

Abstract
Pain is a common and severe symptom in multiple sclerosis (MS), a chronic inflammatory and demyelinating disease of the CNS. The neurobiological mechanism underlying MS pain is poorly understood. In this study, we investigated the role of Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) in driving chronic pain in MS using a mouse experimental autoimmune encephalomyelitis (EAE) model. We found that spinal CaMKIIα activity was enhanced in EAE, correlating with the development of ongoing spontaneous pain and evoked hypersensitivity to mechanical and thermal stimuli. Prophylactic or acute administration of KN93, a CaMKIIα inhibitor, significantly reduced the clinical scores of EAE and attenuated mechanical allodynia and thermal hyperalgesia in EAE. siRNA targeting CaMKIIα reversed established mechanical and thermal hypersensitivity in EAE mice. Furthermore, CaMKIIαT286A point mutation mice showed significantly reduced EAE clinical scores, an absence of evoked pain, and ongoing spontaneous pain when compared with littermate wild-type mice. We found that IL-17 is responsible for inducing but not maintaining mechanical and thermal hyperalgesia that is mediated by CaMKIIα signaling in EAE. Together, these data implicate a critical role of CaMKIIα as a cellular mechanism for pain and neuropathy in multiple sclerosis and IL-17 may act upstream of CaMKIIα in the generation of pain.
SIGNIFICANCE STATEMENT Pain is highly prevalent in patients with multiple sclerosis (MS), significantly reducing patients' quality of life. Using the experimental autoimmune encephalomyelitis (EAE) model, we were able to study not only evoked hyperalgesia, but also for the first time to demonstrate spontaneous pain that is also experienced by patients. Our study identified a role of spinal CaMKIIα in promoting and maintaining persistent ongoing spontaneous pain and evoked hyperalgesia pain in EAE. We further demonstrated that IL-17 contributes to persistent pain in EAE and functions as an upstream regulator of CaMKIIα signaling. These data for the first time implicated CaMKIIα and IL-17 as critical regulators of persistent pain in EAE, which may ultimately offer new therapeutic targets for mitigating pain in multiple sclerosis.
Keywords: CaMKIIα, EAE, kinase, multiple sclerosis, pain, phosphorylation
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease of the CNS with manifestations of neuroinflammation and demyelination, affecting >2.3 million people worldwide. It is the most common disabling neurological disease in people between 20 and 45 years of age (Ragonese et al., 2008). Pain is prevalent in patients with MS, even at the early stage of the disease, with a reported prevalence ranging from 29% to 86% (O'Connor et al., 2008; Solaro and Uccelli, 2011). About one-third of MS patients select pain as one of the worst symptoms, disrupting daily activity, mood, and recreation and severely impacting quality of life (Kalia and O'Connor, 2005; Iannitti et al., 2014). Pain in MS has various types, including musculoskeletal pain and neuropathic pain. The latter commonly presents as dysesthetic extremity pain, trigeminal neuralgia, and Lhermitte's phenomenon (Iannitti et al., 2014; Khan and Smith, 2014).
Pain is also the most commonly treated symptom in MS, accounting for 30% of all symptomatic treatments. However, pain management received low satisfaction among MS patients, with only 61% patients reporting pain relief, the extent of which is <40% (Grau-López et al., 2011). Over the past decades, advances have been made in understanding the neurobiological mechanisms of motor dysfunction in MS, but much fewer advances have been made in understanding those of MS-associated pain. Lacking basic understanding and comprehensive clinical studies, pain management in MS is often driven by anecdotal reports and findings in other forms of chronic pain.
The experimental autoimmune encephalomyelitis (EAE) model is one of the most commonly used rodent models to study MS, mimicking not only the pathological symptoms but also behavioral symptoms of MS, including hyperalgesia (Thibault et al., 2011; Lu et al., 2012). Although several pharmacological studies have been performed studying evoked hyperalgesia (Aicher et al., 2004; Olechowski et al., 2009; Yuan et al., 2012), none has addressed the nonevoked ongoing pain that matters the most to patients (He et al., 2016b).
In this study, we examined the role of spinal Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα), which has been previously implicated in other chronic pain types, in ongoing and evoked pain in the EAE model. CaMKIIα is a serine/threonine protein kinase that is highly expressed in the CNS and is involved in synaptic plasticity and a number of physiological processes including long-term potentiation in the hippocampus (Giese et al., 1998).
Moreover, we studied an upstream mechanism that may turn on CaMKIIα in MS. The T-helper 17 (Th17) cell, a novel CD4+ T-cell subset, and interleukin (IL)-17, the major secretion of Th17 cells, have been implicated as having critical roles in MS pathogenesis. The development of EAE was significantly suppressed in IL-17−/− mice, and adoptive transfer of IL-17−/− CD4+ cells was inefficient in inducing EAE in recipient mice (Komiyama et al., 2006). Furthermore, brain tissue samples from MS patients showed an increased level of IL-17 mRNA, and IL-17 producing cells are significantly increased in active rather than inactive areas of MS (Lock et al., 2002; Tzartos et al., 2008). Dysregulation of IL-17 has also been implicated in the generation of pain in various neuropathic and inflammatory pain states. In a neuropathic pain model, IL-17−/− mice displayed significantly decreased neuroinflammatory responses and also decreased pain hypersensitivity following peripheral nerve injury (Kim and Moalem-Taylor, 2011). In another study of a rat inflammatory pain model (Meng et al., 2013), blockade of IL-17 action using IL-17 antibody was able to attenuate complete Freund's adjuvant (CFA)-induced hyperalgesia. Despite the essential role of IL-17 in the pathogenesis of multiple sclerosis, it is unclear whether IL-17 is also an initiating factor in the development of pain in multiple sclerosis and whether CaMKII contributes to the function of IL-17 in EAE-induced pain. In this current study, we investigated the possible role of CaMKII and IL-17 in the initiation and maintenance of pain in EAE mice.
Materials and Methods
Animals.
Female C57BL/6N mice (10 weeks old) were purchased from Harlan Laboratories and acclimated to the university animal facility for 1 week before the experiments. For CaMKIIαT286A mutant mice, breeders were generously provided by Dr. Alcino Silva, University of California, Los Angeles (Los Angeles, CA; Giese et al., 1998; Chen et al., 2010; He et al., 2016a). These mice have been crossed with C57BL/6J mice for 10 generations. Crossbreeding between heterozygous mice was used to generate female homozygous mutant mice and littermate wild-type mice for this study. Normal growth and reproduction were found in both genotypes. Genotyping was performed by PCR using primers (5′-CTGTACCAGCAGATCAAAGC-3′, 5′-ATCACTAGCACCATGTGGTC-3′; Chen et al., 2010; He et al., 2016a).
Mice were housed in standard conditions with a 14/10 h light/dark cycle (5:00 A.M. to 7:00 P.M.) and allowed free access to food and water. All experiments were performed during the light cycle. Mice were grouped randomly, and the testing investigator was blind to the group information during the behavioral and biochemical experiments. All experiments were performed in accordance with polices and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines after approval by the University of Illinois Institutional Animal Care and Use Committee.
Induction of EAE.
EAE was induced in mice by subcutaneous injections of myelin oligodendrocyte glycoprotein 35–55 (MOG35–55) emulsion (0.1 ml/site) in the upper back and the base of the tail, followed by intraperitoneal injections of pertussis toxin (375 ng in 0.1 ml PBS) at 2 and 24 h after MOG35–55 emulsion. Pertussis toxin and MOG35–55 emulsion (MEVGWYRSPFSRVVHLYRNGK, emulsified to 1 mg/ml in CFA containing 2 mg/ml dead mycobacterium tuberculosis H37Ra, EK-2110) were purchased from Hooke Laboratories. Control group mice received only CFA (CK-2110, Hooke Laboratories) and pertussis toxin.
Clinical assessment of EAE.
Mice were observed daily after immunization. Body weight and clinical scores were recorded for 28 d. Scoring criteria are as follows: 0, normal healthy mouse; 1, limp tail; 2, legs are not spread apart when picked up by base of tail; 3, complete paralysis of hind legs; 4, partial front leg paralysis and mouse is minimally moving around the cage; 5, dead or mouse is killed due to severe paralysis. “In-between” scores (0.5, 1.5, 2.5, 3.5, and 4.5) were given to mice with the clinical signs between two defined scores. The investigator who assigned clinical scores was blind to the treatment and group information.
Rotarod test.
To examine locomotor activity, a rotarod test was conducted as described previously (Chen et al., 2010). Mice were habituated to the rotarod at a fixed speed (4 rpm) for 60 s on 1 day (Model Series 8, IITC Life Science). For testing, mice were placed on a rod accelerating from 4 to 40 rpm over 300 s, and the time to fall off the rotarod was recorded. Three trials were conducted, and times were averaged for data analysis. The baseline level was determined the day before immunization, and mice were tested every 3 d postimmunization (p.i.).
Mechanical sensitivity.
The mechanical threshold was assessed using calibrated von Frey filaments (Stoelting) as previously described (Chaplan et al., 1994; Luo et al., 2008; Chen et al., 2010). Briefly, mice were acclimated in individual Plexiglas containers on wire mesh. The midplantar hindpaw was touched with one of a series of von Frey filaments perpendicularly for 5 s or until a withdrawal response noted. The up-down paradigm was used to determine 50% probability of paw withdrawal threshold (Dixon, 1980; Chaplan et al., 1994; Luo et al., 2008).
Thermal sensitivity.
The sensitivity to thermal stimuli was measured by the use of a plantar tester (model 7372, Ugo Basile) as described previously (Hargreaves et al., 1988; Chen et al., 2010). Mice were placed in a transparent enclosure with a glass floor and allowed to acclimate for 15 min. The radiant heat was applied to the central portion of plantar surface of the mouse hindpaw, and paw withdrawal latency was recorded. A cutoff time of 20 s was applied to prevent tissue damage.
Drug and small interfering RNA administration.
2-[N-(2-hydroxyethyl)-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine; KN93) and 2-[N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-2-propenyl-N-methylbenzylamine (KN92) were purchased from Calbiochem. KN93 or KN92 was administered intrathecally prophylactically on Day 1 p.i. through Day 8 p.i. or acutely on Day 8 p.i. Intrathecal injections were performed as described previously (Chen et al., 2010) by percutaneous puncture through the L5–L6 intervertebral space. CaMKIIα targeting small interfering RNA (siRNA; 5′-CACCACCAUUGAGGACGAAdTdT-3′, 3′-dTdTGUGGUGGUAACUCCUGCUU-5′) or Stealth RNAi-negative control (Invitrogen) was administered on Day 5 p.i. to 7 p.i. (2 μg, twice daily, i.t.). These oligos were mixed with the transfection reagent i-Fect (Neuromics), in a ratio of 1:5 (w/v). Mice were pretreated with IL-17 antibody (5 μg, i.t.; Santa Cruz Biotechnology) 24 and 2 h before immunization and on Day 1, 2, and 3 p.i. or were treated intrathecally acutely on Day 8 p.i. Mice also received recombinant IL-17 protein (100 ng, i.t.; Prospec-Tany TechnoGene) to directly induce pain.
Conditioned place preference.
Conditioned place preference (CPP) testing was performed using the CPP apparatus consisting of three Plexiglas chambers separated by manual doors (San Diego Instruments), as previously described (He et al., 2012; Corder et al., 2013; He and Wang, 2015). Two equal-sized end chambers, connected by a smaller center chamber, were distinguished only by the texture of the floor (rough vs smooth) and wall pattern (vertical vs horizontal stripes). Preconditioning was conducted across 3 d (Days 5 to 7 p.i.) when mice were habituated to the CPP box with free access to all chambers for 30 min. On the last day of preconditioning (Day 7 p.i.), a bias test was performed to determine whether a preexisting chamber preference existed. Those mice that spent >80% or <20% of time in any particular chamber were considered to possess preexisting bias and were excluded from further experiments. On conditioning day (Day 8 p.i.), mice were first given vehicle (saline) intrathecally and were placed immediately in a randomly chosen chamber for 15 min in the morning. Four hours later, mice received lidocaine (0.04% in 5 μl of saline, i.t.) and placed in the other chamber for 15 min in the afternoon. Mice were only allowed to stay in the paired chamber without access to other chambers during the conditioning. Twenty hours after the afternoon pairing, preference tests were performed as mice were placed into the middle chamber and allowed free access to all chambers. The movement of mice and the time spent in each chamber were automatically recorded for 15 min by photobeam breaks. The difference score was calculated as test time − preconditioning time spent in the lidocaine-paired chamber.
Immunoblotting.
For mice treated with prophylactic or acute KN93 administration, tissue samples were collected on day 8 p.i., 2 h after KN93 treatment. For mice treated with CaMKIIα siRNA, tissues were collected on day 8, 12 h after the last dose of siRNA treatment. For consistency, lumbar spinal sections were quickly dissected on ice from mice killed to determine CaMKIIα activation [phosphorylated CaMKIIα (pCaMKII) level] using Western blotting analysis, as described previously (Tang et al., 2006; Chen et al., 2009). In brief, tissues were homogenized in ice-cold RIPA buffer in the presence of phosphatase inhibitors and protease inhibitors. The homogenates were centrifuged, and the protein content in supernatant was measured using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). Samples were then separated by SDS-PAGE and electrotransferred onto polyvinylidene difluoride membrane for Western blotting analysis. Antibodies including a rabbit anti-Thr286-pCaMKIIα antibody (1:1000; Santa Cruz Biotechnology), a mouse anti-β-actin antibody (1:10,000; Thermo Fisher Scientific), and corresponding horseradish peroxidase conjugate secondary antibodies (1:10,000; Santa Cruz Biotechnology) were used. The membrane was developed using an enhanced chemiluminescence detection system (ECL; Pierce Biotechnology). The ECL signals were captured by a ChemiDoc Imaging System (Bio-Rad) and analyzed using the Quantity One program (Bio-Rad). Levels of CaMKIIα activation were expressed as the ratio of the optical densities of pCaMKIIα to those of β-actin.
Immunofluorescence.
Immunofluorescence staining of spinal CaMKIIα was performed as previously described. Mice were deeply anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.) and perfused with 10 ml of ice-cold PBS, pH 7.4, followed by 20 ml of 4% paraformaldehyde solution. The lumbar spinal cords were removed and postfixed in 4% paraformaldehyde at 4°C for 18 h and gradually dehydrated in 10% sucrose for 6 h, 20% sucrose for 12 h, and 30% sucrose for 24 h at 4°C. The tissue was sliced to 20 μm thickness using a cryostat. Floated sections were incubated with the rabbit anti-Thr286-pCaMKIIα antibody (1:1000) at 4°C overnight and were followed by an incubation with Alexa Fluor 488-conjugated secondary antibody (Jackson ImmunoResearch). The fluorescence signals of the spinal cord sections were imaged by an inverted fluorescent microscope (Olympus) and quantified using the MetaMorph Imaging Software (Universal Imaging).
Statistics analysis.
All data are presented as the mean ± SEM. Comparisons between groups were analyzed using a two-way ANOVA followed by the Bonferroni post hoc test. For Western blotting and immunofluorescence results, a one-way ANOVA was followed by the Dunnett's post hoc test. Difference scores were analyzed using the paired t test by comparing the differences between test time and preconditioning time in each chamber for each mouse. Statistical significance was established at the 95% confidence limit.
Results
Prophylactic inhibition of CaMKIIα by KN93 attenuated clinical symptoms and hyperalgesia in EAE
As expected, MOG35–55 delivered in CFA induced EAE in female C57BL/6N mice (Fig. 1). Clinical symptoms were scored on a scale of 0 to 5. On day 12 p.i., EAE mice started to show limp tail and signs of paralysis, and the clinical score peaked on day 16 p.i. (Fig. 1A; 2.8 ± 0.5, p < 0.001, n = 8). The motor impairment revealed by rotarod test was consistent with the clinical score observation [12.2 ± 10.5 vs 105.3 ± 14.1 s (n = 8); p < 0.001; Fig. 1B].
Figure 1.

CaMKIIα inhibition by prophylactic KN93 administration reduced clinical symptoms and attenuated hyperalgesia in EAE. Separate groups of eight mice were treated with KN93 (45 nmol, i.t.) or saline from Day 1 to Day 8 along with the immunization with MOG35-55 on Day 0 to induce EAE. A–D, Prophylactic KN93 administration (45 nmol, i.t.) significantly attenuated clinical symptoms (A), locomotor impairment (B), mechanical allodynia (C), and thermal hyperalgesia (D). Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham immune group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the saline-treated EAE group.
We also determined temporal changes of sensory mechanical and thermal sensitivities using von Frey and Hargreaves tests. EAE mice showed significantly increased mechanical [0.05 ± 0.00 vs 0.78 ± 0.15 g (n = 8); p < 0.001; Fig. 1C] and thermal [5.75 ± 0.88 vs 12.87 ± 0.87 s (n = 8); p < 0.001; Fig. 1D] sensitivities in the early phase of the disease (Day 8 p.i.), compared with sham-immune mice. To avoid interference from motor impairment on the pain test, pain behavioral tests were stopped before the onset of clinical signs of EAE, which provided us a study window of 5–8 d.
We first investigated the prophylactic effects of KN93 on the clinical symptoms and pain behavior in EAE. Mice received KN93 (45 nmol/5 μl, i.t.) or an equal volume of saline once a day from day 1 to day 8. Mice treated with KN93 showed less mechanical hypersensitivity on Day 6 compared with saline-treated EAE mice [0.53 ± 0.07 vs 0.08 ± 0.02 g (n = 8); p < 0.05; Fig. 1C]. Thermal hyperalgesia was also reduced in EAE mice treated with KN93 [Day 6 p.i., 11.48 ± 1.42 vs 6.65 ± 0.93 s (n = 8); p < 0.05; Fig. 1D]. In addition, prophylactic treatment of KN93 reduced the clinical scores of EAE mice [Day 16 p.i.; 1.3 ± 0.3 vs 2.8 ± 0.5 (n = 8); p < 0.001; Fig. 1A] and increased the latencies of falling off the rotarod [Day 15 p.i.; 73.6 ± 16.4 vs 12.2 ± 10.5 s (n = 8); p < 0.05; Fig. 1B]. Therefore, long-term prophylactic inhibition of CaMKIIα by KN93 attenuated the development of both motor dysfunction and sensory hypersensitivity in EAE mice.
Acute inhibition of CaMKIIα by KN93 reversed mechanical allodynia and thermal hyperalgesia in EAE
To investigate whether KN93 can reverse the established mechanical and thermal hyperalgesia in EAE mice, we treated the mice with KN93 (45 nmol/5 μl, i.t.) acutely on day 8 p.i., when mechanical and thermal hyperalgesia had reached the peak level. KN92, an analog of KN93 that does not inhibit CaMKII, was used as a negative control. Treatment with KN93 significantly reduced already established mechanical [0.49 ± 0.12 g vs 0.05 ± 0.01 g (n = 8); p < 0.05; Fig. 2A] and thermal [12.00 ± 1.66 s vs 6.68 ± 0.97 s (n = 8); p < 0.01; Fig. 2B] hypersensitivity, compared with saline-treated EAE mice. In contrast, KN92 did not alter mechanical threshold or thermal latency in EAE mice at any time point tested (Fig. 2), which attributed the effect of KN93 to a CaMKII-mediated effort. These data suggested that acute CaMKII inhibition reversed mechanical and thermal hyperalgesia in EAE mice.
Figure 2.
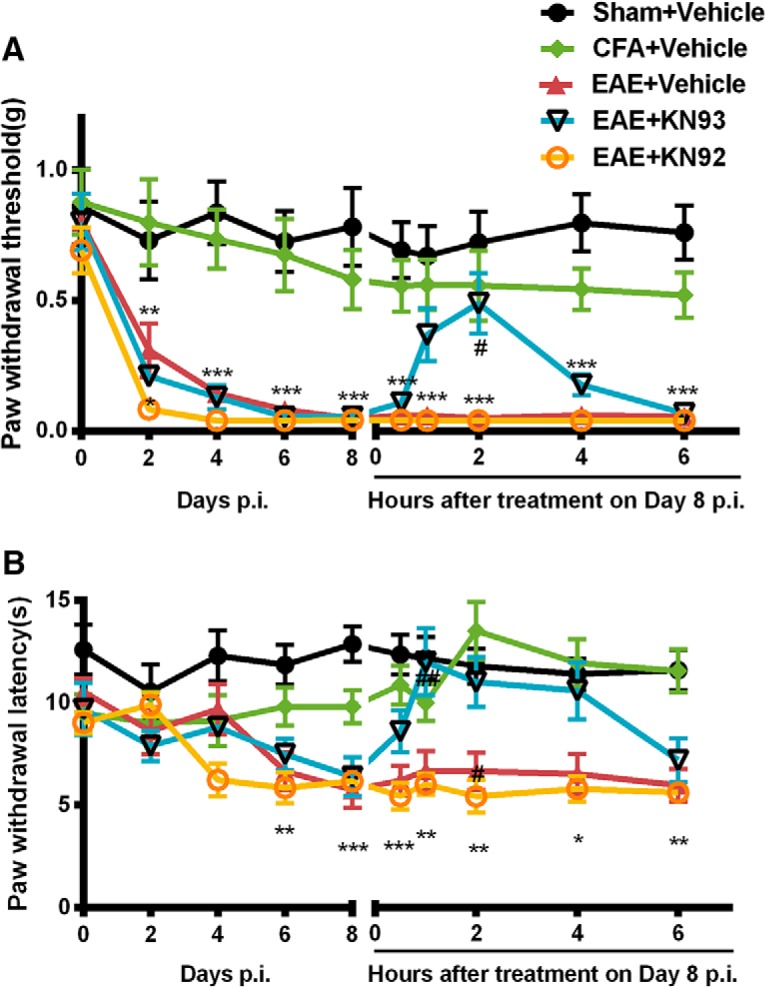
Acute treatment with KN93, but not KN92, reversed mechanical allodynia and thermal hyperalgesia in EAE. Separate groups of 8 mice received KN93 (45 nmol, i.t.), KN92 (45 nmol, i.t.), or saline once on Day 8 after inducing EAE with immunization with MOG35–55 on Day 0. A, B, Acute administration of KN93 (45 nmol, i.t.) significantly reversed the mechanical allodynia (A) and thermal hyperalgesia (B) in EAE mice. KN92 has no effect on the mechanical and thermal sensitivities. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham group; #p < 0.05, ##p < 0.01 compared with the saline-treated EAE group.
Small interfering RNA-mediated CaMKIIα knockdown reduced mechanical and thermal hypersensitivity in EAE
We further knocked down spinal CaMKIIα in EAE mice using CaMKIIα-targeting siRNA (CaMKIIα-siRNA). Starting on day 5 p.i., mice were treated with CaMKIIα-siRNA (2 μg, i.t.) or scrambled siRNA twice a day for 3 consecutive days. Mechanical and thermal sensitivities were tested before injections every day. Compared with vehicle-treated EAE mice, the hypersensitivities to mechanical and thermal stimuli were gradually attenuated by siRNA targeting CaMKIIα (Fig. 3). On day 8, the day after the last dose of siRNA injection, mechanical [0.54 ± 0.07 vs 0.04 ± 0.00 g (n = 8); p < 0.001; Fig. 3C] and thermal [11.31 ± 1.29 vs 5.87 ± 0.85 s (n = 8); p < 0.001; Fig. 3D] hyperalgesia were reversed in EAE mice treated with CaMKIIα siRNA. In contrast, scrambled siRNA did not alter the mechanical and thermal sensitivities in EAE mice (Fig. 3). These data indicated that CaMKIIα is involved in mediating the mechanical and thermal hyperalgesia in EAE.
Figure 3.

siRNA-mediated CaMKIIα knockdown reduced mechanical and thermal hypersensitivity in EAE. Separate groups of 8 mice received (i.t.) CaMKIIα siRNA (2 μg, every 12 h), scrambled siRNA (2 μg, every 12 h), or vehicle for 3 consecutive days from Day 5 to Day 7 postimmunization. A–D, CaMKIIα knockdown by siRNA significantly reduced clinical scores (A), locomotor impairment (B), mechanical allodynia (C), and thermal hyperalgesia (D). Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham immune group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the saline-treated EAE group.
Enhanced activation of spinal CaMKIIα was suppressed by KN93 and CaMKIIα-siRNA in EAE mice
To correlate behavioral effects with biochemical changes, we measured CaMKIIα activity in the spinal cord using Western blot analysis. Activation of CaMKIIα was assessed by the amount of pCaMKIIα (Chen et al., 2010; He et al., 2016a). The phosphorylation level of CaMKIIα in the spinal cord was significantly elevated in EAE mice, compared with that in control mice (Fig. 4). Prophylactic treatment of KN93 significantly prevented an EAE-induced increase of spinal pCaMKIIα [1.17 ± 0.13 vs 1.78 ± 0.15 (n = 3); p < 0.05; Fig. 4A]. Acute treatment with KN93 also significantly reversed the elevated spinal pCaMKIIα level in EAE mice [1.18 ± 0.07 vs 1.56 ± 0.08 (n = 3); p < 0.01; Fig. 4B].
Figure 4.

Enhanced activation of spinal CaMKIIα was suppressed by inhibiting CaMKIIα (by KN93) or knocking down CaMKIIα (by siRNA) in EAE mice. Lumbar spinal cord samples were collected to determine CaMKIIα activity (pCaMKIIα levels), which was elevated in EAE mice. A, Prophylactic KN93 treatment (45 nmol, i.t., daily; sampled on Day 8). B, Acute treatment with KN93 (45 nmol. i.t. at time 0, sampled at 2 h). C, Repeated treatment with CaMKIIα siRNA (2 μg, i.t., twice daily; sampled on Day 8), significantly reduced the enhancement of CaMKIIα activation. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham immune group; #p < 0.05, ##p < 0.01 compared with the saline-treated EAE group.
Application of siRNA targeting of CaMKIIα also suppressed the enhancement of CaMKIIα activity in EAE [1.45 ± 0.12 vs 2.55 ± 0.29 (n = 3); p < 0.01; Fig. 4C], while scrambled siRNA does not alter the enhanced level of pCaMKIIα in the spinal cord of EAE mice (Fig. 4C). These biochemical changes correlated with the behavioral effects of KN93 and CaMKIIα siRNA.
To further correlate biological changes with pain behavioral results, the activation of CaMKIIα was examined using immunofluorescence. The pCaMKIIα immunoreactivity was significantly increased in the superficial spinal dorsal horn, a critical area for pain signaling and processing, in EAE mice, compared with the control group [3840 ± 217 vs 2320 ± 49 (n = 3); p < 0.001; Fig. 5]. Treatment with KN93 and CaMKIIα-siRNA significantly suppressed the elevation of pCaMKIIα immunoreactivity in EAE mice (p < 0.01, n = 3; Fig. 5). These data are in agreement with the Western blot data and also the behavioral results, suggesting an essential role of CaMKIIα in mediating mechanical and thermal hyperalgesia in EAE.
Figure 5.
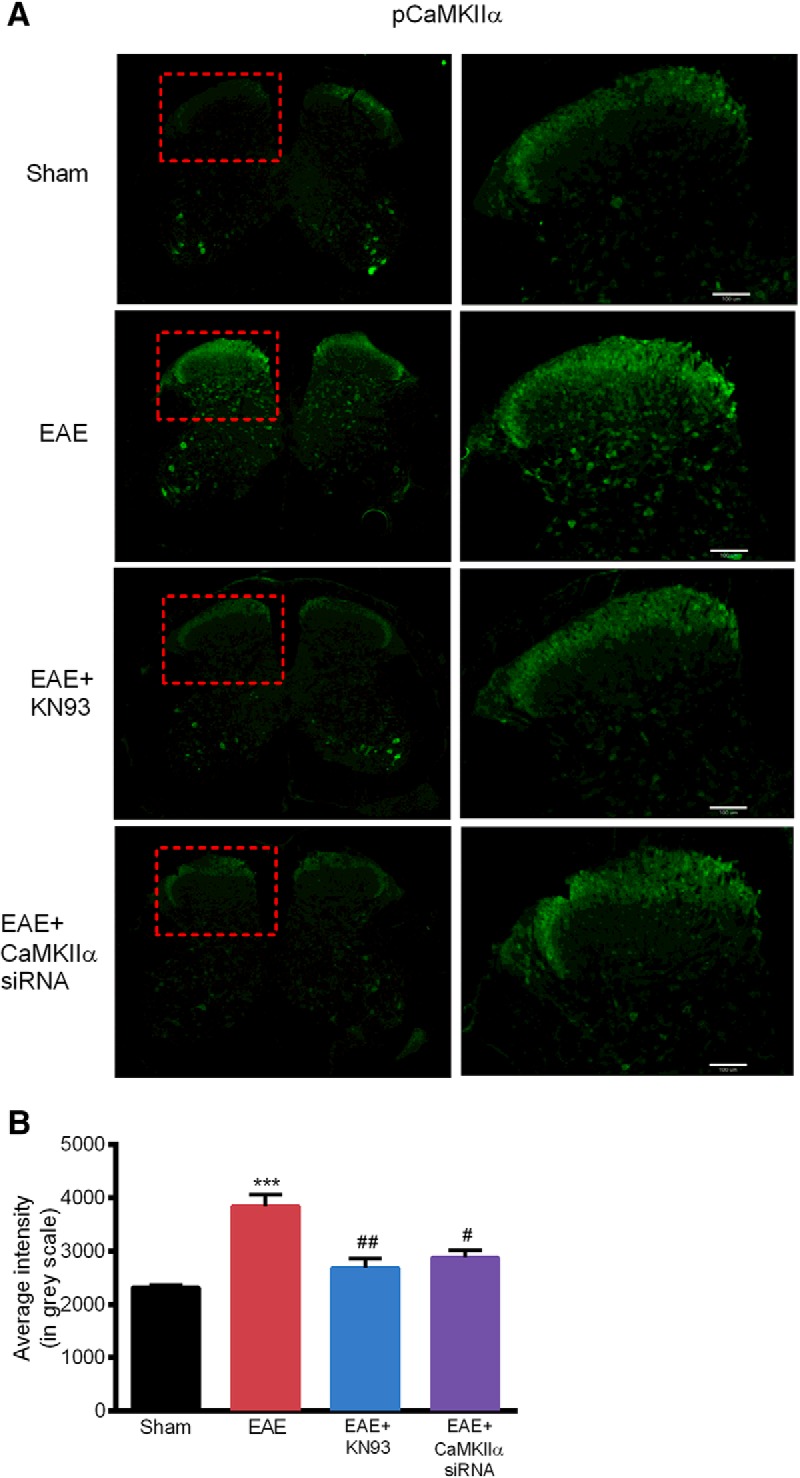
Increased pCaMKIIα immunoreactivity in the spinal dorsal horn of EAE mice was reduced by KN93 and CaMKIIα siRNA. A, pCaMKIIα immunoreactivity in the spinal dorsal horn were observed using immunofluorescence. B, Immunoreactivity of pCaMKIIα was significantly increased in the spinal dorsal horn of EAE mice. Prophylactic KN93 administration or CaMKIIα siRNA significantly reduced the pCaMKIIα immunoreactivity. Data are expressed as the mean ± SEM. ***p < 0.001 compared with the sham immune group; #p < 0.05, ##p < 0.01 compared with the saline-treated EAE group. Scale bar, 100 μm.
Absence of EAE-induced mechanical allodynia and thermal hyperalgesia in CaMKIIαT286A point mutant mice
To ascertain the role of CaMKIIα for the initiation of mechanical and thermal hyperalgesia in EAE, CaMKIIαT286A point mutation mice, which do not undergo CaMKIIα autophosphorylation and further activation, were immunized by MOG35–55 to induce EAE in the study. The baseline mechanical and thermal sensitivities in CaMKIIαT286A point mutation mice were similar to those of wild-type mice (Fig. 6). Separate groups of CaMKIIαT286A point mutation mice and wild-type littermates were treated with MOG35–55 to induce EAE as described above. On Day 8 p.i., when wild-type mice have developed mechanical allodynia [0.06 ± 0.02 vs 0.83 ± 0.18 g for sham controls (n = 8); p < 0.01; Fig. 6B] and thermal hyperalgesia [6.24 ± 0.94 s vs 12.36 ± 1.48 for sham controls (n = 8); p < 0.05; Fig. 6C], neither was detected in CaMKIIαT286A mutation mice, as their paw withdrawal thresholds to von Frey probing (0.83 ± 0.18 g) or paw withdrawal latencies to radiant heat (12.36 ± 1.48 s) were not significantly different from those in sham control mice (p > 0.05). Throughout the 12-day study window (after which sensory testing was stopped to avoid complications from motor dysfunction), mechanical allodynia or thermal hyperalgesia was absent in MOG35–55-treated CaMKIIαT286A mutant mice lacking functional CaMKIIα. The observation was made in comparisons with sham-treated wild-type littermates, CFA-treated littermate wild-type mice (negative control), as well as MOG35–55-treated littermate wild-type mice (positive control). These data indicate that CaMKIIα is required for the development of mechanical and thermal hyperalgesia in EAE.
Figure 6.

Absence of EAE-induced mechanical allodynia and thermal hyperalgesia in CaMKIIαT286A mutant mice. CaMKIIα T286A point mutant mice or wild-type littermates were immunized with MOG35–55 to induce EAE. A, CaMKIIα T286A point mutant mice developed a significantly lower degree of motor dysfunction. B, C, Mechanical allodynia (B) and thermal hyperalgesia (C) were absent in CaMKIIα T286A point mutant mice with EAE. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham immune CaMKIIα wild-type mice; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with CaMKIIα wild-type mice developed with EAE.
Moreover, MOG35–55-treated CaMKIIαT286A mutant mice had a slight delay in developing clinical motor dysfunction. Overall, CaMKIIαT286A mutant mice scored significantly lower in clinical signs, comparing with wild-type mice [Day 16 p.i., 1.4 ± 0.4 vs 2.8 ± 0.3 (n = 8), p < 0.001; Day 18 p.i., 2.1 ± 0.6 vs 3.0 ± 0.3 (n = 8), p < 0.01; Fig. 6A]. Therefore, CaMKIIαT286A mutant mice lacking functional CaMKIIα showed resistance to MOG35–55-induced motor dysfunction.
EAE induced spontaneous pain behaviors that was absent in CaMKIIαT286A point mutant mice.
Most preclinical studies have focused on evoked pain hypersensitivity. Before our study, it was not known whether EAE mice had spontaneous pain behavior such as that experienced by patients with MS. Here we used the CPP testing paradigm that has been previously validated to study ongoing or spontaneous pain in mice (He et al., 2012, 2016a,b; He and Wang, 2015).
Separate groups of CaMKIIα wild-type mice were treated with sham or MOG35–55 immunization. On Day 8 p.i., mice were paired with saline in the morning in a randomly chosen conditioning chamber for 15 min. Four hours later, mice were paired with lidocaine (0.04% in 5 μl of saline, i.t.) in the opposite chamber for 15 min. Chamber preference was tested 20 h later. Before conditioning, wild-type mice showed no preexisting preference for any chambers [372 ± 22 s vs 359 ± 25 s (n = 8); p > 0.05; Fig. 7A]. In the preference test, sham-treated CaMKIIα wild-type mice spent an almost equal amount of time in the saline-paired chamber (399 ± 49 s) or the lidocaine-paired chamber (371 ± 60 s; p > 0.05). However, MOG35–55-treated CaMKIIα wild-type mice spent significantly more time in the lidocaine-paired chamber (551 ± 50 s) than in the saline-paired chamber (222 ± 31 s; p < 0.001, n = 8; Fig. 7A). These results were further supported by the significant difference score (p < 0.01; Fig. 7B), which was absent in sham-treated wild-type mice. These data indicated the presence of lidocaine-induced CPP in wild-type mice with EAE.
Figure 7.
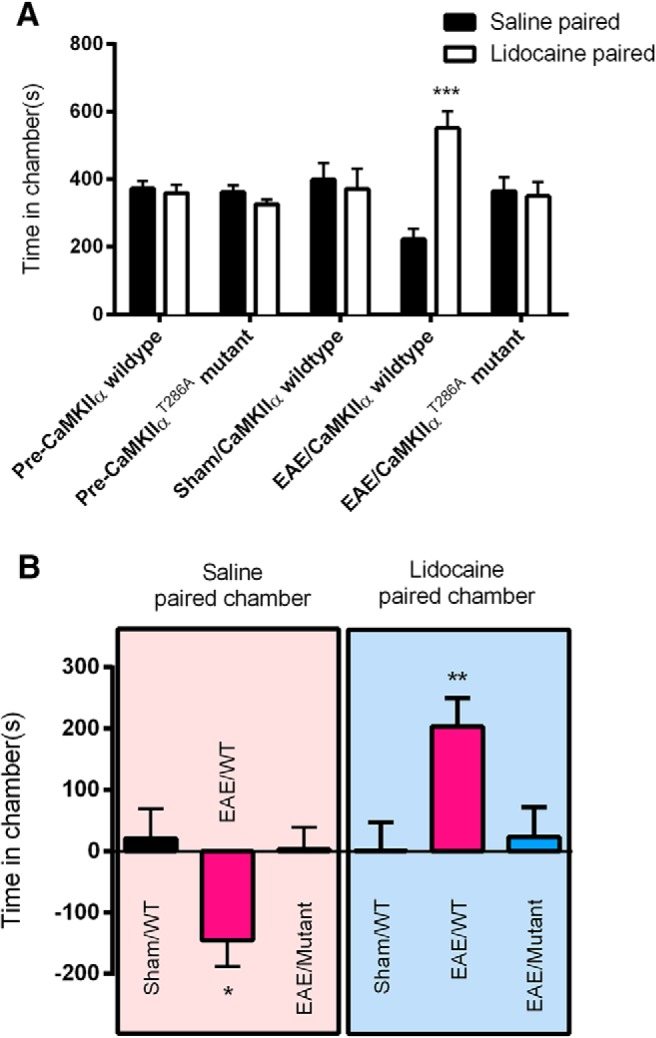
Absence of EAE-induced nonevoked ongoing pain in CaMKIIαT286A point mutant mice. A, Lidocaine produced CPP in wild-type mice with EAE, but CaMKIIαT286A point mutant mice with EAE showed no chamber preference, compared with saline-treated wild-type mice. ***p < 0.001 compared with saline paired chamber. B, Difference scores (test time − preconditioning time in each chamber) confirmed that wild-type mice with EAE, but not CaMKIIαT286A point mutant mice with EAE, exhibited CPP to lidocaine. *p < 0.05, **p < 0.01 test time compared with preconditioning time. Data are expressed as the mean ± SEM.
When CaMKIIαT286A point mutant mice were treated with MOG35–55, chamber-paired with saline and lidocaine and tested in the same manner alongside their littermate wild-type mice, CaMKIIαT286A mutant mice, however, did not show CPP to either lidocaine or saline, as these mice spent similar amounts of time in lidocaine (350 ± 41 s) and saline (365 ± 41 s) chambers (p < 0.05, n = 8; Fig. 7A), and no significant difference score was identified (p > 0.05; Fig. 7B). Therefore, EAE-induced spontaneous pain behavior was present in wild-type mice but was absent in CaMKIIαT286A point mutant mice.
Pretreatment but not acute treatment of IL-17 antiserum alleviated hyperalgesia in EAE mice.
Interleukin-17, the major secretion of Th17 cells, has been studied as a mechanism in MS pathogenesis. Development of EAE was significantly suppressed in IL-17−/− mice, and adoptive transfer of IL-17−/− CD4+ cells was inefficient in inducing EAE in recipient mice (Komiyama et al., 2006). Furthermore, brain tissues from patients with MS have increased levels of IL-17 mRNA and numbers of IL-17-producing cells (Lock et al., 2002; Tzartos et al., 2008). As shown above, EAE produced significant mechanical allodynia and thermal hyperalgesia in MOG35–55-immunized mice. Separate groups of mice were treated with IL-17 antiserum five times (24 h before and 0, 24, 48, and 72 h after the immunization with MOG35–55) to neutralize basal and immunization-induced IL-17. As shown in Figure 8, EAE mice pretreated with IL-17 antiserum presented less mechanical allodynia and thermal hyperalgesia until Day 6 p.i., compared with EAE mice treated with saline [Day 4 p.i.: 0.66 ± 0.16 vs 0.04 ± 0.00 g (n = 8), p < 0.001, Fig. 8A; 7.21 ± 0.90 vs 3.93 ± 0.46 s (n = 8), p < 0.05, Fig. 8B]. The duration of IL-17 antiserum treatment was only until Day 3 p.i., and the IL-17 antiserum-treated mice started to develop mechanical and thermal hypersensitivity around Day 8 p.i. This indicated that pretreatment with IL-17 antiserum effectively prevented the development of both mechanical and thermal hypersensitivities and delayed the onset of hyperalgesia in EAE mice.
Figure 8.

Effects of pretreatment and acute treatment with IL-17 antiserum in EAE-induced hyperalgesia. Groups of mice were injected with IL-17 antiserum or vehicle 24 and 2 h before immunization and on Day 1 to Day 3 p.i. A, B, Mice treated with vehicle developed less and delayed mechanical (A) and thermal (B) hyperalgesia. C, D, Acute treatment of IL-17 antiserum on Day 8 p.i. had no effect on mechanical (C) or thermal (D) hyperalgesia in EAE mice. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the sham immune group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the vehicle-treated EAE group.
To investigate whether IL-17 antiserum can have acute effect, EAE mice received a one-time injection of IL-17 antiserum on Day 8 p.i. when mechanical allodynia and thermal hyperalgesia were both established. IL-17 antiserum-treated EAE mice showed similar hypersensitivity to mechanical and thermal stimuli compared with saline-treated EAE mice (Fig. 8C,D), suggesting that acute treatment of IL-17 antiserum was not able to reverse the established hyperalgesia in EAE mice.
Spinal CaMKIIα mediates IL-17-induced hyperalgesia
When naive mice were treated with recombinant IL-17 (100 ng, i.t.), marked decreases in both paw withdrawal threshold in response to mechanical stimuli and paw withdrawal latencies in response to thermal stimuli were observe within an hour after IL-17 injection and lasted for at least 48 h, indicating that IL-17 alone induced mechanical allodynia and thermal hyperalgesia.
To investigate the involvement of CaMKIIα in IL-17-induced hyperalgesia, mice were given KN93 (45 nmol, i.t.) 24 h after IL-17 injection. Established mechanical and thermal hypersensitivities were both completely reversed by KN93. The effect of KN93 had a rapid onset at 0.5 h after injection [0.66 ± 0.18 vs 0.04 ± 0.00 g (n = 8), p < 0.001, Fig. 9A; 7.07 ± 0.66 vs 4.13 ± 0.53 s (n = 8), p < 0.05, Fig. 9B] and lasted for 2 h. These data indicated that inhibition of spinal CaMKII sufficiently abolished IL-17-induced mechanical and thermal hyperalgesia.
Figure 9.

Spinal CaMKIIα mediates IL-17-induced hyperalgesia. A, B, Groups of mice were intrathecally injected with IL-17 or vehicle and 24 h later were injected with KN93 or vehicle. IL-17-induced mechanical and thermal hyperalgesia in mice and the injection of KN93 reversed the developed hyperalgesia. C, D, Groups of wild-type and CaMKIIα mutant mice were injected with IL-17. Twenty-four hours later, IL-17-induced mechanical and thermal hyperalgesia is absent in CaMKIIα mutant mice. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01 compared with the IL-17-treated CaMKIIα wild-type mice; #p < 0.05, ###p < 0.001 compared with the vehicle-treated IL-17 group.
To further understand the role of CaMKIIα in IL-17-induced hyperalgesia, pain behaviors in CaMKIIα T286A mutant mice and wild-type mice were compared following IL-17 intrathecal injection. The baseline values of mechanical and thermal sensitivities were similar in CaMKIIα T286A mutant mice and wild-type mice (Fig. 9C,D). However, 24 h after IL-17 injection, wild-type mice showed a significantly decreased paw withdrawal threshold in response to mechanical stimuli and shortened paw withdrawal latencies in response to heat stimuli, while CaMKIIα T286A mutant mice retained normal mechanical sensitivity [0.61 ± 0.09 vs 0.75 ± 0.13 g (n = 8), p > 0.05; Fig. 9C] and thermal sensitivity [7.34 ± 0.47 ± 9.85 ± 0.91 s (n = 8), p > 0.05; Fig. 9D] compared with saline-treated mice. These results suggested that CaMKIIα is required for IL-17-induced hyperalgesia.
Intrathecal injection of IL-17 antiserum decreased spinal CaMKIIα activation in EAE mice
To correlate the biochemical and behavioral effects of IL-17, the extent of spinal CaMKIIα activation was measured in EAE mice treated with saline or intrathecal IL-17 antiserum. Pretreatment of IL-17 antiserum significantly decreased the elevation of spinal pCaMKIIα levels in the EAE mice [1.01 ± 0.02 vs 2.01 ± 0.19 (n = 3); p < 0.05, Fig. 10], which was in agreement with significant suppression of pain behaviors. On the other hand, the single acute treatment with IL-17 antiserum on Day 8 had no effect on EAE-induced CaMKIIα activation [2.75 ± 0.45 (n = 3); p > 0.05; Fig. 10], correlating with the lack of behavioral effects by the treatment. These data suggested that the action of spinal IL-17 in generating pain correlates with spinal CaMKIIα activation.
Figure 10.
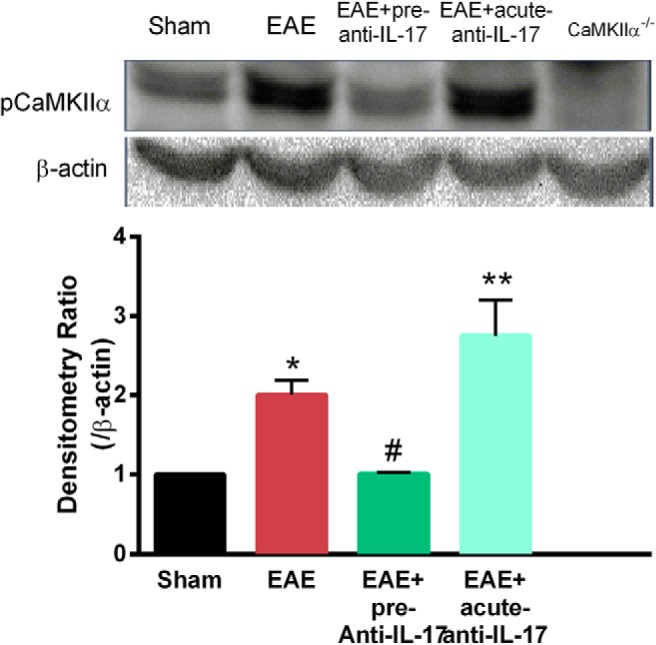
IL-17 antiserum reduced spinal CaMKIIα activation in EAE mice. The level of phosphorylated CaMKIIα was enhanced in the spinal cords of EAE mice. Pretreatment of IL-17 antiserum decreased the CaMKIIα phosphorylation in EAE mice. Acute injection of IL-17 antiserum has no effect on the activation of CaMKIIα. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01 compared with the sham immune group; #p < 0.05 compared with the vehicle-treated EAE group.
Discussion
Patients with MS suffered from dysfunctions of motor, sensory, visual, and autonomic systems (Compston and Coles, 2008). MS is the most common neurological disease in people between 20 and 45 years of age, causing acquired disability (Ragonese et al., 2008). The prevalence of MS is about threefold higher in women compared with that in men (Disanto and Ramagopalan, 2013). In this study, we investigated the roles of IL-17 and CaMKIIα in the generation and maintenance of pain in MS. We established EAE, a widely used preclinical model for MS in mice to study both ongoing spontaneous pain and evoked hypersensitivity.
Patients with MS primarily complaint about ongoing spontaneous pain, which is hard to study and rarely examined in preclinical settings. Here we used the CPP testing paradigm that we have previously validated for studying mice (He et al., 2012, 2016b; He and Wang, 2015). We found that mice with EAE had spontaneous pain behaviors as EAE mice, but not control mice, showed a significant preference for the chamber where the ongoing pain was blocked by lidocaine. This is the first preclinical study demonstrating the presence of ongoing spontaneous pain in MS. We also found that EAE mice exhibited mechanical allodynia and thermal hyperalgesia. The spontaneous and evoked pain occurred earlier than the onset of motor dysfunction and clinical signs. These findings are in agreement with previous reports for the time course of evoked pain behaviors in EAE (Olechowski et al., 2009; Yuan et al., 2012; Dutra et al., 2013; Rodrigues et al., 2016).
Importantly, this time difference provides a window for studying pain in the EAE model without the potential influence of motor impairment. The weakness or paralysis of the hindpaws may interfere with the pain threshold tests, which will not be accurate for distinguishing between reduced tactile and thermal sensitivities and locomotor impairment. Accordingly, sensory tests were performed before the onset of clinical signs of MS. On the other hand, the fact that pain occurs before the onset of clinical signs suggests that MS pain is possibly initiated by factors that appears in the early phase of EAE, rather than having a late onset after demyelination, which happens relatively late in EAE, at which time mice develop motor dysfunction. Some clinical studies reported that ∼20% patients had pain at the onset of their MS, although these studies had unclear descriptions of whether disease onset referred to the diagnosis of MS or the onset of MS symptoms (Stenager et al., 1991; Indaco et al., 1994; O'Connor et al., 2008). More comprehensive clinical studies will be needed to justify whether the pain in MS patients begins before the clinical symptoms.
Many inflammatory factors are involved in triggering the immune response in EAE. Studies have shown that levels of various cytokines and chemokines are elevated in the EAE or MS lesion after the onset of motor dysfunction. However, only the T cells that secrete IL-17, including Th17 cells and Th1/Th17 cells, infiltrate the CNS before the development of clinical symptoms of EAE (Murphy et al., 2010). Combining the fact that pain develops before the onset of motor dysfunctions, IL-17 became a potential contributor to the generation of pain in EAE. Therefore we tested the hypothesis and found that daily intrathecal injection of IL-17 antiserum from the day before immunization for 5 d was able to prevent the development of both thermal and mechanical hyperalgesia. On the other hand, one-time intrathecal administration of IL-17 antiserum (on Day 8 p.i.) to acutely neutralize IL-17 was ineffective in reversing the established hyperalgesia in EAE mice, suggesting that IL-17 plays an essential role in initiating pain development but may not directly contribute to the maintenance of pain in EAE.
In addition to neuroinflammation, glutamate excitotoxicity has recently been found to be an important mechanism involved in MS pathogenesis (Pitt et al., 2000). Studies showed that abnormal overactivation of the NMDA receptor exacerbate the EAE. Glutamate levels were significantly increased in the CSF of MS patients (Kostic et al., 2014). The NMDA receptor inhibitors effectively attenuated the motor dysfunction in EAE (Wallström et al., 1996; Sulkowski et al., 2013a,b; Grasselli et al., 2013). Ceftriaxone upregulated spinal expression of glutamate transporter GLT-1, which is primarily responsible for glutamate clearance, and ameliorated hyperalgesia in EAE mice (Ramos et al., 2010). Excessive glutamate transmission appeared to contribute to the development and maintenance of pain in MS. CaMKIIα has been shown to mediate the activation of NMDA receptors and contributes to the glutamate transmission. Ca2+ influx through activated NMDA receptors can phosphorylate CaMKIIα and also induce its autophosphorylation (Fukunaga et al., 1992; Strack et al., 2000). In turn, activated CaMKIIα can phosphorylate the NMDA receptor and promote its activation (Kitamura et al., 1993; McGlade-McCulloh et al., 1993). Findings from our laboratory and others have revealed a critical role of CaMKIIα in modulating the inflammatory and neuropathic pain. Blocking CaMKIIα signaling reversed CFA-induced inflammatory pain (Luo et al., 2008). Neuropathic pain induced by the spinal nerve ligation can also be reduced by the inhibition of CaMKIIα (Chen et al., 2009). Inhibition of spinal CaMKIIα attenuated opioid-induced hyperalgesia (Chen et al., 2010) and chronic pain in Berkeley sickle cell transgenic mice (He et al., 2016a). It has also been reported that an abnormal CaMKIIα activity level contributes to the shifts of a touch pathway into a pain system (Yu et al., 2015), and CaMKIIα signaling is critical in the transition from acute to chronic pain (Ferrari et al., 2013). In this current study, we tested the hypothesis that CaMKIIα mediates the initiation and maintenance of hyperalgesia in EAE. We demonstrated that hyperalgesia in EAE can be attenuated by suppressing CaMKIIα activity using chemical inhibitors, knocking down CaMKIIα expression by siRNA or genetic deletion of functional CaMKIIα by a point mutation. Both prophylactic and acute treatments of KN93, the most commonly used CaMKIIα inhibitor, reduced mechanical and thermal hypersensitivities in EAE mice. KN92, a chemical analog of KN93 but lacking inhibitory activity at CaMKIIα, did not alter EAE-induced mechanical allodynia or thermal hyperalgesia. Although KN93 is not highly selective for CaMKIIα even in the presence of KN92 as a negative control, experiments using CaMKIIα-siRNA and CaMKIIα T286A point mutant mice pointed to the involvement of CaMKIIα, demonstrating an essential role of CaMKIIα in inducing and maintaining the evoked and nonevoked pain in EAE. We have previously shown that blockade of CaMKIIα activity by KN93 (using KN92 as a negative control) does not affect basal thermal and mechanical nociception in non-EAE mice (He et al., 2016a). In addition depleting CaMKIIα activity, as seen in CaMKIIαT286A mutant mice, did not alter basal thermal and mechanical sensitivities compared with wild-type mice (Fig. 9C,D). The behavioral results were consistent with biochemical findings that pCaMKIIα was significantly increased in superficial spinal dorsal horn of EAE mice, and inhibition of CaMKIIα by KN93 and CaMKIIα siRNA knockdown reduced pCaMKIIα in EAE mice. Studies from our laboratory and other laboratories have demonstrated that CaMKIIα is expressed in dorsal root ganglion (DRG) neurons, and in pain states the pCaMKIIα level is increased in DRG neurons (Kawano et al., 2009; Bangaru et al., 2015; Yu et al., 2015; He et al., 2016a). It has also been reported that intrathecal KN93 decreased pCaMKIIα level in DRG neurons (Hasegawa et al., 2009). These results indicate that spinal, and possibly also nociceptor, CaMKIIα critically contributes to the initiation and maintenance of both evoked and spontaneous ongoing pain in EAE mice.
It has been reported that IL-17 promotes NMDA NR1 phosphorylation and colocalized in NR1-immunoreactive neurons in a rat inflammatory pain model induced by CFA (Meng et al., 2013). A more recent study showed that IL-17 contributes to the persistent neuropathic pain in nerve injury mice via CaMKII signaling (Yao et al., 2016). Here, we found that blockade of IL-17 action reduced CaMKIIα activation in EAE mice. Also, the development of hyperalgesia induced by recombinant IL-17 in naive mice was attenuated by the inhibition of CaMKII and was almost absent in CaMKIIα T286A point mutant mice. We showed that IL-17 is essential in the generation of pain, but does not directly mediate the maintenance of pain, in EAE mice. We further suggested that IL-17 may act through CaMKIIα to maintain pain in EAE.
Together, this is the first report of significant, but differential, roles for IL-17 and CaMKIIα in the development (both IL-17 and CaMKIIα) and maintenance (CaMKIIα) of EAE-induced hyperalgesia and ongoing pain in mice. These findings will not only shed light on the understanding of mechanisms of EAE-induced pain, but may also provide new pharmacological interventions to treat pain in patients with multiple sclerosis. In our parallel studies, we have made efforts to identify and repurpose clinically used drugs that have the property of inhibiting CaMKIIα (Molokie et al., 2014). In addition, we have identified curcumin as a potential inhibitor of CaMKIIα (Hu et al., 2015). These relatively safe drugs can potentially be used for CaMKIIα inhibition that may offer new therapies for treating pain in MS.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grants R01-HL-098141 & R01-DA-041809. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
The authors declare no competing financial interests.
References
- Aicher SA, Silverman MB, Winkler CW, Bebo BF Jr (2004) Hyperalgesia in an animal model of multiple sclerosis. Pain 110:560–570. 10.1016/j.pain.2004.03.025 [DOI] [PubMed] [Google Scholar]
- Bangaru ML, Meng J, Kaiser DJ, Yu H, Fischer G, Hogan QH, Hudmon A (2015) Differential expression of CaMKII isoforms and overall kinase activity in rat dorsal root ganglia after injury. Neuroscience 300:116–127. 10.1016/j.neuroscience.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen Y, Luo F, Yang C, Kirkmire CM, Wang ZJ (2009) Acute inhibition of Ca2+/calmodulin-dependent protein kinase II reverses experimental neuropathic pain in mice. J Pharmacol Exp Ther 330:650–659. 10.1124/jpet.109.152165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang C, Wang ZJ (2010) Ca2+/calmodulin-dependent protein kinase II α is required for the initiation and maintenance of opioid-induced hyperalgesia. J Neurosci 30:38–46. 10.1523/JNEUROSCI.4346-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A, Coles A (2008) Multiple sclerosis. Lancet 372:1502–1517. 10.1016/S0140-6736(08)61620-7 [DOI] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK (2013) Constitutive mu-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science 341:1394–1399. 10.1126/science.1239403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disanto G, Ramagopalan S (2013) On the sex ratio of multiple sclerosis. Mult Scler 19:3–4. 10.1177/1352458512447594 [DOI] [PubMed] [Google Scholar]
- Dixon WJ. (1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20:441–462. 10.1146/annurev.pa.20.040180.002301 [DOI] [PubMed] [Google Scholar]
- Dutra RC, Bento AF, Leite DF, Manjavachi MN, Marcon R, Bicca MA, Pesquero JB, Calixto JB (2013) The role of kinin B1 and B2 receptors in the persistent pain induced by experimental autoimmune encephalomyelitis (EAE) in mice: evidence for the involvement of astrocytes. Neurobiol Dis 54:82–93. 10.1016/j.nbd.2013.02.007 [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD (2013) Role of nociceptor αCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J Neurosci 33:11002–11011. 10.1523/JNEUROSCI.1785-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Soderling TR, Miyamoto E (1992) Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem 267:22527–22533. [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ (1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279:870–873. 10.1126/science.279.5352.870 [DOI] [PubMed] [Google Scholar]
- Grasselli G, Rossi S, Musella A, Gentile A, Loizzo S, Muzio L, Di Sanza C, Errico F, Musumeci G, Haji N, Fresegna D, Sepman H, De Chiara V, Furlan R, Martino G, Usiello A, Mandolesi G, Centonze D (2013) Abnormal NMDA receptor function exacerbates experimental autoimmune encephalomyelitis. Br J Pharmacol 168:502–517. 10.1111/j.1476-5381.2012.02178.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau-López L, Sierra S, Martínez-Cáceres E, Ramo-Tello C (2011) Analysis of the pain in multiple sclerosis patients. Neurología (Barcelona) 26:208–213. 10.1016/j.nrl.2010.07.014 [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:77–88. 10.1016/0304-3959(88)90026-7 [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Kohro Y, Tsuda M, Inoue K (2009) Activation of cytosolic phospholipase A2 in dorsal root ganglion neurons by Ca2+/calmodulin-dependent protein kinase II after peripheral nerve injury. Mol Pain 5:22. 10.1186/1744-8069-5-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wang ZJ (2015) Nociceptor β II, δ, and ε isoforms of PKC differentially mediate paclitaxel-induced spontaneous and evoked pain. J Neurosci 35:4614–4625. 10.1523/JNEUROSCI.1580-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Tian X, Hu X, Porreca F, Wang ZJ (2012) Negative reinforcement reveals non-evoked ongoing pain in mice with tissue or nerve injury. J Pain 13:598–607. 10.1016/j.jpain.2012.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Chen Y, Tian X, Yang C, Lu J, Xiao C, DeSimone J, Wilkie DJ, Molokie RE, Wang ZJ (2016a) CaMKIIalpha underlies spontaneous and evoked pain behaviors in Berkeley sickle cell transgenic mice. Pain 157:2798–2806. 10.1097/j.pain.0000000000000704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wilkie DJ, Nazari J, Wang R, Messing RO, DeSimone J, Molokie RE, Wang ZJ (2016b) PKCdelta-targeted intervention relieves chronic pain in a murine sickle cell disease model. J Clin Invest 126:3053–3057. 10.1172/JCI86165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Huang F, Szymusiak M, Liu Y, Wang ZJ (2015) Curcumin attenuates opioid tolerance and dependence by inhibiting Ca2+/calmodulin-dependent protein kinase II alpha activity. J Pharmacol Exp Ther 352:420–428. 10.1124/jpet.114.219303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannitti T, Kerr BJ, Taylor BK (2014) Mechanisms and pharmacology of neuropathic pain in multiple sclerosis. Curr Top Behav Neurosci 20:75–97. 10.1007/7854_2014_288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indaco A, Iachetta C, Nappi C, Socci L, Carrieri PB (1994) Chronic and acute pain syndromes in patients with multiple sclerosis. Acta Neurol (Napoli) 16:97–102. [PubMed] [Google Scholar]
- Kalia LV, O'Connor PW (2005) Severity of chronic pain and its relationship to quality of life in multiple sclerosis. Mult Scler 11:322–327. 10.1191/1352458505ms1168oa [DOI] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, Liang MY, Kwok WM, Hogan Q, Sarantopoulos C (2009) Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci U S A 106:8725–8730. 10.1073/pnas.0901815106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Smith MT (2014) Multiple sclerosis-induced neuropathic pain: pharmacological management and pathophysiological insights from rodent EAE models. Inflammopharmacology 22:1–22. 10.1007/s10787-013-0195-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CF, Moalem-Taylor G (2011) Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain 12:370–383. 10.1016/j.jpain.2010.08.003 [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Miyazaki A, Yamanaka Y, Nomura Y (1993) Stimulatory effects of protein kinase C and calmodulin kinase II on N-methyl-D-aspartate receptor/channels in the postsynaptic density of rat brain. J Neurochem 61:100–109. 10.1111/j.1471-4159.1993.tb03542.x [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177:566–573. 10.4049/jimmunol.177.1.566 [DOI] [PubMed] [Google Scholar]
- Kostic M, Dzopalic T, Zivanovic S, Zivkovic N, Cvetanovic A, Stojanovic I, Vojinovic S, Marjanovic G, Savic V, Colic M (2014) IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol 79:181–186. 10.1111/sji.12147 [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8:500–508. 10.1038/nm0502-500 [DOI] [PubMed] [Google Scholar]
- Lu J, Kurejova M, Wirotanseng LN, Linker RA, Kuner R, Tappe-Theodor A (2012) Pain in experimental autoimmune encephalitis: a comparative study between different mouse models. J Neuroinflammation 9:233. 10.1186/1742-2094-9-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Yang C, Chen Y, Shukla P, Tang L, Wang LX, Wang ZJ (2008) Reversal of chronic inflammatory pain by acute inhibition of Ca2+/calmodulin-dependent protein kinase II. J Pharmacol Exp Ther 325:267–275. 10.1124/jpet.107.132167 [DOI] [PubMed] [Google Scholar]
- McGlade-McCulloh E, Yamamoto H, Tan SE, Brickey DA, Soderling TR (1993) Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature 362:640–642. 10.1038/362640a0 [DOI] [PubMed] [Google Scholar]
- Meng X, Zhang Y, Lao L, Saito R, Li A, Bäckman CM, Berman BM, Ren K, Wei PK, Zhang RX (2013) Spinal interleukin-17 promotes thermal hyperalgesia and NMDA NR1 phosphorylation in an inflammatory pain rat model. Pain 154:294–305. 10.1016/j.pain.2012.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molokie RE, Wilkie DJ, Wittert H, Suarez ML, Yao Y, Zhao Z, He Y, Wang ZJ (2014) Mechanism-driven phase I translational study of trifluoperazine in adults with sickle cell disease. Eur J Pharmacol 723:419–424. 10.1016/j.ejphar.2013.10.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AC, Lalor SJ, Lynch MA, Mills KH (2010) Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun 24:641–651. 10.1016/j.bbi.2010.01.014 [DOI] [PubMed] [Google Scholar]
- O'Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH (2008) Pain associated with multiple sclerosis: systematic review and proposed classification. Pain 137:96–111. 10.1016/j.pain.2007.08.024 [DOI] [PubMed] [Google Scholar]
- Olechowski CJ, Truong JJ, Kerr BJ (2009) Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE). Pain 141:156–164. 10.1016/j.pain.2008.11.002 [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS (2000) Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 6:67–70. 10.1038/71555 [DOI] [PubMed] [Google Scholar]
- Ragonese P, Aridon P, Salemi G, D'Amelio M, Savettieri G (2008) Mortality in multiple sclerosis: a review. Eur J Neurol 15:123–127. 10.1111/j.1468-1331.2007.02019.x [DOI] [PubMed] [Google Scholar]
- Ramos KM, Lewis MT, Morgan KN, Crysdale NY, Kroll JL, Taylor FR, Harrison JA, Sloane EM, Maier SF, Watkins LR (2010) Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience 169:1888–1900. 10.1016/j.neuroscience.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues DH, Leles BP, Costa VV, Miranda AS, Cisalpino D, Gomes DA, de Souza DG, Teixeira AL (2016) IL-1β is involved with the generation of pain in experimental autoimmune encephalomyelitis. Mol Neurobiol 53:6540–6547. 10.1007/s12035-015-9552-0 [DOI] [PubMed] [Google Scholar]
- Solaro C, Uccelli MM (2011) Management of pain in multiple sclerosis: a pharmacological approach. Nat Rev Neurol 7:519–527. 10.1038/nrneurol.2011.120 [DOI] [PubMed] [Google Scholar]
- Stenager E, Knudsen L, Jensen K (1991) Acute and chronic pain syndromes in multiple sclerosis. Acta Neurol Scand 84:197–200. 10.1111/j.1600-0404.1991.tb04937.x [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem 275:23798–23806. 10.1074/jbc.M001471200 [DOI] [PubMed] [Google Scholar]
- Sulkowski G, Dąbrowska-Bouta B, Strużzyńska L (2013a) Modulation of neurological deficits and expression of glutamate receptors during experimental autoimmune encephalomyelitis after treatment with selected antagonists of glutamate receptors. Biomed Res Int 2013:186068. 10.1155/2013/186068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski G, Dąbrowska-Bouta B, Chalimoniuk M, Strużzyńska L (2013b) Effects of antagonists of glutamate receptors on pro-inflammatory cytokines in the brain cortex of rats subjected to experimental autoimmune encephalomyelitis. J Neuroimmunol 261:67–76. 10.1016/j.jneuroim.2013.05.006 [DOI] [PubMed] [Google Scholar]
- Tang L, Shukla PK, Wang LX, Wang ZJ (2006) Reversal of morphine antinociceptive tolerance and dependence by the acute supraspinal inhibition of Ca(2+)/calmodulin-dependent protein kinase II. J Pharmacol Exp Ther 317:901–909. 10.1124/jpet.105.097733 [DOI] [PubMed] [Google Scholar]
- Thibault K, Calvino B, Pezet S (2011) Characterisation of sensory abnormalities observed in an animal model of multiple sclerosis: a behavioural and pharmacological study. Eur J Pain 15:231.e1–16. 10.1016/j.ejpain.2010.07.010 [DOI] [PubMed] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172:146–155. 10.2353/ajpath.2008.070690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallström E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T (1996) Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci 137:89–96. 10.1016/0022-510X(95)00339-4 [DOI] [PubMed] [Google Scholar]
- Yao CY, Weng ZL, Zhang JC, Feng T, Lin Y, Yao S (2016) Interleukin-17A acts to maintain neuropathic pain through activation of CaMKII/CREB signaling in spinal neurons. Mol Neurobiol 53:3914–3926. 10.1007/s12035-015-9322-z [DOI] [PubMed] [Google Scholar]
- Yu H, Pan B, Weyer A, Wu HE, Meng J, Fischer G, Vilceanu D, Light AR, Stucky C, Rice FL, Hudmon A, Hogan Q (2015) CaMKII controls whether touch is painful. J Neurosci 35:14086–14102. 10.1523/JNEUROSCI.1969-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Shi Y, Tang SJ (2012) Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J Neuroimmune Pharmacol 7:904–913. 10.1007/s11481-012-9370-3 [DOI] [PubMed] [Google Scholar]