1998) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 8:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB (2007a) Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell 18:5060–5068. 10.1091/mbc.E07-04-0327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Mulkearns EE, Shulman JM, Biernat J, Mandelkow EM, Feany MB (2007b) S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res 85:1271–1278. 10.1002/jnr.21232 [DOI] [PubMed] [Google Scholar]
- Szendrei GI, Lee VM, Otvos L Jr (1993) Recognition of the minimal epitope of monoclonal antibody Tau-1 depends upon the presence of a phosphate group but not its location. J Neurosci Res 34:243–249. 10.1002/jnr.490340212 [DOI] [PubMed] [Google Scholar]
- Voss K, Combs B, Patterson KR, Binder LI, Gamblin TC (2012) Hsp70 alters tau function and aggregation in an isoform specific manner. Biochemistry 51:888–898. 10.1021/bi2018078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hagemann TL, Kalwa H, Michel T, Messing A, Feany MB (2015) Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat Commun 6:8966. 10.1038/ncomms9966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hagemann TL, Messing A, Feany MB (2016) An in vivo pharmacological screen identifies cholinergic signaling as a therapeutic target in glial-based nervous system disease. J Neurosci 36:1445–1455. 10.1523/JNEUROSCI.0256-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW (2006) Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci 9:1234–1236. 10.1038/nn1774 [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- Yamada K, Patel TK, Hochgräfe K, Mahan TE, Jiang H, Stewart FR, Mandelkow EM, Holtzman DM (2015) Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol Neurodegener 10:55. 10.1186/s13024-015-0052-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, Wang Y-C, Swerts J, Beyens J, Miskiewicz K, Vilain S, Dewachter I, Moechars D, De Strooper B, Spires-Jones TL, De Wit J, Verstreken P (2017) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun 8:15295. 10.1038/ncomms15295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Lee HG, Chao M, Nunomura A, Tabaton M, Petersen RB, Perry G, Smith MA (2003) Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxid Redox Signal 5:571–576. 10.1089/152308603770310220 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full genotypes. Download Figure 1-1, DOC file (47.5KB, doc)
Full genotypes and statistical analyses. Download Figure 2-1, DOC file (153KB, doc)
Full genotypes and statistical analysis. Download Figure 3-1, DOC file (75KB, doc)
Full genotypes and statistical analysis. Download Figure 4-1, DOC file (75KB, doc)
Full genotypes and statistical analysis. Download Figure 5-1, DOC file (178KB, doc)
Full genotypes and statistical analysis. Download Figure 6-1, DOC file (100.5KB, doc)
Full genotypes and statistical analysis. Download Figure 7-1, DOC file (74.5KB, doc)
Full genotypes and statistical analysis. Download Figure 8-1, DOC file (74.5KB, doc)