

Abstract
While cholinergic receptor activation has long been known to dramatically enhance the excitability of cortical neurons, the cellular mechanisms responsible for this effect are not well understood. We used intracellular recordings in rat (both sexes) neocortical brain slices to assess the ionic mechanisms supporting persistent firing modes triggered by depolarizing stimuli following cholinergic receptor activation. We found multiple lines of evidence suggesting that a component of the underlying hyperexcitability associated with persistent firing reflects a reduction in the standing (leak) K+ current mediated by Ether-a-go-go-Related Gene (ERG) channels. Three chemically diverse ERG channel blockers (terfenadine, ErgToxin-1, and E-4031) abolished persistent firing and the underlying increase in input resistance in deep pyramidal cells in temporal and prefrontal association neocortex. Calcium accumulation during triggering stimuli appears to attenuate ERG currents, leading to membrane potential depolarization and increased input resistance, two critical elements generating persistent firing. Our results also suggest that ERG current normally governs cortical neuron responses to depolarizing stimuli by opposing prolonged discharges and by enhancing the poststimulus repolarization. The broad expression of ERG channels and the ability of ERG blocks to abolish persistent firing evoked by both synaptic and intracellular step stimuli suggest that modulation of ERG channels may underlie many forms of persistent activity observed in vivo.
SIGNIFICANCE STATEMENT Persistent activity, where spiking continues beyond the triggering stimulus, is a common phenomenon observed in many types of neurons. Identifying the mechanism underlying this elementary process of memory is a step forward in understanding higher cognitive function including short-term memory. Our results suggest that a reduction in the currents normally mediated by Ether-a-go-go-Related Gene (ERG) K+ channels contributes to persistent firing in neocortical pyramidal cells. ERG currents have been previously studied primarily in the heart; relatively little is known about ERG function in the brain, although mutations in ERG channels have recently been linked to schizophrenia. The present study is among the first to describe its role in neocortex in relation to biophysical correlates of memory function.
Keywords: afterdepolarization, brain slice, cholinergic modulation, intrinsic persistent activity, patch clamp
Introduction
Many brain regions contain neurons that generate long-lasting spiking responses to brief stimuli. In some of these areas, such as the brainstem nuclei that mediate the vestibulo-ocular reflex (VOR; Major et al., 2004), persistent responses play a central role in maintaining a sensory-motor system in a stable state. In cortical brain regions, persistent activity is associated with encoding of short-term memories (e.g., delay period firing during working memory tasks; Fuster and Alexander, 1971) and in neuronal ensembles that represent time intervals (Eichenbaum, 2014). Recent work also demonstrates that persistent activity also occurs in hippocampal brain slices where mossy cells receive long-lasting synaptic barrages following brief stimuli (Larimer and Strowbridge, 2010; Hyde and Strowbridge, 2012). Despite the widespread nature of persistent firing, the underlying mechanisms responsible for this firing mode have remained mysterious.
While synaptic reverberation has long been proposed to underlie persistent spiking responses recorded in vivo, especially in cortical regions (Hebb, 1949; Hopfield, 1982; Wang, 1999), there is relatively little experimental support for this mechanism. By contrast, several decades of work using brain slices has demonstrated the ability of many cortical (Andrade, 1991; Haj-Dahmane and Andrade, 1996; Rahman and Berger, 2011) and olfactory (Pressler et al., 2007) neurons to generate persistent firing through cell-autonomous biophysical mechanisms. These intrinsic forms of persistent activity can be initiated by bursts of action potentials (APs; Krnjević et al., 1971; Andrade, 1991) and are typically studied using brain slices exposed to muscarinic receptor agonists to enhance excitability (Krnjević et al., 1971; Andrade, 1991; Haj-Dahmane and Andrade, 1996; Haj-Dahmane and Andrade, 1998; Rahman and Berger, 2011) that likely mimic the increase in the firing rate of cholinergic basal forebrain neurons during periods of heightened attention (Richardson and DeLong, 1986). Understanding the biophysical basis of persistent firing in brain slices would likely provide new tools that could be used to determine the relative roles of intrinsic and circuit mechanisms to persistent activity recorded in vivo.
While most experimental work on the underlying mechanism is in agreement that an increase in intracellular Ca2+ is required to trigger intrinsic persistent firing (Egorov et al., 2002; Fransén et al., 2006; Pressler et al., 2007), the critical downstream ion channels modulated by Ca2+ have not been identified. Many studies (Egorov et al., 2002; Rahman and Berger, 2011; Jochems and Yoshida, 2013) have suggested that inward current mediated by Ca2+-activated nonselective cation channels (ICAN) underlies intrinsic persistent firing. Since the molecular basis of ICAN has remained elusive, this hypothesis has been difficult to test definitively. Transient receptor potential (TRP) channels, a likely component of ICAN (Haj-Dahmane and Andrade, 1996; Zhang et al., 2011), have few selective antagonists and there have been no reports to date of TRP channel knockouts abolishing persistent activity though expression of dominant negative canonical TRP (TRPC) subunits can attenuate neocortical afterdepolarization (ADP) responses (Yan et al., 2009; but see Dasari et al., 2013). The ability of TRP proteins to form multimers (Nilius and Owsianik, 2011) complicates testing this hypothesis.
In addition to ICAN-mediated Ca2+-sensitive currents, the modulation of inward or outward currents active near rest have been suggested to underlie intrinsic persistent activity. Yamada-Hanff and Bean (2013) demonstrated that the biophysical properties of subthreshold Na+ current could enable persistent firing. However, the absence of selective blockers of subthreshold Na+ channel activity had precluded directly testing this model. Alternatively, a decrease in a “leak” K+ current could underlie the enhanced excitability associated with persistent firing. Presumably, many muscarinic receptor agonists work through this mechanism to enhance overall excitability by inhibiting KCNQ channels that form the M current (IM; Schnee and Brown, 1998; Delmas and Brown, 2005). Similarly, Ether-a-go-go-related gene (ERG) channels can be modulated by second messenger cascades that converge on protein kinase C (PKC; Cockerill et al., 2007). ERG is highly expressed in neurons within the deep layers of neocortex (Saganich et al., 2001; Papa et al., 2003), among other regions, and is known to influence the excitability of a variety of neurons including midbrain dopaminergic neurons (Ji et al., 2012), brainstem neurons (Hardman and Forsythe, 2009), and hippocampal CA1 pyramidal cells (Fano et al., 2012).
Mutations in ERG have been linked to schizophrenia (Huffaker et al., 2009; Atalar et al., 2010) and many common antipsychotic medications are potent ERG blockers (Wible et al., 2005), suggesting that altered ERG function may underlie some types of cognitive dysfunction. Whether the increase in excitability that enables persistent responses to transient depolarizing stimuli is mediated by ICAN currents, the activation of subthreshold Na+ channels or attenuation of outward currents active near rest is not known. In the present study, we used electrophysiological methods define the mechanism responsible for intrinsic persistent activity in rodent neocortical slices. We find that ERG channel blockers abolish persistent firing in pyramidal cells in both temporal association neocortical neurons as well as in prefrontal neurons. Blockade of ERG channels also greatly attenuates the increase in input resistance that underlies persistent firing, presumably reflecting a Ca2+-dependent attenuation in the leak ERG current. The robustness of ERG-mediated persistent firing and the widespread expression of ERG channels across diverse cortical regions suggest that modulation of ERG current may underlie many forms of persistent firing reported in vivo.
Materials and Methods
Slice preparation.
Sprague Dawley rats of either sex from postnatal day 14 (P14) to P25 were used in all experiments. Rats were anesthetized with ketamine and decapitated. The brain was then dissected and transferred into ice-cold (∼0°C) artificial CSF (ACSF) composed of the following (in mm): 124 NaCl, 2.54 KCl, 1.23 NaH2PO4, 6.2 MgSO4, 26 NaHCO3, 10 dextrose, and 1 CaCl2, equilibrated with 95% O2/5% CO2. Horizontal slices (300 μm) were prepared from temporal association cortex (TeA; at the same dorsal–ventral level as the ventral hippocampus) using a Leica VT1200 Vibratome. Recordings from prefrontal cortex (PFC) were performed on 300-μm-thick coronal slices that included the medial PFC. Slices were incubated at 30°C for ∼30 min and then maintained at room temperature (∼25°C) until use. All experiments were performed under guidelines approved by the Case Western Reserve University Animal Care and Use Committee. This study, including pilot and ancillary experiments, was based on intracellular recordings from 382 neurons from 92 animals. The experimental data presented in the Results section were based on recordings from 158 L5 neocortical neurons.
Electrophysiology.
Intracellular and cell-attached recordings were performed in a submerged chamber maintained at 30°C and perfused continuously (∼2 ml/min) with ACSF containing the following (in mm): 124 NaCl, 3 KCl, 1.23 NaH2PO4, 1.2 MgSO4, 26 NaHCO3, 10 dextrose, 2.5 CaCl2, equilibrated with 95% O2/5% CO2. Whole-cell and cell-attached recordings were made using an Axopatch 1D Amplifier (Molecular Devices/Molecular Dynamics). Patch-clamp recording electrodes with resistances of 3–8 MΩ were pulled from 1.5 mm outer diameter thin wall borosilicate glass tubing (WPI), using a micropipette puller (P-97, Sutter Instruments). The pipettes contained the following (in mm): 140 K-methylsulfate (MP Biomedicals), 4 NaCl, 10 HEPES, 0.2 EGTA, 4 MgATP, 0.3 Na3GTP, and 10 phosphocreatine. In some experiments, 10 mm bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) was substituted for EGTA in the internal solution to enhance Ca2+ buffering. In several sets of experiments, 10 μm E-4031 was added to the internal solution. Individual neurons were visualized under infrared differential interference contrast (IR-DIC) video microscopy (Axioskop FS1, Zeiss). Recordings were low-pass filtered at 5 kHz (FLA-01, Cygnus Technology) and acquired at 10 kHz using a simultaneously sampling 16-bit data acquisition system (ITC-18, InstruTech) operated by custom software written in VB.NET on a Windows-based PC. Unless specified otherwise, all membrane potentials presented in the text and contained within the illustrations are not corrected for the liquid junction potential. We estimated this junction potential using standard software (Barry, 1994) to be 10.3 mV using ACSF with 3 mm K+ and 10.7 mV with the ACSF used for voltage-clamp experiments (containing 10 mm K+; see below). Synaptic stimulation was performed using an insulated tungsten monopolar electrode placed in layer 3 (L3) near and connected to a constant current stimulus isolation unit (A360, WPI).
In this study, we recorded exclusively from L5 neocortical pyramidal cells that generated “regular spiking” responses to 2 s duration depolarizing currents, as defined by previous neocortical studies (Connors and Gutnick, 1990; Schubert et al., 2001; Dégenètais et al., 2002). Cells were selected using IR-DIC visualization and were confirmed to be pyramidal cells using two-photon imaging in a subset of recordings. The average resting membrane potential of TeA L5 neurons was −66.1 ± 0.25 (N = 149; range, −60 to −70 mV). The average input resistance (RIn) value of these neurons was 107 ± 3.7 MΩ. Recordings from neurons with resting membrane potentials of more than −60 mV or RIn < 60 MΩ were excluded. Persistent firing was evoked in most experiments in this study using a standardized protocol described in the study by Yoshida and Hasselmo (2009) (2 s duration depolarizing step that generated ∼10 Hz continuous firing from −70 mV). While several recent studies (Dembrow et al., 2010; Groh et al., 2010; Kim et al., 2015) have identified subtypes of L5 regular spiking pyramidal cells, prefrontal cortex-based axonal targets, and intrinsic properties, we are not aware of any comparable analysis performed in temporal association cortex. From their pronounced spike frequency adaptation, both the TeA and mPFC cells included in our study resemble more closely the commissural-projecting subtype L5 pyramidal cells more than the corticopontine projection cells (Dembrow et al., 2010; Dembrow and Johnston, 2014).
Except for experiments using intracellularly applied E-4031, all drugs were applied by switching the bath perfusion reservoir. Unless specified, all drugs were purchased from Tocris Bioscience. Drugs used included the following: carbamoylcholine chloride [CCh (carbachol)] used at 2 μm (10 μm in one experiment) with 10 mm stock solutions of CCh in water were prepared each day; gabazine (SR 95531, used at 10 μm); 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; used at 10 μm); 4-aminopyridine (4-AP; used at 100 μm and purchased from Sigma-Aldrich); tetraethylammonium (TEA; used at 1 mm and purchased from Sigma-Aldrich); ZD7288 (used at 10 μm); nimodipine (20 μm); d-APV (used at 10 μm); N-(3,4-difluorophenyl)-N′-(3-methyl-1-phenyl-1H-pyrazol-5-yl)urea (ML297 used at 0.67 μM); terfenadine (Terf; used at 10 μm); E-4031 dihydrochloride (used at 10 μm); fexofenadine (used 30 μm); linopirdine dihydrochloride (used at 30 μm); XE991 (used at 10 μm); pirenzepine dihydrochloride (used at 10 μm); and ErgToxin-1 (used at 50 nm) was purchased from Alomone Labs and dissolved in ACSF.
Experimental design and statistical analysis.
We estimated input resistance under both static and time-varying conditions. For static tests, which were performed when we were able to hold the membrane at a constant potential for extended periods, we estimated RIn from the maximal voltage deflection elicited by a standard (typically −50 pA for 2 s) injected current step. To estimate RIn changes underlying persistent activity (see Fig. 6B,C), we hyperpolarized the membrane potential 500 ms following the offset of a depolarizing step to abolish persistent firing and then injected a series of small hyperpolarizing test pulses (−50 pA, 300 ms duration) to assay input resistance every 600 ms. By delaying the RIn assay procedure by 500 ms, we could ensure that the depolarizing step was effective in triggering persistent activity since in all experiments the first poststep AP was elicited within 500 ms. We then used a two-stage correction process to account for the effect of the changing membrane potential during the ADP and the voltage dependence of input resistance in neocortical pyramidal cells (McCormick and Prince, 1985). First, we detrended each response to the 300 ms hyperpolarizing pulses by subtracting the regression line fit between the mean potential during 10 ms periods acquired immediately before and 290 ms following each step. We then calculated a raw input resistance estimate from the voltage deflection elicited by each step calculated from the mean potential during the final 50 ms of the detrended step response. Finally, we compared this RIn estimate to calibration RIn measurements acquired in interleaved trials where RIn was measured under static conditions. We used linear regression fits of this calibration data to determine the expected input resistance at any arbitrary voltage within the calibrated range (−75 to −60 mV; R2 = 0.76 ± 0.029 for RIn estimates in ACSF and 0.83 ± 0.063 in CCh). Our final “corrected” delta input resistance measurement reflected the difference between the measured RIn and the expected RIn at that particular membrane potential. For example, in one experiment the static input resistance recorded at −70 mV was 100 MΩ, which increased to 110 MΩ at a −65 mV holding potential. If the peak ADP response occurred at −65 mV in that cell, generating a detrended RIn estimate of 150 MΩ, we would then report a corrected change in input resistance of +40 MΩ (150–110 MΩ) at that time point. We repeated this procedure using at least three ADP trials in each cell and at each time point, and report the mean corrected delta RIn values. Summary time plots show the mean ± SEM across multiple experiments of these single-cell corrected delta RIn estimates.
Figure 6.
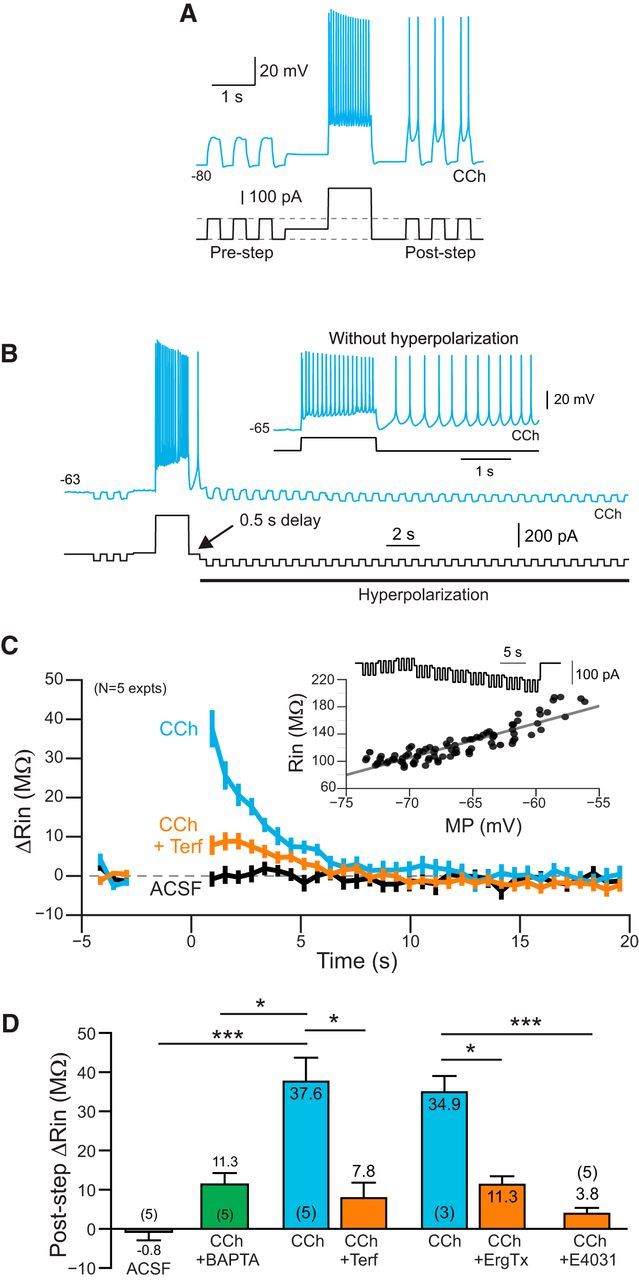
Time course of ERG-mediated change in input resistance. A, Demonstration of increased excitability following a depolarizing conditioning step in CCh. Brief test pulses that were subthreshold before the conditioning step become suprathreshold following the step. Neuron maintained at −80 mV to prevent persistent firing between test pulses. B, Example response to a train of hyperpolarizing test pulses used to assay input resistance. Additional continuous hyperpolarizing bias current was applied 500 ms following the offset of the depolarizing conditioning step. Without this bias current, the neuron fired persistently in response to the conditioning step (inset). C, Plot of the change in input resistance assayed by hyperpolarizing pulses in ACSF (black plot), CCh (blue), and CCh + Terf (orange). See Materials and Methods for details. Inset shows example input resistance calibration response. D, Summary of change input resistance following the depolarizing conditioning step. ***ACSF/CCh: p = 1.29E-04, T = 6.23, df = 11, two-sample t test; *CCh/BAPTA vs CCh: p = 0.0171, T = 3.46, df = 8, two sample t test; *CCh/CCh+Terf: p = 0.022, T = 4.49, df = 4, paired t test; *CCh/CCh+ErgTx: p = 0.029, T = 5.79, df = 2, paired t test; ***CCh vs CCh/E4031: p = 3.29E-04, T = 5.58, df = 11, two-sample t test. Two independent CCh datasets (N = 3 and 5) were combined when computing two-sample t test statistics.
We calculated the probability of triggering persistent firing by examining three to five consecutive responses to standard 2 s duration depolarizing conditioning steps (1–2 min between trials; persistent firing was terminated by manually hyperpolarizing the bias current—moving the membrane potential to less than −80 mV and then gradually reducing the bias current to restore the standard −70 mV holding potential). Except for experiments designed to measure dynamic changes in RIn, we determined whether the neuron continued to fire for at least 5 s following the offset of the depolarizing conditioning step to assess whether persistent firing occurred. Summary persistent firing probabilities presented in the figures (see Fig. 2E) reflect the average of the probabilities computed from each neuron tested. Membrane potential sag ratios were assessed from responses to 2 s duration current steps that evoked ∼20 mV hyperpolarization from a −60 mV holding potential. Sag ratio was computed by dividing the estimated RIn at the end of the step from the RIn estimated 50 ms following step onset. The action potential threshold was calculated as the voltage at which the rate of voltage change over time (dV/dt) was >10% of maximum dV/dt during the rising phase of the AP. The AP half-width was calculated from the duration at the voltage halfway between AP threshold and AP peak. We the calculated I–V relationships shown in Figure 8 using current-clamp recordings by determining the injected current required to reach each membrane potential during ramp stimuli acquired in control and experimental conditions. Data presented represent the difference in estimated whole-cell current between the experimental and control conditions at each voltage.
Figure 2.

ERG blockers abolish persistent firing in neocortical neurons. A, Terf (10 μm; orange traces) abolished persistent firing evoked by depolarizing steps in CCh + ML297. Terfenadine exposure time are indicated above each trace. B, The peptide ERG channel blocker ErgToxin1 (50 nm) also abolished persistent firing. C, Extracellular application of E-4031 (10 μm) blocked persistent firing recorded under the same conditions as in A and B. D, Intracellular perfusion with E-4031 (10 μm) abolished persistent firing within 12 min. Persistent firing continued to be evoked by test stimuli in interleaved control experiments without intracellular E-4031 for >40 min (N = 6). E, Left, Plot of the probability of evoking persistent firing >10 s before and after exposure to terfenadine. At 20 min: **p = 0.0029, T = 6.50, df = 4, paired t test; at 40 min: p = 0.0079, Fisher's exact test. Right, Persistent activity could be stably evoked in parallel experiments extending through the same duration without Terf (0 min vs 20 min: p = 0.44; 20 min vs 40 min: p = 0.98; 0 min vs 40 min: p = 0.39, Tukey's HSD test for multiple comparison). F, Terfenadine abolished persistent firing assayed in a cell-attached recording from an L5 neocortical neuron. Response triggered by two extracellular stimuli in L3 (asterisks). Terfenadine abolished the synaptically triggered persistent firing (orange trace). Intracellular recordings from the same neuron following a breakthrough to whole-cell mode demonstrated physiological normal step responses after synaptically evoked persistent firing was abolished (inset).
Figure 8.
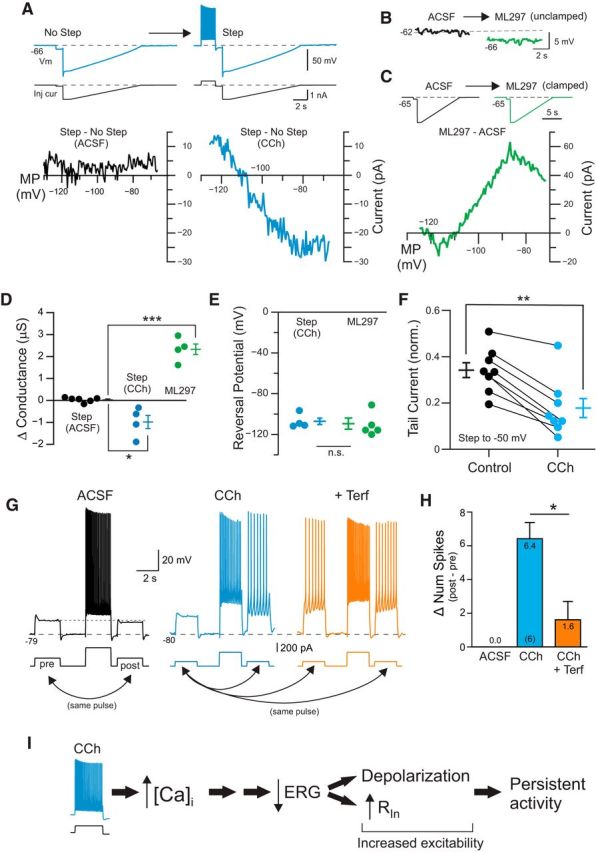
Reduction in leak ERG current contributes to increased excitability following depolarizing step. A, Example responses to ramp current injections with and without a preceding depolarizing current step. Bottom, Difference in I–V responses obtained by subtracting the step + ramp response from the ramp response alone (for details, see Materials and Methods). Black curve recorded under control (ACSF) conditions, and blue curve in CCh. B, Bath application of the GIRK activator ML297 (0.67 μM) hyperpolarizes L5 pyramidal cells. C, Difference I–V response calculated by subtracting ramp response in ML297 from ramp response recorded in ACSF. Initial holding potential adjusted to −65 mV under both conditions. Current injection protocol shown above I–V plot. D, Summary of estimated conductance evoked by depolarizing current steps in control (black symbols), in CCh (blue) and following ML297 application (green). *Step ACSF/CCh: p = 0.0123, T = 3.69, df = 8; ***ML297: p = 1.45E-05, T = 10.21, df = 8; two-sample t test. E, Plot of reversal potential of difference I–V plots. n.s., p = 0.77, two-sample t test. F, Plot of the reduction in ERG-related tail current by CCh. Responses normalized to maximal tail current response evoked by 4 s duration depolarizing steps from −90 to −10 mV (tail current reduction p = 9.1 × 10−4, T = 5.50, df = 7). We found a similar reduction in raw (not normalized) tail current (120 ± 25 to 76.5 ± 27.0 pA; p = 2.9 × 10−4, T = 6.67, df = 7). G, Modulation of responses to weak test depolarizing pulses by conditioning depolarizing step response. Modulation in ACSF assayed in separate experiments than CCh/CCh+Terf. H, Summary plot of the change in the number of spikes evoked by two test pulses under each condition (poststep − pre-step). *p = 0.0199, T = 3.37, df = 5; paired t test. I, Diagram of potential cascade evoked by the conditioning depolarizing step that leads to a reduction in the component of the leak K+ current mediated by ERG channels. The loss of part of the standing K+ current can account for both the depolarization of pyramidal cells and the transient increase in input resistance.
Voltage-clamp analysis of ERG current (see Figs. 4, 8F) used a modified ACSF with higher [K+] (10 mm, replacing equimolar Na+; Sturm et al., 2005) and a set of pharmacological blockers to attenuate other K+ currents in cortical pyramidal cells. This drug cocktail included 1 mm TEA, 100 μm 4-AP, 10 μm ZD7288 (to block hyperpolarization-activated current [IH]), 10 μm XE991 (to block IM), and 1 μm tetrodotoxin (TTX). (XE991 was not included in the experiment shown in Fig. 4A.) ERG-related tail currents were assayed using the protocol in Figure 4B except in the experiments shown in Figure 4E–G where the tail step potential was varied. We quantified the ERG-related tail current by subtracting steady state (the mean current from 1750 to 2000 ms within the tail response) from the mean current measured between 25 and 30 ms (slightly delayed from the tail step onset to attenuate contributions from the capacitive transient). Half-maximal activation voltages were estimated by fitting to the Boltzmann equation. Series resistance was compensated by 80% (using 10 μs lag) in voltage-clamp recording experiments. We monitored access resistance continuously through each experiment by analyzing transient responses to weak hyperpolarizing steps. We excluded experiments with initial access resistances >5 MΩ or if the access resistance increased by >1 MΩ during the experiment. The average access resistance in the voltage-clamp experiments presented in this study was 4.05 ± 0.12 MΩ.
Figure 4.
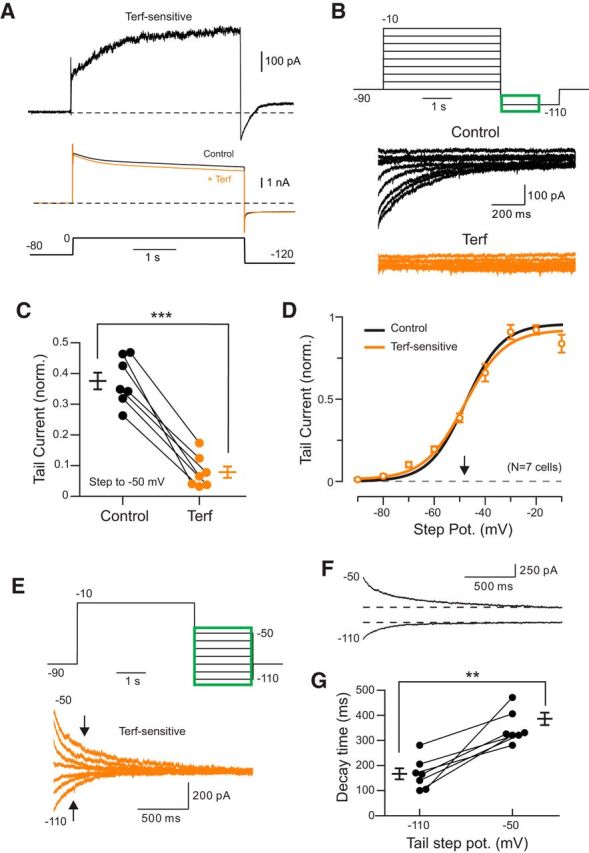
Voltage-clamp analysis of ERG currents in neocortical pyramidal cells. A, Isolation of Terf-sensitive current in response to steps to 0 mV. Raw responses (not leak subtracted; shown in middle); the top trace represents subtraction of the Terf response from the control response shown below. Responses acquired with external K+ increased from 3 to 10 mm. B, Terfenadine blocked most of the tail current response assayed in steps from −90 to −10 mV [responses acquired in Terf (orange traces) shown below control responses (black traces)]. ERG responses were recorded in 1 μm TTX, 1 mm TEA, 100 μm 4-AP, 10 μm ZD7288, and 10 μm XE991 (and in subsequent panels). C, Estimation of voltage required for half-maximal Erg current activation (V1/2). Each orange symbol represents the mean ± SEM from 7 experiments in which responses in Terf were subtracted from control responses. The V1/2 estimated from Terf-sensitive tail currents (−48.1 mV, slope = 7.9, orange curve) was similar to the V1/2 estimated from tail current responses under control conditions (without subtract responses in Terf; −47.7 mV; slope = 7.2; black curve). D, Plot of the reduction in ERG tail current by Terf; **p = 4.66 × 10−4, T = 6.877, df = 6. Results plotted reflect ERG current evoked by depolarizing steps to −50 mV normalized to the maximal ERG current elicited by steps to −10 mV, but a similar reduction (by 68%) also was evident following the raw tail current without normalization (p = 3.59 × 10−5, T = 10.87). Access resistance changed by <15% during Terf exposure. E, Voltage sensitivity of ERG tail currents. Top traces illustrate the voltage-clamp protocol; green area represents response time period shown in orange Terf-sensitive (Terf − control) traces below. Arrows indicate decay time estimates (time required for ERG tail current to decline to 1–1/e). F, Slower, nonexponential decay kinetics of ERG currents assayed at −50 vs −110 mV also were apparent in raw control traces (before Terf was applied). G, Plot of ERG current decay time (time to 1–1/e) in tail current responses recorded at −50 and −110 mV in control conditions; ***p = 0.0015, T = 5.483, df = 6. Similar results observed when analyzing Terf-sensitive tail currents; 244.9 ms (−110) vs 334.2 ms (−50 mV), p = 4.75 × 10−4, T = 6.854.
Data were expressed as the mean ± SEM. Significance level with p < 0.05 was used. Multiple comparisons using t tests were Bonferroni corrected; comparisons among large groups of data (>5) were based on the Tukey's HSD test. We used the Fisher's exact test for comparisons of binary proportions. We indicate the statistical test that was used within the figure legends. We also conducted an additional analysis of comparisons that did not appear to be statistically significant to verify that the specific N values included in the experiment were sufficient to reach a power of at least 0.8. Data analysis used custom programs written in Python 3.6 and Matlab 2015b. Statistical tests were performed in Python and R.
Results
As reported by others [Egorov et al., 2002 (entorhinal cortical neurons); Rahman and Berger, 2011 (neocortical neurons)], the activation of muscarinic receptors attenuated the hyperpolarizing afterpotential that normally follows a burst of APs and revealed an ADP that can trigger persistent firing (Fig. 1A). Throughout this study, we refer to the depolarizing current injection that initiates the persistent firing as the “conditioning” step to differentiate it from other current stimuli used to measure input resistance and to assay intrinsic excitability in experiments described below. In addition to enabling persistent firing, the cholinergic receptor agonist CCh (2 μm) depolarized pyramidal cells and triggered spontaneous firing by itself in most cells within 20–30 min. To compensate for this second effect of CCh, we included a low concentration (0.67 μm) of the GIRK channel activator ML297 in a subset of experiments. (Drugs used in each experiment are specified in the related figure legend.) At this concentration, the tonic hyperpolarization produced by ML297 compensated for the depolarizing effect of CCh, leading to relatively stable membrane potential while retaining the ability of CCh to enable persistent firing in response to depolarizing current steps over multiple trials (Fig. 1A). As reported by others (Egorov et al., 2002; Gulledge et al., 2009), the ability of cholinergic receptor agonists to facilitate persistent firing responses to depolarizing stimuli was largely mediated by actions of the m1 subclass of muscarinic receptors. In our experiments, the m1 receptor antagonist pirenzepine abolished persistent firing responses (Fig. 1B,C). The effectiveness of standard depolarizing step responses (2 s duration triggering firing at ∼8–12 Hz from −70 mV) was similar in CCh and CCh + ML297 (Fig. 1C). Persistent firing responses did not occur in control ACSF solution (0 of 5 experiments interleaved with CCh experiments where persistent firing was reliably evoked) and never occurred when ML297 was presented without CCh (0 of 8 cells).
Figure 1.
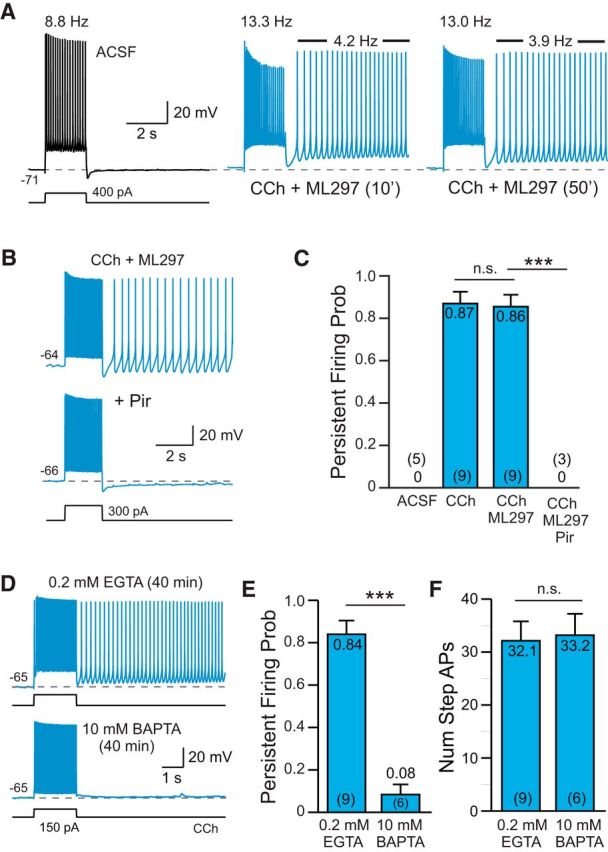
Cholinergic receptor activation enables persistent firing following depolarizing stimuli A, Bath application of 2 μm CCh (blue traces) reveals persistent spiking following depolarizing steps in L5 pyramidal cells from rat TeA neocortex. Firing rates during the step response and during the poststep persistent spiking period were stable >45 min (the right panel acquired following 50 min exposure to CCh; firing rates indicated above traces). The GIRK activator ML297 (0.67 μm) was included in the bath solution to compensate for the tendency of CCh to depolarize pyramidal cells (for details, see Materials and Methods). B, Blockade of persistent firing by 10 μm pirenzepine (Pir). C, Summary of the probability of triggering persistent firing in ACSF, CCh, CCh + ML297, and Pir (and CCh + ML297). ***p = 6.49E-05, T = 7.62, df = 9, two-sample t test; n.s., p = 1.00, T = 0.0879, df = 15, two-sample t test. Group means and N values are indicated inside each bar. D, Persistent firing in response to depolarizing step stimuli could be evoked using an internal solution containing 0.2 mm EGTA (with CCh + ML297, top trace) but not with an internal solution containing 10 mm BAPTA (bottom trace). E, Summary plot of the probability of triggering persistent firing in 0.2 mm EGTA and 10 mm BAPTA. ***p = 2.36E-06, T = 7.96, df = 13, two-sample t test. F, Plot of the number of APs evoked by the depolarizing step in EGTA- and BAPTA-based internal solution. n.s., p = 0.862, T = −0.178, df = 13, two-sample t test.
Persistent firing in L5 neocortical neurons following cholinergic stimulation required an increase in intracellular calcium. We rarely observed persistent firing following depolarizing test stimuli when the intracellular Ca2+ concentration was strongly buffered using 10 mm BAPTA (8% of experiments; Fig. 1D–F). In parallel experiments in which the time since breakthrough to whole-cell recording mode was similar (40 min), we were able to evoke persistent firing in >80% of experiments using our standard internal solution that contained 0.2 mm EGTA (Fig. 1D). The spiking response to the conditioning depolarizing step was similar in both recording conditions (Fig. 1F). These results suggest that persistent firing is triggered by an increase in intracellular Ca2+ and are consistent with previous work testing Ca2+ chelators in neocortical neurons (Rahman and Berger, 2011). Persistent firing also reflected cell-autonomous processes since this response could be evoked following blockade of ionotropic glutamate and GABA receptors with NBQX (10 μm), APV (25 μm), and gabazine (10 μm; N = 3 experiments; data not shown).
ERG channel blockers abolish persistent firing
While previous reports (Winograd et al., 2008; Rahman and Berger, 2011; Knauer et al., 2013; Yamada-Hanff and Bean, 2013) have suggested multiple potential biophysical channels that could contribute to the increased excitability responsible for persistent firing in a variety of cortical pyramidal cells, the underlying mechanism has not been clearly demonstrated. A common explanation for increased excitability associated with an increase in input resistance is a reduction in a subpopulation of K+ channels that are open near the resting membrane potential (McCormick and Prince, 1986; Greene et al., 1994). As part of a survey of potential leak K+ channel mechanisms, we found that three chemically diverse ERG (comprising Kv11/KCNH2 channels; Shepard et al., 2007; Atalar et al., 2010) channel antagonists abolished persistent firing in L5 neocortical pyramidal cells. ERG currents help set the resting membrane potential in many CNS cells (Sacco et al., 2003; Hardman and Forsythe, 2009; Fano et al., 2012; Ji et al., 2012) and ERG1 and ERG3 subunits are expressed at high levels in neocortical neurons (Saganich et al., 2001).
Terfenadine (10 μm), a histamine H1 receptor antagonist that also commonly is used to attenuate ERG currents (Wible et al., 2005; Hill et al., 2007) eliminated persistent firing evoked by depolarizing step stimuli (5 of 5 experiments in CCh + ML297 and 6 of 6 experiments in CCh alone; Fig. 2A; tested at −70 mV in all experiments). This effect of Terf appeared to be independent of its action on histamine receptors since another H1 receptor-specific antagonist that is structurally similar to Terf (fexofenadine; Scherer et al., 2002) had no effect on persistent firing even when tested at a three times higher concentration (30 μm; N = 3 experiments). Terfenadine also did not appear to abolish persistent firing by attenuating the depolarizing stimulus since there was no significant reduction in the number of spikes evoked by the depolarizing step in CCh (22.8 ± 1.4 vs 23.0 ± 1.4 spikes in Terf; p = 0.945; paired t test; N = 10 experiments). Terfenadine also abolished persistent firing evoked in higher concentrations of CCh (10 μm; N = 1) without decreasing the number of APs triggered by the depolarizing step.
While Terf required at least 20 min to block persistent firing in our experiments, this time course is consistent with the intracellular binding site on ERG channels for this antagonist (Spector et al., 1996; Kamiya et al., 2006). ErgToxin1, by contrast, blocked persistent firing in <20 min (Fig. 2B; 4 of 4 experiments). This ERG-specific peptide toxin produced endogenously by scorpions binds to extracellular sites on ERG channel subunits (Pardo-López et al., 2002; Milnes et al., 2003; Vandenberg et al., 2004; Chtcheglova et al., 2008), accounting for the more rapid time course of blockade. We also tested a third ERG channel antagonist, E-4031, which binds to an intracellular domain on ERG channel subunits (Spector et al., 1996; Kamiya et al., 2006; Hardman and Forsythe, 2009). This compound was effective when applied both in the extracellular bathing media (Fig. 2C; 5 of 5 experiments) and rapidly blocked persistent firing when we recorded from L5 pyramidal cells using an internal solution containing 10 μm E-4031 (Fig. 2D; 5 of 5 experiments). The ability of ERG channel blockers to abolish persistent firing was unlikely to be attributable to run-down in these experiments because Terf abolished persistent firing within the 20–30 min time frame when persistent firing could be reliably evoked in interleaved control experiments (Fig. 2E). Terfenadine also abolished persistent firing evoked by extracellular synaptic stimulation and assayed through cell-attached recordings from L5 pyramidal cells (Fig. 2F), demonstrating that ERG channel antagonists were effective under relatively physiological conditions that that did not involve a whole-cell recording configuration.
ERG-mediated responses in L5 pyramidal cells
The ability of three different ERG channel blockers to abolish persistent firing suggests that this subtype of K+ channel may play a critical role in regulating excitability in pyramidal cells. To our knowledge, there are no published reports addressing properties of ERG currents in neocortical neurons. We tested the role of ERG currents in these neurons by using a set of previously established tests of ERG channel function (Hardman and Forsythe, 2009; Ji et al., 2012; Niculescu et al., 2013). First, we found that blockade of ERG channels with Terf increased the number of spikes evoked by a standard depolarizing step when tested in control ACSF (Fig. 3A). Over 14 similar experiments, attenuation of ERG currents with Terf increased the number of spikes evoked by 2 s depolarizing steps by 23.5% (Fig. 3B). The increased excitability generated by Terf likely reflected a reduction in the resting (leak) K+ conductance mediated by ERG channels since Terf increased the apparent input resistance by 21.1% in L5 pyramidal cells maintained at −70 mV (via adjusting the bias current injected; Fig. 3C). In separate experiments where the membrane potential was not manually clamped, Terf depolarized L5 pyramidal cells by ∼10 mV (Fig. 3D), consistent with a reduction in a tonically open ERG channels. Activation of muscarinic receptors did not occlude the ability of Terf to attenuate a component of the leak K+ current since Terf was still able to depolarize pyramidal cells (Fig. 3E) and increased apparent input resistance (Fig. 3F) even in the presence of 2 μm CCh. With cholinergic receptor stimulation via CCh, Terf was able to depolarize L5 pyramidal cells sufficiently to induce spontaneous firing (Fig. 3E, orange trace), demonstrating that a reduction in the steady-state ERG current is sufficient to induce persistent firing.
Figure 3.

ERG-mediated responses in neocortical pyramidal cells. A, Terfenadine increases the number of spikes evoked by depolarizing current stimuli (comparison from ACSF to Terf). B, Plot of results from 14 experiments similar to A. ***p = 2.93E-04, T = 4.89, df = 13, paired t test. C, Terfenadine increases input resistance assayed from similar reference membrane potentials (approximately −70 mV). ***p = 1.50E-04, T = 5.28, df = 13, paired t test. D, Plot of mean membrane depolarization evoked by Terf in three pyramidal cells. E, Following cholinergic stimulation with CCh (blue trace), the same concentration of Terf elicited spontaneous firing (orange trace). F, Plot of input resistance assayed at −70 mV in ACSF, following CCh and with CCh + Terf. *p = 0.027, T = 3.74, df = 5; **p = 0.0058, T = 5.4253, df = 5, paired t test. G, Terfenadine attenuates poststep repolarization to both subthreshold (top) and suprathreshold (bottom) responses to depolarizing steps. Enlargements of repolarization shown on the right. H, Plot of the attenuation of the poststep repolarization. Subthreshold: **p = 0.00787, T = 4.93, df = 4; **Suprathreshold: p = 0.00167, T = 7.53, df = 4. Both paired t tests.
ERG current functions to help repolarize APs in cardiac cells, where Ether-a-go-go-Related Gene channels are most frequently studied (Vandenberg et al., 2004). ERG currents appear to play a similar role in neocortical neurons since Terf reduced the membrane potential repolarization following both subthreshold (Fig. 3G, top) and suprathreshold depolarizing steps (Fig. 3G, bottom). In five pyramidal cells tested, Terf eliminated approximately one-third of the repolarization following both types of steps (Fig. 3H, repolarization measured relative to the prestep membrane potential). These results suggest that neocortical pyramidal cells express ERG channels that are tonically active near the normal resting potential, which function both to dampen excitability in response to depolarizing stimuli and to enhance the poststimulus repolarization.
Using previously established voltage-clamp recording protocols for assaying ERG currents (Sacco et al., 2003; Hardman and Forsythe, 2009; Niculescu et al., 2013), Terf selectively attenuated a late-developing, voltage-sensitive outward current (Fig. 4A). We analyzed the voltage dependence and kinetics of ERG currents using tail current protocols developed in these previous studies with slightly elevated extracellular [K+] and a series of pharmacological blockers to attenuate other K+ currents active at similar membrane potentials. Under these conditions, Terf typically blocked almost all of the tail current responses evoked by steps from −90 to −10 mV (Fig. 4B). Both the peak steady-state Terf-sensitive outward current and the maximal tail current amplitude increased with larger amplitude depolarizing steps as expected for ERG currents. When assayed using steps from −50 mV, Terf blocked 79% of the tail current recorded under our experimental conditions (including IM and IH blockers; Fig. 4C). We obtained estimates of the voltage required to elicit the half-maximal ERG current analyzing Terf-sensitive difference currents evoked by steps from −90 to −10 mV (−47.7 mV; Fig. 4D, orange symbols and Boltzmann fit) and also applying the same methods to the control traces (without subtracting Terf responses, −48.1 mV; Fig. 4D, black curve). After correcting for the 10.7 mV liquid junction potential using ACSF containing 10 mm [K+], the V1/2 for neocortical ERG current estimated from our recordings was −37 mV. This estimate falls between the previously reported V1/2 estimates for the two ERG subunits expressed in neocortical neurons (ERG1, −23.0 mV; ERG3, −45.1 mV; Papa et al., 2003; Sturm et al., 2005; Gustina and Trudeau, 2013), raising the possibility that L5 pyramidal cells express heteromultimers containing both ERG1 and ERG3.
We confirmed that ERG current is active near the resting potential of L5 neurons by comparing the magnitude of tail current responses following steps to −70 mV versus maintaining a −90 mV holding potential. Both the tail current recorded under control conditions and Terf-sensitive tail currents were significantly larger following steps to −70 than to −90 mV (control ACSF: p = 2.72 × 10−4, T = 7.59, df = 6; Terf-sensitive: p = 0.0016, T = 5.43; both paired t tests). Consistent with previous voltage-clamp studies of ERG current in other neurons (Hardman and Forsythe, 2009) and in expression systems (Wang et al., 1997; Zhou et al., 1998), we find relatively slow deactivation kinetics, especially when assayed at moderately depolarizing potentials (approximately −50 mV). While ERG-related tail currents relaxed with a time course that was well fit by a single decaying exponential function (τ = 236 ± 28 ms at −110 mV, N = 7 based on Terf-subtracted responses), the decay kinetics of responses recorded at more depolarizing potentials (−50 mV) were more complex and did not recover completely until >1 s [observed in both Terf-sensitive tail currents (Fig. 4E) and in control responses recorded in ACSF (Fig. 4F)]. Over seven experiments, empirical estimates of decay kinetics consistently demonstrated slower kinetics in tail current responses recorded at −50 mV than those recorded at −110 mV (decay time to 1–1/e: 351 ± 25 vs 167 ± 24 ms, Fig. 4G; percentage of initial tail current remaining after 300 ms: 38.7 ± 1.7% vs 20.8 ± 2.5%; p = 0.0015, T = 5.48, df = 6). The relatively slow kinetics of ERG currents we and other investigators are markedly slower that observed for IM, another intrinsic current active at subthreshold membrane potentials in CNS neurons [∼20 ms decay τ at −100 mV (equivalent to our uncorrected −110 mV membrane potential) and 125–140 ms decay τ at −40 mV; Wang et al., 1998; Prole et al., 2003; Pattnaik and Hughes, 2012].
Neocortical pyramidal cells express a wide variety of K+ channels, which also could affect excitability following depolarizing conditioning steps in CCh. IH often triggers rebound hyperexcitability (Robinson and Siegelbaum, 2003) and is expressed in L5 pyramidal cells (Fig. 5A–C; Notomi and Shigemoto, 2004). IH therefore could potentially contribute to persistent activity. However, the selective IH blocker ZD7288 failed to abolish persistent firing in six of six cells tested (Fig. 5B) while consistently attenuating the membrane potential “sag” in responses to hyperpolarizing steps mediated by IH (Fig. 5C). In a separate set of experiments, we were able to evoke typical persistent firing responses from our standard depolarizing test responses in slices pretreated with ZD7288 (Fig. 5D) or in the nonselective IH blocker Cs+ (10 mm; N = 3; data not shown), suggesting that modulation of IH was not responsible for persistent firing in L5 neurons. Even with IH blocked with ZD7288, Terf was still able to abolish persistent firing (Fig. 5D; N = 4). The selective IM blockers XE991 (10 μm; Wang et al., 1998; Fig. 5E) and linopirdine (30 μm; Schnee and Brown, 1998; Fig. 5F) also failed to abolish persistent firing in four of four cells tested independently. We verified that pharmacological M current attenuation was effective by determining that the number of APs evoked by the depolarizing step was increased by treatment, both under control conditions (Fig. 5F) and in CCh (Fig. 5G). The dihydropyridine Ca2+ channel antagonist nimodipine also did not prevent persistent firing (N = 6; 20 μm), suggesting that persistent firing did not result from an interaction between K+ and l-type Ca2+ channels.
Figure 5.

IH and IM currents are not required for persistent firing. A, Bath application of the specific IH blocker ZD7288 (10 μm) hyperpolarized L5 pyramidal cells. Symbols represent the mean ± SEM. B, Blockade of IH by ZD7288 eliminated the membrane potential sag evident in responses to hyperpolarizing current steps (top) but did not abolish persistent firing evoked by a depolarizing step (bottom, both representative of 5 of 5 cells tested). C, ZD7288 significantly reduced sag ratio. *p = 0.0257, T = 3.464, df = 4, paired t test; for details of sag ratio analysis, see Materials and Methods). ZD7288 did not block persistent firing in 5 of 5 cells tested (n.s., p = 1.00, Fisher's exact test). D, Blockade of IH by ZD7288 did not occlude the ability of Terf to abolish persistent firing (representative of 4 of 4 experiments). E, Attenuation of IM current with XE991 (10 μm) did not occlude persistent firing in CCh. F, + Linopirdine (30 μm), another M current blocker, also failed to abolish persistent firing. G, Plot of the increase in number of spikes evoked during the depolarizing step before and after + Linopirdine. **p = 0.0087, T = 4.797, df = 4. H, Plot of the increase in number of spikes with linopridine following CCh treatment. *p = 0.020, T = 7.024, df = 2. Both paired t tests.
Transient increase in input resistance associated with persistent activity
The results presented thus far suggest that the hyperexcitability underlying persistent activity could reflect a transient attenuation in the component of the leak K+ current mediated by ERG channels. We next tested this hypothesis by assaying the change in intrinsic properties immediately following depolarizing step stimuli. When assayed using brief depolarizing steps, the depolarizing conditioning step converted just subthreshold responses into suprathreshold responses (Fig. 6A), which is consistent with either an increase in inward current (e.g., ICAN) or a reduction in a leak K+ current like ERG. We assayed input resistance using trains of brief (300 ms) hyperpolarizing current pulses to discriminate between these possibilities. In these experiments, we injected a steady hyperpolarizing current starting 0.5 s after the offset of the conditioning depolarizing step to prevent continuous persistent firing; the train of hyperpolarizing test pulses was applied on top of this steady hyperpolarization (Fig. 6B). By delaying the steady hyperpolarization by 0.5 s, we could verify that the conditioning step stimulus was effective in initiating persistent firing (see example trace in Fig. 6B with a single spike following the conditioning step).
Simply assaying RIn based on the membrane potential change elicited by hyperpolarizing current pulses following the conditioning step suggested that the input resistance increased by ∼34 MΩ during the ADP (from 130 ± 25.6 to 164 ± 30.3 MΩ; p = 4 × 10−4, T = 8.32, df = 5; paired t test; N = 6). This estimate, however, is subject to several potential artifacts. First, the membrane potential is continuously hyperpolarizing following the conditioning step, especially during first few RIn test pulses. We corrected for this effect by detrending the membrane potential before calculating the voltage deflection elicited by each test pulse (for details, see Materials and Methods). The second complication is that input resistance in L5 pyramidal cells varies depending on the membrane potential, with higher RIn estimates at more depolarized membrane potentials. To compensate for this effect, we determined the RIn vs. membrane potential relationship over the relevant voltage range in each neuron (and in each drug condition; Fig. 6C, inset) and present our results as changes in RIn following the conditioning step relative to the steady-state RIn measured at that particular membrane potential before the conditioning step. Following these two correction procedures, we still find a large (35–38 MΩ) increase in input resistance following the conditioning step that decays with a time constant of 4.3 s (Fig. 6C, blue trace). (The similarity between RIn estimates with and without correction procedures reflects the opposing effects of the two types of artifacts when using hyperpolarizing test pulses.) During the same time period, we find no change in apparent RIn following conditioning depolarizing step responses in control ACSF (Fig. 6C, black trace).
In CCh, the ERG channel blocker Terf attenuated most of the increase in input resistance following the conditioning step (Fig. 6C, orange trace). We found similar results in three experiments using ErgToxin1 where most of the increase in RIn was eliminated after ERG channels were blocked. Figure 6D summarizes the reduction in elevation in apparent RIn values in separate sets of experiments with Terf and ErgToxin1. We also repeated the same procedure in five neurons recorded with the intracellular ERG channel blocker E-4031 added to the internal solution and observed only a small (∼4 MΩ) increase in apparent RIn. The modest residual increase in apparent RIn following ERG channel blockade was similar to (or less than) the peak RIn increase observed when intracellular Ca2+ was strongly buffered with a BAPTA-containing internal solution (Fig. 6D, green bar).
Neocortical pyramidal cells express large subthreshold Na+ currents that can contribute to persistent firing (Yamada-Hanff and Bean, 2013; Crill, 1996) and can influence RIn estimates. To determine whether the effects of ERG blockers were independent of voltage-gated Na+ channels, we applied TTX (1 μm) along with 4-AP and CCh to increase excitability. In this drug combination, depolarizing conditioning steps reliably triggered a series of Ca2+-mediated spikes (Fig. 7A). At similar membrane potentials used throughout this study (approximately −70 mV), conditioning steps that evoked repeated Ca2+ spikes triggered persistent firing (spontaneous Ca2+ spikes following of the offset of the depolarizing current injection), which were abolished by Terf (Fig. 7B,C). Even with voltage-gated Na+ channels blocked with TTX, Terf increased steady-state input resistance (Fig. 7D), which is consistent with a primary action on ERG channels rather than indirectly affecting excitability via interactions with Na+ channels. The effect of Terf on persistent Ca2+ spiking did not reflect a diminished stimulus since there was no reduction in the number of Ca2+ spikes evoked by the conditioning step (Fig. 7E). In TTX and CCh, depolarizing conditioning steps still evoked a large transient increase in input resistance that was greatly attenuated by Terf (Fig. 7F,G). We found a similar effect of Terf in reducing the transient increase in input resistance assayed with trains of both positive and negative current test pulses (Fig. 7H), arguing that this experiment reflected the underlying neuronal input resistance rather than rectification properties of other currents triggered by the conditioning step (Inoue and Isenberg, 1990a,b) or artifacts related to the detrending procedure we used.
Figure 7.
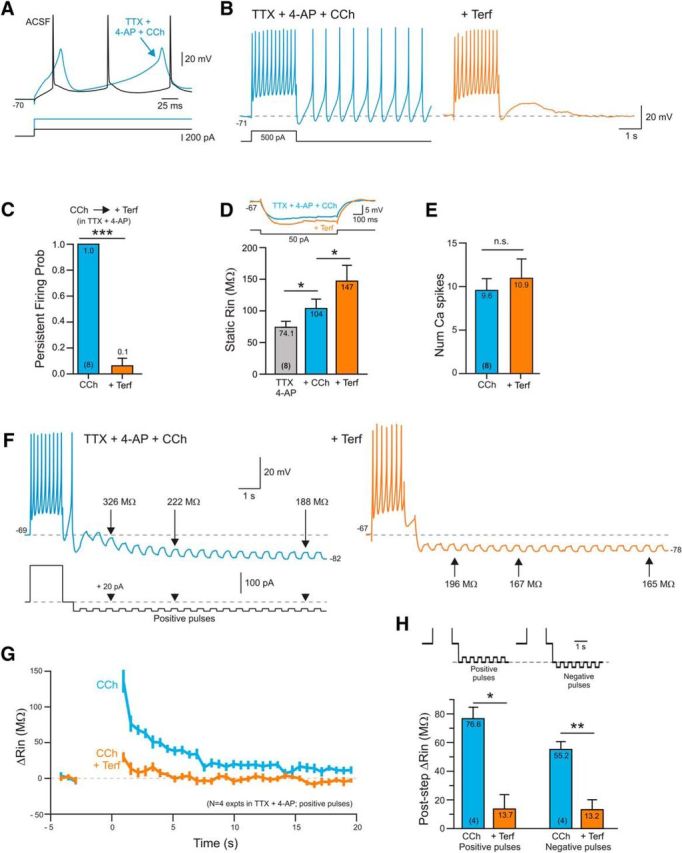
Terfenadine-sensitive persistent firing in the absence of voltage-gated Na+ channels. A, Ca2+ spikes evoked by depolarizing steps following application of 1 μm TTX, 100 μm 4-AP, and 2 μm CCh. B, Terfenadine blocks persistent Ca2+ spiking activity triggered by a depolarizing step. C, Plot of the probability of triggering persistent Ca2+ spikes before and after Terf treatment. ***p = 1.41E-06, T = 15, df = 7, paired t test. D, Plot of input resistance assayed using a single hyperpolarizing step in TTX + 4-AP, following CCh treatment (blue) and following the subsequent addition of Terf (orange). Example step responses shown above plot. *TTX/CCh: p = 0.0463, T = 2.89, df = 7; *CCh/CCh+Terf: p = 0.0458, T = 2.90, df = 7; paired t test. E, Plot of the number of Ca2+ spikes evoked by the conditioning step before and after Terf (p = 0.36; paired t test). F, The underlying ADP response in TTX + 4-AP + CCh is associated with an increase in input resistance assayed using trains of positive current pulses. Terfenadine attenuates both the ADP and the related increase in input resistance. Example RIn estimates indicated in F are detrended. G, Summary plot of the change in input resistance from 4 experiments similar to F using positive current test pulses. H, Summary of change in input resistance following conditioning depolarizing step using both trains of positive and negative current pulses. *Positive pulses: p = 0.023, T = 4.31, df = 3; **negative pulses: p = 0.015, T = 5.12, df = 3; paired t test.
Our results suggest that ERG channel blockers are effective in abolishing persistent firing because the conditioning depolarizing step functioned to reduce the steady-state ERG current, leading to a transient hyperexcitable period following the step. This hypothesis would explain the Terf-sensitive increase in input resistance following the conditioning step (Figs. 6B,C, 7F–H) and the enhanced responses to depolarizing test pulses in Figure 6A. We next sought to test this hypothesis by using a slow current-clamp ramp protocol (Fig. 8A–E) to reveal how the I–V relationship was altered by the conditioning depolarizing step. In control ACSF, we found essentially no difference in the I–V relationship following the conditioning step (Fig. 8A, black trace) shows the subtraction of the ramp response following the conditioning step (“step”) from the response to an identical ramp stimulus presented in isolation; “no step”). In CCh (and without TTX/TEA), the same protocol revealed a negative slope in the I–V relationship in four of four experiments. This negative slope response likely reflected a reduction in a leak K+ current—rather than an inward current—given its reversal near the K+ equilibrium potential. In separate experiments, we applied the GIRK activator ML297 (0.67 μm; without CCh), which generated an outward current in the difference I/V relationship that reversed polarity at approximately the same potential. Results from these experiments are summarized in Figure 8, D and E, and are consistent with a transient reduction in ERG current contributing to the hyperexcitability following the conditioning depolarizing step in CCh. The ability of depolarizing conditioning steps to transiently reduce ERG current only in the presence of CCh and not in control ACSF suggests that the activation of muscarinic receptors is a critical step both to increase overall excitability and to enable modulatory mechanisms. One prediction of this model is that tonic activation of mAChRs by CCh should decrease the ERG current active at subthreshold membrane potentials. We tested that prediction by determining the effect of CCh ERG-mediated tail currents following moderate depolarizing voltage-clamp steps. As shown in Figure 8F, CCh reduced the leak ERG current activated by steps to −50 mV by ∼50% without affecting access resistance (mean change in access resistance was 0.12 ± 0.17 MΩ (2.8% change).
As a final test of the role of ERG modulation in contributing to poststep hyperexcitability, we applied two identical just-subthreshold depolarizing test pulses, one before the conditioning step and the other 0.5 s following the conditioning step. In control conditions (ACSF), the response to the second test step (“post”) was slightly diminished compared with the first test step (“pre”; Fig. 8G, black trace). In CCh, the response to the second test step was enhanced and generated a train of APs (Fig. 8G, blue trace). When Terf was applied in combination with CCh, the previously subthreshold first test step triggered a train of APs, which was only slightly increased following the conditioning step (8 spikes on the poststep vs 7 spikes in response to the initial test stimulus; Fig. 8G, orange trace). Figure 8G summarizes six experiments similar to the one shown in Figure 8G and suggests that blockade of ERG current with Terf not only increases the steady-state excitability of neocortical pyramidal cells (accounting for the increased response to the first test step) but also occludes the ability of the conditioning depolarizing step to transiently increase intrinsic excitability.
A proposed mechanism for postconditioning step hyperexcitability is diagrammed in Figure 8I and postulates that increases in intracellular Ca2+ generated in response to the conditioning step leads to a reduction in the component of leak K+ current mediated by ERG channels. The increased postconditioning step excitability results from both the steady-state depolarization as well as the increased input resistance, reflecting decreased ERG current (when muscarinic receptors are activated). Pharmacological blockage of ERG increases steady-state excitability (neocortical neurons depolarize and have higher input resistance) whether or not muscarinic receptors are activated (Fig. 3), likely due to the reduction in the ERG component of the leak conductance. When tested with both muscarinic receptor agonists and ERG blockers, conditioning steps no longer can increase excitability because the steady-state ERG current is already attenuated (by the ERG blocker) and likely cannot be substantially reduced further. In most of our experiments, we applied a hyperpolarizing bias current soon after the conditioning step to prevent prolonged periods of persistent firing. Without this intervention, the period of postconditioning step hyperexcitability presumably would last longer as Ca2+ accumulations triggered by additional Na+ spikes re-engages the same modulatory mechanism, likely leading to the persistently attenuated ERG current.
ERG-mediated hyperexcitability in prefrontal cortical neurons
Finally, we tested whether leak ERG channels contribute to the intrinsic excitability of pyramidal cells in medial prefrontal cortex, where persistent firing is commonly recorded during working memory tasks in both rodents and primates (Fuster and Alexander, 1971; Miyashita and Chang, 1988; Young et al., 1997; Liu et al., 2014). Blockade of ERG current with Terf increased the number of APs evoked by depolarizing steps in L5 mPFC pyramidal cells held at approximately −70 mV (Fig. 9A). As with TeA neocortical neurons, Terf increased both the average number of spikes (by 40%; Fig. 9B) and the resting input resistance (by 36%; Fig. 9C) in L5 mPFC pyramidal cells. Terfenadine also abolished persistent firing evoked in 2 μm CCh in six of six experiments (Fig. 9D,E). Using the same protocol presented in Figure 6, B and C, we found a similar increase in input resistance following the conditioning depolarizing step, which was greatly reduced by Terf in four of four experiments (Fig. 9F,G; results reflecting the same dual RIn correction procedure outlined above). The maximal increase in apparent input resistance was somewhat smaller in mPFC than in TeA L5 pyramidal cells (∼36 vs 25 MΩ), suggesting that ERG-mediated persistent firing may be more robust in TeA than PFC neurons. In both TeA and PFC neurons, Terf abolished persistent firing without affecting either the AP threshold (Fig. 9H) or the AP half-width (Fig. 9I), which is consistent with a relatively specific action of this agent on a slowly activating K+ current.
Figure 9.
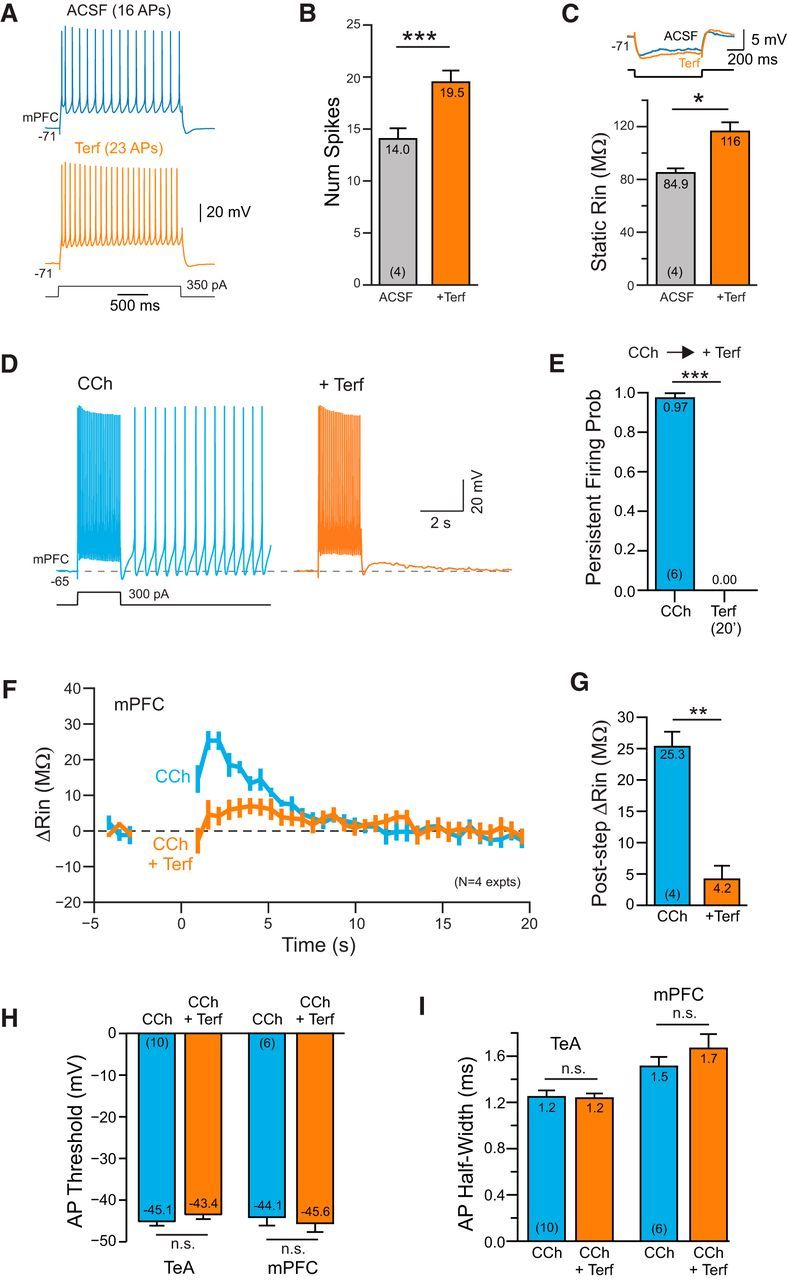
ERG-mediated persistent activity in prefrontal cortical neurons. A, Terfenadine increases the number of APs evoked by depolarizing steps in L5 mPFC pyramidal cells. B, Summary plot of number of APs evoked by the conditioning step in ACSF and Terf. *p = 0.0131, T = 5.30, df = 3, paired t test. C, Plot of the increase in input resistance with Terf. Example responses above plot. ***p = 8.52E-05, T = 29.54, df = 3, paired t test. D, Terfenadine abolishes persistent firing evoked in CCh. E, Summary plot of effectiveness of Terf in blocking persistent firing in CCh in 6 experiments. ***p = 3.62E-07, T = 34.93, df = 5, paired t test. F, Plot of the change in input resistance in 4 mPFC pyramidal cells following conditioning depolarizing steps in CCh and CCh + Terf. Experimental protocol is the same as shown in Figure 6, B and C. G, Summary plot of maximal increase in input resistance observed during ADP response. **p = 0.00163, T = 10.95, df = 3; paired t test. H, Plot of AP threshold in CCh before and after Terf. Left column pair in TeA neocortex and right pair in medial prefrontal cortex. TeA: p = 0.25; PFC: p = 0.40; paired t test. I, Plot of AP half-width calculated at the half-maximal AP amplitude. Same column order as H. TeA: p = 0.78; PFC: p = 0.33, paired t test.
Discussion
We make three principal conclusions in this report, all of which we believe have not been reported previously. First, we find that tonically active ERG currents contribute to both setting the resting membrane potential and regulating the number of APs triggered in response to depolarizing stimuli in neocortical neurons. This result, observed in both TeA and PFC pyramidal cells, supports the hypothesis that leak ERG K+ currents play an important role in shaping the intrinsic physiology of cortical neurons. Second, using both biophysical and pharmacological assays, we find that a reduction in leak ERG current appears to play a central role in mediating persistent firing in neocortical neurons. Apparent input resistance increases during the ADP that underlies persistent firing—a result that is inconsistent with the expected decrease in input resistance in ICAN-mediated afterdepolarizations (Haj-Dahmane and Andrade, 1998; Egorov et al., 2002; Fransén et al., 2006). Finally, we find that depolarizing stimuli when presented in combination with m1 receptor activation triggers a transient increase in excitability by attenuating leak ERG currents through a Ca2+-dependent mechanism. Together, these results point to the modulation of leak ERG current as a central underlying mechanism responsible for persistent firing in neocortical neurons and a novel therapeutic target for neurological and psychiatric diseases.
Potential mechanism of poststimulus hyperexcitability
Our results suggests that a component of the increased excitability responsible for persistent firing modes in neocortical neurons arises from a rapid attenuation in ERG K+ currents that contribute to the leak conductance in pyramidal cells. Following cholinergic receptor activation, depolarizing stimuli appear to attenuate leak ERG current, leading to both membrane depolarizing and an increase in input resistance. Our results provide the following three principal lines of evidence for ERG contributing to the ADP that underlies persistent firing in neocortical neurons: (1) we directly assayed input resistance during the ADP response and observed a large (30–40%) increase even after compensating for artifacts related to variable RIn estimates at different voltages and the ability of subthreshold Na+ channels to influence RIn measurements; (2) the I–V relationship assayed during the ADP reversed near the K+ equilibrium potential; and (3) three chemically diverse ERG blockers abolished persistent firing while attenuating both the underlying ADP and the increase in apparent input resistance following the conditioning step.
The ERG-based model of persistent firing is reminiscent of classic studies of the mechanism of cholinergic hyperexcitability (Krnjević et al., 1971; McCormick and Prince, 1985, 1986), which suggested a central role for attenuated leak K+ current in promoting persistent firing. This explanation was supplanted by the ICAN model when Egorov et al. (2002) and Rahman and Berger (2011) found that the TRP blocker flufenamic acid (FFA) abolished persistent firing while it attenuated the underlying ADP in a variety of neurons. Unfortunately, FFA affects many ionic currents, including ERG (Malykhina et al., 2002; Guinamard et al., 2013), making it less useful in discriminating between potential mechanisms. Another commonly used TRP channel blocker, SKF-96365, blocks heterologously expressed ERG channels (Liu et al., 2016).
Our results from assays of input resistance and the I–V properties of the ADP response are inconsistent with recent ICAN-based models of persistent activity (Fransén et al., 2006; Rahman and Berger, 2011; Zhang et al., 2011; Jochems and Yoshida, 2013; Knauer et al., 2013), which predict that persistent firing should be accompanied by a decrease (rather than an increase) in input resistance during the ADP. However, several previous investigators observed increases in apparent input resistance during the ADP (Haj-Dahmane and Andrade, 1998), which they attributed to rectification properties of TRP channels (Inoue and Isenberg, 1990a,b). This prior result provided the motivation for our two-step RIn correction procedure and for assays of input resistance using both positive- and negative-going current steps that were feasible once voltage-gated Na+ channels were blocked, as in the experiments shown in Figure 7. Haj-Dahmane and Andrade (1998) observed that Cs+ ions failed to abolish persistent activity. While we confirmed this result, this finding is difficult to interpret since ERG channels—unlike most voltage-gated K+ channels—are not blocked by Cs+ (Zhang et al., 2003). Nevertheless, without further experiments we cannot exclude that subthreshold Na+ channel and ICAN mechanisms contribute to persistent firing, along with ERG, since we consistently find a residual small ADP following treatment with ERG blockers. However, our results suggest that in 2 μm CCh, attenuating ERG using Terf is sufficient to trigger persistent firing from −70 mV. Based on previous studies using heterologously-expressed ERG channels, we expect that our pharmacological treatments blocked 50–80% of whole-cell ERG current (e.g., the IC50 for ErgToxin1 is 10–60 nm; Pardo-López et al., 2002; Hill et al., 2007). Until new molecular or pharmacological tools become available, we cannot determine whether the residual ADP represented unblocked ERG current or contributions from other mechanisms.
While our results suggest that attenuation of leak ERG current appears to be a critical component in intrinsic persistent spiking responses, we did not attempt to define the specific biophysical mechanisms responsible for ERG modulation. In other systems, ERG currents are inhibited both by PKC-mediated phosphorylation (Cockerill et al., 2007) and by depleting phosphatidylinositol 4,5-bisphosphate (PIP2; Bian et al., 2001). Activation of muscarinic m1 receptors activates phospholipase C, which hydrolyzes PIP2 into diacylglycerol and inositol-1,4,5-triphosphate, potentially enabling both PIP2 and PKC mechanisms. Understanding how the transient elevation of intracellular Ca2+ concentration during the conditioning step decreases ERG current is a central but difficult question to resolve. The rapid onset of persistent firing (and the underlying ADP) would suggest a dominant role of PIP2-mediated modulation since FRET-based measurements suggest that that process can operate within ∼1 s (Jensen et al., 2009). It is possible that PIP2 depletion leads to the initial postconditioning step increase in excitability, which is reinforced by subsequent PKC-mediated phosphorylation of ERG channels. Alternatively, the normally slow actions of protein kinases could be accelerated if they were “primed” via scaffolding proteins such as caveolin (Isshiki and Anderson, 1999; Gallego et al., 2014). Human ERG channels can interact with caveolin (Lin et al., 2008; Ma et al., 2014), providing another potentially rapid pathway for G-protein-stimulated phosphorylation. It is also possible that the modulation of ERG affects primarily gating properties rather than reducing channel conductance, as suggested by previous work (Bian et al., 2001; Hirdes et al., 2004; Cockerill et al., 2007). Discriminating between these potential mechanisms and revealing how Ca2+ transients function to trigger persistent firing when combined with muscarinic receptor activation will likely require the development of new rapid FRET-based tools that are beyond the scope of the present study.
Molecular genetic tests of the role of ERG channels in neocortical neurons will likely require conditional knockouts of one or more ERG genes as permanent deletion of these proteins is lethal in mice at embryonic day 11.5 (Teng et al., 2008; Vijayaraj et al., 2012). To our knowledge, conditional ERG knock-out mice have not been generated. ERG channels are also a frequent nonspecific target of pharmacological agents, including many antipsychotic agents (Wible et al., 2005). ERG channels are permeable to Cs+ ions (Zhang et al., 2003), further complicating conventional voltage clamp-based current analysis.
Functional significance of ERG-mediated intrinsic persistent firing
While the origin of persistent activity associated with short-term memory and other cognitive functions (Fuster and Alexander, 1971; Eichenbaum, 2014; Zylberberg and Strowbridge, 2017) has not been determined, the identification of ERG modulation as a potential intrinsic mechanism should facilitate determination of the role of intrinsic versus recurrent network mechanisms. The availability of highly specific ERG channel blockers (e.g., ErgToxin1) makes it feasible to directly test to what extent intrinsic biophysical properties contribute to commonly observed persistent firing modes such as the delay period firing working memory tasks. ERG blocking agents also could be used to determine whether persistently active subcortical circuits (e.g., the VOR) rely on the modulation of ERG current.
Cholinergic receptor stimulation has long been known to increase neuronal excitability and promote intrinsic persistent firing modes (Krnjević et al., 1971). Since cholinergic agents strongly influence cognitive processes associated with persistent firing, a fundamental question arising from previous in vitro studies is how intrinsic and circuit mechanisms could be integrated to generate stimulus-specific persistent firing. Modulating most types of intrinsic leak currents would be expected to dramatically alter the tuning of synaptic weights within recurrent networks (Zylberberg and Strowbridge, 2017) and yet endogenous cholinergic tone is likely increased during periods of heightened attention, such as during a working memory task (Fuster and Alexander, 1971; Richardson and DeLong, 1986). Because of its slow activation kinetics (Hirdes et al., 2004; Cockerill et al., 2007), ERG-mediated persistence is an attractive intrinsic mechanism to coexist with precisely tuned synaptic networks. Presumably brief synaptically evoked discharges will be only weakly affected by ERG currents—leaving delicate network tuning unaffected—while stronger, or more sustained, synaptic excitation would be preferentially amplified by intrinsic currents. Through this mechanism, ERG-mediated intrinsic persistent activity could function to “tag” the most active subset of cells within a larger neuronal ensemble driven by a stimulus. A central predication of this hypothesis is that ERG blockers should preferentially attenuate late phases of neuronal discharges as well as reducing poststimulus persistent activity.
Recent genetic studies have suggested an association between single nucleotide polymorphism in ERG channels and schizophrenia (Atalar et al., 2010; Hashimoto et al., 2013). The mutated ERG channel, Kv11.1–3.1, has altered gating properties compared to its wild-type counterpart (Heide et al., 2012) and is highly expressed in cortical neurons within a subpopulation of schizophrenic patients (Huffaker et al., 2009). Since schizophrenia is often associated with altered PFC activity and impaired working memory function (Weinberger and Berman, 1996), it is possible that a component of this disease reflects abnormal ERG function, which could result in changes in both the average discharge rate of cortical neurons and the regularity of their firing. Many common second-generation antipsychotic mediations, such as risperidone, are potent ERG blockers (Wible et al., 2005) and patients with ERG mutations appear to be preferentially responsive to ERG-blocking antipsychotic agents (Apud et al., 2012). Since our study suggests that ERG is an important component of the normal constellation of leak K+ channels in at least a subset of neocortical neurons (regular spiking deep pyramidal cells), our results provide a novel cellular mechanism for the actions of many second-generation antipsychotic agents that could help explain cognitive dysfunction associated with schizophrenia.
Footnotes
This work was supported by National Institutes of Health Grant R01-DC-04285 to B.W.S. We thank Todd Pressler and Hannah Arnson and for helpful discussions; and Chris Ford, Rodrigo Andrade, and Diana Kunze for providing constructive comments on this manuscript.
References
- Andrade R. (1991) Cell excitation enhances muscarinic cholinergic responses in rat association cortex. Brain Res 548:81–93. 10.1016/0006-8993(91)91109-E [DOI] [PubMed] [Google Scholar]
- Apud JA, Zhang F, Decot H, Bigos KL, Weinberger DR (2012) Genetic variation in KCNH2 associated with expression in the brain of a unique hERG isoform modulates treatment response in patients with schizophrenia. Am J Psychiatry 169:725–734. 10.1176/appi.ajp.2012.11081214 [DOI] [PubMed] [Google Scholar]
- Atalar F, Acuner TT, Cine N, Oncu F, Yesilbursa D, Ozbek U, Turkcan S (2010) Two four-marker haplotypes on 7q36.1 region indicate that the potassium channel gene HERG1 (KCNH2, Kv11.1) is related to schizophrenia: a case control study. Behav Brain Funct 6:27. 10.1186/1744-9081-6-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry PH. (1994) JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods 51:107–116. 10.1016/0165-0270(94)90031-0 [DOI] [PubMed] [Google Scholar]
- Bian J, Cui J, McDonald TV (2001) HERG K+ channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circ Res 89:1168–1176. 10.1161/hh2401.101375 [DOI] [PubMed] [Google Scholar]
- Chtcheglova LA, Atalar F, Ozbek U, Wildling L, Ebner A, Hinterdorfer P (2008) Localization of the ergtoxin-1 receptors on the voltage sensing domain of hERG K+ channel by AFM recognition imaging. Pflugers Arch 456:247–254. 10.1007/s00424-007-0418-9 [DOI] [PubMed] [Google Scholar]
- Cockerill SL, Tobin AB, Torrecilla I, Willars GB, Standen NB, Mitcheson JS (2007) Modulation of hERG potassium currents in HEK-293 cells by protein kinase C. Evidence for direct phosphorylation of pore forming subunits. J Physiol 581:479–493. 10.1113/jphysiol.2006.123414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ (1990) Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci 13:99–104. 10.1016/0166-2236(90)90185-D [DOI] [PubMed] [Google Scholar]
- Crill WE. (1996) Persistent sodium current in mammalian central neurons. Annu Rev Physiol 58:349–362. 10.1146/annurev.ph.58.030196.002025 [DOI] [PubMed] [Google Scholar]
- Dasari S, Abramowitz J, Birnbaumer L, Gulledge AT (2013) Do canonical transient receptor potential channels mediate cholinergic excitation of cortical pyramidal neurons? Neuroreport 24:550–554. 10.1097/WNR.0b013e3283621344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dégenètais E, Thierry AM, Glowinski J, Gioanni Y (2002) Electrophysiological properties of pyramidal neurons in the rat prefrontal cortex: an in vivo intracellular recording study. Cereb Cortex 12:1–16. 10.1093/cercor/12.1.1 [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6:850–862. 10.1038/nrn1785 [DOI] [PubMed] [Google Scholar]
- Dembrow N, Johnston D (2014) Subcircuit-specific neuromodulation in the prefrontal cortex. Front Neural Circuits 8:54. 10.3389/fncir.2014.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, Johnston D (2010) Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci 30:16922–16937. 10.1523/JNEUROSCI.3644-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA (2002) Graded Persistent activity in entorhinal cortex neurons. Nature 420:173–178. 10.1038/nature01171 [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. (2014) Time cells in the hippocampus: a new dimension for mapping memories. Nat Rev Neurosci 15:732–744. 10.1038/nrn3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fano S, Çalişkan G, Heinemann U (2012) Differential effects of blockade of ERG channels on gamma oscillations and excitability in rat hippocampal slices. Eur J Neurosci 36:3628–3635. 10.1111/ejn.12015 [DOI] [PubMed] [Google Scholar]
- Fransén E, Tahvildari B, Egorov AV, Hasselmo ME, Alonso AA (2006) Mechanism of graded persistent cellular activity of entorhinal cortex layer V neurons. Neuron 49:735–746. 10.1016/j.neuron.2006.01.036 [DOI] [PubMed] [Google Scholar]
- Fuster JM, Alexander GE (1971) Neuron activity related to short-term memory. Science 173:652–654. 10.1126/science.173.3997.652 [DOI] [PubMed] [Google Scholar]
- Gallego M, Alday A, Alonso H, Casis O (2014) Adrenergic regulation of cardiac ionic channels: role of membrane microdomains in the regulation of kv4 channels. Biochim Biophys Acta 1838:692–699. 10.1016/j.bbamem.2013.06.025 [DOI] [PubMed] [Google Scholar]
- Greene CC, Schwindt PC, Crill WE (1994) Properties and ionic mechanisms of a metabotropic glutamate receptor-mediated slow afterdepolarization in neocortical neurons. J Neurophysiol 72:693–704. [DOI] [PubMed] [Google Scholar]
- Groh A, Meyer HS, Schmidt EF, Heintz N, Sakmann B, Krieger P (2010) Cell-type specific properties of pyramidal neurons in neocortex underlying a layout that is modifiable depending on the cortical area. Cereb Cortex 20:826–836. 10.1093/cercor/bhp152 [DOI] [PubMed] [Google Scholar]
- Guinamard R, Simard C, Del Negro C (2013) Flufenamic acid as an ion channel modulator. Pharmacol Ther 138:272–284. 10.1016/j.pharmthera.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH (2009) M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci 29:9888–9902. 10.1523/JNEUROSCI.1366-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina AS, Trudeau MC (2013) The eag domain regulates hERG channel inactivation gating via a direct interaction. J Gen Physiol 141:229–241. 10.1085/jgp.201210870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R (1996) Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J Neurosci 16:3848–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R (1998) Ionic mechanism of the slow afterdepolarization induced by muscarinic receptor activation in rat prefrontal cortex. J Neurophysiol 80:1197–1210. [DOI] [PubMed] [Google Scholar]
- Hardman RM, Forsythe ID (2009) Ether-à-go-go-related gene K+ channels contribute to threshold excitability of mouse auditory brainstem neurons. J Physiol 587:2487–2497. 10.1113/jphysiol.2009.170548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Ohi K, Yasuda Y, Fukumoto M, Yamamori H, Kamino K, Morihara T, Iwase M, Kazui H, Takeda M (2013) The KCNH2 gene is associated with neurocognition and the risk of schizophrenia. World J Biol Psychiatry 14:114–120. 10.3109/15622975.2011.604350 [DOI] [PubMed] [Google Scholar]
- Hebb D. (1949) The organization of behavior: a neuropsychological theory. New York: Wiley. [Google Scholar]
- Heide J, Mann SA, Vandenberg JI (2012) The schizophrenia-associated Kv11.1–3.1 isoform results in reduced current accumulation during repetitive brief depolarizations. PLoS One 7:e45624. 10.1371/journal.pone.0045624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AP, Sunde M, Campbell TJ, Vandenberg JI (2007) Mechanism of block of the hERG K+ channel by the scorpion toxin CnErg1. Biophys J 92:3915–3929. 10.1529/biophysj.106.101956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirdes W, Horowitz LF, Hille B (2004) Muscarinic modulation of erg potassium current. J Physiol 559:67–84. 10.1113/jphysiol.2004.066944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield JJ. (1982) Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci U S A 79:2554–2558. 10.1073/pnas.79.8.2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffaker SJ, Chen J, Nicodemus KK, Sambataro F, Yang F, Mattay V, Lipska BK, Hyde TM, Song J, Rujescu D, Giegling I, Mayilyan K, Proust MJ, Soghoyan A, Caforio G, Callicott JH, Bertolino A, Meyer-Lindenberg A, Chang J, Ji Y, et al. (2009) A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med 15:509–518. 10.1038/nm.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde RA, Strowbridge BW (2012) Mnemonic representations of transient stimuli and temporal sequences in the rodent hippocampus in vitro. Nat Neurosci 15:1430–1438. 10.1038/nn.3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Isenberg G (1990a) Intracellular calcium ions modulate acetylcholine-induced inward current in guinea-pig ileum. J Physiol 424:73–92. 10.1113/jphysiol.1990.sp018056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Isenberg G (1990b) Effect of membrane potential on acetylcholine-induced inward current in guinea-pig ileum. J Physiol 424:57–71. 10.1113/jphysiol.1990.sp018055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki M, Anderson RG (1999) Calcium signal transduction from caveolae. Cell Calcium 26:201–208. 10.1054/ceca.1999.0073 [DOI] [PubMed] [Google Scholar]
- Jensen JB, Lyssand JS, Hague C, Hille B (2009) Fluorescence changes reveal kinetic steps of muscarinic receptor-mediated modulation of phosphoinositides and Kv7.2/7.3 K+ channels. J Gen Physiol 133:347–359. 10.1085/jgp.200810075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Tucker KR, Putzier I, Huertas MA, Horn JP, Canavier CC, Levitan ES, Shepard PD (2012) Functional characterization of ether-a-go-go-related gene potassium channels in midbrain dopamine neurons—implications for a role in depolarization block. Eur J Neurosci 36:2906–2916. 10.1111/j.1460-9568.2012.08190.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochems A, Yoshida M (2013) Persistent firing supported by an intrinsic cellular mechanism in hippocampal CA3 pyramidal cells. Eur J Neurosci 38:2250–2259. 10.1111/ejn.12236 [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC (2006) Molecular determinants of hERG channel block. Mol Pharmacol 69:1709–1716. 10.1124/mol.105.020990 [DOI] [PubMed] [Google Scholar]
- Kim EJ, Juavinett AL, Kyubwa EM, Jacobs MW, Callaway EM (2015) Three types of cortical layer 5 neurons that differ in brain-wide connectivity and function. Neuron 88:1253–1267. 10.1016/j.neuron.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer B, Jochems A, Valero-Aracama MJ, Yoshida M (2013) Long-lasting intrinsic persistent firing in rat CA1 pyramidal cells: a possible mechanism for active maintenance of memory. Hippocampus 23:820–831. 10.1002/hipo.22136 [DOI] [PubMed] [Google Scholar]
- Krnjević K, Pumain R, Renaud L (1971) The mechanism of excitation by acetylcholine in the cerebral cortex. J Physiol 215:247–268. 10.1113/jphysiol.1971.sp009467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer P, Strowbridge BW (2010) Representing information in cell assemblies: persistent activity mediated by semilunar granule cells. Nat Neurosci 13:213–222. 10.1038/nn.2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Lin S, Choy PC, Shen X, Deng C, Kuang S, Wu J, Xu W (2008) The regulation of the cardiac potassium channel (HERG) by caveolin-1. Biochem Cell Biol 86:405–415. 10.1139/O08-118 [DOI] [PubMed] [Google Scholar]
- Liu D, Gu X, Zhu J, Zhang X, Han Z, Yan W, Cheng Q, Hao J, Fan H, Hou R, Chen Z, Chen Y, Li CT (2014) Medial prefrontal activity during delay period contributes to learning of a working memory task. Science 346:458–463. 10.1126/science.1256573 [DOI] [PubMed] [Google Scholar]
- Liu H, Yang L, Chen KH, Sun HY, Jin MW, Xiao GS, Wang Y, Li GR (2016) SKF-96365 blocks human ether-a-go-go-related gene potassium channels stably expressed in HEK 293 cells. Pharmacol Res 104:61–69. 10.1016/j.phrs.2015.12.012 [DOI] [PubMed] [Google Scholar]
- Ma Q, Yu H, Lin J, Sun Y, Shen X, Ren L (2014) Screening for cardiac HERG potassium channel interacting proteins using the yeast two-hybrid technique. Cell Biol Int 38:239–245. 10.1002/cbin.10196 [DOI] [PubMed] [Google Scholar]
- Major G, Baker R, Aksay E, Seung HS, Tank DW (2004) Plasticity and tuning of the time course of analog persistent firing in a neural integrator. Proc Natl Acad Sci U S A 101:7745–7750. 10.1073/pnas.0401992101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malykhina AP, Shoeb F, Akbarali HI (2002) Fenamate-induced enhancement of heterologously expressed HERG currents in Xenopus oocytes. Eur J Pharmacol 452:269–277. 10.1016/S0014-2999(02)02330-0 [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA (1985) Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci U S A 82:6344–6348. 10.1073/pnas.82.18.6344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA (1986) Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol 375:169–194. 10.1113/jphysiol.1986.sp016112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnes JT, Dempsey CE, Ridley JM, Crociani O, Arcangeli A, Hancox JC, Witchel HJ (2003) Preferential closed channel blockade of HERG potassium currents by chemically synthesised BeKm-1 scorpion toxin. FEBS Lett 547:20–26. 10.1016/S0014-5793(03)00662-8 [DOI] [PubMed] [Google Scholar]
- Miyashita Y, Chang HS (1988) Neuronal correlate of pictorial short-term memory in the primate temporal cortex. Nature 331:68–70. 10.1038/331068a0 [DOI] [PubMed] [Google Scholar]
- Niculescu D, Hirdes W, Hornig S, Pongs O, Schwarz JR (2013) Erg potassium currents of neonatal mouse Purkinje cells exhibit fast gating kinetics and are inhibited by mGluR1 activation. J Neurosci 33:16729–16740. 10.1523/JNEUROSCI.5523-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Owsianik G (2011) The transient receptor potential family of ion channels. Genome Biol 12:218. 10.1186/gb-2011-12-3-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R (2004) Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol 471:241–276. 10.1002/cne.11039 [DOI] [PubMed] [Google Scholar]
- Papa M, Boscia F, Canitano A, Castaldo P, Sellitti S, Annunziato L, Taglialatela M (2003) Expression pattern of the ether-a-gogo-related (ERG) K+ channel-encoding genes ERG1, ERG2, and ERG3 in the adult rat central nervous system. J Comp Neurol 466:119–135. 10.1002/cne.10886 [DOI] [PubMed] [Google Scholar]
- Pardo-López L, García-Valdés J, Gurrola GB, Robertson GA, Possani LD (2002) Mapping the receptor site for ergtoxin, a specific blocker of ERG channels. FEBS Lett 510:45–49. 10.1016/S0014-5793(01)03218-5 [DOI] [PubMed] [Google Scholar]
- Pattnaik BR, Hughes BA (2012) Effects of KCNQ channel modulators on the M-type potassium current in primate retinal pigment epithelium. Am J Physiol Cell Physiol 302:C821–C833. 10.1152/ajpcell.00269.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressler RT, Inoue T, Strowbridge BW (2007) Muscarinic receptor activation modulates granule cell excitability and potentiates inhibition onto mitral cells in the rat olfactory bulb. J Neurosci 27:10969–10981. 10.1523/JNEUROSCI.2961-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prole DL, Lima PA, Marrion NV (2003) Mechanisms underlying modulation of neuronal KCNQ2/KCNQ3 potassium channels by extracellular protons. J Gen Physiol 122:775–793. 10.1085/jgp.200308897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman J, Berger T (2011) Persistent activity in layer 5 pyramidal neurons following cholinergic activation of mouse primary cortices. Eur J Neurosci 34:22–30. 10.1111/j.1460-9568.2011.07736.x [DOI] [PubMed] [Google Scholar]
- Richardson RT, DeLong MR (1986) Nucleus basalis of Meynert neuronal activity during a delayed response task in monkey. Brain Res 399:364–368. 10.1016/0006-8993(86)91529-5 [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA (2003) Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol 65:453–480. 10.1146/annurev.physiol.65.092101.142734 [DOI] [PubMed] [Google Scholar]
- Sacco T, Bruno A, Wanke E, Tempia F (2003) Functional roles of an ERG current isolated in cerebellar Purkinje neurons. J Neurophysiol 90:1817–1828. 10.1152/jn.00104.2003 [DOI] [PubMed] [Google Scholar]
- Saganich MJ, Machado E, Rudy B (2001) Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci 21:4609–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer CR, Lerche C, Decher N, Dennis AT, Maier P, Ficker E, Busch AE, Wollnik B, Steinmeyer K (2002) The antihistamine fexofenadine does not affect I(Kr) currents in a case report of drug-induced cardiac arrhythmia. Br J Pharmacol 137:892–900. 10.1038/sj.bjp.0704873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnee ME, Brown BS (1998) Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther 286:709–717. [PubMed] [Google Scholar]
- Schubert D, Staiger JF, Cho N, Kötter R, Zilles K, Luhmann HJ (2001) Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J Neurosci 21:3580–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PD, Canavier CC, Levitan ES (2007) Ether-a-go-go-related gene potassium channels: what's all the buzz about? Schizophr Bull 33:1263–1269. 10.1093/schbul/sbm106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Keating MT, Sanguinetti MC (1996) Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Open-channel block by methanesulfonanilides. Circ Res 78:499–503. 10.1161/01.RES.78.3.499 [DOI] [PubMed] [Google Scholar]
- Sturm P, Wimmers S, Schwarz JR, Bauer CK (2005) Extracellular potassium effects are conserved within the rat erg K+ channel family. J Physiol 564:329–345. 10.1113/jphysiol.2004.078840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng GQ, Zhao X, Lees-Miller JP, Quinn FR, Li P, Rancourt DE, London B, Cross JC, Duff HJ (2008) Homozygous missense N629D hERG (KCNH2) potassium channel mutation causes developmental defects in the right ventricle and its outflow tract and embryonic lethality. Circ Res 103:1483–1491. 10.1161/CIRCRESAHA.108.177055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Torres AM, Campbell TJ, Kuchel PW (2004) The HERG K+ channel: progress in understanding the molecular basis of its unusual gating kinetics. Eur Biophys J 33:89–97. 10.1007/s00249-003-0338-3 [DOI] [PubMed] [Google Scholar]
- Vijayaraj P, Le Bras A, Mitchell N, Kondo M, Juliao S, Wasserman M, Beeler D, Spokes K, Aird WC, Baldwin HS, Oettgen P (2012) Erg is a crucial regulator of endocardial-mesenchymal transformation during cardiac valve morphogenesis. Development 139:3973–3985. 10.1242/dev.081596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282:1890–1893. 10.1126/science.282.5395.1890 [DOI] [PubMed] [Google Scholar]
- Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL (1997) A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol 502:45–60. 10.1111/j.1469-7793.1997.045bl.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ. (1999) Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci 19:9587–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF (1996) Prefrontal function in schizophrenia: confounds and controversies. Philos Trans R Soc Lond B Biol Sci 351:1495–1503. 10.1098/rstb.1996.0135 [DOI] [PubMed] [Google Scholar]
- Wible BA, Hawryluk P, Ficker E, Kuryshev YA, Kirsch G, Brown AM (2005) HERG-Lite(R): a novel comprehensive high-throughput screen for drug-induced hERG risk. J Pharmacol Toxicol Methods 52:136–145. 10.1016/j.vascn.2005.03.008 [DOI] [PubMed] [Google Scholar]
- Winograd M, Destexhe A, Sanchez-Vives MV (2008) Hyperpolarization-activated graded persistent activity in the prefrontal cortex. Proc Natl Acad Sci U S A 105:7298–7303. 10.1073/pnas.0800360105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada-Hanff J, Bean BP (2013) Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J Neurosci 33:15011–15021. 10.1523/JNEUROSCI.0577-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HD, Villalobos C, Andrade R (2009) TRPC channels mediate a muscarinic receptor-induced afterdepolarization in cerebral cortex. J Neurosci 29:10038–10046. 10.1523/JNEUROSCI.1042-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Hasselmo ME (2009) Persistent firing supported by an intrinsic cellular mechanism in a component of the head direction system. J Neurosci 29:4945–4952. 10.1523/JNEUROSCI.5154-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BJ, Otto T, Fox GD, Eichenbaum H (1997) Memory representation within the parahippocampal region. J Neurosci 17:5183–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kehl SJ, Fedida D (2003) Modulation of human ether-à-go-go-related K+ (HERG) channel inactivation by Cs+ and K+. J Physiol 548:691–702. 10.1113/jphysiol.2003.039198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Reboreda A, Alonso A, Barker PA, Séguéla P (2011) TRPC channels underlie cholinergic plateau potentials and persistent activity in entorhinal cortex. Hippocampus 21:386–397. 10.1002/hipo.20755 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT (1998) Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74:230–241. 10.1016/S0006-3495(98)77782-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylberberg J, Strowbridge BW (2017) Mechanisms of persistent activity in cortical circuits: possible neural substrates for working memory. Annu Rev Neurosci 40:603–627. 10.1146/annurev-neuro-070815-014006 [DOI] [PMC free article] [PubMed] [Google Scholar]