

Abstract
Background
Oncogenic activation of phosphatidylinositol-3 kinase (PI3K) signaling plays a pivotal role in the development of glioblastoma (GBM). However, pharmacological inhibition of PI3K has so far not been therapeutically successful due to adaptive resistance through a rapid rewiring of cancer cell signaling. Here we identified that WEE1 is activated after transient exposure to PI3K inhibition and confers resistance to PI3K inhibition in GBM.
Methods
Patient-derived glioma-initiating cells and established GBM cells were treated with PI3K inhibitor or WEE1 inhibitor alone or in combination, and cell proliferation was evaluated by CellTiter-Blue assay. Cell apoptosis was analyzed by TUNEL, annexin V staining, and blotting of cleaved caspase-3 and cleaved poly(ADP-ribose) polymerase. Both subcutaneous xenograft and orthotropic xenograft studies were conducted to evaluate the effects of the combination on tumorigenesis; the tumor growth was monitored by bioluminescence imaging, and tumor tissue was analyzed by immunohistochemistry to validate signaling changes.
Results
PI3K inhibition activates WEE1 kinase, which in turn phosphorylates cell division control protein 2 homolog (Cdc2) at Tyr15 and inhibits Cdc2 activity, leading to G2/M arrest in a p53-independent manner. WEE1 inhibition abrogated the G2/M arrest and propelled cells to prematurely enter into mitosis and consequent cell death through mitotic catastrophe and apoptosis. Additionally, combination treatment significantly suppressed tumor growth in a subcutaneous model but not in an intracranial model due to limited blood–brain barrier penetration.
Conclusions
Our findings highlight WEE1 as an adaptive resistant gene activated after PI3K inhibition, and inhibition of WEE1 potentiated the effectiveness of PI3K targeted inhibition, suggesting that a combinational inhibition of WEE1 and PI3K might allow successful targeted therapy in GBM.
Keywords: mitotic catastrophic cell death, PI3K inhibition resistance, WEE1
Importance of the study
GBMs are driven by PI3K and over 50% of human GBMs have mutations of phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1)/phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA)/phosphatase and tensin homolog (PTEN). Given the genetic instability of cancer cells, genetic modifications could enable them to acquire a resistant phenotype to anti-PI3K therapies. Adaptive resistance can occur immediately after starting targeted therapy through a rapid rewiring of cancer cell signaling. Here we demonstrated that WEE1 kinase acting as a key G2/M checkpoint regulator is activated after transient exposure to the PI3K inhibitor buparlisib (BKM120), which in turn phosphorylates Cdc2 and inactivates and inhibits Cdc2 activity, leading to G2/M arrest. WEE1 inhibition by a small-molecule inhibitor abrogated PI3K inhibition–induced G2/M arrest and propelled cells into premature mitosis and consequent cell death. Our findings identify WEE1 as an adaptive resistant gene upregulated after PI3K inhibition, and inhibition of WEE1 potentiated the effectiveness of PI3K targeted inhibition, suggesting that a combinational inhibition of WEE1 and PI3K might allow successful GBM targeted therapy.
Glioblastoma (GBM) is the most common primary tumor of the central nervous system in adults, with a mean survival of 15 months after diagnosis.1 One of the most frequently dysregulated pathways in GBM is the phosphatidylinositol-3 kinase (PI3K)/Akt molecular cascade, which regulates various cellular processes, including protein synthesis, metabolism, cell proliferation, growth, and apoptosis.2 PI3K/Akt signaling is constitutively activated by genetic alterations in several proteins of this pathway in GBM. Amplification of epidermal growth factor receptor leads to activation of the PI3K pathway and has been noted in approximately 50% of GBM cases.3 PIK3CA (the p110α subunit of PI3K) or PIK3R1 (the p85 regulatory subunit of PI3K) is amplified or mutated in approximately 15% of GBM.4–6 Phosphatase and tensin homolog (PTEN), the negative regulator of the PI3K pathway, is inactivated by mutations, chromosomal deletions, or epigenetic gene silencing in about 40% of GBM cases.7 Total aberrant hyperactivation of PI3K/Akt accounts for 63%–86% of primary and 31% of secondary GBM.6–8 PI3K inhibitors produce a partial response, but complete response is rare. However, given the genetic instability of cancer cells, genetic modifications could enable them to acquire a resistant phenotype to anti-PI3K therapies. A common mechanism of resistance is unknown, and understanding the molecular mechanisms affecting cancer cell sensitivity or resistance to such inhibitors is an utter need. Inhibition of the PI3K/Akt pathway can lead to upregulation and activation of parallel pathways through engagement of several homeostatic feedback loops to maintain PI3K pathway equilibrium. Inhibition of the PI3K pathway has been shown to relieve negative feedback inhibition of upstream pathways. Inhibition of the mammalian target of rapamycin (mTOR) complex 1 by rapamycin has shown to relieve a negative feedback loop from S6 kinase to insulin receptor substrate 1, leading to activation of insulin-like growth factor 1 receptor, PI3K, and Akt.9–11 Expressions of ribosomal protein (RP) S6 kinases RPS6KA2 (RSK3), RPS6KA6 (RSK4), and RPS6KA5 (MSK-1) have been shown to be upregulated after PI3K/mTOR blockade and are potential mechanisms of resistance to PI3K/mTOR pathway inhibitors.12,13
Our previous study14 showed that p53-mutant GBM cells are more resistant to PI3K inhibition than p53-wildtype (wt) cells. PI3K inhibition induced apoptosis in p53-wt GBM cells. However, the apoptosis induction in p53-mutant cells is minimal. As PI3K inhibition also induces G2/M arrest in p53-mutant GBM cells, an alternative G2/M transition checkpoint is activated in a p53-independent manner, which allows cells to recover from PI3K inhibition–induced stress and render adaptive resistance to PI3K inhibition. A critical mediator for this checkpoint is WEE1 kinase, which is also reported to be directly phosphorylated by Akt.15 Therefore, it is of great interest to investigate the role of WEE1 kinase in PI3K inhibition–induced G2/M arrest as well as the interference of WEE1 activity to enhance PI3K inhibition efficacy.
WEE1 kinase is a key molecule in maintaining G2 cell-cycle checkpoint arrest for premitotic DNA repair. Activated WEE1 induces inhibitory phosphorylation at the Tyr15 residue of cell division control protein 2 homolog (Cdc2), leading to G2 arrest and allowing tumor cells a chance for repair of damaged DNA, thus conferring a survival advantage. MK1775 is a selective WEE1 kinase inhibitor being actively investigated in phase I/II clinical trials. Inhibition of WEE1 by MK1775 activates Cdc2 by suppressing Cdc2 Tyr15 phosphorylation, leads to abrogation of G2/M check, and pushes the cells to enter unscheduled mitosis and undergo apoptosis.16 In fact, MK1775 has been shown to enhance the activity of chemotherapy agents against a variety of malignancies.17,18
In this study, we identified that WEE1 kinase was activated as an adaptive resistant kinase after PI3K inhibition and that inhibiting WEE1 by MK1775 together with PI3K inhibition by buparlisib (BKM120, hereafter BKM) showed a synergistic cytotoxic effect. This effect was more pronounced in p53-mutant cells. This study provides a basis for clinical investigation of combining MK1775 with BKM to improve PI3K targeted therapy.
Materials and Methods
Cell Lines and Reagents
GBM cell lines and patient-derived glioma-initiating cell (GIC) lines with various p53 and PTEN statuses were used for this study. The GIC lines were established by isolating neurosphere-forming cells from fresh surgical specimens of human GBM tissue between 2005 and 2008, as described previously.19 This study was approved by the institutional review board of The University of Texas MD Anderson Cancer Center, Houston, Texas. Cells were authenticated by testing short tandem repeats using the Applied Biosystems AmpFISTR Identifier kit. The last authentication testing was done in March 2014. GBM cells were maintained as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% fetal bovine serum and penicillin-streptomycin (all from Life Technologies). The GICs were cultured as GBM neurospheres in DMEM/F12 containing B27 supplement (Invitrogen) and 20 ng/mL each of basic fibroblast growth factor and epidermal growth factor.
Cell Proliferation Assay
Cells were treated in triplicate for 72 h with the PI3K inhibitor BKM (62.5 nM–4 μM) and the WEE1 inhibitor MK1775 (31.25 nM–2 μM). Cell proliferation was estimated using the CellTiter-Blue (Promega) viability assay. The half-maximal inhibitory concentration value was calculated, as the mean drug concentration required to inhibit cell proliferation by 50% compared with vehicle-treated controls. The extent and direction of MK1775 and BKM anti-proliferation interactions were determined by standard fixed-ratio isobologram analyses, and combination index values were calculated using CompuSyn software (ComboSyn).
Western Blot Analysis
Cells were harvested in lysis solution19 and subjected to western blotting. Membranes were probed with the following primary antibodies: phospho-Ser473-Akt, phospho-Y15-cdc2, Akt, cleaved poly(ADP-ribose) polymerase (PARP), cleaved caspase-3, cleaved caspase-9, caspase-8, WEE1, and phospho-S642-WEE1 (all from Cell Signaling). Anti–β-actin antibody was purchased from Sigma and used as loading control.
Plasmids and Transfection
Precision LentiORF TP53 was from GE Healthcare Dharmacon. Lentiviral vector pLKO.1-mediated expression of short hairpin (sh)RNAs for WEE1 (clone ID TRCN0000001702) and pGIPZ-mediated expression of shRNA of p53 (clone ID V3LHS333919) were purchased from GE Healthcare Dharmacon. Lentiviral particles were produced in human embryonic kidney 293FT cells with the mixed set of packing plasmids, and the viruses were concentrated and titered as previously described.19
Immunofluorescence Staining of Ki-67
Cells were seeded into Lab-Tek II tissue culture slides (Thermo Fisher) and treated with BKM and MK1775. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% goat serum in phosphate buffered saline (PBS) and then stained overnight at 4°C with mouse anti–Ki-67 antibody (BD Pharmingen). Cells were washed with PBS and stained with secondary antibody (Alexa Fluor 488 goat anti-mouse immunoglobulin G; Invitrogen) for 2 hours. The cells were counterstained with Vecta shield sealant containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). The percentage of cells shown to be Ki-67 positive was quantified by counting 5 random fields in 3 independent experiments.
Mitotic Catastrophe
Cells cultured in Lab-Tek II tissue culture slides (Thermo Fisher) were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with 5% goat serum in PBS, and stained with Alexa Fluor 488 phalloidin (Invitrogen‒Molecular Probes) for 30 minutes at room temperature and then mounted with anti-fade containing 4′,6-diamidino-2-phenylindole (Vector Laboratories). The criterion for defining cells undergoing mitotic catastrophe was the presence of nuclei fragmented with 2 or more lobes within a single cell.
Flow Cytometric Analysis of Cell Cycle and Apoptosis
For cell cycle analysis, cells were pelleted and washed by PBS and fixed in 70% ethanol at −20°C. Immediately before flow cytometry, the cells were washed in cold PBS and incubated with PBS containing 1 mg/mL propidium iodide (Sigma-Aldrich) and 0.1 mg/mL RNase (Sigma-Aldrich) at room temperature in the dark for 30 minutes. Samples were measured (10000 events collected from each) in a BD Pharmingen FACScan. Cell apoptosis was detected using the Annexin-V-Fluos staining kit (Roche) according to the manufacturer’s instructions.
TUNEL Assay
To detect DNA fragmentations, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) assay was performed using the In Situ Cell Death Detection Kit (Roche Applied Science) according to the manufacturer’s instructions. Briefly, cells were fixed with 4% paraformaldehyde in PBS for 1 hour at room temperature and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 minutes on ice. Cells were labeled with TUNEL reaction mixture at 37°C for 1 hour and counterstained with Vecta shield sealant containing DAPI (Vector Laboratories). The percentage of cells shown to be TUNEL positive was quantified by counting 5 random fields in 3 independent experiments.
Animal Studies
All animal studies were conducted in the veterinary facilities of MD Anderson in accordance with institutional rules. To create the intracranial disease model, the GIC line GSC23 expressing luciferase (GSC23-luc) (5 × 105 cells) was implanted intracranially into nude mice using a previously described guide-screw system.14 For the subcutaneous tumor model, GSC23-luc (5 × 106) cells were implanted in the hind flank of nu/nu mice. Animals were randomly divided into 4 groups (10 mice per group for the intracranial model and 5 per group for the subcutaneous model). Starting on day 4 after tumor cell implantation, mice were treated with 20 mg/kg BKM and 30 mg/kg MK1775 alone or in combination, or vehicle (control). Both compounds were dissolved in 0.5% methylcellulose solution, and mice in the control group were given an equal volume of vehicle by oral gavage. The treatment frequency was once a day for 5 days, with 2 days off between treatments, for a total duration of 5 weeks. Tumor growth and development were visualized and quantified by an IVIS Spectrum in vivo imaging system. Mice were monitored daily and euthanized when they became moribund. Whole brains were extracted, rapidly frozen in liquid nitrogen, and stored at −70°C.
Immunohistochemical Staining
Sections (5-μm thick) of formalin-fixed, paraffin-embedded whole brains from animals were stained with anti–cleaved caspase-3, anti–pSer473-Akt, anti–γH2AX (Cell Signaling Technology), Ki-67 (BD Pharmingen), and anti–pY15-Cdc2. The sections were visualized using a diaminobenzidine substrate kit, and the slides were examined under a bright field microscope.
Statistical Analysis
Statistical analysis was performed using Student’s t-test. The results are presented as the mean of at least 3 independent experiments. Survival curves were plotted using the Kaplan–Meier method, and log-rank tests were used to compare curves between groups.
Results
BKM Activated WEE1 Signaling and Induced G2 Arrest in p53-Mutant GBM Cells
Our previous study has shown that PI3K inhibition by BKM induced G2 arrest in p53-mutant GBM cell lines, suggesting that an alternative G2/M transition checkpoint is activated in p53-mutant cells in a p53-independent manner, which allows cells to recover from PI3K inhibition–induced stress and render resistance to PI3K inhibition. To understand the mechanism of PI3K inhibition–induced resistance and G2 arrest in p53-mutant cells, we tested the effect of PI3K inhibition on cell cycle distribution in p53-mutant patient-derived GICs. As shown in Fig. 1A, BKM treatment for 72 hours caused G2 arrest in GICs (GSC25, GCS23, and GSC240) as well as established GBM cells (U251). As WEE1 and its downstream kinase Cdc2 play a key role in G2/M transition and WEE1 kinase was reported to be directly phosphorylated and regulated by Akt,15 we asked if BKM treatment affects WEE1 and Cdc2 activity. We showed that WEE1 is activated after 24 hours of treatment by BKM, as demonstrated by decreased phosphorylation of WEE1-Ser642. This in turn led to phosphorylation of Cdc2 at Tyr,15 inactivating Cdc2 and inducing G2/M arrest (Fig. 1B). This result indicates that WEE1 acts as an adaptive resistant gene induced by transient exposure to PI3K inhibition by BKM in GICs and GBM cells.
Fig. 1.

WEE1 is activated by PI3K inhibition and regulates PI3K inhibition–induced G2/M arrest. (A) GSC25, GSC240, GSC23, and U251 cells were treated with BKM at 1 μM for 72 hours. Cell cycle distribution was analyzed by flow cytometry. Statistical analysis showed significant difference of G2/M distribution between control and BKM treatment. (B) Activity of pAkt, pWEE1, and pCdc2 was detected by specific phosphorylation antibodies after BKM treatment. (C) GSC23 cells were treated with vehicle control, BKM alone (1 μM), MK1775 alone (0.5 μM), or combination for 24 hours, and pCdc2 and pAkt were detected by western blot. (D) GSC23 cells were treated with vehicle control, BKM alone (1 μM), MK1775 alone (0.5 μM), or combination for 72 hours, and cell cycle distribution was analyzed by flow cytometry. P-value for G2 phase cell fraction.
To study if WEE1-Cdc2 activity plays an important role in BKM-induced G2 arrest, GSC23 cells (p53 mutant) were treated with the WEE1 inhibitor MK1775. Western blot study showed that BKM-induced Cdc2-Y15 phosphorylation was blocked by MK1775 treatment (Fig. 1C). Correspondingly, BKM-induced G2 arrest was abrogated by MK1775 treatment (Fig. 1D), suggesting that WEE1-Cdc2 indeed plays a critical role in regulating BKM-induced G2 transition in p53-mutant GBM cells.
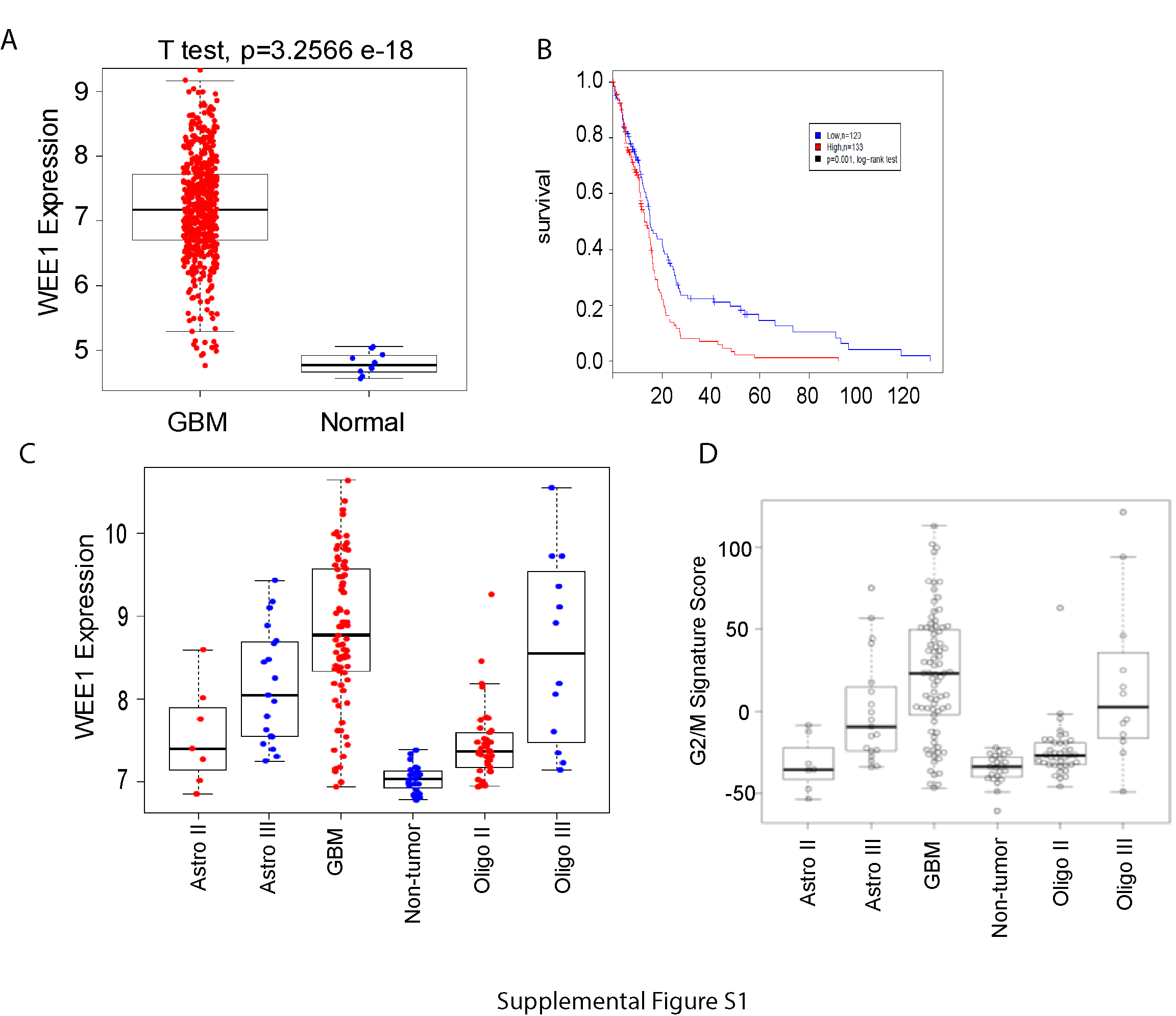
To support the association of WEE1 kinase signaling and PI3K signaling, we analyzed the expression of WEE1 in primary GBM samples from 2 different databases. The results from The Cancer Genome Atlas showed increased expression of WEE1 in GBM samples compared with normal brain (Supplementary Figure S1A). Interestingly, in GBM samples, WEE1 expression is significantly correlated with poor patient survival (Supplementary Figure S1B). Analysis of WEE1 expression in the GSE4290 database also showed high expression of WEE1 in GBM compared with that in nontumor tissue. Notably, WEE1 expression correlated with glioma grade, with the highest in GBM (World Health Organization [WHO] grade IV, the most aggressive glioma), lowest in astrocytoma, and moderate expression in anaplastic astrocytoma, and oligodendroglioma (Supplementary Figure S1C). Overall, the high expression of WEE1 in GBM and extremely low expression in normal brain suggests that inhibiting WEE1 is a specific target for GBM therapy with minimum effect on normal brain tissue.
Interestingly, applying a recently published G2/M-specific signature20 to the dataset GSE4290, we identified that GBM had significantly higher G2/M scores than all other pathohistological groups (P < 0.05, Student’s t-test), except for WHO grade III oligodendroglioma (P = 0.7) (Supplementary Figure S1D), suggesting a link between WEE1 expression and G2/M arrest in gliomas.
Combination of BKM and MK1775 Suppressed GBM Cell Proliferation
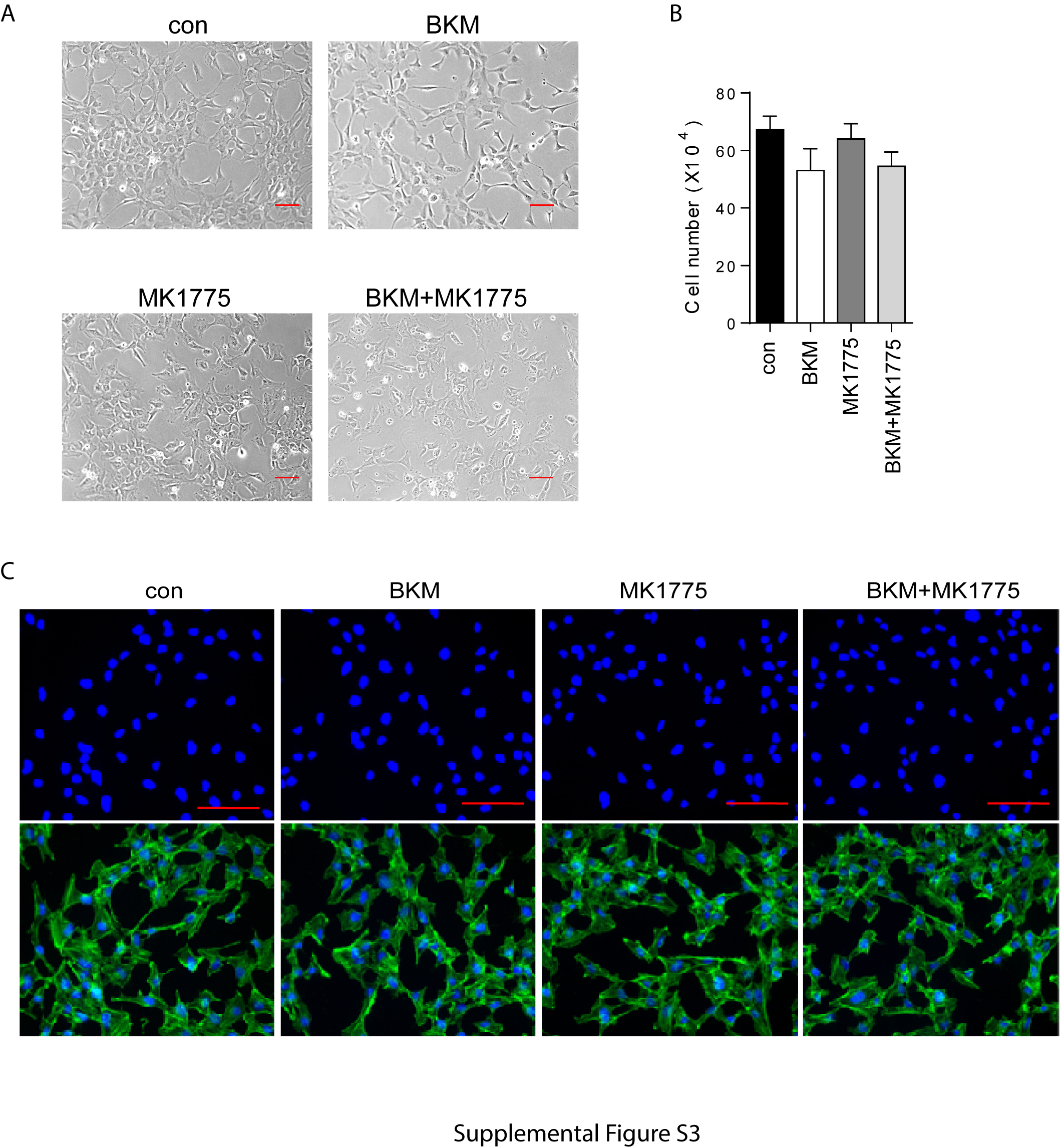
To test our hypothesis that WEE1 activation–induced G2/M arrest allows cells to recover from stress, thus conferring a survival escape, we examined the combined effect of BKM and the WEE1 inhibitor MK1775 on GBM cell proliferation. GSC23 cells cultured as neurospheres were treated with BKM and MK1775 for 72 hours. As shown in Fig. 2A, BKM alone or MK1775 alone decreased sphere size and number, while combination of BKM and MK1775 completely inhibited sphere formation. Growth curve analysis showed that combination treatment completely inhibited cell proliferation (P < 0.0001 compared with control and BKM; P = 0.0008 compared with MK1775) (Fig. 2B). Combination of BKM and WEE1 shRNA also inhibited cell growth and proliferation (Supplementary Figure 2A–C). Similarly, in the established GBM cell line U251, BKM or MK1775 suppressed cell proliferation, whereas combination treatment completely blocked cell growth as demonstrated by cell number counting (Fig. 2C and D). Combination treatment also greatly decreased the Ki-67 index compared with either BKM or MK1775 treatment alone (Fig. 2E). Notably, MK1775 has no significant effect on normal human astrocyte (NHA) cell growth, and no combination effect was observed in NHA cells (Supplementary Figure S3A and B).
Fig. 2.

Combination of BKM and MK1775 suppressed GBM cell proliferation. (A) GSC23 cells were treated with BKM, MK1775, or combination for 72 hours. Neurosphere formation was observed by microscope. (B) GSC23 cells were treated with 1 μM BKM, 0.5 μM MK1775, or combination. Cell numbers were counted every day and a cell proliferation curve was plotted. P < 0.0001 for combination vs control and BKM; P = 0.0008 for combination vs MK1775. (C–D) U251 cells were treated with BKM, MK1775, or combination for 72 hours, cell morphology was observed under microscope (C) and cell number was counted (D). (E) U251 cells in chamber slides were stained with Ki-67. Representative immunofluorescent images of Ki-67 staining were shown and a bar figure was presented with mean + SD. Scale bars, 100 microns for (A) and (C), 50 microns for (E).
MK1775 Selectively Synergizes with BKM to Inhibit Cell Growth in p53-Mutant Cells but Not in p53-wt Cells
Next we extended the combinational test to more GICs and GBM cells and asked if the combination activity is associated with p53 and PTEN status. Eighteen GIC lines and 4 established GBM cell lines with various p53 and PTEN statuses were treated with MK1775, BKM, or combination (representative response is shown in Fig. 3A and B). For the combination index (CI) as calculated by CompuSyn, a CI = 1 would suggest no interaction between MK1775 and BKM; a CI > 1 would suggest antagonistic interaction between MK1775 and BKM; a CI < 1 would suggest a synergistic or supra-additive interaction between MK1775 and BKM. The p53 and PTEN statuses and combination indexes at fraction affected = 0.5 for all 22 cell lines are listed in Fig. 3C. Interestingly, the average CI is significantly lower in p53-mutant cell lines than in p53-wt cell lines (Fig. 3D). In contrast, PTEN status did not have a significant effect on the CI (Fig. 3E). These results suggest that MK1775 selectively synergizes with BKM to inhibit cell proliferation in p53-mutant cells. To validate this result, we then inactivated p53 expression by shRNA in p53-wt U87 cells (Fig. 3F). We found that inactivation of p53 enhanced the synergistic effect as indicated by CI (Fig. 3H). In contrast, overexpression of p53 in mutant cells GSC25 and U251 abrogated the synergistic effects (Fig. 3G and H).
Fig. 3.

Specific synergist of MK1775 with BKM to inhibited cell growth in p53-mutant cells but not p53-wt GBM cells. (A–B) Assessment of the degree of synergy between BKM and MK1775 in GSC23 (p53-mutant cells) and GSC7-10 (p53-wt cells) using the Chou–Talalay method. Drug interactions are expressed as fraction affected (Fa) curves and combination index (CI) plots. (C) The CI values (Fa = 0.5) of 22 cell lines were generated using the method depicted in (A) and (B) and the genetic background of p53 and PTEN was checked. (D–E) Dot plot of correlation between p53 and PTEN status and synergistic effect by showing CI of individual cell line. (F–H) P53 was either knocked down in wt U87 with shRNA or overexpressed in mutant GSC25 and U251, and CI was evaluated.
Combination of MK1775 and BKM Leads to Increased Mitotic Catastrophe
Inhibition of WEE1 by MK1775 abrogates the G2 checkpoint and pushes cells into premature mitosis. We next determined if the combination of BKM and MK1775 induces mitotic catastrophe. Cells were treated with BKM alone or MK1775 alone or the combination was stained with phalloidin and DAPI and scored for the presence of multinucleated cells. Representative immunofluorescence images illustrating the presence of multilobulated nuclei in U251 cells following drug treatments are presented in Fig. 4A. The combination of MK1775 with BKM resulted in a supra-additive increase in multinucleated cells, statistically greater than either treatment alone in tumor cells. As a control, combination treatment failed to induce mitotic catastrophe in NHA cells (Supplementary Figure S3C).
Fig. 4 .

Combination of MK1775 and BKM leads to increased mitotic catastrophe and induced apoptotic cell death in p53-mutant cells. (A) P53-mutant cell GSC23 was treated with BKM, MK1775, or combination for 48 hours. Cells were stained with phalloidin and DAPI to examine multinucleated cells. Representative fields were shown and multinucleated cells were quantified by counting 3 fields of each treatment. (B) SCR cells and WEE1-sh cells were treated with BKM for 48 hours. The multinucleated cells were examined by phalloidin and DAPI staining, observed under microscope, and quantified by counting 3 fields of each treatment. Apoptosis was evaluated by cleavage of PARP and caspase-3 and caspase-9 (C), TUNEL staining (D and G), annexin V (E and H) sub-G1 population (F). Scale bars, 50 microns.
To exclude the off-target effects of MK1775, inhibition of WEE1 was also achieved by WEE1 shRNA. Similarly, combination of WEE1 shRNA and BKM treatment triggered greater increase of multinucleated cells than either treatment alone (Fig. 4B).
Combination of PI3K and WEE1 Inhibition Induced Apoptotic Cell Death
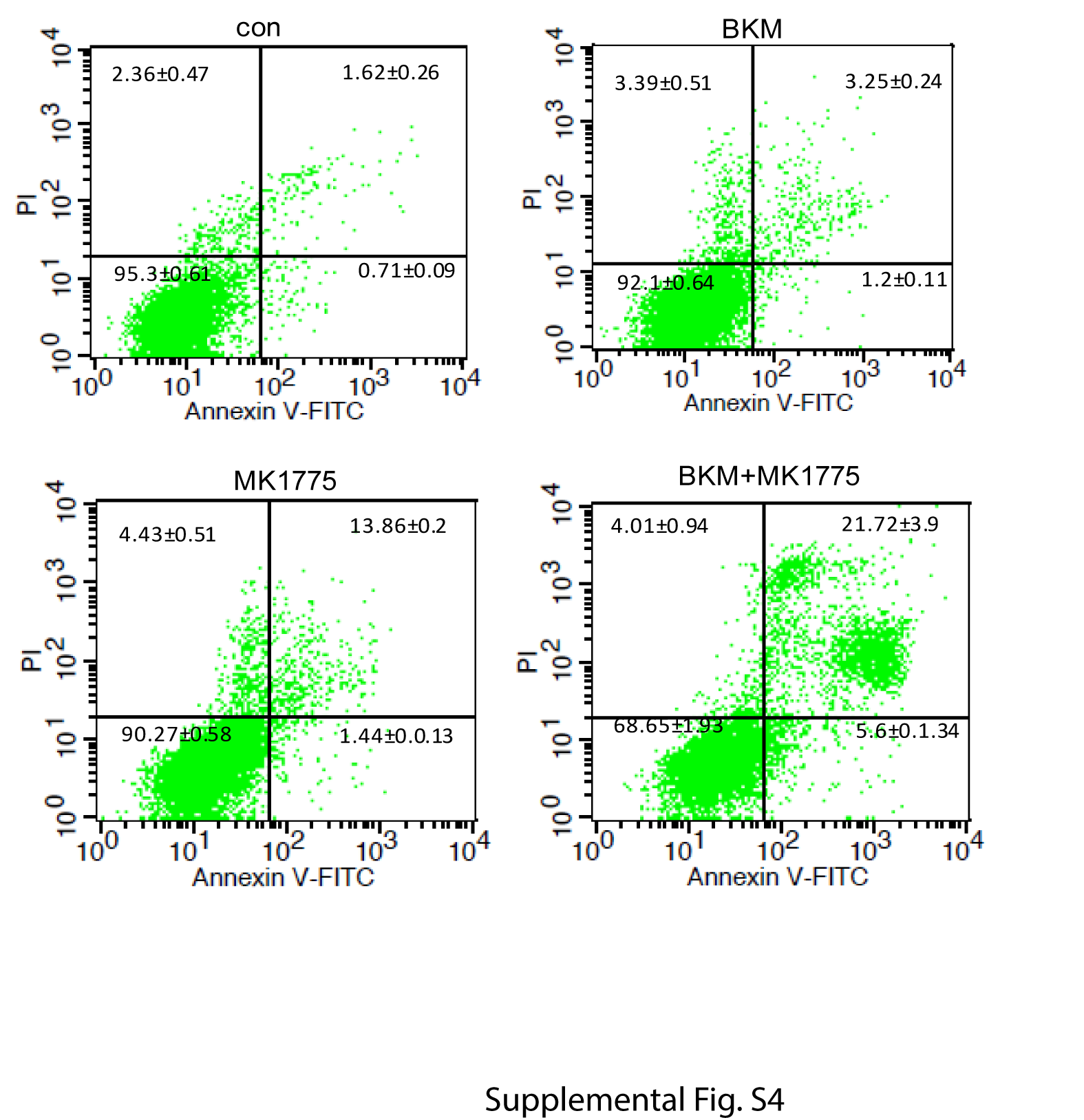
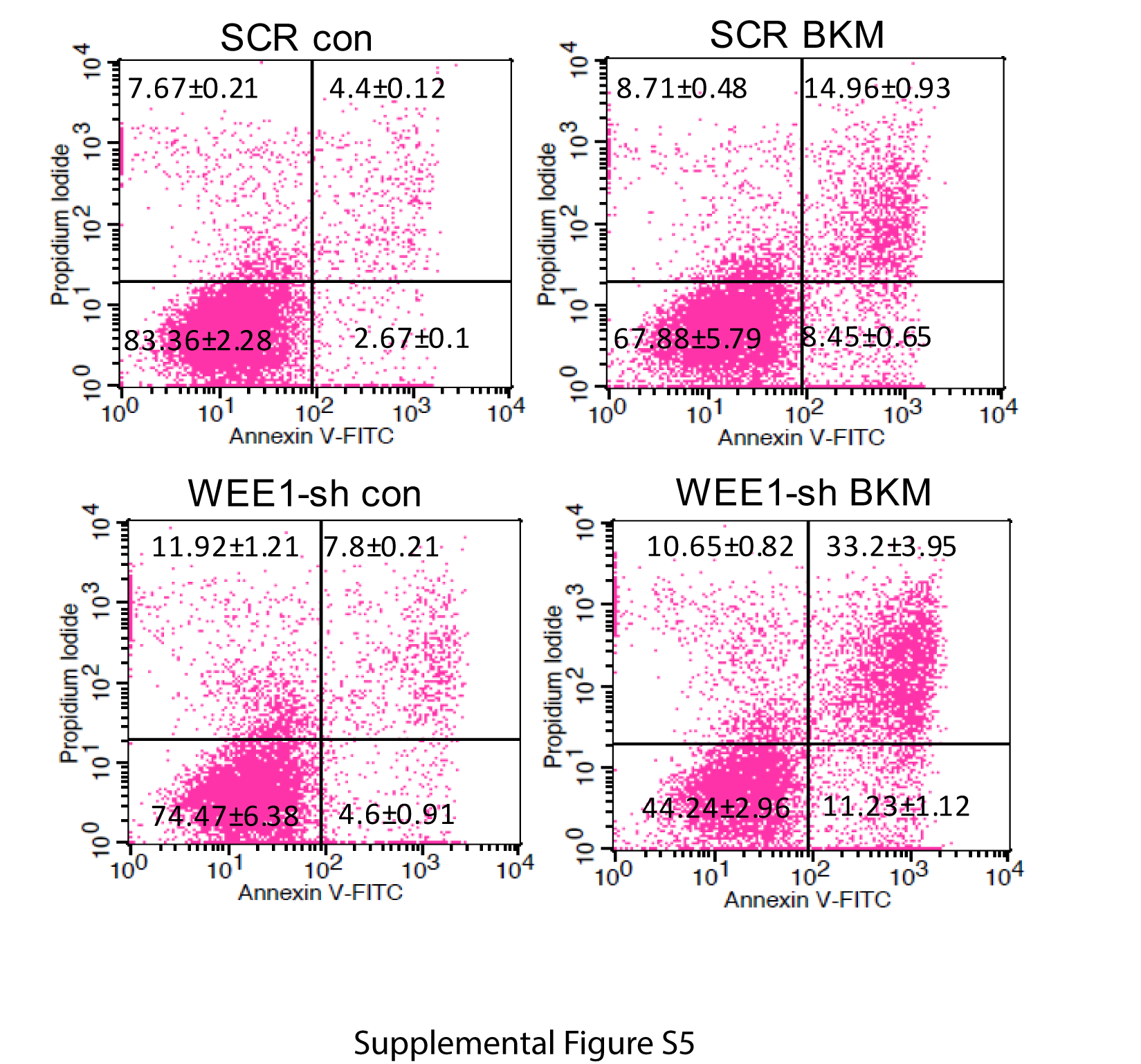
Mitotic catastrophe triggers an attempt at aberrant chromosome segregation, which culminates in the activation of the apoptotic pathway and cellular demise.21 We therefore determined if the combination of BKM and MK1775 induced apoptosis in GBM cells. The apoptosis was first evaluated by examining caspase-9, caspase-3, and PARP cleavage. While inhibition of PI3K or WEE1 alone induces no or slight cleavage of caspase-9, caspase-3, and PARP, the combination of BKM and MK1775 induced a strong cleavage of caspase-9, caspase-3, and PARP in a time-dependent manner (Fig. 4C). In line with this observation, combination of BKM and MK1775 resulted in a supra-additive increase of TUNEL-positive cells than either treatment alone (Fig. 4D). The apoptosis was also measured by annexin V staining. Similarly, combination of BKM and MK1775 resulted in a significant annexin V–positive cell percentage than either treatment alone (Fig. 4E and Supplementary Figure S4). Further, we knocked down WEE1 expression by shRNA and tested its combination with BKM treatment. Consistently, combination of BKM and WEE1 shRNA also induced a significant apoptosis as indicated by increased sub-G1 cells (Fig. 4F), TUNEL-positive cells (Fig. 4G), and annexin V–positive cell percentage (Fig. 4H and Supplementary Figure S5).
Efficacy of MK1775 in Combination with BKM in Subcutaneous Tumors
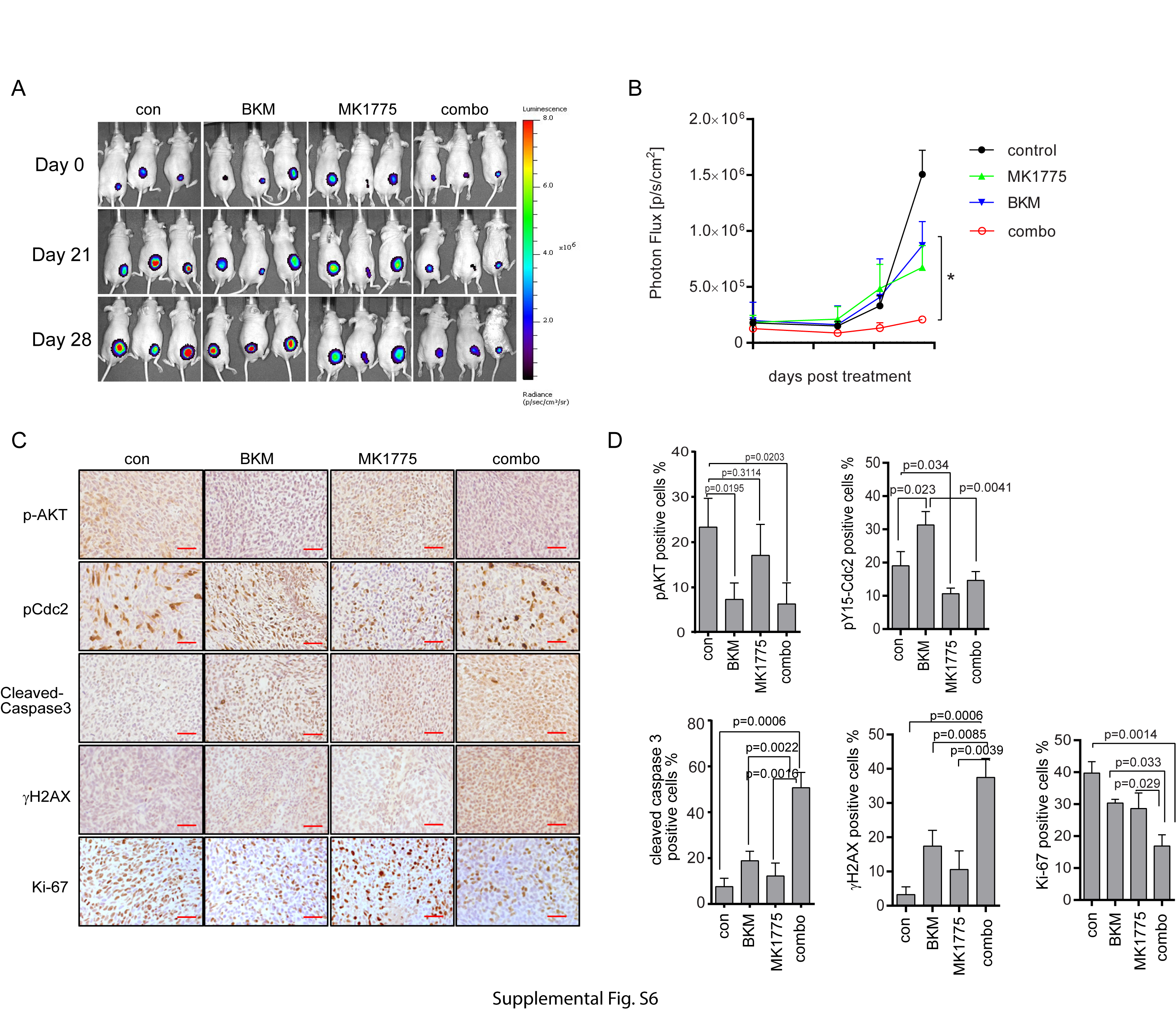
To evaluate the in vivo efficacy of MK1775 and BKM, subcutaneous models were generated by injecting GSC23-luc (Fig. 5) and GSC25-luc cells (Supplementary Figure S6) into the hind legs of nude mice. Mice were treated with single modality or combination for 4 weeks. The growth of tumors was monitored weekly by bioluminescence imaging. The BKM or MK1775 single treatment has no significant effect to inhibit tumor growth (BKM vs control P = 0.789; and MK1775 vs control P = 0.599; Fig. 5A and B). However, combinational treatment greatly suppressed tumor growth compared with BKM alone (P = 0.017), MK1775 alone (P = 0.006), or control treatment (P = 0.038) (Fig. 5A and B; also see Supplementary Figure S6A and B). Tumor size was also measured by caliper. Consistent with the imaging results, the single treatment failed to inhibit tumor growth, whereas combination treatment significantly inhibited tumor growth (Fig. 5C).
Fig. 5.

Combination of BKM and MK1775 inhibited tumor growth in subcutaneous model. Mice with established flank xenografts were administered BKM (20 mg/kg/d), MK1775 (30 mg/kg/d), or combination. Tumor size was monitored by bioluminescence imaging (A–B) as well as being measured by caliper (C). The tissue sections were incubated with antibodies against pS473-Akt, pY15-Cdc2, γH2AX, cleaved caspase-3, and Ki-67 (D) and quantified by Image-Plus pro (E). Scale bars, 50 microns.
An immunohistochemical analysis of the tumor tissue showed that Akt phosphorylation was reduced by BKM treatment (Fig. 5D for GSC23 and Supplementary Figure S6C for GSC25), confirming the efficacy of BKM at inhibiting PI3K/Akt signaling in vivo. Consistent with the in vitro analysis, immunohistochemical staining further proved that BKM treatment increased Cdc2-Y15 phosphorylation in vivo, which was blocked by WEE1 inhibition (Fig. 5D and Supplementary Figure S6C). In addition, combination of BKM and MK1775 in vivo decreased Ki-67–positive cells compared with BKM (P = 0.0174), MK1775 (P = 0.0112), or control treatment (P = 0.0001). Combination also increased cleaved caspase-3 to a greater extent than BKM alone (P = 0.007), MK1775 (P = 0.0008), or control treatment (P = 0.0006). Gamma-H2AX was increased by combination treatment compared with BKM (P = 0.0024), MK1775 (P = 0.0009), or control treatment (P = 0.0008), suggesting that the combination treatment compromised DNA damage repair and induced apoptosis in vivo.
Lack of MK1775 Efficacy in Intracranial Tumors
Antitumor efficacy of combination treatment was evaluated in an intracranial GSC23-luc tumor model. BKM alone moderately suppressed tumor size (vs control, P = 0.028). MK1775 treatment did not suppress tumor growth (vs control, P = 0.208), and addition of MK1775 to BKM did not impact tumor growth compared with BKM alone (P = 0.38) (Fig. 6A and B). The median survival times for the control group, the MK1775-treated group, the BKM-treated group, and the combination treatment group were 67.5 days, 70.5 days, 75 days, and 80.5 days, respectively (Fig. 6C). Although combination treatment showed survival advantage over control treatment (P = 0.0126) and MK1775 alone (P = 0.045), there is no significant difference between combination treatment and BKM treatment in regard to median survival time (P = 0.978). These data demonstrated a lack of efficacy of MK1775 alone or in combination with BKM in brain.
Fig. 6.

Evaluation of efficacy of MK1775 and BKM in an intracranial animal model. (A) Mice were administered BKM (20 mg/kg/d), MK1775 (30 mg/kg/d), or combination by gavage 5 times a week for 5 weeks. Representative bioluminescence imaging of tumor at day 39 is shown. (B) Quantitative assessments of tumor growth following implantation. Data expressed as mean + SD, n = 6 per group. P = 0.208 for control vs MK1775; P = 0.028 for control vs BKM; P = 0.0169 for control vs combination; P = 0.079 for MK1775 vs combination; P = 0.38 for BKM vs combination. (C) Mice were sacrificed at morbidity, survival curves were plotted in Kaplan–Meier graphs, and differences evaluated using the log-rank test (P = 0.0126 for control vs combination; P = 0.045 for MK1775 vs combination; P = 0.978 for BKM vs combination). (D) Immunostaining of the brain sections of animals that had been treated for 5 weeks. The tissue sections were incubated with antibodies against pS473-Akt, pY15-Cdc2, and cleaved caspase-3. Scale bars, 50 microns.
MK1775 has a very poor blood–brain barrier (BBB) penetration; the ratio of concentration of MK1775 in brain to blood was reported to be about 4% to 5%.22 To evaluate if there is functional concentration of MK1775 in our intracranial tumors, we performed immunohistochemistry to detect pY15-Cdc2, the direct substrate of WEE1 kinase. Consistent with the in vitro and subcutaneous results, BKM induced phosphorylation of Y15-Cdc2 (P = 0.04, compared with control); however, unlike the in vitro and subcutaneous results, this phosphorylation was not blocked by addition of MK1775 (P = 0.12, combination vs BKM) (Fig. 6D and E), suggesting that there is no functional concentration of MK1775 in our intracranial tumors.
Discussion
Advances in the development of selective therapeutic agents have resulted in exciting changes to the therapeutic landscape for solid tumors; however, success in treating GBM patients has remained limited. Activation of the PI3K/Akt/mTOR pathway through different mechanisms has been reported in GBMs, and recent clinical trials with selective PI3K and/or mTOR inhibitors have reported limited efficacy,23,24 which is believed to be due to negative feedback loops and activation of bypass signaling pathways, thus highlighting the importance to target multiple effectors simultaneously. In the present study, we investigated the therapeutic sensitizing abilities of PI3K inhibition along with WEE1 kinase inhibition in GBM.
WEE1 kinase is a key molecule in maintaining G2 cell-cycle checkpoint arrest for premitotic DNA repair. Whereas normal cells repair damaged DNA during G1 arrest, cancer cells often have a deficient G1 arrest and largely depend on G2 arrest. The molecular switch for the G2/M transition is held by WEE1 and is pushed forward by Cdc25. Although PI3K/Akt inhibition alone has been reported to promote apoptosis in Hodgkin lymphoma,25 in GBM it only suppresses proliferation through G2/M arrest.26 We have shown previously that BKM induced G2/M arrest in established GBM cell lines14 as well as in GICs. How PI3K/Akt regulates G2/M transition remains unclear. In this study, we showed that PI3K inhibition by BKM activated WEE1 by decreasing the inhibitory phosphorylation of WEE1 at Ser642. This result is consistent with previous reports showing that Akt directly binds to and inactivates WEE1 by phosphorylating WEE1 at Ser642, leading to cell cycle arrest.15 Although molecules other than WEE1 might also contribute to PI3K/Akt inhibition–induced G2/M arrest,27 in this study we show that WEE1 inhibition by shRNA or by WEE1 inhibitor MK1775 abrogated BKM-induced G2/M arrest, suggesting that WEE1 plays an important role in the PI3K/Akt-regulated cell cycle.
Our hypothesis was based on the fact that the failure to induce apoptosis by BKM might be due to PI3K/Akt inhibition–induced G2/M arrest, which allows transformed cells the time needed to recover from stress and thus confers a survival advantage.24 We tested this by combining PI3K inhibition with MK1775. We show that MK1775 abrogated BKM-induced G2/M arrest, forced the glioma cells to enter premature mitosis, and induced mitotic catastrophe. The culmination of aberrant chromosome segregation triggered by combination treatment ultimately activated apoptotic death in GICs. The apoptotic response was observed in p53-mutant cell lines.
Cell cycle is tightly regulated by multiple checkpoints to facilitate DNA repair and promote cell death in unrepaired cells. P53 is a key regulator of the G1 checkpoint; however, p53 is mutated in 33% of GBM, and p53-deficient cells are predicted to be more dependent on G2 checkpoint.28 WEE1 was suggested to play a dominant role in controlling the G2 checkpoint.29 Consistent with this concept, multiple studies have reported that WEE1 inhibition sensitized p53-deficient tumors to DNA-damaging agents for several types of cancer, like pancreatic cancer,18 sarcomas,30 ovarian cancer,31 and nasopharyngeal carcinoma.32 Here we show that PI3K inhibition synergized by WEE1 inhibition was specifically observed in p53-mutant GBM cells but not in p53-wt cells. Therefore a large population of GBM patients might benefit from combination treatment given the fact that p53 is mutated in 33% of GBM.
The in vivo efficacy of the combination of BKM and MK1775 was tested in a subcutaneous xenograft model as well as in an intracranial xenograft model. Combination treatment substantially suppressed subcutaneous tumor growth, suggesting a promising therapeutic strategy to overcome BKM resistance. In the intracranial model, combination treatment showed only a moderate decrease of tumor size compared with either the control group (P = 0.017) or the MK1775-treated group (P = 0.039); the difference was not significant compared with the BKM-treated group (P = 0.38) (Fig. 6A and B). Similarly, combination treatment failed to extend animal survival compared with the BKM-treated group (P = 0.9778). One of the reasons may be inefficient delivery of MK1775 to tumor cells due to poor blood–brain penetration of MK1775. Indeed, Pokorny et al used matrix-assisted laser desorption/ionization/mass spectrometry imaging to demonstrate that the accumulation of MK1775 is limited and heterogeneous in the GBM orthotopic xenograft model.22 To evaluate if there is functional concentration of MK1775 in our intracranial model, we analyzed the tumor tissues by immunohistochemistry and showed that MK1775 failed to block Cdc2-Y15 phosphorylation in brain tumor (Fig. 6D), while it completely blocked both basal and BKM-induced Cdc2-Y15 phosphorylation in vitro (Fig. 1C) and in the subcutaneous model (Fig. 5D and E), confirming the limited delivery of MK1775 to brain tumors. However, MK1775 was reported to enhance radiosensitivity in orthotopic GBM xenografts,33,34 suggesting that radiation disrupted the BBB,35 and resulted in increased MK1775 accumulation within brain tumor. Therefore, the combination of BKM and MK1775 as a treatment for GBM may be most effective following standard surgical and radiation therapy which will disrupt BBB and increase penetration. Overall, it is critically important to improve the BBB penetration of MK1775 for its successful application in brain tumor.
Our rationale for combining WEE1 inhibition with PI3K inhibition in GBM is supported by several arguments. First of all, as mentioned above, a large fraction of GBM cells harbor p53 mutation, making these GBM cells particularly dependent on G2 checkpoint. In addition, recent reports29,33,34 and our current study using either different databases or array-based gene expression analysis indicated that WEE1 is overexpressed in GBM and correlates with survival, suggesting that WEE1 would be a tumor-specific target and that GBMs rely on WEE1 to escape fatal DNA damage. Last, aberrant hyperactivation of PI3K/Akt signaling in GBM and direct crosstalk between Akt and WEE1 support the necessity of combination inhibition. Our study showed that combined inhibition of WEE1 and PI3K signaling sensitizes p53-deficient glioblastoma to apoptotic cell death and provides a compelling basis for clinical investigation that improves efficacy of PI3K targeting in GBM.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This study was funded by a CPRIT grant (RP120256 to W.K.A.Y.), a National Brain Tumor Society grant (Defeat GBM), a National Foundation for Cancer Research (NFCR) grant (to W.K.A.Y.), a SPORE grant (P50 CA127001 to F. F. Lang), and a Cancer Center Support grant (CA016672 to Ronald A. DePinho).
Conflict of interest statement. W.K.A.Y. discloses a conflict of interest as a consultant with DNATrix. All other authors have no conflict of interest to disclose.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Behin A, Hoang-Xuan K, Carpentier AF, Delattre JY. Primary brain tumours in adults. Lancet. 2003;361(9354):323–331. [DOI] [PubMed] [Google Scholar]
- 2. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. [DOI] [PubMed] [Google Scholar]
- 3. Mellinghoff IK, Wang MY, Vivanco I et al. . Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–2024. [DOI] [PubMed] [Google Scholar]
- 4. Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma–animal models and therapeutic challenges. Brain Pathol. 2009;19(1):112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Broderick DK, Di C, Parrett TJ et al. . Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64(15):5048–5050. [DOI] [PubMed] [Google Scholar]
- 6. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71(13):4550–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kita D, Yonekawa Y, Weller M, Ohgaki H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007;113(3):295–302. [DOI] [PubMed] [Google Scholar]
- 9. Cloughesy TF, Yoshimoto K, Nghiemphu P et al. . Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harrington LS, Findlay GM, Gray A et al. . The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O’Reilly KE, Rojo F, She QB et al. . mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Serra V, Eichhorn PJ, García-García C et al. . RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest. 2013;123(6):2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu S, Wang S, Zheng S, Verhaak R, Koul D, Yung WK. MSK1-mediated β-catenin phosphorylation confers resistance to PI3K/mTOR inhibitors in glioblastoma. Mol Cancer Ther. 2016;15(7):1656–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koul D, Fu J, Shen R et al. . Antitumor activity of NVP-BKM120—a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18(1):184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katayama K, Fujita N, Tsuruo T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol Cell Biol. 2005;25(13):5725–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Würdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17(13):4200–4207. [DOI] [PubMed] [Google Scholar]
- 17. Hirai H, Arai T, Okada M et al. . MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9(7):514–522. [DOI] [PubMed] [Google Scholar]
- 18. Rajeshkumar NV, De Oliveira E, Ottenhof N et al. . MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17(9):2799–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saito N, Fu J, Zheng S et al. . A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells. 2014;32(1):301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tirosh I, Venteicher AS, Hebert C et al. . Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539(7628):309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–2837. [DOI] [PubMed] [Google Scholar]
- 22. Pokorny JL, Calligaris D, Gupta SK et al. . The efficacy of the wee1 inhibitor MK-1775 combined with temozolomide is limited by heterogeneous distribution across the blood-brain barrier in glioblastoma. Clin Cancer Res. 2015;21(8):1916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wen PY, Lee EQ, Reardon DA, Ligon KL, Alfred Yung WK. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro Oncol. 2012;14(7):819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Westhoff MA, Karpel-Massler G, Brühl O et al. . A critical evaluation of PI3K inhibition in glioblastoma and neuroblastoma therapy. Mol Cell Ther. 2014;2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Mills GB, Younes A. Inhibition of the phosphatidylinositol-3 kinase/Akt promotes G1 cell cycle arrest and apoptosis in Hodgkin lymphoma. Br J Haematol. 2006;132(4):503–511. [DOI] [PubMed] [Google Scholar]
- 26. Kandel ES, Skeen J, Majewski N et al. . Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22(22):7831–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okumura E, Fukuhara T, Yoshida H et al. . Akt inhibits Myt1 in the signalling pathway that leads to meiotic G2/M-phase transition. Nat Cell Biol. 2002;4(2):111–116. [DOI] [PubMed] [Google Scholar]
- 28. Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98(3):523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mir SE, De Witt Hamer PC, Krawczyk PM et al. . In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18(3):244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kreahling JM, Foroutan P, Reed D et al. . Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS One. 2013;8(3):e57523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mizuarai S, Yamanaka K, Itadani H et al. . Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol Cancer. 2009;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mak JP, Man WY, Chow JP, Ma HT, Poon RY. Pharmacological inactivation of CHK1 and WEE1 induces mitotic catastrophe in nasopharyngeal carcinoma cells. Oncotarget. 2015;6(25):21074–21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mueller S, Hashizume R, Yang X et al. . Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014;16(3):352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sarcar B, Kahali S, Prabhu AH et al. . Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol Cancer Ther. 2011;10(12):2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cao Y, Tsien CI, Shen Z et al. . Use of magnetic resonance imaging to assess blood-brain/blood-glioma barrier opening during conformal radiotherapy. J Clin Oncol. 2005;23(18):4127–4136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.